
Abstract
Among the most well-known members of the mitotic protein kinase family are polo-like kinases. Polo-like kinase 4 as a type of this family is used as a therapeutic target in the treatment of proliferative diseases. A three-dimensional quantitative structure activity relationship study with comparative molecular field analysis and comparative molecular similarity indices analysis was carried out on a data set of 47 molecules consisting of (E)-3-((1H-indazol-6-yl)methylene) indolin-2-ones derivatives of polo-like kinase 4 inhibitors to rational design of new drug. The validity of model was tested with a data set divided into training and test set. All constructed models show good statistical reliability in terms of predicting polo-like kinase inhibitory activity of the molecules, based on molecular property fields like steric, electrostatic, hydrophobic, hydrogen bond donor and hydrogen bond acceptor fields. Moreover, molecular docking with CDOCKER algorithm was done to investigate interactions of between ligand and protein and to achieve bioactive ligand conformer. The energy difference between the highest occupied molecular orbital and the lowest unoccupied molecular orbital (gap) implicitly stated the high reactivity of the most active molecule in the active site of protein. Furthermore, the molecular electrostatic potential energy at density functional theorylevel confirm the results from molecular docking. The identified key features obtained from the quantitative structure activity relationship modeling enabled us to design novel indolinone derivatives. In silico absorption, distribution, metabolism and excretion and toxicity risk assessment analyses were carried out on the new molecules to investigate compliance with the standard ranges.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polo-like kinases (PLKs) are a family of serine/threonine kinases which play a significant part in the regulation of mitosis and DNA damage pathways (Barr et al., 2004). Having barrel-shaped and microtubule-based structures, centrioles are duplicated exactly once in every cell cycle and serve as platforms in order to assemble centrosomes and cilium (Mónica Bettencourt-Dias et al., 2011; Bornens, 2012; Gönczy, 2012; Lüders and Stearns, 2007; Nigg and Raff, 2009). Centrosomes make up the major microtubule-organizing centers in animal cells. In each centrosome, two centrioles are embedded in a protein matrix recognized as the pericentriolar material (PCM). In quiescent and differentiated cells, centrioles function as basal bodies for the formation of cilia and flagella. Anomalies in centrosome number and/or structure have long been implicated in tumorigenesis (Basto et al., 2008; Ganem et al., 2009; Nigg, 2002). Genetic studies have revealed that mutations in centriolar and centrosomal proteins are in charge of a different range of developmental diseases, namely remarkably ciliopathies, microcephaly and dwarfism (Bettencourt-Dias et al., 2011; Davis and Katsanis, 2012; Megraw et al., 2011). PLK4 also known as SAK, has a central role in confining centriole biogenesis and duplication PLK4 has only one polo-box and an active site with high homology to the Aurora kinases (Habedanck et al., 2005; Kleylein-Sohn et al., 2007; Nigg, 2007); the linking mechanism between PLK4 activity and centriole formation is unknown, though. PLK4 localizes to the centriole and is also essential for controlling centriole duplication and mitotic progression (Bettencourt-Dias et al., 2005; Hudson et al., 2001). PLK4-induced centriole biogenesis in human cells involves the sequential assembly of several essential proteins, including human Sas-6, Cep135, CPAP (human Sas-4) and CP110 (Kleylein-Sohn et al., 2007). This type of centriole is not much abundant in normal adult tissues and can automatically regulate its own stability. Overexpression of PLK4 in human cells not only induces centrosome amplification through simultaneous generation of multiple procentrioles adjoining each parental centriole (Kleylein-Sohn et al., 2007) but it is also able to produce procentriole formation which results in the recruitment of electron-dense material onto the proximal walls of parental centrioles. It is known that the PLK4 depletion can be resulted in the death of breast cancer cell lines but not in the normal breast cells (Mak, 2012; Mason et al., 2011). The researchers have found that RNAi-mediated depletion of PLK4 in breast cancer cells prevents centriole duplication which causes mitotic defects and cell death and growth suppression of breast cancer xenografts in vivo (Mak, 2012; Mason et al., 2011).
Computer-aided drug design approaches make a positive contribution in the design of potential PLK4 inhibitors and would help more to further understanding of the role of this enzyme in cancer cell proliferation. Herein, selective molecular field approaches in designing new selective PLK4 inhibitors were employed. In this research, three-dimensional-quantitative structure activity relationship (3D-QSAR) studies using comparative molecular field analysis (CoMFA) (Cramer et al., 1988) and comparative molecular similarity indices analysis (CoMSIA) (Klebe et al., 1994) descriptors on (E)-3-((1H-indazol-6-yl)methylene) indolin-2-ones derivatives are reported. A partial least square (PLS) (Kubinyi, 1993) based on statistical analysis was used to find the correlation between the biological activity and descriptors generated based on aligned molecules. Both CoMFA and CoMSIA contour maps revealed some key factors affecting the activities of the inhibitors and provided us with some guidelines to design some new potent dual PLK4 inhibitors.
Materials and methods
Data set and structures
A set of 47 (E)-3-((1H-indazol-6-yl) methylene) indolin-2-ones derivatives of PLK4 inhibitors as anti-proliferative agents with associated activity data were collected from literature. The potent anti-proliferative activity against the MDA-MB-468 breast cancer cell line, the PLK4 inhibitors were selected testing in the corresponding mouse xenograft model. Nanomolar activity against PLK4 and concomitant anti-proliferative effects against a panel of breast cancer cell lines was measured using an indirect ELISA detection system (Johnson et al., 2007; Laufer et al., 2013). The inhibitory activity values (IC50) were converted into negative logarithm scale value in mole (pIC50) as a dependent variable for all the models subsequently developed. The pIC50 values of the data set extent from 4.5 to 9.5. The dataset was randomly partitioned into training and test set compounds by considering activity range. The structure of compound and their biological activities are shown in Table 1.
CoMFA and CoMSIA
CoMFA and CoMSIA were done using SYBYL 7.3 molecular modeling software package from Tripos, Inc., St. Louis, MO. The 3D structures of dataset compounds were drawn in SYBYL and the Gasteigere–Hückel method was applied to calculate partial atomic charges.
The energy minimizations were processed by means of the Tripos force field with a distance-dependent dielectric and the Powell conjugate gradient algorithm convergence criterion of 0.01 kcal/mol Å. The most active compound 32 was selected as the template for aligning other compounds. Rigid body alignment (using Distill) of molecules was done on the maximum common substructure without involving bond types in ring. A sp3 hybridized carbon atom with +1 charge was chosen and interaction energies between the probe atom and molecules were calculated in each intersection as descriptors. The steric (van der Waals) interaction is modeled using the Lennard–Jones (6–12) potential and the electrostatic interaction is modeled using coulombic, by the Tripos force field. The CoMSIA approach was performed with the same 3D cubic box and the descriptors involving steric, electrostatic, hydrogen bond donor, hydrogen bond acceptor and hydrophobic fields were extracted using a probe atom with +1.0 charge, radius 1.0 Å, hydrophobic and hydrogen bond properties of +1.0.
Partial least squares and validation of models
PLS analysis that is a method multiple regression analysis used to studies 3D-QSAR where CoMFA and CoMSIA descriptors were used as independent variables and the pIC50 values as dependent variables. Cross validation as an internal validation technique was used to assess the prediction quality of the models. In a standard internal validation method, named leave one out cross validation (LOOCV), one compound is excluded from the original training set, and a new model is built based on new training set and this model is used to predict the activity of the excluded one. For each model, this procedure is repeated for whole compounds of data set, and each compound is excluded once, then cross validated q 2 which is considered as a criterion of robustness and predictive ability of the models, is calculated by Eq. 1.
where \(\bar y\) presents average activity value of the entire dataset and y i and \(\hat y\) are observed and predicted activity values, respectively. A high q 2 value (q 2 > 0.5) is used as an evidence of high predictive ability of the model.
Golbarikh and Tropsha reported that the high value of q 2 is essential and important but not adequate for a predictive model. So to investigate the prediction of model, an external analysis by test set of molecules should be employed. According to Golbarikh and Tropsha, a QSAR model is predictive if it fulfills the following conditions:
where R 2 is squared correlation coefficient values between the observed and predicted values of the test set compounds.
\(R_0^2\) and \(R_0^{\prime 2}\) are squared correlation coefficient values for observed versus predicted and predicted versus observed activities, through origin, respectively and k′ is the slope of regression lines through the origin.
The R pred 2 value was calculated according to the Eq. 2:
where \({\hat y_{i}}\) and y i indicate predicted and observed activity values of the test set, respectively, and \(\bar y\) is the average value of training set activities and m is the number of compounds in the test set.
Molecular docking
To find the best binding conformation of the inhibitors to the active site of PLK4, docked inhibitor conformations were generated. Molecular docking by CDOCKER algorithm was done in Discovery Studio 2.5 (AccelrysInc, San Diego, CA, USA) (Studio, 2009). Compound 32 was typed with CHARMm force field and partial charges were calculated by Momany–Rone (Momany and Rone, 1992) option. The resulting structure was minimized with Smart Minimizer algorithm which performs 1000 steps of steepest descent with a RMS gradient tolerance of 3, followed by conjugate gradient minimization. The crystal structures of the PLK4 were downloaded from the RCSB protein data bank (PDB code: 3COK). The protein preparation and minimization were performed in Discovery Studio 2.5 to investigate interactions of ligand and protein and to achieve bioactive ligand conformer (Ghasemi and Shiri, 2012). The complex was typed with CHARMm force field, hydrogen atoms were added, proteins’ ligands and all water molecules were removed and pH of protein was adjusted to almost neutral, 7.4, using protein preparation. A 15 Å radius sphere was defined around the bounded ligand to compose the active site of the X-ray structure, then the most active compound 32 was docked into the protein structure using CDOCKER algorithm.
Quantum chemical calculations
In this study, the geometry optimization and calculations were made at the density functional theory (DFT) level on a personal computer (PC) by energy optimization, using the Gaussian 09 (Frisch et al., 2009) program package. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies at B3LYP/6-31G (d, p) level were calculated for bioactive conformer obtained in the docking studies. The molecular electrostatic potential was also studied at the same level. The MEP surface was created using Molekel (Varetto, 2009).
ADME and toxicity risk assessment studies
Four important agents in pharmacokinetics are absorption, distribution, metabolism and excretion (ADME) (Van de Waterbeemd and Gifford, 2003) and their significance has been thoroughly recognized and predicted in drug design. These factors include pharmacokinetics issues that determine whether the drug molecules reach the target protein in the body or not and how long the drug molecules will remain in the body. There is a relationship between chemical structures and physiological parameters; thus, some chemical descriptors were used to calculate pharmacokinetic parameters by the QikProp v. 3.2 programs (Schrödinger, Portland, OR, USA, 2009) and were checked with the related standard ranges. 12 molecules designed in SYBYL 7.3 molecular modeling package (Tripos Inc., St. Louis, USA) were submitted to Schrödinger 9.0 to predict ADMET properties such as polar surface area (PSA). PSA is a surface descriptor which applies to measure permeability of drugs and is defined as a part of the surface area contributed by nitrogen, oxygen, and connected hydrogen atoms. Molecular weight is another important descriptor. Larger molecules invariably having too many functional groups are able to form hydrogen bonds, and compounds with a molecular weight more than 500 Da are quite likely to have more than ten rotatable bonds. We computed the blood-brain barrier (log BB). Some distribution descriptors in the body have the volume of distribution that can afford a measurement of restrictive or non-restrictive function of plasma binding, and also log K hsa for serum protein binding. Another distribution descriptors are permeability (such as the apparent Caco-2 or MDCK permeability), and skin-permeability coefficient (log Kp). The octanol–water partition coefficient (log P) is a physicochemical parameter of drug’s hydrophobicity. Compounds with a higher lipophilicity have an increased metabolism and poor absorption, and a high probability of binding to unwanted hydrophobic macromolecules hence an increased potential of toxicity. These parameters are used for an acceptable oral activity. Topological polar surface area (TPSA) was calculated by OSIRIS property explorer that could be ≤140 Å2 of a molecule correlating well with the passive molecular transport through membranes. To assess the toxicity risk assessment including mutagenicity, tumorigenicity, irritant and reproductive effects for the compounds designed, Osiris program was employed. Also, their various drug relevant values such as drug-likeness and drug score value were determined.
Results and discussion
CoMFA and CoMSIA results
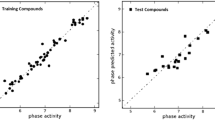
Statistical quality parameters of CoMFA and CoMSIA methods are summarized in Table 2 and the experimental versus predicted activities of the training and test set compounds are presented in Fig. 1. PLS analysis shows a high q 2 value of 0.629 with six components for CoMFA model. The non-cross-validated PLS analysis results in a conventional r 2 of 0.953, F = 94, and a standard error of estimation (SEE) of 0.373, with a column filtering of 2.0. The good r 2 pred values of 0.911 from CoMFA show that the model has acceptable predictive power. For the CoMSIA model, the highest cross-validated q 2 was obtained by using a combination of steric, electrostatic, hydrophobic and H-bond acceptor fields (CoMSIA, q 2 = 0.630, r 2 ncv = 0.961, r 2 pred = 0.889, F = 120.6, SEE = 0.331, SEP = 0.073) with six components. Also, the ability of prediction and the strength of the models evaluated by bootstrapping r 2 boots for CoMFA (0.965) and CoMSIA (0.965) (Smith and Gemperline, 2002) indicates degree of confidence in analysis. In addition, the QSAR model is considered to be predictive if the following conditions are satisfied: q 2 > 0.6 and r 2 pred > 0.5 (Tropsha et al., 2003). The results of the external validation are shown in Table 3.

Predicted against observed activities for training and test sets based on the a CoMFA model b CoMSIA model
CoMFA and CoMSIA contour maps analysis
The ability of CoMFA model for graphical representation of the results (PLS regression coefficients) is an advantage of CoMFA model over other classic QSAR methods. These maps show regions where differences in molecular fields are associated with differences in biological activity (Mao et al., 2012). The CoMFA steric and electrostatic contour maps are shown in Fig. 2. In the CoMFA steric contour maps, green contours show sterically favorable regions and yellow contours show sterically unfavorable regions. These contours represent 80 and 20 % contributions, respectively. In the CoMFA electrostatic contour map, blue contours show electropositive charge favorable areas and red contours show electronegative charge favorable regions with 80 and 20 % contributions, respectively. The most active compound (compound 32) was chosen to be the reference molecule.

Contour maps of a steric and b electrostatic fields of CoMFA based on the most active compound 32
A large yellow area near substitute Aril (Ar) indicates that small groups are more desirable. To justify this, we could say that the activities of the compounds M33, M34 with piperazine substituent attached to the pyridine ring and compound M35 with substituent –CO2H attached to the phenyl ring are lower than those of the compounds M24, M28 with substituent H.
In addition, the substituent R with yellow area shows that the bigger group is undesirable. This corresponds with the experimental results, and it could be explained why the activities of the compounds M17 and M24 with the group –OMe are higher than the compound M23 with substituent Et. The Red area near substituent Ar indicates that high electron density is preferred. The terminal O atom of compound M35 has higher electron density than the N atom in NMe2 of compound M31, so this confirms why compound M31 has a lower activity than compound M35. One blue contour near substituent R indicating electron donating groups like –OMe attached to the benzene ring will increase biological activity. Thus, the compounds M32, M24, M28 and M34 have high activities. The electrostatic contour maps of CoMFA field are in a large part due to the diversity of structures. Also, the blue contour maps which cover a separate area showing electron donating group (NH2) in compounds M36 and M39 is effective for activity.
The CoMSIA contour maps are shown in Fig. 3. It can be seen that CoMSIA steric and electrostatic contour maps are similar to the corresponding CoMFA ones. The hydrophobic contour maps of CoMSIA are shown by Fig. 3c. The yellow (hydrophobic favorable) and white (hydrophobic unfavorable) contours represent 80 and 20 % contributions, respectively. The yellow contour near phenyl ring substituent Ar indicating hydrophobic groups in this area are preferred for PLK4 inhibitory activity. According to docking results, there are some interactions with hydrophobic residues of receptor such as LYS40 confirming hydrophobic contour of CoMSIA. This is a sensible reason why compound M35 with hydrophobic phenyl ring has higher activity than compound M33 with hydrophilic pyridine ring.

Contour maps of steric (a), electrostatic (b), hydrophobic (c), hydrogen bond donor (d) and hydrogen bond acceptor (e) based on the most active compound 32
White contour covers the substituent Ar so hydrophilic groups that were attached to the phenyl ring will increase activity. That is why compound M35 with substituent –CO2H has more activity than compounds M30 and M33 with piperazine and hydrogen substituents.
Figure 3d depicts H-bond donor field distribution of CoMSIA model which is represented by cyan and purple contours. Cyan and purple contours indicate regions where hydrogen bond donor groups on ligand are favoured and disfavored, respectively. There is one purple contour near the CO group in indolinone ring and –OMe group indicating that hydrogen bond donor groups in the receptor enhance the activity of inhibitor. The hydrogen bond donor group of ARG98 and GLU95 of the docking results complements the purple contour in the ligand. Based on hydrogen bond acceptor field in Fig. 3e, magenta contours show regions where hydrogen bond acceptor groups are favoured and red contours indicate regions where hydrogen bond acceptor groups are unfavoured for increasing the activity. There is one magenta contour covering –NMe2 substituent in phenyl ring suggesting hydrogen bond acceptor group is favoured. The hydrogen bond donor group of LYS40 complements the magenta contour in the ligand.
Design for new active compounds
The structure–activity relationship revealed by 3D-QSAR and molecular docking studies are illustrated in Fig. 4. To propose new active compounds, the resulting CoMFA contour maps were used. These molecules activities were predicted by the CoMFA model which had previously been recognized. The structures and predicted pIC50 values of designed compounds in SYBYL 7.3 illustrated in Table 4 shows that electron-donor substituent at Ar position are essential to increase activity designed molecules. Compounds DM1, DM2 and DM10 which display case better predicted pIC50 values contain electron- donating substituent at the terminal of the Aromatic ring side chain. From the comparison of compound DM1 and DM2, with the increase of electron donation and the distance between the aromatic ring and NMe2 group; the activity increases as well.

Structure−activity relationship revealed by 3D-QSAR and docking studies
Docking results
To validate the docking reliability, root-mean square distance (RMSD) value was calculated between bounded inhibitor and redocked ligand which were 1.839 Å in this method. This value shows a high reliability of CDOCKER method to reproduce the known binding mode of these inhibitors. Docking results show a hydrogen bond between NMe2 substituent and LYS40, and other hydrogen bonds was found between the CO substituent of indolinone and ARG98, GLU95 (Fig. 5). The phenyl ring of the compound M32 has a p-cation interaction to NH3+of LYS40 in accordance with phenyl ring. Fig. 5 shows the key residues, pi interaction and hydrogen bond binding between the compound M32 and the binding site of receptor.

The best docked conformation of the most active compounds (compound 32), in the binding site of PLK4
Frontier molecular orbital
According to the frontier molecular orbitals theory, HOMO and LUMO energy are two significant indicators of chemical reactivity. The character electron donor and electron acceptor of compound were measured by the HOMO and LUMO energies respectively. The energy difference between the HOMO and LUMO (gap) is an important factor to represent a simple measure of molecule stability. A small gap value implies high reactivity of molecules in reactions while a large gap value implies high stability of molecules and low reactivity of molecules in reactions.
HOMO represents the ability to donate an electron while LUMO as an electron acceptor represents the ability to obtain an electron. In Fig. 6, HOMO and LUMO orbitals of the conformer obtained from docking with HOMO–LUMO gap are given. As seen in the figure, in the HOMO, the electrons are mainly delocalized on the nitrogen of the indolinone and indazole ring, and partially on the nitrogen of Ar substituted; in the LUMO the electrons are completely delocalized on the indolinone and indazole rings. These observations confirm the obtained results from molecular docking.

The HOMO and LUMO orbitals and the energy levels for the most active molecule (32)
Molecular electrostatic potential
The molecular electrostatic potential (MEP), V(r), at a given point r(x, y, z) located in the neighborhood of a molecule can be defined in terms of the interaction energy between the electrical charge generated from the molecule electrons and nuclei as well as a positive test charge (a proton) placed at r. The V(r) values for the system studied were calculated employing the equation (Politzer and Murray, 2002)
where Z A is the charge of nucleus A located at R A, ρ(r′) is the electronic density function of the molecule, and r′ is the dummy integration variable.
The molecular electrostatic potential is related to the electronic density and can also be used as a highly beneficial descriptor for the determination of sites for electrophilic attack and nucleophilic reactions as well as hydrogen-bonding interactions (Kaufman, 1979; Pomelli et al., 2001). The electrostatic potential V(r) is also well-suited for analyzing processes based on the recognition of one molecule from another one as in drug–receptor and enzyme substrate interactions, since it is through their potentials that the two species first ‘see’ each other (Politzer et al., 1985). Defined as a real physical property, V(r) can be determined experimentally by diffraction or by computational methods (Politzer et al., 1981).
MEP was calculated at the B3LYP/6-31G (d, p) optimized geometry so that it was possible to anticipate reactive sites for electrophilic and nucleophilic attack for the title molecule. As shown in Fig. 7, the two regions namely negative (red) and positive (blue) were related to electrophilic and nucleophilic reactivity respectively. As can be seen from the figure, the negative region for electrophilic attack is on the oxygen of indolinone with red color. This oxygen is H-bond acceptor from GLU95 and ARG98 in molecular docking.

Molecular electrostatic potential map (in a.u.) of the most active molecule (32)
ADMET analysis
Analyzing the predicted ADMET properties provided the chance for their more optimization. Therefore, we should modify the structure of indolinone to improve the solubility and permeability. Analyzing pharmacokinetic parameters necessary for ADMET (skin-permeability coefficient (log Kp), apparent Caco-2 and MDCK permeability (the higher the value of MDCK cell, the higher the cell permeability), log BB, aqueous solubility (log S), maximum of transdermal transport rate (Jm), human oral absorption in the gastrointestinal tract (GI), log K hsa for serum protein binding, log P for octanol/water) were computed using QikProp 3.2 shown in acceptable range (Table 5). Furthermore, they all show drug-like properties according to Lipinski’s rule of five (Lipinski et al., 2012). Log P for octanol/water shows the partition coefficient, which is important for the estimation of absorption and distribution of drugs within the body. Partition coefficient for the compounds design ranged from 3.761 to 6.225, which is in the acceptable range of −2.0 to 6.5. Apparent Caco-2 permeability as a main parameter governing drug metabolism and its access to biological membranes, ranged from 82 to 355. The percentage of human oral absorption calculated for the design molecules was average to high. Also, the parameters for the risk assessment of toxicity and pharmacological properties of compounds design are in the acceptable range and considering these compounds as drugs is valid. As it can be seen in Table 6, none of the designed compounds had the risk of mutagenicity, tumorigenesis and irritating effects.
Conclusion
In this study, molecular docking and 3D-QSAR methods were performed on a series of (E)-3-((1H-indazol-6-yl) methylene) indolin-2-ones derivatives as PLK4 inhibitors to explore the structure–activity relationship. The good predictive ability of CoMFA and CoMSIA observed for the test set of compounds indicates that these models could be successfully used for predicting the pIC50 values. In addition, the CoMFA and CoMSIA contour maps along with the docking results can provide us with more useful insight into understanding the interaction between ligand and target. They assist to design new potent candidates.
References
Barr FA, Silljé HH, Nigg EA (2004) Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 5(6):429–441
Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW (2008) Centrosome amplification can initiate tumorigenesis in flies. Cell 133(6):1032–1042
Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA (2011) Centrosomes and cilia in human disease. Trends Genet 27(8):307–315
Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt M, Glover D (2005) SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 15(24):2199–2207
Bornens M (2012) The centrosome in cells and organisms. Science 335(6067):422–426
Cramer RD, Patterson DE, Bunce JD (1988) Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. JAm Chem Soc 110(18):5959–5967
Davis EE, Katsanis N (2012) The ciliopathies: a transitional model into systems biology of human genetic disease. Curr Opin Genet Dev 22(3):290–303
Frisch M, Trucks G, Schlegel HB, Scuseria G, Robb M, Cheeseman J, Petersson G (2009) Gaussian 09, revision A. 02, vol 19. Gaussian, Inc., Wallingford, pp 227–238
Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460(7252):278–282
Ghasemi JB, Shiri F (2012) Molecular docking and 3D-QSAR studies of falcipain inhibitors using CoMFA, CoMSIA, and Open3DQSAR. Med Chem Res 21(10):2788–2806
Gönczy P (2012) Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol 13(7):425–435
Habedanck R, Stierhof Y-D, Wilkinson CJ, Nigg EA (2005) The polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 7(11):1140–1146
Hudson J, Kozarova A, Cheung P, Macmillan J, Swallow C, Cross J, Dennis J (2001) Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr Biol 11(6):441–446
Johnson EF, Stewart KD, Woods KW, Giranda VL, Luo Y (2007) Pharmacological and functional comparison of the polo-like kinase family: insight into inhibitor and substrate specificity. Biochemistry 46(33):9551–9563
Kaufman JJ (1979) Quantum chemical and physicochemical influences on structure–activity relations and drug design. Int J Quant Chem 16(2):221–241
Klebe G, Abraham U, Mietzner T (1994) Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem 37(24):4130–4146
Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof Y-D, Nigg EA (2007) Plk4-induced centriole biogenesis in human cells. Dev Cell 13(2):190–202
Kubinyi H (1993) 3D QSAR in drug design: theory methods and applications, vol 1. Springer Science & Business Media, New York
Laufer R, Forrest B, Li S-W, Liu Y, Sampson P, Edwards L, Plotnikova O (2013) The discovery of PLK4 inhibitors:(E)-3-((1H-Indazol-6-yl) methylene) indolin-2-ones as novel antiproliferative agents. J Med Chem 56(15):6069−6087
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2012) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 64:4–17
Lüders J, Stearns T (2007) Microtubule-organizing centres: a re-evaluation. Nat Rev Mol Cell Biol 8(2):161–167
Mak T (2012) Targeting the cell cycle in cancer: TTK (MPS1) and PLK4 as novel mitotic targets, book of abstracts. In: Paper presented at the AACR 103rd annual meeting, Chicago, April 1–5
Mao Y, Li Y, Hao M, Zhang S, Ai C (2012) Docking, molecular dynamics and quantitative structure-activity relationship studies for HEPTs and DABOs as HIV-1 reverse transcriptase inhibitors. J Mol Model 18(5):2185–2198
Mason J, Wei S, Luo S, Nadeem V, Kiarash R, Huang P, Feher M (2011) Inhibition of polo-like kinase 4 as an anti-cancer strategy, book of abstracts. In: Paper presented at the AACR 102nd annual meeting, Orlando, Florida, 2–6 April 2011
Megraw TL, Sharkey JT, Nowakowski RS (2011) Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol 21(8):470–480
Momany F, Rone R (1992) Validation of the general purpose QUANTA® 3.2/CHARMm® force field. J Comp Chem 13:888
Nigg EA (2002) Centrosome aberrations: cause or consequence of cancer progression?. Nat Rev Cancer 2(11):815–825
Nigg EA (2007) Centrosome duplication: of rules and licenses. Trends Cell Biol 17(5):215–221
Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139(4):663–678
Politzer P, Kammeyer CW, Bauer J, Hedges WL (1981) Polar properties of carbon monoxide. J Phys Chem 85(26):4057–4060
Politzer P, Laurence P, Jayasuriya K (1985) Structure−activity correlation in mechanism studies and predictive toxicology. Spec Issue Environ Health Perspect 61:191
Politzer P, Murray JS (2002) The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor Chem Acc 108(3):134–142
Pomelli CS, Tomasi J, Barone V (2001) An improved iterative solution to solve the electrostatic problem in the polarizable continuum model. Theor Chem Acc 105(6):446–451
Smith BM, Gemperline PJ (2002) Bootstrap methods for assessing the performance of near‐infrared pattern classification techniques. J Chemom 16(5):241–246
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci 22(1):69–77
Van de Waterbeemd H, Gifford E (2003) ADMET in silico modelling: towards prediction paradise?. Nat Rev Drug Discov 2(3):192–204
Varetto U (2009) Molekel 5.4. 0.8. Swiss National Supercomputing Centre, Manno
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Shiri, F., Rakhshani-morad, S., Samzadeh-kermani, A. et al. Computer-aided molecular design of some indolinone derivatives of PLK4 inhibitors as novel anti-proliferative agents. Med Chem Res 25, 2643–2665 (2016). https://doi.org/10.1007/s00044-016-1638-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1638-3