
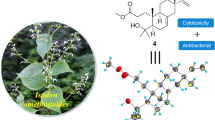
Abstract
Four known metabolites, macrophin (1), rosellisin (2), 2-(2-hydroxy-5-6-methoxy-3-methylene-1,4-benzodioxin-2(3H)-one (3), and methoxyphenoxyacrylic acid (4) were isolated, for the first time, from an endophytic fungus, Phoma macrostoma inhabiting the inner tissue of medicinal plant Glycyrrhiza glabra Linn. Their structures were characterized by comparison of their NMR data with literature data, and X-ray diffraction data of macrophin (1) as described herein for the first time. The compounds (1–4) were evaluated for their growth-inhibitory activities against a panel of cancer cell lines. Of these substances, macrophin (1) showed prominent cytotoxic activity against the MDA-MB-231, T47D, MCF-7, and MIAPaCa-2 cancer-cell lines with IC50 values of 14.8, 8.12, 13.0, and 0.9 μM, respectively. This biological significance has prompted us to generate a series of five acylated analogs (1a–1e) of 1 to conduct structure–activity relationship (SAR) study. The result illustrates all the analogues (1a–1e) were more potent than 1 against MDMB-231 and MIAPaCa-2 cancer cell lines. The compound 1d, an n-butanoyl substituent, has been proven to be three-fold more potent than 1 with IC50 values of 0.3 and 3.7 µM against MIAPaCa-2 and MDMB-231 cell lines, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In addition to basic needs like air, water, nutrients, etc., micro-organisms need to have special capabilities to combat their living environment. They get these special capabilities exclusively by expressing their silent biosynthetic genes encoded for specific metabolic pathways (Wang et al. 2018). Therefore, every endophytic fungus that grows inside the living plant tissue must have a unique set of biosynthetic metabolic pathways, which make them one of the rich sources of secondary metabolites (Kusari et al. 2012; Pusztahelyi et al. 2015). Thus, the exploitation of this endophytic fungi represents a new opportunity for the discovery of bioactive secondary metabolites (Alurappa et al. 2018; Jia et al. 2016). During the last decade, microbial secondary metabolites have played a pivotal role in the discovery of clinically important molecules (Butler 2004, 2005). In the course of our continuing exploration of secondary metabolites of endophytic fungi derived from Indian medicinal plants (Arora et al. 2016; Nalli et al. 2017, 2015), a fungal strain GG3F49, isolated from Glycyrrhiza glabra Linn., and identified as Phoma macrostoma, was selected as its extract showed significant cytotoxic activity.
In plants, Phoma species may exist as both pathogenic as well as endophytic in nature (Qadri et al. 2014; Strobel et al. 2011). The fungus P. macrostoma is a ubiquitous organism with a wide distribution and board host range (Aveskamp et al. 2010). It exists as a weak or wound pathogen occurring mostly on woody hosts and thus considered as a rare phyto-pathogen of significance (Bennett et al. 2018). Strains of P. macrostoma have not been extensively explored for bioactive natural products while the investigation of one culture has led to the isolation of macrocidins A and B (Graupner et al. 2003). Thus, in the current study, in an effort to characterize the metabolites from GG3F49, we report the isolation of four known metabolites (1–4) and the characterization of 1 by X-ray diffraction data. Further, cytotoxic activities of 1 and its synthetic acylated analogs (1a–1e) against MDMB-231, T47D, MCF-7, and MIAPaCa-2 cell lines and death mechanism induced by 1 in MIA PaCa-2 cells are reported.
Results and discussion
P. macrostoma GG3F49 was cultured in PD broth for 15 days at 25 °C, and the broth and mycelia extracted with EtOAc. In the crude extract, four major spots (metabolites) were noticed on TLC (CHCl3–MeOH, 9:1) at Rf values 0.77, 0.55, 0.22, 0. All the four metabolites were purified by silica gel column chromatography and characterized spectroscopically as macrophin (1) (Crombie et al. 1988), rosellisin (2) (Nair 1976), 2-(2-hydroxy-5-6-methoxy-3-methylene-1,4-benzodioxin-2(3H)-one (3) (Assante et al. 1981), and methoxyphenoxyacrylic acid (4), respectively (Fig. 1). The structure of 1 was further confirmed, for the first time, by X-ray crystallography data analysis (Fig. 2). All the isolated compounds (1–4) were evaluated for their cytotoxic activities against four cancer cell lines viz MDA-MB-231, T47D, MCF-7, and MIAPaCa-2 (Table 1). The investigation illustrates only macrophin (1) exhibited cytotoxicity against the MDA-MB-231, T47D, MCF-7, and MIAPaCa-2 cancer cell lines with the IC50 values of 14.8, 8.12, 13.0, and 0.9 μM, respectively (Table 1). This biological significance has prompted us to generate a structural diversity of 1 to conduct structure–activity relationship (SAR) study (Scheme 1). Therefore, a series of five acylated analogues (1a–1e) of 1 was synthesized by adding corresponding acid anhydrides (0.5 mL) to a solution of 1 (30 mg) and pyridine (0.5 mL) in 10 mL in a round bottom flask at room temperature for 4 h. Further evaluation of these analogues (1a–1e) for their cytotoxic activities showed that almost all the analogues were more potent than 1 against MDA-MB-231 and MIA PaCa-2. The compound 1d, an n-butanoyl substituent of 1, has been proven to be three fold more potent than parent molecule 1 with the IC50 values of 3.7 and 0.3 µM against MDA-MB-231 and MIA PaCa-2 cell lines, respectively (Table 1). Further, we have explored the death mechanism induced by 1 in MIA PaCa-2 cells. Cancer cells have dysregulated programmed cell death machinery (Fulda and Debatin 2006). Therefore, that is capable of inducing non-pathological cancer cell death could be of clinical significance (Kumar et al. 2015). In animals, there are two types of programmed cell death mechanisms, apoptosis and autophagy, also known as type-1 and type-2, respectively. With this view, we have explored the molecular mechanism of cell death induced by 1 in MIA PaCa-2 cells by various experiments like typical morphological changes, acidic compartment (like lysosome) staining, and cell cycle analysis. It was evident from fluorescent microscopy that there is no induction of autophagy in MIA PaCa-2 cells when treated with macrophin (1). It was stained with acridine orange, a lysotropic dye that accumulates in acid compartments such as lysosome or autophagosome. Rapamycin (200 nM) (well-known autophagy inducer) (Rubinsztein et al. 2007) was taken as a positive control (Fig. 3). Therefore, this data suggested that cell death induced by macrophin (1) is not autophagic in nature. The events of apoptosis were characterized by a series of typical morphological changes like cell shrinkage as well as the formation of apoptotic bodies (Gerl and Vaux 2005). Cells treated with 1 at a concentration of 0.5 and 1 μM for 24 h in which ultrastructure, blebbing phenotype was observed, which is a characteristic feature of apoptosis and camptothecin (1 μM for 24 h), were taken as a positive control (Fig. 4). Therefore, the above data taken together suggested that cell death in pancreatic cancer cell induced by 1 could be apoptotic in nature. Furthermore, several anticancer drugs have been shown to induce apoptosis by targeting cell cycle specific events (Almasan et al. 1995). Alternatively, efforts to increase S phase arrest have also been proven to enhance apoptosis and such strategies have their synergistic effect on cytotoxicity with conventional cytotoxic drugs (Estrov et al. 2003). Surprisingly, macrophin (1) (600 nM) treated cells exhibited a significant increase in the cell population at S-phase arrest, 40.70% compared to 17.41% non-treated cells, analyzed by flow cytometer (Fig. 5). Together, these results demonstrate that macrophin (1) induces apoptosis through S-phase arrest. Further, detailed mechanistic pharmacological activities regarding growth inhibitory potential against MDA-MB-231 (IC50 3.7 µM) and MIAPaCa-2 (IC50 0.3 µM) cell lines of the most potent analogue (1d) of 1 are in progress.

Chemical structures of isolated compounds 1–4

ORTEP diagram (CCDC 1832008) of compound 1

Reagents and conditions: pyridine (0.5 mL), corresponding acid anhydride (a–e) (0.5 mL), at RT, 4 h

Pancreatic cancer cell line (MIAPaCa-2) was treated with different concentrations of macrophin (1) (0.5 and 1 μM) for 24 h, then cells were stained with acridine orange (1 μg/mL) for 15 min at 37 °C, and visualized under fluorescent microscope. Rapamycin (200 nM) was used as the positive control. Experiment was repeated three times (p < 0.005)

Effect of macrophin (1) on pancreatic cancer cells through scanning electron microscopy: cells were treated with macrophin (1) at a concentration of 0.5 and 1 μM, camptothecin (1 μM) was taken as a positive control and analyzed under a scanning electron microscope. Red and white arrow represents membrane blebbing and cell shrinkage. Experiments were repeated three times (p < 0.005)

a Macrophin (1) causes cell cycle arrest from G2 to S-Phase in pancreatic cancer cells (MiaPaca2): cells were treated with macrophin (1) (100, 200, and 600 nM) for 48 h. After 48 h, RNase was added for 90 min followed by propidium-iodide for 1 h and cells were analyzed by flow cytometer. Data are presented as relative fluorescence intensity in a 2-dimensional FACS profile (ModFit LT software). b Comprehensive bar diagram of cell cycle. Bar diagrams represented are mean of three independent experiments (p < 0.005) and standardized gating was used for all samples. Ten thousand events were analyzed for each sample
Finally, it concludes that the present study highlights the characterization of 1 through X-ray diffraction data, and cytotoxic activities of 1 and its semi-synthetic acyl analogues (1a–1e) against four cancer cell lines MDA-MB-231, T47D, MCF-7, and MIAPaCa-2. The present study also proves the compound 1d is the most potent analogue of 1 with the IC50 values of 3.7 and 0.3 µM against MDA-MB-231 and MIAPaCa-2 cell lines, respectively. Overall, the present study supports the importance of the plant-associated endophytic fungi as a source of secondary metabolites with promising anticancer activity.
Material and methods
General experimental procedures
High-resolution mass spectra were obtained on Agilent 6540 (Q-TOF) high-resolution mass spectrometer, in the electrospray (ESIMS) mode. 1H NMR spectra were recorded (Brucker Avance) DPX FT-NMR at 400 and 13C NMR at 100 MHz in CDCl3 and MeOD, chemical shifts values are reported in δ (ppm) units and coupling constants values in Hertz. Tetramethylsilane (TMS) was used as internal standard. Column chromatography was performed using silica gel (100–200 mesh; Merck). All reactions were monitored by thin layer chromatography (TLC) on silica gel 60F254 (0.25 mm thick, Merck) with spots visualized by UV (254 and 366 nm) and anisaldehyde reagent was used as the development agent. HPLC was performed on an Agilent 1260 series with a photodiode array detector. HPLC-grade acetonitrile and water used in the study were purchased from Merck, India. Rest of the chemicals and solvents used were of analytical grade procured from Sigma-Aldrich, St. Louis, USA.
Isolation and fermentation
The endophytic fungus was isolated as described previously (Qadri et al. 2014). Briefly, rhizome of G. glabra, collected from Jammu (J&K), Jammu (32° 43′ 48″; 74° 50′ 58″), were thoroughly washed with running tap water and surface sterilized with 1% sodium hypochlorite (Sigma-Aldrich, St. Louis, MO, USA) for 5 min followed by 70% ethanol for 3 min. Before the treatment with alcohol, traces of sodium hypochlorite were removed by washing in sterile distilled water. The tissues were cut into small pieces of 0.5–1 cm and plated on water agar. The plates were incubated at 25 °C for 3 weeks. Hyphal tips of the fungus, emerging out of the plant tissue, were picked and grown on potato dextrose agar in pure culture. This isolate was recovered from only one plant tissue and subcultured on PDA to obtain a pure culture. For the extraction of secondary metabolites, the culture was grown in shake flasks in PD broth at 25 °C and 200 rpm for 15 days, in an incubator shaker. For morphological analysis, microscopic slides of the endophyte were prepared by staining with lactophenol—cotton blue and examined under a light microscope (Olympus BX51). All media, antibiotics, and stains used were from HiMedia, unless stated otherwise.
Phylogenetic analysis by ITS1-5.8S-ITS2 ribosomal gene sequencing
Phylogenetic analysis of GG3F49 was carried out by the acquisition of the ITS1-5.8S-ITS2 ribosomal gene sequencing. The ITS regions of the fungi were amplified with the universal ITS primers, ITS4 (5′TCCTCCGCTTATTGATATGC3′) and ITS5 (5′GGAAGTAAAAGTCGTAACAA3′), using the polymerase chain reaction (PCR). The amplified product was sequenced and aligned with the sequences in the GenBank by BLASTN program to find out the sequence homology with closely related organisms. Sequences from the closely related organisms were downloaded and a phylogenetic tree was constructed.
Extraction and purification
After 15 days of cultivation, the whole broth (5 L) was extracted three times with EtOAc, and all EtOAc solutions were combined and concentrated under reduced pressure to obtain the whole crude extract (2.6 g). The crude extract was applied to a silica gel (100–200 mesh) column with a step gradient elution of CHCl3–MeOH. Compounds 1 (210.0 mg; Rf 0.77), 2 (55.3 mg, Rf 0.55), 3 (18.6 mg, Rf 0.22), and 4 (13.0 mg, Rf 0.02) were eluted at 2%, 4%, 6%, and 10% MeOH in CHCl3, respectively. The purity of the compounds was >95% by HPLC/UV analysis. The HPLC used was equipped with a RP-18 column (100 × 4.6 mm, 3 μm; Agilent). The elution performed with water (A) and acetonitrile (B) as mobile phase (0–8 min, 5% B; 8–30 min, 5–95% B; 30–35 min, 5% B; 35–40 min, 5% B).
Macrophin (1)
Colorless crystals; molecular formula: C16H20O7; (+) HR-ESI-MS m/z 324.1209; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 15.4 Hz, 1H), 6.81 (d, J = 15.4 Hz, 1H), 5.77–5.57 (m, 1H), 5.09 (s, 2H), 4.61 (s, 2H), 4.12 (s, 3H), 3.81 (s, 3H), 2.18 (d, J = 1.1 Hz, 3H), 1.89 (d, J = 1.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 169.41, 166.29, 166.15, 158.16, 154.25, 130.51, 125.71, 118.14, 115.35, 109.74, 62.82, 56.05, 54.74, 52.26, 27.46, 20.36; HPLC purity 97.5%.
Rosellisin (2)
White solid; molecular formula: C12H14O7; (+) HR-ESI-MS m/z 270.0740; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 15.4 Hz, 1H), 6.67 (d, J = 15.4 Hz, 1H), 4.50 (s, 2H), 4.46 (s, 2H), 4.10 (s, 3H), 3.74 (s, 3H); 13C NMR (100 MHz, MeOD) δ 168.88, 166.92, 164.50, 153.69, 131.30, 124.80, 119.61, 113.91, 63.14, 54.89, 53.91, 52.39.
6-Methoxy-3-methylenebenzo[b][1,4]dioxin-2(3H)-one (3)
White solid; molecular formula: C10H8O4; (+) HR-ESI-MS m/z 192.0423; 1H NMR (400 MHz, CDCl3) δ 6.97 (d, J = 9.7 Hz, 1H), 6.55 (dd, J = 4.7, 2.2 Hz, 2H), 5.83 (d, J = 2.0 Hz, 1H), 5.31 (d, J = 2.0 Hz, 1H), 3.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.07, 155.36, 143.25, 139.97, 133.09, 117.51, 108.93, 104.80, 101.49, 55.70.
2-(2-Hydroxy-5-methoxyphenoxy)-acrylic acid (4)
Colorless solid; molecular formula: C10H10O5; (+) HR-ESI-MS m/z 210.0528; 1H NMR (400 MHz, MeOD) δ 6.86–6.83 (m, 1H), 6.64 (dd, J = 8.8, 3.0 Hz, 1H), 6.56 (t, J = 3.8 Hz, 1H), 5.55 (d, J = 2.2 Hz, 1H), 4.68 (d, J = 2.2 Hz, 1H), 3.71 (s, 3H); 13C NMR (100 MHz, MeOD) δ 164.49, 153.45, 150.73, 142.27, 142.21, 117.33, 110.75, 106.98, 99.66, 54.87.
General procedure for preparation of compounds (1a–1e)
To the solution of macrophin (1) (30 mg) and pyridine (0.5 mL) in 10 mL round bottom flask, acid anhydride (0.5 mL) was added. The reaction mixture was stirred at room temperature for 4 h. After completion of reaction, the aqueous saturated CaSO4 (50 mL) solution was added to quench pyridine. Finally, the aqueous layer was extracted with ethyl acetate (3 × 50 mL), and the combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo afforded gummy viscous. TLC (Hex:EtOAc 9:1 v/v) exhibited the single spot which was further purified by silica (100–200 mesh) gel column chromatography. The column was eluted with mixtures of hexane and ethyl acetate with increasing polarity [100% (0.5 L) hexane, and 1–20% (0.5 L each) EtOAc] to afford pure acylated macrophin analogs (1a–1e).
1a
Colorless oil; molecular formula: C19H22O9; (+) HR-ESI-MS m/z 394.1262; 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 15.4 Hz, 1H), 6.80 (d, J = 15.4 Hz, 1H), 5.66 (s, 1H), 5.06 (s, 2H), 5.01 (s, 2H), 4.05 (s, 3H), 3.80 (s, 3H), 2.15 (s, 3H), 2.05 (m, 3H), 1.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.31, 169.06, 166.01, 165.95, 162.10, 158.04, 155.41, 130.27, 126.37, 115.31, 114.05, 109.47, 62.74, 56.03, 55.71, 52.21, 27.37, 20.69, 20.29; HPLC purity 98.7%.
1b
Colorless oil; molecular formula: C20H24O9; (+) HR-ESI-MS m/z 408.1423; 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 15.3 Hz, 1H), 6.81 (d, J = 15.4 Hz, 1H), 5.67 (s, 1H), 5.07 (s, 2H), 5.03 (s, 2H), 4.05 (s, 3H), 3.81 (s, 3H), 2.33 (dd, J = 15.0, 7.5 Hz, 2H), 2.16 (s, 3H), 1.88 (s, 3H), 1.13 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.76, 169.10, 166.03, 165.96, 162.10, 158.00, 155.40, 130.32, 126.33, 115.33, 114.18, 109.62, 62.75, 56.04, 55.63, 52.20, 27.41, 27.36, 20.30, 8.98; HPLC purity 98.6%.
1c
Colorless oil; molecular formula: C21H26O9; (+) HR-ESI-MS m/z 422.1579; 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 15.4 Hz, 1H), 6.82 (d, J = 15.4 Hz, 1H), 5.75–5.52 (m, 1H), 5.08 (s, 2H), 5.02 (s, 2H), 4.05 (s, 3H), 3.81 (s, 3H), 2.55 (dt, J = 14.0, 7.0 Hz, 1H), 2.17 (d, J = 1.0 Hz, 3H), 1.88 (d, J = 1.0 Hz, 3H), 1.17 (s, 3H), 1.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.45, 169.09, 166.05, 165.96, 162.16, 158.11, 155.38, 130.36, 126.27, 115.31, 114.26, 109.54, 62.73, 56.07, 55.67, 52.23, 33.91, 27.42, 20.33, 18.86; HPLC purity 97.4%.
1d
Colorless oil; molecular formula: C21H26O9; (+) HR-ESI-MS m/z 422.1577; 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 15.4 Hz, 1H), 6.76 (d, J = 15.4 Hz, 1H), 5.61 (s, 1H), 5.02 (s, 2H), 4.97 (s, 2H), 3.99 (s, 3H), 3.75 (s, 3H), 2.23 (d, J = 7.4 Hz, 2H), 2.11 (d, J = 0.7 Hz, 3H), 1.82 (d, J = 0.8 Hz, 3H), 1.61–1.56 (m, 2H), 0.86 (d, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.00, 169.09, 166.06, 165.97, 162.14, 158.09, 155.42, 130.29, 126.37, 115.33, 114.18, 109.53, 62.73, 56.07, 55.53, 52.23, 35.98, 27.41, 20.33, 18.37, 13.57; HPLC purity 98.3%.
1e
Colorless oil; molecular formula: C23H30O9; (+) HR-ESI-MS m/z 450.1877; 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 15.4 Hz, 1H), 6.81 (d, J = 15.4 Hz, 1H), 5.67 (s, 1H), 5.08 (s, 2H), 5.02 (s, 2H), 4.05 (s, 3H), 3.81 (s, 3H), 2.29 (dd, J = 14.3, 6.8 Hz, 2H), 2.17 (s, 3H), 1.88 (s, 3H), 1.62 (dd, J = 14.6, 7.3 Hz, 2H), 1.29 (dd, J = 7.3, 3.9 Hz, 4H), 0.87 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.20, 169.10, 166.06, 165.96, 162.16, 158.11, 155.39, 130.31, 126.34, 115.31, 114.20, 109.46, 62.73, 56.06, 55.54, 52.23, 34.08, 31.22, 27.41, 24.55, 22.23, 20.32, 13.82; HPLC purity 97.5%.
Cell culture, growth conditions, and treatment
MDMB-231 breast cancer, T47D breast cancer, MIAPACA-2 pancreatic cancer, and MCF-7 breast cancer cells were obtained from the National Cancer Institute (NCI), Bethesda, USA. The cells were grown in RPMI-1640 or DMEM medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 units mL−1), streptomycin (100 µg mL−1), L-glutamine (0.3 mg mL−1), pyruvic acid (0.11 mg mL−1), and 0.37% NaHCO3. Cells were grown in a CO2 incubator (Thermocon Electron Corporation, MA, USA) at 37 °C under an atmosphere of 95% air and 5% CO2 with 98% humidity.
Cell proliferation assay
The MTT assay was performed to determine the cell viability. Cells were seeded in 96-well plates and exposed to different concentrations of the synthesized compounds for 48 h. The MTT dye (10 μL of 2.5 mg mL−1 in PBS) was added to each well 4 h prior to experiment termination. The plates were then centrifuged at 1500 rpm for 15 min and the supernatant was discarded, while the MTT formazan crystals were dissolved in 150 µL of DMSO. The OD was measured at 570 nm.
Statistical analysis
Data are presented as means of three similar experiments and the error bars represent the standard deviation (SD) between the experiments. Statistical analysis was done using the Bonferroni method and a p-value < 0.05 was considered to be significant (***p < 0.001, **p < 0.01, *p < 0.05).
References
Almasan A, Yin Y, Kelly RE, Lee EY, Bradley A, Li W, Bertino JR, Wahl GM (1995) Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci USA 92:5436–5440
Alurappa RCS, Narayanaswamy R, Sinniah UR, Mohanty SK, Swamy MK (2018) Endophytic fungi and bioactive metabolites production: an update. In: Patra J, Das G, Shin HS(eds) Microbial Biotechnology. Springer, Singapore
Arora P, Wani ZA, Nalli Y, Ali A, Riyaz-Ul-Hassan S (2016) Antimicrobial potential of Thiodiketopiperazine derivatives produced by Phoma sp., an endophyte of Glycyrrhiza glabra Linn. Microb Ecol 72:802–812
Assante G, Camarda L, Merlini L, Nasini G (1981) Secondary metabolites from Mycosphaerella ligulicola. Phytochemistry 20:1955–1957
Aveskamp MM, de Gruyter J, Woudenberg JHC, Verkley GJM, Crous PW (2010) Highlights of the Didymellaceae: a polyphasic approach to characterise Phoma and related pleosporalean genera. Stud Mycol 65:1–60
Bennett A, Ponder M, Garcia-Diaz J (2018) Phoma infections: classification, potential food sources, and their clinical impact. Microorganisms 6:58
Butler MS (2004) The role of natural product chemistry in drug discovery. J Nat Prod 67:2141–2153
Butler MS (2005) Natural products to drugs: natural product derived compounds in clinical trials. Nat Prod Rep 22:162–195
Crombie L, Crombie WML, Jamieson SV, Palmer CJ (1988) Acid catalyzed terpenylations of olivetol in the synthesis of cannabinoids. J Chem Soc Perkin Trans 1:1243–1250
Estrov Z, Shishodia S, Faderl S, Harris D, Van Q, Kantarjian HM, Talpaz M, Aggarwal BB (2003) Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood 102:987–995
Fulda S, Debatin KM (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25:4798
Gerl R, Vaux DL (2005) Apoptosis in the development and treatment of cancer. Carcinogenesis 26:263–270
Graupner PR, Carr A, Clancy E, Gilbert J, Bailey KL, Derby JA, Gerwick BC (2003) The macrocidins: novel cyclic tetramic acids with herbicidal activity produced by Phoma macrostoma. J Nat Prod 66:1558–1561
Jia M, Chen L, Xin H-L, Zheng C-J, Rahman K, Han T, Qin L-P (2016) A friendly relationship between endophytic fungi and medicinal plants: a systematic review. Front Microbiol 7:906
Kumar S, Guru SK, Venkateswarlu V, Malik F, Vishwakarma RA, Sawant SD, Bhushan S (2015) A novel quinoline based second-generation mTOR inhibitor that induces apoptosis and disrupts PI3K-Akt-mTOR signaling in human leukemia HL-60 cells. Anti-Cancer Agents Med Chem 15:1297–1304
Kusari S, Hertweck C, Spiteller M (2012) Chemical ecology of endophytic fungi: origins of secondary metabolites. Chem Biol 19:792–798
Nair MSR (1976) Biogenesis and revised structure of rosellisin; structure of rosellisin aldehyde. Phytochemistry 15:1090–1091
Nalli Y, Arora P, Wadhwa B, Malik FA, Vishwakarma RA, Gupta VK, Riyaz-Ul-Hassan S, Ali A (2017) Diapolic acid A-B from an endophytic fungus, Diaporthe terebinthifolii depicting antimicrobial and cytotoxic activity. J Antibiot 70:212–215
Nalli Y, Mirza DN, Wani ZA, Wadhwa B, Mallik FA, Raina C, chaubey A, Riyaz-Ul-Hassan S, Ali A (2015) Phialomustin A–D, new antimicrobial and cytotoxic metabolites from an endophytic fungus, Phialophora mustea. RSC Adv 5:95307–95312
Pusztahelyi T, Holb I, Pócsi I (2015) Secondary metabolites in fungus–plant interactions. Front Plant Sci 6:573
Qadri M, Rajput R, Abdin MZ, Vishwakarma RA, Riyaz-Ul-Hassan S (2014) Diversity, molecular phylogeny, and bioactive potential of fungal endophytes associated with the Himalayan blue pine (Pinus wallichiana). Microb Ecol 67:877–887
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ (2007) Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6:304–312
Strobel G, Singh SK, Riyaz-Ul-Hassan S, Mitchell AM, Geary B, Sears J (2011) An endophytic/pathogenic Phoma sp. from creosote bush producing biologically active volatile compounds having fuel potential. FEMS Microbiol Lett 320:87–94
Wang X, Zhou H, Chen H, Jing X, Zheng W, Li R, Sun T, Liu J, Fu J, Huo L, Li Y-z, Shen Y, Ding X, Müller R, Bian X, Zhang Y (2018) Discovery of recombinases enables genome mining of cryptic biosynthetic gene clusters in Burkholderiales species. Proc Natl Acad Sci USA 115:E4255–E4263
Acknowledgements
PA is supported by the Department of Science and Technology, New Delhi, India through INSPIRE Senior Research Fellowship. PA and SK acknowledge AcSIR for their enrollment in the Ph.D. program. The authors would like to acknowledge the Ministry of AYUSH (New Delhi) for providing financial support for the work of YN (Z.28015/229/2015-HCP EMR). The article bears the institutional manuscript no. IIIM/2271/2018.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nalli, Y., Arora, P., Khan, S. et al. Isolation, structural modification of macrophin from endophytic fungus Phoma macrostoma and their cytotoxic potential. Med Chem Res 28, 260–266 (2019). https://doi.org/10.1007/s00044-018-2281-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-018-2281-y