
Summary
Today, pancreatic cancer (PC) is a major health problem in the United States. It remains a challenge to develop efficacious clinically useful PC therapies. New avenues, based on translational approaches and innovative validated biomarkers could be a preclinical option to evaluate PC drug candidates or drug combinations before clinical trials. Herein, we describe evaluation of combination therapies by incorporating a novel pathway modulator, p53-Activator Wnt Inhibitor-2 (PAWI-2) with other FDA-approved cancer drugs that have been used in PC clinical trials. PAWI-2 is a potent inhibitor of drug-resistant PC cells that has been shown to selectively ameliorate human pancreatic cancer stem cells (i.e., hPCSCs, FGβ3 cells). In the present study, we showed PAWI-2 produced therapeutic synergism with certain types of anti-cancer drugs. These drugs themselves oftentimes do not ameliorate PC cells (especially PCSCs) due to high levels of drug-resistance. PAWI-2 has the ability to rescue the potency of drugs (i.e., erlotinib, trametinib) and inhibit PC cell growth. Key molecular regulators of PAWI-2 could be used to predict synergistic/antagonistic effects between PAWI-2 and other anti-cancer drugs. Anti-cancer results showed potency could be quite accurately correlated to phosphorylation of optineurin (OPTN) in PC cells. Synergism/antagonism was also associated with inhibition of PCSC marker SOX2 that was observed in FGβ3 cells. Synergism broadens the potential use of PAWI-2 as an adjunct chemotherapy in patients with PC that have developed resistance to first-line targeted therapies or chemotherapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer (PC) is a major health problem in the United States and is the third leading cause of cancer-related deaths [1, 2]. PC is one of the most lethal diagnoses that oncology patients face. PC is known to be highly resistant to currently available treatments. Surgical resection with negative margins is the only potentially curative treatment for PC, but only 15%–20% of patients with PC are eligible for resection at initial diagnosis [3]. The remaining PC patients usually have metastatic or locally advanced disease that generally is considered incurable [4].
Most of the drugs approved by the United States Food and Drug Administration (FDA) for PC including capecitabine, erlotinib, 5-fluorouracil, gemcitabine, irinotecan, nab-paclitaxel, oxaliplatin, etc., are generally chemotherapies [5]. Unfortunately, PC oftentimes becomes resistant to these therapies. Increased administration of drugs is limited by somatic toxicity and serious side effects [6,7,8]. Combination of two or more standard therapies (i.e., chemotherapies) has the potential to revolutionize the treatment and care of PC. For example, a large number of PC patients do not respond to gemcitabine due to high levels of intrinsic and/or acquired chemo-resistance [9]. To improve clinical efficacy, gemcitabine-based therapy, is often combined with a second cytotoxic agent (e.g., platinum agents, fluoropyrimidine, etc.) [10,11,12,13,14,15]. This has been extensively investigated and showed superior efficacy over single-agent treatment in several prospective clinical trials [9, 13,14,15]. However, due to increased treatment-related toxicity, most trials to date have failed to show a significant improvement in overall survival.
Lack of effective PC treatments underscores an inadequate understanding of the biological complexity of PC. It is still unclear why PC often becomes resistant to therapies that work against other types of cancer. Understanding the underlying mechanism and impact of inhibiting PC signaling pathways could lead to new treatment strategies. Combination therapy of two or more cancer drugs is a mainstay of anticancer treatment in PC, with optimal combinations producing synergistic anti-tumor responses. Currently, most PC agents work at combining different S-phase targeting agents affecting DNA synthesis or function [16, 17]. Additional incremental increases in survival benefit may be achieved by combining agents with established safety profiles and non-overlapping mechanisms of action. However, testing drug alternatives in clinical trials is challenging unless they work with standard treatments (e.g., gemcitabine) [16]. New approaches, based on translational work and innovative validated biomarkers could be a potential solution to address this problem.
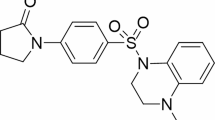
Recently, characteristics of inherent resistance to cancer therapy have been linked to a small subpopulation of cells classified as cancer stem cells (CSCs). CSCs are believed to be the hallmark of cancer responsible for tumor initiation, maintenance, dormancy, metastasis and relapse [18,19,20]. Accordingly, we focused on a human pancreatic cancer stem cell (hPCSC) reported previously (i.e., FGβ3 cells; a validated hPCSC model [21,22,23]). Previously, we identified and developed a novel pathway modulator (p53-Activator Wnt Inhibitor-2, PAWI-2; Fig. 1a) [24,25,26,27,28] that inhibited in vitro PC cell growth regardless of PC types [27]. PAWI-2 was shown to selectively ameliorate hPCSCs [29, 30]. Herein, we evaluate new combination therapies by incorporating PAWI-2 with other FDA-approved anti-cancer drugs that have been in PC clinical trials. We investigated whether key regulator(s) that are associated with the mechanism of action of PAWI-2 could be used to predict synergistic, additive or antagonistic effects between PAWI-2 and other anti-cancer drugs. Effects of drugs and PAWI-2 were evaluated against FGβ3 and with other bulk PC cells (i.e., FG, MIA PaCa-2 cells) for comparison. Results showed that PAWI-2 produced potent synergism with certain types of anti-cancer drugs. Synergism was correlated to the phosphorylation of optineurin (OPTN) and also associated with inhibition of a CSC biomarker - Sex-determining region Y-Box2 (SOX2) that was observed in hPCSCs (i.e., FGβ3 cells).

a Structure of PAWI-2. b–d Correlations between IC50s to inhibit cell viability of MIA PaCa-2, FG and FGβ3 cells by anti-cancer drugs. b There was no significant correlation between ratios of IC50s for inhibition of cell viability by anti-cancer drugs in FG cells versus FGβ3 cells and the ratio of IC50s for anti-cancer drugs in MIA PaCa-2 versus FGβ3 cells (Spearman’s rank-correlation coefficient (ρ): −0.252, P = 0.346). c There was also no significant correlation between ratios of IC50s for inhibition of cell viability by anti-cancer drugs in FGβ3 cells in the presence or absence of treatment with PAWI-2 (10 nM; <IC50) and ratios of IC50s for anti-cancer drugs in FG cells versus FGβ3 cells (ρ: −0.216, P = 0.421). d There was a significant correlation between ratios of IC50s for inhibition of cell viability by anti-cancer drugs in FGβ3 cells in the presence or absence of treatment with PAWI-2 versus the ratio of IC50s for anti-cancer drugs in MIA PaCa-2 versus FGβ3 cells (ρ: 0.542, P = 0.0300*). The 16 anti-cancer drugs were chosen from 22 cancer drugs because they were potent below 50 μM based on Table 1 (i.e., 5-FU, CAPE, OXP, Rib, Pal and Tra excluded). Correlations in b, c, d were analyzed by Spearman’s rank-correlation test and P < 0.05 was considered a significant correlation
Materials and methods
Cell lines
MIA PaCa-2 (CRL-1420), HPAC (CRL-2199), AsPC-1 (CRL-1682) and BxPC-3 (CRL-1687) PC cell lines were purchased from American Type Culture Collection (ATCC). 779E is a patient-derived, low-passage primary PC cell line from Dr. Andrew Lowy (UC San Diego) [31]. FG and FGβ3 cells were provided by Dr. David Cheresh (UC San Diego and the Scripps Research Institute) [21,22,23]. Commercial cell lines were grown according to ATCC recommendations and authenticated by short tandem repeat (STR) DNA profiling at ATCC. 779E cells were characterized and cultured as reported [31]. FG and FGβ3 cells were grown in DMEM with 10% FBS. After thawing, cell lines were cultured at 37 °C in a humidified 5% CO2 atmosphere and routinely screened for mycoplasma contamination.
Compounds
Synthesis and pharmaceutical properties of PAWI-2 (Fig. 1a) were reported previously [24, 25]. FDA-approved clinical cancer drugs used in this study were from the National Cancer Institute (NCI) DTP Plated Compounds (10 mM in DMSO) [32].
Cell viability and apoptosis assays
Cells were seeded onto plates and treated with test compounds (vehicle: 0.5% DMSO; PAWI-2 or other drugs: 0.1 nM to 50 μM) for 72 h. Cell viability was quantified using CellTiter-Glo (Promega). Data were expressed as percentage of survival compared to survival of vehicle-treated cells. Chou-Talalay analysis (ComboSyn) was used to test synergy of PAWI-2 in the presence of clinical cancer drugs [33].
Stem cell biomarker array
FG and FGβ3 cells were seeded onto plates and treated with test compounds (vehicle: 0.5% DMSO; PAWI-2: 50 nM) for 8 or 16 h. Stem cell marker array tests were carried out according to the manufacturer’s protocol (R&D systems). Briefly, whole-cell extracts were obtained after lysis with non-denaturing buffer and incubated with array membranes (containing 15 different anti-stem cell marker antibodies) overnight at 4 °C. Signals were detected using detection antibody cocktail and chemiluminescence reagents after exposure. Densities of immunoblot spots were quantified using ImageJ (NIH).
Immunoblotting
Immunoblot experiments were carried out as before [27]. Briefly, whole-cell extracts were obtained after lysis with RIPA buffer (Supplementary Materials and Methods). Protein extracts were resolved by SDS-PAGE followed by immunoblotting using antibodies specific for target proteins (Supplementary Materials and Methods). Densities of immunoblot bands were quantified using ImageJ (NIH).
Statistical analysis
Statistical analyses and graphical plots were done using GraphPad Prism. IC50 values were calculated using nonlinear regression analysis of the mean and standard deviation (SD) of at least triplicate samples for each biological assay. Student t tests were used to calculate statistical significance for comparison between two groups. P-values <0.05 were considered significant. Spearman’s rank-correlation test was used to analyze the correlation between two ranked variables. The strength and direction of association between two ranked variables was defined by Spearman’s Rank correlation coefficient (ρ). P value <0.05 was considered a significant correlation.
Results
Effect of clinically used cancer drugs on cell viability in different PC cells
Integrin β3/KRAS-driven cancer stemness and drug resistance have been reported [21,22,23]. FGβ3 cells were generated by stable transfection of fast-growing (FG) human PC cells with human integrin β3/pcDNA3.1. FGβ3 cells have been thoroughly documented as an aggressive cell line showing CSC-like properties and cancer drug resistance [21,22,23]. In order to systematically characterize stem-like properties of these cells (i.e., FGβ3 cells), a stem cell biomarker array test was applied. FGβ3 cells and parental FG cells were compared. Several stem cell biomarkers were observed overexpressed in FGβ3 cells (i.e., Oct-3/4, SOX2, Nanog) but expressed less in FG cells (Supplemental Fig. S1A,B). This showed FGβ3 cells possessed stem-like properties.
We used these cells to investigate how PAWI-2 re-sensitized clinical cancer drugs that normally have limited effects to inhibit PC cell viability. First, IC50 values were determined for 22 clinical anti-cancer drugs examined in three cell lines (i.e., MIA PaCa-2, FG and FGβ3 cells; Table 1). The ratios of IC50 values represent the loss of drug-sensitivity between bulk PC cells to PCSCs (i.e., the ratio of IC50s in MIA PaCa-2 versus FGβ3 cells or the ratio of IC50s in FG cells versus FGβ3 cells). These ratios were plotted against one another. There was no significant correlation between IC50 ratios for drugs in FG cells (integrin β3− cells) versus FGβ3 cells (integrin β3+ cells; hPCSCs) and IC50 ratios for drugs in MIA PaCa-2 cells (bulk PC cells) versus FGβ3 cells (Fig. 1b; Spearman’s rank-correlation coefficient (ρ): −0.252, P = 0.346). This showed that IC50 values for this cohort of drugs did not possess a similar trend from drug-sensitive PC cells (low IC50 values) to drug-resistant PC cells (large IC50 values). Second, we quantified IC50 values of 22 drugs in the presence of PAWI-2 (co-treated with 10 nM PAWI-2; < IC50) in FGβ3 cells (Table 1). The IC50 ratios (i.e., the ratio of IC50s in FG versus FGβ3 cells or the ratio of IC50s in MIA PaCa-2 versus FGβ3 cells) for 22 drugs were plotted versus IC50 ratios in the presence and absence of PAWI-2 (Fig. 1c, d). The result showed no significant correlation between IC50 ratios in the presence or absence of PAWI-2 (+PAWI-2/-PAWI-2) in FGβ3 cells and the ratio of IC50s in FG cells (integrin β3−) versus FGβ3 (integrin β3+) cells (Fig. 1c; ρ: −0.216, P = 0.421). This data showed the ability of PAWI-2 to facilitate sensitization of anti-cancer drugs was not mainly associated with overexpression of integrin β3 in FGβ3 cells. In contrast, there was a significant correlation between IC50 ratios in the presence or absence of PAWI-2 (+PAWI-2/-PAWI-2) in FGβ3 cells and IC50 ratios for drugs in MIA PaCa-2 cells (bulk PC cells) versus FGβ3 cells (Fig. 1d; ρ: 0.542, P = 0.03). The data showed the ability of PAWI-2 to overcome anti-cancer drug resistance in PCSCs was mainly correlated to recover the sensitivity of these clinical anti-cancer drugs in PCSCs. This effect could rescue the drug sensitivity back to a comparable level as that in bulk PC cells (i.e., MIA PaCa-2).
Effect of PAWI-2 on PC cell viability in the presence of clinically used anti-cancer drug classes
Many of the drugs used in this study have been used in clinical trials of PC treatment [16, 34]. Drugs were separated into different classes for evaluation. Their ability to overcome drug-resistance was determined by testing the effect of each drug to inhibit FGβ3 cell viability in the presence of PAWI-2 (10 nM; <IC50). We expanded this study to include class-specific clinically used drugs (i.e., 55 drugs; Fig. 2). This list included DNA repair/damage agents, epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs), vascular endothelial growth factor receptor (VEGFR)-TKI, breakpoint cluster region protein (BCR)/Abelson murine leukemia viral oncogene homolog 1 (ABL) inhibitors, other kinase inhibitors (i.e., mTOR, CDK4/6, MEK, PI3K, ALK), microtubule (MT) disturbance agents and other pathway inhibitors (i.e., HDAC, PARP, Proteasome, Hedgehog pathway, Bcl-2). PAWI-2 significantly enhanced (P < 0.05) inhibition of FGβ3 cell viability in the presence of certain classes of inhibitors (Fig. 2). Included in this list are most EGFR-TKIs (Fig. 2b), VEGFR-TKIs (Fig. 2c), mTOR inhibitors (Fig. 2e), MT disturbance agents (Fig. 2f), HDAC inhibitors (Fig. 2g). PAWI-2 also significantly enhanced (P < 0.05) inhibition of cell viability in the presence of Bcl-2 inhibitor Venetoclax (i.e., currently the only FDA-approved Bcl-2 inhibitor, Fig. 2g). The data for Venetoclax was consistent with previous results that showed PAWI-2 consistently acted at antagonizing a pro-survival role of Bcl-2 and Bcl-2-like proteins (i.e., Bcl-xL, Mcl-1) [27, 28].

Percent inhibition of FGβ3 cell viability by clinically used anti-cancer drugs in the presence of PAWI-2 compared to single agent treatment. a DNA repair/damage agents, b EGFR-TKIs, c VEGFR-TKIs, d BCR/ABL inhibitors, e other kinase inhibitors (i.e., mTOR, CDK4/6, MEK, PI3K, ALK), f microtubule disturbance agents and g other pathway inhibitors (i.e., HDAC, PARP, Proteasome, Hedgehog pathway, Bcl-2). Concentration of PAWI-2 used was 10 nM. A dashed baseline in each figure stands for the level of % inhibition on cell viability by the treatment of PAWI-2 alone; treatment time used was 72 h; vehicle control (0.5% DMSO). Data are mean ± SD (n = 3); P-values were estimated by Student t tests (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001)
Chou-Talalay analysis of synergism/antagonism of PAWI-2 with 22 clinically used anti-cancer drugs
Twenty-two drugs that have been used in clinical trials of PC treatment (including DNA repair/damage agents, EGFR-TKI, VEGFR-TKI, mTOR, CDK4/6, MEK, ALK inhibitors, MT disturbance agents and HDAC, Proteasome inhibitors, as shown in Table 2) [16, 34] were examined in synergism analysis. Chou-Talalay analysis was done based on dose-dependent studies with PAWI-2. Chou-Talalay analysis (in vitro) is a well-established synergism analysis method based on comparison of dose-dependent inhibition of cell viability for drugs alone and drugs in combination [33]. Synergism or antagonism between drugs was defined by combination index (CI) values [33].
In FGβ3 cells, PAWI-2 synergized (CI < 1, Table 2) EGFR-TKI (i.e., Erlotinib), VEGFR-TKI (i.e., Sorafenib), mTOR inhibitor (i.e., Sirolimus), MEK inhibitor (i.e., Trametinib), ALK inhibitor (i.e., Ceritinib), MT disturbance agents (i.e., Cabazitaxel, Docetaxel, Paclitaxel, Vinorelbine) and HDAC inhibitor (i.e., Vorinostat). PAWI-2 antagonized (CI > 1, Table 2) DNA repair/damage agents examined (i.e., 5-Fluorouracil, Capecitabine, Oxaliplatin, Irinotecan, Mitomycin C, Epirubicin, Gemcitabine), CDK4/6 inhibitors (i.e., Ribociclib, Palbociclib) and proteasome inhibitor (i.e., Bortezomib). PAWI-2 strongly antagonized MT destabilizer - Vinblastine (CI at ED95, >8). Strong antagonism was also observed when FGβ3 cells were co-treated with PAWI-2 and another MT destabilizer – Colchicine (CI at ED95, >15). The antagonism between PAWI-2 and MT destabilizer may be attributed to the fact that PAWI-2 binds to a similar binding site as MT destabilizer [26].
The effect of PAWI-2 on Chou-Talalay synergism/antagonism of 22 clinically used anti-cancer drugs was evaluated in the bulk PC cell line (i.e., MIA PaCa-2). The results showed comparable trends on CI values for different classes of clinically used anti-cancer drugs as that observed in FGβ3 cells (Supplemental Table S1). However, in MIA PaCa-2 cells, PAWI-2 synergized additional drugs (e.g., Mitomycin C, Gemcitabine, Palbociclib; see Supplemental Table S1). In a parallel study in PCSCs (i.e., FGβ3 cells), gemcitabine only showed good synergism with 5 of 21 drugs tested (Supplemental Table S2).
Correlation between combination index (CI) values and biomarker protein expression
The expression of biomarker proteins involved in the mechanism of action of PAWI-2 in FGβ3 cells includes modulation of OPTN and phosphorylation of OPTN at Ser177 (P-OPTN), modulation of p62/Sequestosome-1 (p62/SQSTM1, abbreviated as p62) and phosphorylation of p62 at Ser403 (P-p62), modulation of serine/threonine kinase Tank-binding kinase 1 (TBK1, IκB kinase (IKK)-related kinase) and phosphorylation of TBK1 at Ser172 (P-TBK1), modulation of cyclin-dependent kinase inhibitor p21 (also known as p21WAF1/Cip1, abbreviated as p21) and modulation of cyclin D3 (CCND3) [29]. After individual administration of 22 clinically used anti-cancer drugs to FGβ3 cells, PAWI-2 modulated biomarker protein expression P-OPTN, P-OPTN/OPTN, P-p62, P-p62/p62, P-TBK1, p21, and CCND3 (Supplemental Fig. S2). Fold-change of protein biomarker expression in FGβ3 cells treated with anti-cancer drugs and PAWI-2 compared to drug alone were plotted versus CI values calculated for combination treatment (as shown in Table 2). Results showed a significant correlation between CI values and fold-change for phospho-Ser177-OPTN (P-OPTN) (Fig. 3a and Table 3; ρ: −0.929, P < 0.0001 for CI values at ED90) and relative phosphorylation of OPTN (P-OPTN/OPTN) (Fig. 3b and Table 3; ρ: −0.916, P < 0.0001 for CI values at ED90). In FGβ3 cells, data showed that in the presence of anti-cancer drugs greater synergism with PAWI-2 (lower CI values) was associated with increased phosphorylation of OPTN. In MIA PaCa-2 cells, CI values showed similar trends as that observed for FGβ3 cells for different classes of clinically used anti-cancer drugs (Supplemental Table S3; ρ: −0.842, P < 0.0001 for P-OPTN/OPTN versus CI values at ED90). The phosphorylation of OPTN (expression level of P-OPTN or the ratio of P-OPTN/OPTN) induced by the 22 anti-cancer drugs examined in the presence or absence of PAWI-2 was useful for predicting drug synergism sensitivity in PC cells. In addition, observations about phosphorylation of OPTN was also very useful because it was broadly predictive of synergy by PAWI-2 regardless of PC cell types (Supplemental Fig. S3). In contrast, other relevant protein biomarkers (i.e., p62 and P-p62, TBK1 and P-TBK1, p21, CCND3), showed no significant correlation between biomarker expression ratios and CI values (Table 3; ρ: −0.30 to 0.48, P > 0.05).

Correlations between fold-change of protein biomarkers ± PAWI-2 and combination index (CI) values for 22 anti-cancer drugs in FGβ3 cells. There was a significant correlation for fold-change of a phospho-S177-optineurin versus CI (P-OPTN; ρ: −0.929, P < 0.0001****), b relative phosphorylation of optineurin versus CI (P-OPTN/OPTN; ρ: −0.916, P < 0.0001****), c SOX2 (ρ: 0.905, P < 0.0001****) versus CI values. Fold-change of biomarkers in a, b, c where the ratio of protein levels in cells with co-treatment of 22 anti-cancer drugs and PAWI-2 was expressed relative to that in cells treated with anti-cancer drugs alone. These ratios represent an enhanced inhibition or activation of each specific protein biomarker by comparing combination of PAWI-2 with tested clinically used anti-cancer drugs to drugs alone. CI values in a, b, c were calculated at ED90. Synergistic, additive or antagonistic effects of cancer drugs ± PAWI-2 (i.e., CI values of <1, = 1 or > 1, respectively) were examined in FGβ3 cells. Correlations in a, b, c were analyzed by Spearman’s rank-correlation test and P < 0.05 was considered a significant correlation (****P < 0.0001)
As determined by a stem cell biomarker array test (Supplemental Fig. S1) and further confirmed by immunoblots of individual biomarkers (Supplemental Fig. S2), PAWI-2 primarily inhibited expression of three stem cell biomarkers (i.e., Oct-4A, Nanog, and SOX2). Similarly, fold-changes of stem cell biomarker expression in FGβ3 cells treated with combinations of anti-cancer drug and PAWI-2 compared to drug alone were plotted versus CI values calculated for anti-cancer drug combination treatment (as shown in Table 2). There was no significant correlation between fold-changes of stem cell biomarker expression (i.e., Nanog and Oct-4A) for the 22 drugs examined and the CI values for synergism or antagonism (Table 3; ρ: 0.01 to 0.18, P > 0.4). However, a significant correlation was found between expression level ratios of SOX2 and CI values of drugs synergized or antagonized by PAWI-2 (Fig. 3c and Table 3; ρ: 0.905, P < 0.0001). This suggested that certain types of stem cell markers could also be used to predict drug-sensitivity in PC cells with stem-like properties. However, this was only observed in FGβ3 cells and not in bulk PC cells (i.e., FG cells had 2-fold less expression and MIA PaCa-2 cells had undetectable expression of those stem cell markers).
Finally, there was no significant correlation between biomarker expression ratios (i.e., P-OPTN, P-p62, CCND3 and SOX2) and CI values evaluated by the same analysis of gemcitabine (Supplemental Table S4; ρ: −0.3 to 0.2, P > 0.1). In summary, increases in drug-sensitivity predicted by correlation of CI values and phosphorylation of OPTN or SOX2 expression is anti-cancer drug class specific and apparently unique for PAWI-2.
Discussion
Although chemotherapies (i.e., gemcitabine-based therapy, often combined with a second cytotoxic agent such as platinum agents [10], fluoropyrimidine [11, 12] or a targeted cytotoxic agent [13,14,15]) is still the standard of care for patients with PC, recent clinical studies have not shown striking results with combination therapies involving two or more drugs [9, 35]. For example, only a combination of erlotinib with gemcitabine improved patients’ survival, albeit not in a clinically meaningful way [13]. Considering the pivotal role of CSCs in the processes of tumorigenesis, progression, invasion, and metastasis, several CSC regulatory agents have been developed as PC therapies. While these agents initially showed encouraging results in clinical studies (e.g., tarextumab) they eventually failed due to a lack of patient benefit [34]. Targeting PCSCs by CSC regulatory agents applied in combination PC therapies (i.e., napabucasin with gemcitabine and nab-paclitaxel) also failed to significantly benefit patients [34]. Therefore, further studies are needed to identify optimal agents or best combination regimens for efficient therapies for PC especially for highly aggressive drug-resistant PC types (with stem-like properties).
In the present study, most of the 22 clinically used anti-cancer drugs for PC trials and used herein showed 5- to 20-fold less potent IC50 values in PCSCs than that observed in bulk PC cells. In one case, the IC50 value of Trametinib (i.e., a MEK1/2 inhibitor) in FGβ3 cells was >104 fold-less potent than that in MIA PaCa-2 cells (Table 1). This illustrates the fact that FGβ3 cells are relatively more drug-resistant than other PC cells. We observed that poorly potent drugs against PCSCs were “rescued” by PAWI-2. In some cases, drug potency was returned to that observed in bulk PC cells. This effect was not specifically related to stemness induced by overexpression of integrin β3 because the ability of PAWI-2 to facilitate increased sensitization of anti-cancer drugs was not correlated to drug-resistance specifically induced by overexpression of integrin β3 (shown in Fig. 1c). The conclusion is that PAWI-2 does not work mainly through affecting dysregulated KRAS-NFκB signaling as we reported previously [29]. PAWI-2 is not dependent on one specific PCSC signaling pathway. Thus, the breadth of mechanism for PAWI-2 for inhibition of stem-like properties broadens the use of PAWI-2 in combination therapy applications to overcome drug-resistance of PCSCs.
Herein, the effect of PAWI-2 to induce anti-PC cell growth interactions was evaluated by Chou-Talalay synergism analysis with 22 clinically used anti-cancer drugs. These data showed PAWI-2 selectively synergized almost half (10 of 22) of these first-line therapies. Synergism between PAWI-2 and other clinically used drugs was class-specific but not single-drug specific (Fig. 2), indicating the effect may be attributed to cross-talk between multiple pathways and not an exclusive effect for certain drugs. Compared to FGβ3 cells, bulk PC cells (i.e., MIA PaCa-2 cells) showed similar sensitivity to some but not all classes of drug combinations. The data showed that PAWI-2 could re-sensitize inhibition of clinically used PC drugs and overcome certain types of drug resistance in highly aggressive PC types (i.e., PCSCs). In a parallel study of gemcitabine, less apparent synergism was observed (6 of 21 drugs). Chou-Talalay analysis showed there was only limited additive effects between gemcitabine and erlotinib in aggressive hPCSCs (i.e., FGβ3 cells, Supplemental Table S2). This may help explain the modest effect of gemcitabine to increase overall survival in combination regimens for the treatment of PC in the clinic.
Molecular targeting of specific signaling pathways have proven to be a useful strategy to overcome drug resistance. For example, increased expression of thymidine phosphorylase (TP) was reported to correlate with a poor response to 5-FU-based treatment in PC patients [36]. Additionally, gemcitabine-resistance is associated with elevated levels of cytidine deaminase (CDA) [37]. Development of efficient combination therapies based on certain protein expression profiles could be a helpful strategy for selecting agents that are likely to synergize and maximize the response to treatment. Our studies of the mechanism of PAWI-2 showed that certain proteins (e.g., P-OPTN, P-p62, P-TBK1, p21, CCND3, etc.) are associated with sensitivity to PAWI-2 [29]. To evaluate whether any changes occurred in cells that initiate/induce drug synergy or antagonism, we also analyzed expression levels of proteins associated with the mechanism of PAWI-2. The present data suggest that elevated levels of phosphorylation of OPTN, coupled with low expression of SOX2 (a stem cell biomarker) may be important determinants for developing combination regimens with PAWI-2.
As reported previously, phosphorylation of conserved OPTN residue (Ser177) induced by PAWI-2 promotes OPTN translocation into the nucleus and causes G2/M arrest [29]. Concomitantly, OPTN phosphorylation induced by PAWI-2 has negative feedback regulation of TBK1 functional activity to reverse tumor stemness and drug resistance in FGβ3 cells. Herein, phosphorylation of OPTN was observed to be broadly associated with determining control of PAWI-2 in increasing drug-sensitivity of PC. This effect was observed for PAWI-2 but not for other standard treatments (i.e., gemcitabine, Supplemental Table S4). In addition, certain biomarkers may also explain the differential response of PAWI-2 in synergizing other drugs in different PC types. For example, low stimulation of phosphorylation of OPTN by PAWI-2 in BxPC-3 cells (Supplemental Fig. S3) may account for less apparent synergism observed previously [27].
Transcription factor SOX2 is involved in CSC maintenance and dysregulation of SOX2 expression is highly associated with the capacity of cancer cell proliferation and metastasis. Moreover, there is evidence that SOX2 mediates resistance towards established cancer therapies. However, SOX2 is aberrantly expressed in PC and the function of SOX2 in PC is unclear [38]. Recently, SOX2 was shown to contribute to PC stemness/de-differentiation via regulation of G1/S transition. SOX2 overexpression promotes S-phase entry and cell proliferation associated with cyclin D3 induction [39]. SOX2 expression is also associated with increased levels of other PCSC biomarkers (e.g., ALDH1, ESA and CD44) [39]. Targeting and negatively regulating expression of SOX2 by PAWI-2 could further contribute to the ability of PAWI-2 to overcome cancer stemness in PCSCs. SOX2 protein expression could also be a useful biomarker for predicting drug-sensitivity of clinical drugs with PAWI-2 for SOX2-positive PC types (i.e., PCSCs).
Genetic heterogeneity of PC confers characteristics of invasion, metastatic migration and multiple treatment resistance [40, 41]. PC has been described as a complex molecular landscape with no predominant phenotype and several core molecular pathways (e.g., DNA damage repair, cell cycle regulation, etc.) [42]. These molecular pathways may be considered druggable but only affect a limited percentage of patients with PC. Personalized therapeutic strategies have not made a significant improvement for PC [43, 44]. In the preclinical setting, results herein suggest that a combined evaluation and quantification of several protein markers could help develop individualized medicines by exploring biomarker expression patterns in patients with different PC genotypes.
In conclusion, PAWI-2 is a potent drug for treating drug-resistant PCs. PC drug sensitivity can be predicted by correlations between synergism induced by PAWI-2 and biomarker expression (i.e., P-OPTN, SOX2). No cross-relationship for this type of correlation for other drugs (e.g., gemcitabine) was observed pointing to the novelty of PAWI-2. The results suggest that PAWI-2 could be used alone or as adjunct chemotherapy in patients with PC that have developed resistance to one of the first-line chemotherapies or targeted therapies. In addition, quantitative analyses of certain biomarkers in different PC types point to a potent strategy for developing individualized chemo-therapeutic regimens.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM (2014) Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74(11):2913–2921. https://doi.org/10.1158/0008-5472.CAN-14-0155
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin 68(1):7–30. https://doi.org/10.3322/caac.21387
Li D, Xie K, Wolff R, Abbruzzese JL (2004) Pancreatic cancer. Lancet. 363(9414):1049–1057. https://doi.org/10.1016/S0140-6736(04)15841-8
Ansari D, Tingstedt B, Andersson B, Holmquist F, Sturesson C, Williamsson C, Sasor A, Borg D, Bauden M, Andersson R (2016) Pancreatic cancer: yesterday, today and tomorrow. Future Oncol 12(16):1929–1946. https://doi.org/10.2217/fon-2016-0010
National Cancer Institute at the National Institutes of Health: Drugs Approved for Pancreatic Cancer. https://www.cancer.gov/about-cancer/treatment/drugs/pancreatic. Accessed 21 Apr 2020
Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, von Hoff DD (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15(6):2403–2413. https://doi.org/10.1200/jco.1997.15.6.2403
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y et al (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364(19):1817–1825. https://doi.org/10.1056/NEJMoa1011923
Conroy T, Gavoille C, Adenis A (2011) Metastatic pancreatic cancer: old drugs, new paradigms. Curr Opin Oncol 23(4):390–395. https://doi.org/10.1097/CCO.0b013e3283473610
Jin J, Teng C, Li T (2018) Combination therapy versus gemcitabine monotherapy in the treatment of elderly pancreatic cancer: a meta-analysis of randomized controlled trials. Drug Des Devel Ther 12:475–480. https://doi.org/10.2147/DDDT.S156766
Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, Andre T et al (2005) Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol 23(15):3509–3516. https://doi.org/10.1200/JCO.2005.06.023
Berlin JD, Catalano P, Thomas JP, Kugler JW, Haller DG, Benson AB 3rd (2002) Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: eastern cooperative oncology group trial E2297. J Clin Oncol 20(15):3270–3275. https://doi.org/10.1200/JCO.2002.11.149
Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, Harper PG, Dunn J, Tudur-Smith C, West J, Falk S, Crellin A, Adab F, Thompson J, Leonard P, Ostrowski J, Eatock M, Scheithauer W, Herrmann R, Neoptolemos JP (2009) Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 27(33):5513–5518. https://doi.org/10.1200/JCO.2009.24.2446
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W, National Cancer Institute of Canada Clinical Trials Group (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada clinical trials group. J Clin Oncol 25(15):1960–1966. https://doi.org/10.1200/JCO.2006.07.9525
Spano JP, Chodkiewicz C, Maurel J, Wong R, Wasan H, Barone C, Létourneau R, Bajetta E, Pithavala Y, Bycott P, Trask P, Liau K, Ricart AD, Kim S, Rixe O (2008) Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: an open-label randomised phase II study. Lancet. 371(9630):2101–2108. https://doi.org/10.1016/S0140-6736(08)60661-3
Philip PA, Benedetti J, Corless CL, Wong R, O'Reilly EM, Flynn PJ, Rowland KM, Atkins JN, Mirtsching BC, Rivkin SE, Khorana AA, Goldman B, Fenoglio-Preiser CM, Abbruzzese JL, Blanke CD (2010) Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: southwest oncology group-directed intergroup trial S0205. J Clin Oncol 28(22):3605–3610. https://doi.org/10.1200/JCO.2009.25.7550
Nevala-Plagemann C, Hidalgo M, Garrido-Laguna I (2020) From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat Rev Clin Oncol 17(2):108–123. https://doi.org/10.1038/s41571-019-0281-6
Shapiro GI, Harper JW (1999) Anticancer drug targets: cell cycle and checkpoint control. J Clin Invest 104(12):1645–1653. https://doi.org/10.1172/JCI9054
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5(4):275–284. https://doi.org/10.1038/nrc1590
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell. 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1(3):313–323. https://doi.org/10.1016/j.stem.2007.06.002
Desgrosellier JS, Barnes LA, Shields DJ, Huang M, Lau SK, Prevost N et al (2009) An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med 15(10):1163–1169. https://doi.org/10.1038/nm.2009
Seguin L, Camargo MF, Wettersten HI, Kato S, Desgrosellier JS, von Schalscha T, Elliott KC, Cosset E, Lesperance J, Weis SM, Cheresh DA (2017) Galectin-3, a Druggable vulnerability for KRAS-addicted cancers. Cancer Discov 7(12):1464–1479. https://doi.org/10.1158/2159-8290.CD-17-0539
Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H, Cascone T, Diao L, Wang J, Wistuba II, Heymach JV, Lippman SM, Desgrosellier JS, Anand S, Weis SM, Cheresh DA (2014) An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol 16(5):457–468. https://doi.org/10.1038/ncb2953
Cashman JR, Mercola M, Schade D, Tsuda M (2013) Compounds for inhibition of cancer cell proliferation. Google Patents. US 13/748,70
Okolotowicz KJ, Dwyer M, Ryan D, Cheng J, Cashman EA, Moore S, Mercola M, Cashman JR (2018) Novel tertiary sulfonamides as potent anti-cancer agents. Bioorg Med Chem 26(15):4441–4451. https://doi.org/10.1016/j.bmc.2018.07.042
Cheng J, Dwyer M, Okolotowicz KJ, Mercola M, Cashman JR (2018) A novel inhibitor targets both Wnt signaling and ATM/p53 in colorectal Cancer. Cancer Res 78(17):5072–5083. https://doi.org/10.1158/0008-5472.CAN-17-2642
Cheng J, Okolotowicz KJ, Ryan D, Mose E, Lowy AM, Cashman JR (2019) Inhibition of invasive pancreatic cancer: restoring cell apoptosis by activating mitochondrial p53. Am J Cancer Res 9(2):390–405
Cheng J, Moore S, Gomez-Galeno J, Lee DH, Okolotowicz KJ, Cashman JR (2019) A novel small molecule inhibits tumor growth and synergizes effects of enzalutamide on prostate cancer. J Pharmacol Exp Ther 371(3):703–712. https://doi.org/10.1124/jpet.119.261040
Cheng J, Cashman JR (2020) PAWI-2 overcomes tumor stemness and drug resistance via cell cycle arrest in integrin β3-KRAS dependent pancreatic cancer stem cells. Sci Rep 10:9162. https://doi.org/10.1038/s41598-020-65804-5
Cheng J, Cashman JR (2020) PAWI-2: a novel inhibitor for eradication of Cancer. Med Chem Res 29:1147–1159. https://doi.org/10.1007/s00044-020-02575-8
Fujimura K, Wright T, Strnadel J, Kaushal S, Metildi C, Lowy AM, Bouvet M, Kelber JA, Klemke RL (2014) A hypusine-eIF5A-PEAK1 switch regulates the pathogenesis of pancreatic cancer. Cancer Res 74(22):6671–6681. https://doi.org/10.1158/0008-5472.CAN-14-1031
National Cancer Institute at the National Institutes of Health: National Cancer Institute Developmental Therapeutics Program. https://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm. Accessed 10 Apr 2020
Chou TC (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70(2):440–446. https://doi.org/10.1158/0008-5472.CAN-09-1947
Zhang Y, Yang C, Cheng H, Fan Z, Huang Q, Lu Y, Fan K, Luo G, Jin K, Wang Z, Liu C, Yu X (2018) Novel agents for pancreatic ductal adenocarcinoma: emerging therapeutics and future directions. J Hematol Oncol 11(1):14. https://doi.org/10.1186/s13045-017-0551-7
Ciliberto D, Botta C, Correale P, Rossi M, Caraglia M, Tassone P, Tagliaferri P (2013) Role of gemcitabine-based combination therapy in the management of advanced pancreatic cancer: a meta-analysis of randomised trials. Eur J Cancer 49(3):593–603. https://doi.org/10.1016/j.ejca.2012.08.019
Miyake K, Imura S, Yoshizumi T, Ikemoto T, Morine Y, Shimada M (2007) Role of thymidine phosphorylase and orotate phosphoribosyltransferase mRNA expression and its ratio to dihydropyrimidine dehydrogenase in the prognosis and clinicopathological features of patients with pancreatic cancer. Int J Clin Oncol 12(2):111–119. https://doi.org/10.1007/s10147-006-0634-x
Eda H, Ura M, FO K, Tanaka Y, Miwa M, Ishitsuka H (1998) The antiproliferative activity of DMDC is modulated by inhibition of cytidine deaminase. Cancer Res 58(6):1165–1169
Novak D, Huser L, Elton JJ, Umansky V, Altevogt P, Utikal J (2019) SOX2 in development and cancer biology. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2019.08.007
Herreros-Villanueva M, Zhang JS, Koenig A, Abel EV, Smyrk TC, Bamlet WR, de Narvajas AAM, Gomez TS, Simeone DM, Bujanda L, Billadeau DD (2013) SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis. 2:e61. https://doi.org/10.1038/oncsis.2013.23
Samuel N, Hudson TJ (2011) The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 9(2):77–87. https://doi.org/10.1038/nrgastro.2011.215
Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang SB, Brown AMK, Kim JC, Wang T, Simpson JT, Beck T, Borgida A, Buchner N, Chadwick D, Hafezi-Bakhtiari S, Dick JE, Heisler L, Hollingsworth MA, Ibrahimov E, Jang GH, Johns J, Jorgensen LGT, Law C, Ludkovski O, Lungu I, Ng K, Pasternack D, Petersen GM, Shlush LI, Timms L, Tsao MS, Wilson JM, Yung CK, Zogopoulos G, Bartlett JMS, Alexandrov LB, Real FX, Cleary SP, Roehrl MH, McPherson JD, Stein LD, Hudson TJ, Campbell PJ, Gallinger S (2016) A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 538(7625):378–382. https://doi.org/10.1038/nature19823
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P et al (2015) Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 518(7540):495–501. https://doi.org/10.1038/nature14169
Biankin AV, Piantadosi S, Hollingsworth SJ (2015) Patient-centric trials for therapeutic development in precision oncology. Nature. 526(7573):361–370. https://doi.org/10.1038/nature15819
Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, Mead S, Jones MD, Samra JS, Gill AJ, Watson N, Chin VT, Humphris JL, Chou A, Brown B, Morey A, Pajic M, Grimmond SM, Chang DK, Thomas D, Sebastian L, Sjoquist K, Yip S, Pavlakis N, Asghari R, Harvey S, Grimison P, Simes J, Biankin AV, Australian Pancreatic Cancer Genome Initiative (APGI), Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial Management Committee of the Australasian Gastrointestinal Trials Group (AGITG) (2015) Precision medicine for advanced pancreas Cancer: the individualized molecular pancreatic Cancer therapy (IMPaCT) trial. Clin Cancer Res 21(9):2029–2037. https://doi.org/10.1158/1078-0432.CCR-15-0426
Acknowledgements
We thank Dr. Andrew Lowy of the University of California, San Diego for 779E cells. We also thank Dr. David Cheresh of the University of California, San Diego and The Scripps Research Institute for FG and FGβ3 cells. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of CIRM or any other agency of the State of California.
Funding
This work was supported by an Inception Award from the California Institute for Regenerative Medicine (CIRM) (DISC1–10583; J. R. Cashman) and by funds from the Human BioMolecular Research Institute and ChemRegen, Inc. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of CIRM or any other agency of the State of California.
Author information
Authors and Affiliations
Contributions
J. C. and J. R. C. conceived the study. J. C. conducted and carried out all cell-based studies, data analysis and statistical analysis. Both authors contributed to drafting and revising the manuscript. Both authors approved the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 874 kb)
Rights and permissions
About this article

Cite this article
Cheng, J., Cashman, J.R. Pancreatic cancer drug-sensitivity predicted by synergy of p53-Activator Wnt Inhibitor-2 (PAWI-2) and protein biomarker expression. Invest New Drugs 39, 131–141 (2021). https://doi.org/10.1007/s10637-020-00998-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-020-00998-z