
Abstract
The Monte Carlo method was used for quantitative structure–activity relationship modeling of bis-quinolinium and bis-isoquinolinium compounds as acetylcholine esterase inhibitors for a series of 36 compounds. quantitative structure–activity relationship models were calculated with the representation of the molecular structure by the Simplified Molecular Input-Line Entry System. One split into the training and test set was examined. The statistical quality of the developed model was good. The best calculated quantitative structure–activity relationship model had following statistical parameters: r 2 = 0.8097 for the training set and r 2 = 0.9372 for the test set. Structural indicators defined as molecular fragments responsible for increases and decreases in the inhibition activity were calculated. The computer-aided design of new compounds as potential acetylcholine esterase inhibitors with the application of defined structural alerts was presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myasthenia gravis (MG) is an autoimmune disorder which results from the destruction of the post-synaptic membrane in the neuromuscular junction (Conti-Fine et al., 2006; Vincent et al., 2001). In most MG cases, human antibodies are produced to the nicotinic acetylcholine receptor (nAChR) (Tzartos et al., 1998; Vincent et al., 2003). An autoimmune attack to the endplate region of the neuromuscular junction resulting in reduced density of nAChR is initiated with these antibodies, which further leads to the destruction of the synaptic folds and the general simplification of the post-synaptic membrane. Reduced transmission in the neuromuscular junction results in a characteristic symptom of MG, that is, weakness of the striated muscles is caused by the decreased probability of the acetylcholine (ACh)–nAChR interaction (Santa et al., 1972). Acetylcholinesterase inhibitors can be used in the treatment of disorders with impaired cholinergic transmission, therefore the first-line treatment in early stages and in the mild forms of MG is based on peripheral inhibitors of AChE since they work by increasing the concentration of ACh in the synaptic junction and thus enhance the cholinergic transmission in spite of the nAChR depletion (Drachman, 1994; Lindstrom, 2000; Richman and Agius, 2003). It has to be noted that the application of AChE inhibitors is only a symptomatic approach and it does not resolve the original cause of the disease. The treatment of patients with MG is a complex task. The quality of life of patients with MG can be improved with the application of mild physical exercise. Therefore, physicians specialized in sports medicine, physiatrists and experts in sports in general can ease lives of MG patients and help them in continuous fight against this severe disorder (Lucia et al., 2007; Rubin and Hentschel, 2007). Most common drugs used in current MG therapy are presented in Fig. 1. Unfortunately, the use of these compounds often leads to the development of serious gastrointestinal side effects, increased bronchial secretion, cardiac arrhythmia. Moreover, high intake of these compounds may lead to cholinergic crisis, characterized by even more severe weakness (Froelich and Eagle, 1996; Juel and Massey, 2007; Leigh et al., 2003). Considering the above-stated facts, the search is being continued for a far more effective AChE inhibitor that can be used in the treatment of MG.

Drugs used in Myasthenia gravis therapy
In modern drug design, quantitative structure–activity relationship (QSAR) modeling is widely used due to the fact that this powerful computational approach can make an early prediction of activity-related characteristics of drug candidates and eliminate compounds with undesired properties (Cherkasov et al., 2014; Dearden, 2016; Roy et al., 2015; Tropsha and Golbraikh, 2007). The main aim of QSAR modeling is to develop a simple mathematical equation that correlates a molecule’s studied biological activity with its properties and molecular characteristics calculated as quantitative parameters–descriptors. Therefore, descriptors can be defined as entities that characterize specific information of a studied molecule in terms of numerical values associated with the chemical constitution for the correlation of the chemical structure with the biological activity (Cronin and Schultz, 2003). When a QSAR model is built on geometry-dependent molecular descriptors, then the model development usually involves a relatively difficult calculation of the optimal molecular geometry which encompasses the application of high computational resources and a long time for computational experiments. For this reason, the conformation-independent 0D, 1D and 2D-QSPR methods emerge as an alternative approach for developing models based on the constitutional and topological molecular features of molecules (Duchowicz et al., 2012; Talevi et al., 2012). Descriptors calculated on the basis of molecular graphs are often used in QSAR modeling (Ivanciuc, 2013; Talevi et al., 2012). The simplified molecular input-line entry system (SMILES) can be considered as an alternative to molecular graphs and it can be used for defining of molecular structures (Toropov and Benfenati, 2007; Veselinović et al., 2015). One-variable QSAR models built up by the Monte Carlo optimization method based on SMILES descriptors have been published recently. One of the main advantages of this method in comparison to most commonly used QSAR models is the fact that it is not dependent on molecule conformation since it is based on constitutional and topological features of molecules. Moreover, QSAR models based on SMILES notation descriptors have similar or better statistical characteristics in comparison to 3D descriptors based QSAR models. All the above stated facts make QSAR modeling based on SMILES notation descriptors an attractive alternative to commonly used methods in drug design and discovery (Veselinović et al., 2015).
The aim of this study is to build a QSAR model based on the SMILES notation optimal descriptors using the Monte Carlo method for bis-quinolinium and bis-isoquinolinium compounds as AChE inhibitors and to make an attempt to define the molecular fragments responsible for the stated inhibitory effect. Furthermore, we used the built model and defined molecular fragments for the computer aided drug design of new potentially promising AChE inhibitors.
Method
The data set
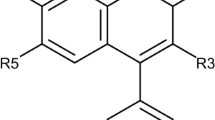
A QSAR model was developed for a series of 36 bis-quinolinium and bis-isoquinolinium compounds acting as AChE inhibitors (Komloova et al., 2011; Musilek et al., 2011). General structures of these compounds are presented in Fig. 2. Molecular structures of the studied compounds were transformed into the canonical SMILES with the ACD/ChemSketch program (ACD/ChemSketch v. 11.0). To represent the pharmacological activity, pIC50 (−log IC50) was used as a dependent variable for building both QSAR models (Supplementary Table S1). Molecules with reported zero activity were discarded from the model development. The QSAR model for was built up for one random split (20 % of the compounds were used in the test set). The normality distribution plot of the whole dataset was checked according to literature (Ojha and Roy, 2011).

General molecular structures of studied compounds
QSAR model development and validation
The main concept of the QSAR modeling can be defined as the following:
The SMILES notation is one of the most convenient molecule structure representations. In the presented QSAR modeling, the SMILES notation is used for defining a molecular optimal descriptor (DCW). DCW is calculated as a function of the molecule’s SMILES notation as shown in Eq. 2:
Based on the SMILES notation, two types of optimal descriptors defined can be defined. The first type of optimal descriptors is a local SMILES attribute, defined as SMILES atoms, a fragment of the SMILES notation which contains one (‘C’, ‘O’, ‘#‘, etc.) or two symbols (‘Cl’, ‘Br’, ‘@@‘, etc.) which cannot be examined separately. The simplest way of defining a molecular DCW as a mathematical function of SMILES atoms can be achieved with the application of local SMILES attributes, as a mathematical function of each character of the SMILES. Therefore, the first and the simplest one SMILES notation based descriptor is Sk (SMILES atom), a descriptor related to one SMILES symbol (or two that cannot be separated). A linear combination of two or three SMILES atoms are descriptors defined as SSk and SSSk (Veselinović et al., 2013, 2015). The linear combination of the presented SMILES notation based descriptors can be summarized in Eq. 3.
The inclusion of SMILES based descriptors in the QSAR model development can be achieved with the application of a simple rule—if the value of a coefficient is 1, then an appropriate descriptor is included or if it is 0, then an appropriate descriptor is discarded form the model building. For this reason, coefficients α, β and γ from Eq. 3 are either 1 (yes) or 0 (no). In the presented QSAR model for AChE inhibition all coefficients were 1, therefore all local SMILES notation based descriptors were used in the model development.
Each calculated optimal descriptor receives a numerical value defined as the correlation weight (CW) with the application of the Monte Carlo method. The applied Monte Carlo method is based on a principle that suitable random numbers are generated and further observed to see how that fraction of numbers obeys some property/properties. The application of iterative algorithms is used for the computation of obtaining and distribution of an unknown probabilistic entity (Toropov et al., 2013). Therefore, the main purpose of the Monte Carlo optimization process is the calculation of the numerical data for the CW which gives the maximal value of the correlation coefficient between the endpoint and the optimal descriptor. The Monte Carlo method is based on two parameters for the computation of stated numerical values—Threshold (T) and the Number of epochs (N epoch). Threshold can be defined as the separator of calculated SMILES based descriptors into active and inactive with the application of the following principle: if the descriptor is active, then T has some numerical value; however, if the descriptor is inactive, then T has a fixed value of zero. The number of epochs (N epoch) is related to the computational iterative process with one aim—to obtain the best statistical quality for the training set (Veselinović et al., 2013, 2015).
The linear regression approach is used as a method for developing a QSAR model after all CW are calculated (Eq. 4)
CORAL software was used for the development of the presented QSAR model (http://www.insilico.eu/coral). All local SMILES descriptors and global attributes (Sk, SSk and SSSk) were taken into consideration during the building of QSAR models. The search for the most predictive combination of T and N epoch for all splits was performed from values 0–10 for T and 0–60 for N epoch.
The main purpose of any QSAR modeling is to develop a robust model capable of predicting the properties of new molecules in an objective, reliable and precise manner (Roy, 2007). The application of three methods can be used for the assessment of robustness and reliability of a developed QSAR model (Roy et al., 2008): (a) internal validation or cross-validation using the training set compounds, (b) external validation using the test set compounds and (c) data randomization or Y-scrambling. This methodology was successfully applied for the validation of SMILES notation optimal descriptor based QSAR models (Veselinović et al., 2015).
Several standard statistical metrics were used for validating the developed QSAR model, including the correlation coefficient (r 2), cross-validated correlation coefficient (q 2), standard error of estimation (s), mean absolute error (MAE), Fischer ratio (F) and root-mean-square error (RMSE). In addition, novel metrics (R m 2 and MAE based metric) were used for further validation. Novel statistical metric (R m 2) can be used to estimate true predictive potential of a developed QSAR model (Ojha et al., 2011; Roy et al., 2012). The calculation of this metric is based on the correlation between the observed and predicted values with (R 2) and without (R 0 2) intercept for the least squares regression lines as shown in eq. 5:
The main advantage of this metric is that it avoids the overestimation of the quality of prediction due to a wide response (Y-range) since it does not consider the differences between individual responses and the training set mean. In addition, a scaled version of R m 2 metric can be used in order to have a better estimation of QSAR model predictability. For calculating both R m 2 and scaled version of R m 2 metrics, an open-access web application “Rm2 calculator” is available at http://aptsoftware.co.in/rmsquare/. Finally, the mean absolute error (MAE) metric was used for the final estimation of the developed QSAR model prediction quality according to the published methodology (Roy et al., 2016). A software from http://teqip.jdvu.ac.in/QSAR_Tools/ web link was used for the calculation of the MAE based metric.
One of the most important features of all developed QSAR models is its applicability domain (AD). Molecules from the training set are used for this purpose. For all developed QSAR models, the AD is defined as biological, structural, or physicochemical space, knowledge, or information on which the model of the training set is developed and which can be used for predicting whether a developed QSAR model can be used on compounds which are not used in the model developing. Therefore, a defined AD can be applied for the assessment of the reliability of a developed QSAR model. If the application of the AD defines a studied compound as very different in comparison to all compounds from the training set, a reliable prediction of its property/activity is uncertain (Gadaleta et al., 2016; Gramatica, 2007). Considering QSAR models based on SMILES based descriptors, the difference between experimental and calculated values for a studied endpoint can be used for defining the AD with the application of Delta(obs), d and \(\bar d\) (Toropov et al., 2011). For each molecule used in the QSAR study, Delta(obs) is calculated as the difference between experimental and calculated values from a studied endpoint. d and \(\overline{\overline d}\) are defined in Eqs. 6 and 7, respectively, where n is the number of studied compounds.
If Delta(obs) of a studied compound belongs within the range \(\bar d\) − d and \(\bar d\) + d, then it falls in the defined model AD. However, if compound’s Delta(obs) does not belong inside a defined range, then that compound does not belong in the defined AD. For this reason, the compound is considered as outlier, and it is discarded from the development of a QSAR model. The relation used for defining the AD is given in Eq. 8:
A defined methodology for the determination of the AD was successfully applied in the development of various QSAR models based on the Monte Carlo method and SMILES based descriptors (Veselinović et al., 2015).
After defining the AD, the molecule 14 from the research reported by Musilek et al. was defined as outlier and it was discarded from the model development.
Results and discussion
Table 1 shows the statistical quality of the built QSAR model for bis-quinolinium and bis-isoquinolinium compounds acting as AChE inhibitors. The presented results reveal that there is the reproduction of the statistical quality for the calculated model in three independent runs of the Monte Carlo optimization. Furthermore, the results from Table 1 show that the predictability is good. In order to further assess the quality of the developed QSAR model, novel statistical metric was used according to published methodologies and the results are presented in Table 2. The presented QSAR model for IC50 is satisfactory from the point of view of new criteria (Ojha et al., 2011; Roy et al., 2012). Supplementary Table S2 shows Y-randomization (Ojha and Roy, 2011) which also confirms the robustness of the suggested models. MAE based metric revealed that the developed QSAR model is “moderate”, what classified this model as valid. The search for preferable T and N epoch revealed that preferable T is 2 and preferable N epoch 10. Figure 3 graphically presents the best Monte Carlo optimization run (the highest value for r 2) for the developed QSAR model.

Graphical representation of the developed QSAR model for bis-quinolinium and bis-isoquinolinium derivates as acetylcholine esterase inhibitors
DCW(T,N epoch) for compounds in the training and test sets are calculated as described in the Method section of this manuscript. The application of the above-mentioned T and the N epoch gives the following model for the pIC50 calculated according to Eq. 4:
According to the calculated correlation weights (CW), the values of molecular features (SAk) from three Monte Carlo optimization runs, SAk can be classified as promoters of increase or promoters of decrease of a studied activity, and as undefined molecular features. A simple rule is applied for this classification: if the CW(SAk) obtained from three independent Monte Carlo optimization runs is positive then that SAk is the promoter of increase, likewise if the CW(SAk) from three independent Monte Carlo optimization runs is negative then that SAk is the promoter of decrease; if there are both positive and negative values of the CW(Sk) in three runs of the Monte Carlo optimization process, then that SAk is undefined (Veselinović et al., 2015). The list of all SAk, with the correlation weights for three runs of the Monte Carlo optimization process of the built QSAR model for maleimide derivatives is given in Supplementary Table S3. The built QSAR model represented with Eq. 5 has the mechanistic interpretation as well as calculated SAk which can be further defined as molecular fragments. According to the presented data, several important SAks that can be classified as promoters of pIC50 value decrease are O…C…C… (ethoxy group) and C……….. (methyl group) while several important SAks that can be classified as promoters of pIC50 value increase are = ……….. (double bond), C… = ……. and C… = …C… (double bond on a carbon atom), C…/……., C…/…C…, /……….., \……….. and \…C……. both cis and trans conformations, O……….. (sp3 hybridized oxygen atom), O…C……. (methoxy group), C…C……. (ethyl group), etc. The obtained findings are in accordance with the results of SAR findings from published data (Komloova et al., 2011; Musilek et al., 2011).
The analysis of SAk presented in Supplementary Table. S3 can be useful in the search and computer aided design of novel derivatives with desired pIC50 values. The structures of novel compounds obtained by molecular modeling are presented in Fig. 4.

The molecular design of perspective acetylcholine esterase inhibitors using the QSAR model calculated with Eq. (5) and SAks calculated using the Monte Carlo method
The molecule 37 from dataset was selected as a template for the molecular design. One of the goals of the presented molecular design was to obtain molecules with higher pIC50 values in comparison to the template molecule, since higher pIC50 values mean that a lower concentration is needed to inhibit 50 % of an enzyme. Molecular structures presented as the SMILES notation of designed molecules and their calculated pIC50 values obtained with the application of the built QSAR model for compounds (Eq. 5) are presented in Table 3.
Table 4 presents an example of the calculation of DCW(2,10) for the template molecule A. When Eq. 5 is applied for calculating DCW, the resulting endpoint value (pIC50) is 6.6140. In the molecule A1+ two more carbon atoms were added to the molecule. In the molecule A1+ one more SAk was added in comparison to the molecule A, C…C……. (ethyl group), defined as the promoter of Ac increase. For this reason, the molecule A1+ has the calculated Ac value of 7.6291. The molecule A2+ has two more carbon atoms in comparison to the molecule A1+ and also a higher value of Ac (8.5491). The molecule A3+ has substituted one carbon atom with oxygen which leads to the introduction of O…C……. SAk, also defined as the promoter of increase. The calculated value for the molecule A3+ Ac was 7.8063. Molecules A4+ and A5+ represent two stereo isomers. Since both have promoters of Ac increase, their values for the calculated Ac were 7.9879 and 7.7828, respectively. Results suggest that trans isomer is favorable in comparison to cis since it has a higher value for the calculated Ac.
Conclusion
QSAR models for bis-quinolinium and bis-isoquinolinium compounds as acetylcholine esterase inhibitors were built. The Monte Carlo optimization process incorporated within CORAL software was capable to be an efficient tool to build up a robust model of good statistical quality. The predictive potential of the applied approach was tested and the robustness of the model was proven with different methods. The SMILES attributes, defined as SMILES notation based molecular descriptors, which are promoters of IC50 increase/decrease were identified. The suggested modeling process and computer aided drug design were based on computational experiments with the application of statistically stable structural alerts (promoters of increase or decrease of IC50). This approach can be applied in the search for new potential acetylcholine esterase inhibitors.
References
Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’Min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A (2014) QSAR modeling: where have you been? Where are you going to? J Med Chem 57:4977–5010
Conti-Fine BM, Milani M, Kaminski HJ (2006) Myasthenia gravis: past, present, and future. J Clin Invest 116:2843–2854
Cronin MTD, Schultz TW (2003) Pitfalls in QSAR. J Mol Struct-THEOCHEM 622:39–51
Dearden JC (2016) The History and Development of Quantitative Structure-Activity Relationships (QSARs). IJQSPR 1:1–44
Drachman DB (1994) Myasthenia gravis. N Engl J Med 330:1797–1810
Duchowicz PR, Comelli NC, Ortiz EV, Castro EA (2012) QSAR study for carcinogenicity in a large set of organic compounds. Curr Drug Saf 7:282–288
Froelich J, Eagle CJ (1996) Anaesthetic management of a patient with myasthenia gravis and tracheal stenosis. Can J Anaesth 43:84–89
Gadaleta D, Mangiatordi GF, Catto M, Carotti A, Nicolotti O (2016) Applicability domain for QSAR models: where theory meets reality. IJQSPR 1:45–63
Gramatica P (2007) Principles of QSAR models validation: internal and external. QSAR Comb Sci 26:694–701
Ivanciuc O (2013) Chemical graphs, molecular matrices and topological indices in chemoinformatics and quantitative structure–activity relationships. Curr Comput Aided Drug Des 9:153–163
Juel VC, Massey JM (2007) Myasthenia gravis. Orphanet J Rare Dis 2:44
Komloova M, Musilek K, Horova A, Holas O, Dohnal V, Gunn-Moore F, Kuca K (2011) Preparation, in vitro screening and molecular modelling of symmetrical bis-quinolinium cholinesterase inhibitors—implications for early Myasthenia gravis treatment. Bioorg Med Chem Lett 21:2505–2509
Leigh P, Abrahams S, Al-Chalabi A, Ampong M, Goldstein L, Johnson J, Lyall R, Moxham J, Mustfa N, Rio A, Shaw C, Willey E (2003) The management of motor neurone disease. J Neurol Neurosurg Psychiatry 74(Suppl 4):iv32-iv47
Lindstrom JM (2000) Acetylcholine receptors and myasthenia. Muscle Nerve 23:453–477
Lucia A, Maté-Muñoz JL, Pérez M, Foster C, Gutiérrez-Rivas E, Arenas J (2007) Double trouble (McArdle’s disease and myasthenia gravis): how can exercise help? Muscle Nerve 35:125–128
Musilek K, Komloova M, Holas O, Hrabinova M, Pohanka M, Dohnal V, Nachon F, Dolezal M, Kuca K (2011) Preparation and in vitro screening of symmetrical bis-isoquinolinium cholinesterase inhibitors bearing various connecting linkage - Implications for early Myasthenia gravis treatment. Eur J Med Chem 46:811–818
Ojha PK, Mitra I, Das R, Roy K (2011) Further exploring rm2 metrics for validation of QSPR models. Chemom Intell Lab Syst 107:194–205
Ojha PK, Roy K (2011) Comparative QSARs for Antimalarial Endochins: importance of descriptor-thinning and noise reduction prior to feature selection. Chemom Intell Lab Sys 109:146–161
Richman DP, Agius MA (2003) Treatment of autoimmune myasthenia gravis. Neurology 61:1652–1661
Roy K (2007) On some aspects of validation of predictive quantitative structure activity relationship models. Expert Opin Drug Dis 2:1567–1577
Roy K, Das RN, Ambure P, Aher RB (2016) Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom Intell Lab Syst 152:18–33
Roy K, Kar S, Das RN (2015) Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment. Academic Press, USA
Roy K, Mitra I, Kar S, Ojha PK, Das RN, Kabir H (2012) Comparative studies on some metrics for external validation of QSPR models. J Chem Inf Model 52:396–408
Roy PP, Leonard JT, Roy K (2008) Exploring the impact of the size of training sets for the development of predictive QSAR models. Chemom Intell Lab Syst 90:31–42
Rubin DI, Hentschel K (2007) Is exercise necessary with repetitive nerve stimulation in evaluating patients with suspected myasthenia gravis?. Muscle Nerve 35:103–106
Santa T, Engel AG, Lambert EH (1972) Histometric study of neuromuscular junction ultrastructure. I. Myasthenia gravis. Neurology 22:71–82
Talevi A, Bellera CL, Ianni MD, Duchowicz PR, Bruno-Blanch LE, Castro EA (2012) An integrated drug development approach applying topological descriptors. Curr Comput Aided Drug Des 8:172–181
Toropov AA, Benfenati E (2007) SMILES as an alternative to the graph in QSAR modelling of bee toxicity. Comput Biol Chem 31:57–60
Toropov AA, Toropova AP, Lombardo A, Roncaglioni A, Benfenati E, Gini G (2011) CORAL: building up the model for bioconcentration factor and defining it’s applicability domain. Eur J Med Chem 46:1400–1403
Toropov AA, Toropova AP, Puzyn T, Benfenati E, Gini G, Leszczynska D, Leszczynski J (2013) QSAR as a random event: modeling of nanoparticles uptake in PaCa2 cancer cells. Chemosphere 92:31–37
Tropsha A, Golbraikh A (2007) Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr Pharm Des 13:3494–3504
Tzartos SJ, Barkas T, Cung MT (1998) Anatomy of the antigenic structure of a large membrane autoantigen, the muscle-type nicotinic acetylcholine receptor. Immunol Rev 163:89–120
Veselinović AM, Milosavljević JB, Toropov AA, Nikolić GM (2013) SMILES-based QSAR model for arylpiperazines as high-affinity 5-HT1A receptor ligands using CORAL. Eur J Pharm Sci 48:532–541
Veselinović AM, Veselinović JB, Živković JV, Nikolić GM (2015) Application of SMILES notation based optimal descriptors in drug discovery and design. Curr Top Med Chem 15:1768–1779
Vincent A, Bowen J, Newsom-Davis J (2003) Seronegative generalised myasthenia gravis: clinical features, antibodies, and their targets. Lancet Neurol 2:99–106
Vincent A, Palace J, Hilton-Jones D (2001) Myasthenia gravis. Lancet 357:2122–2128
Acknowledgments
The authors would like to thank anonymous reviewers whose comments and suggestions made a significant improvement to the manuscript. This work has been supported by the Ministry of Education and Science, the Republic of Serbia, under Project Number 43012.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sokolović, D., Aleksić, D., Milenković, V. et al. QSAR modeling of bis-quinolinium and bis-isoquinolinium compounds as acetylcholine esterase inhibitors based on the Monte Carlo method—the implication for Myasthenia gravis treatment. Med Chem Res 25, 2989–2998 (2016). https://doi.org/10.1007/s00044-016-1720-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1720-x