
Abstract
A library of ring-A-monosubstituted chalcone derivatives (4a–4i, 5a and 5b) was designed and synthesised. The structures as well as the identities of these compounds were established on the basis of spectral (1H NMR, 13C NMR, FT-IR and Mass) and elemental (C, H, N) analyses. All the derivatives were evaluated for their anti-inflammatory and antioxidant activities in vitro using the inhibition of protein denaturation and 2,2-diphenyl-1-picrylhydrazyl radical scavenging assays, respectively. The results indicated a promising anti-inflammatory activity for most of the synthesised compounds with many derivatives showing activities similar to or greater than that of the standard. The sulphonamide-substituted chalcones 4h, 4i, 5a and 5b were found to be the most active derivatives across the concentration range tested. However, all the derivatives exhibited rather mild antioxidant activity compared to the ascorbic acid standard. Interestingly, it was observed that the unsubstituted parent chalcone was one of the optimal compounds with only the trifluoromethyl analogue 4a showing better activity as an antioxidant. The two regioisomeric aminochalcones and 4′-cyanochalcone 4b also seemed to possess decent antioxidant potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
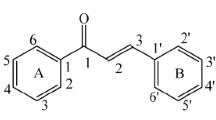
The chalcone moiety remains a popular scaffold amongst medicinal chemists owing to its structural simplicity. Not only are chalcones abundantly found in nature, but also they are key intermediates for many other medicinally important natural products like flavonoids, isoflavonoids and aurones (Detsi et al., 2009; Venkatachalam et al., 2012). The therapeutic activities of chalcones are attributed to the presence of the key pharmacophoric α,β-unsaturated keto group, more specifically a 2-propen-1-one chain (Orlikova et al., 2011). It is proved that the inherent electrophilicity of this α,β-unsaturated carbonyl functionality is involved in the antioxidant and anti-inflammatory properties of chalcones (Kumar et al., 2011; Maydt et al., 2013). Both the terminal carbons of the 2-propen-1-one fragment are attached to a phenyl ring, conventionally referred to as ring A and ring B on each of which a multitude of functional groups may be appended. Besides such substituted phenyl groups, the synthetically derived chalcones may also carry heterocyclic and condensed ring systems as their aryl substituents (Meng et al., 2007; Solomon and Lee, 2012; Tran et al., 2012). While the presence of a double bond allows these molecules to exist as cis or trans geometric isomers, the latter configuration has been proven to be thermodynamically as well as biologically favourable (Cheng et al., 2000). The structure of the simplest unsubstituted chalcone (1) is illustrated in Fig. 1.

Prototypical structure of chalcone
The diverse pharmacological applications of chalcones include their potential utility as antioxidant, antibacterial, antileishmanial, antiangiogenic, analgesic, antifungal, antiprotozoal, gastric protectant, antimutagenic, antitumorigenic and anti-inflammatory agents amongst others (Gacche et al., 2008; Wu et al., 2011; Yadav et al., 2011; Reddy et al., 2012). Some of the well-known natural anti-inflammatory chalcones also possessing good antioxidant potential are butein, xanthohumol, isoliquiritigenin, cardamonin, licochalcone A, flavokawain A and B, all of which represent predominantly hydroxylated, methoxylated and alkylated versions of the basic chalcone scaffold (Nowakowska, 2007; Srinivasan et al., 2009; Vogel et al., 2010). Even though many chalcones found in nature are known to be traditional folk remedies for inflammatory conditions, the effects of substituting various functional groups at different positions on the rings can be studied systematically only if the target molecules are synthesised chemically. The availability of a simple method to prepare chalcones via Claisen–Schmidt reaction, involving the condensation of substituted acetophenone precursors with substituted benzaldehydes in the presence of a base, has made it easier to assemble large numbers of derivatives within a short amount of time (Bandgar et al., 2010; Sing et al., 2012).
Many of the reports pertaining to synthetic chalcones are limited in their ability to establish a detailed structure activity relationship (SAR) footprint with respect to anti-inflammatory properties of these molecules. The crucial aspect in our ongoing search for a potent yet safe anti-inflammatory compound is to bridge such a wide information gap that currently exists between structure and anti-inflammatory potential of this scaffold. A closer look at the various mechanisms of inflammatory responses points out that the presence of antioxidants capable of protecting oxidative stress-related injury and inflammatory disease will improve the anti-inflammatory activity (Isa et al., 2012; Schinella et al., 2002). The notion of this correlation between antioxidant and anti-inflammatory activity is strengthened by the findings of research groups that attribute the importance of free radicals in the progression of oxidative as well as inflammatory damage (Roome et al., 2008; Saldanha et al., 1990). It is, therefore, felt that the estimation of antioxidant potential is a logical extension of studying a compound’s anti-inflammatory activity and accordingly, the study presented herein deals with the rational design of a focused library of ring-A-monosubstituted chalcones, their synthesis and subsequent in vitro evaluation for anti-inflammatory and antioxidant activities. Apart from helping elucidate SAR, estimation of the antioxidant activity of these chalcones also represents our attempt to probe structural attributes that would confer dual pharmacological activity to the hits identified.
Experimental
Materials and methods
All commercial reagents were used as provided unless otherwise indicated. All reactions were performed in oven-dried glassware. TLC was performed on silica gel G 40 µm particle size-coated glass plates, and spots were visualized by UV and/or I2 chamber. Melting points were determined using a Roy capillary melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 or Bruker AV III 500 MHz spectrometer. 19F NMR was recorded on Bruker AV III 500 MHz spectrometer. Proton, carbon and fluorine chemical shifts are reported in ppm. The internal standard for proton and carbon was residual CHCl3 (7.26 and 77.16 ppm, respectively). Proton chemical data are reported as follows: chemical shift, multiplicity (ovlp. = overlapping, s = single, d = doublet, t = triplet, dt = doublet of triplet, tt = triplet of triplet and m = multiplet), coupling constant and integration. IR spectra were recorded on a Shimadzu FT-IR spectrophotometer or Perkin Elmer spectrum RXI FT-IR spectrophotometer. LC–MS chromatograms and mass spectra were obtained on a Waters e 2695-Waters 3100 instrument with ESI-PMT arrangement as the mode of ionisation and type of detector, respectively. Elemental analyses were determined on an Elementar Vario EL III elemental analyser.
For studying the anti-inflammatory activity, ibuprofen standard was obtained as a gift from Porus Labs Pvt. Ltd., Hyderabad. Protein used in this assay was bovine serum albumin fraction V (98 %, Nice Chemicals). Analytical grade DMF (99.5 %, Otto Chemei, India) was used to solubilise the test compounds. Absorbance readings were taken from Schimadzu UV–Vis spectrophotometer. With regard to the antioxidant assay using DPPH, all chemicals were purchased from SDFCL, Mumbai unless specified otherwise. DPPH (90 %) was procured from Sigma Aldrich (Germany). A Shimadzu UV-1800 Spectrophotometer was used for measuring the optical density of the samples.
Design and in silico analysis of target chalcones
The design of ring-A-monosubstituted chalcones was primarily based on the electronic properties of the substituents. The selected functional groups are placed at 2′-(ortho), 3′-(meta) or 4′-(para) position of ring A of the scaffold while ring B is left intact. Significant groups like amino and cyano groups were selected for their mesomeric (+M/−M) effect; similarly groups like trifluoromethyl and azido were selected for their inductive (+I/−I) effect. To test the effect of lipophilicity on the pharmacological activities of chalcones, sulphonamide functional groups including methanesulphonamide and benzenesulphonamide at ortho and para (5a, 4h, 5b and 4i; Scheme 1) positions were employed (Balasubramanian and Vijayagopal, 2012). The proposed molecules were then subjected to in silico Lipinski Rule of Five (Ro5) analysis for assessing drug-likeness using Molsoft (molsoft.com/mprop/), an online academic software wherein properties like molecular weight, hydrogen bond donors as well as acceptors and calculated partition coefficient of the molecules were determined.

Synthesis of relevant acetophenone precursors and target chalcones
Synthesis
A total of eleven monosubstituted chalcones were sought out in this study of which ten were successfully prepared. While the synthetic route(s) used to access them are presented in Scheme 1, experimental procedures and characterisation data of the respective compounds are detailed herein. Typically, the classic base-catalysed aldol-type condensation was employed to assemble the target compounds 4a, 4b, 4d–4i as well as the congener 4c that acted as a precursor to the two other target sulphonamides 5a and 5b. Spectral characterisation following the synthesis unambiguously confirmed the structure of the compounds.
General procedure for synthesis of 4a–4e
To a solution of substituted acetophenone (10 mmol) in rectified spirit (30 mL), benzaldehyde (1.01 mL, 10 mmol) was added followed by an aqueous solution of 10 % KOH (10 mL). The mixture was stirred and kept overnight at room temperature. The contents of the reaction mixture were poured into crushed ice and acidified with dil. HCl (0.1–0.2 N). The precipitated chalcone derivative was filtered off and recrystallised from rectified spirit. Column chromatography (10–20 % EtOAc/cyclohexane) was performed whenever recrystallisation failed to sufficiently purify the target compound.
4′-Trifluoromethylchalcone 4a (Wilhelm et al., 2012)
Off-white crystals; yield: 77 %; R f = 0.50 (10 % EtOAc/cyclohexane); mp 94–98 °C; 1H NMR (CDCl3, 500 MHz) δ 8.10 (d, J = 8 Hz, 2H), 7.83 (d, J = 15.5 Hz, 1H), 7.77 (d, J = 8 Hz, 2H), 7.64–7.66 (m, 2H), 7.49 (d, J = 15.5 Hz, 1H), 7.42–7.45 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 189.7, 146.2, 141.1, 134.5, 134.2, 133.9, 131.0, 129.1, 128.9, 128.7, 125.8, 125.7, 125.6, 124.8, 122.6, 121.6; 19F NMR (CDCl3, 470 MHz) δ −63.00 (s, 3F); IR (KBr) 3060 (arom. –CH str.), 1943 (C–F3 str.), 1666 (α,β-unsaturated keto group, –C=O str.), 1608, 1574 (C=C arom. str., C=C olefinic str.), 1321 (ip arom. C–H bend.), 985 (oop –CH bend. vibration of alkene), 840 (1,4 disubstitution), 772 (arom. bend.) cm−1; Anal. calcd. for C16H11F3O: C 69.56, H 4.01. Found: C 69.42, H 3.99; ESI MS (m/z, relative abundance) 277 [(M+H)+, 100], 298 [(M+Na)+, 44], 320 [(M+2Na)+, 25], [C32H20O2 2+, 7].
4′-Cyanochalcone 4b (Kumar et al., 1985)
White crystals; yield: 23 %; Rf = 0.57 (20 % EtOAc/cyclohexane); mp 92–96 °C; 1H NMR (CDCl3, 500 MHz) δ 8.08 (d, J = 8.5 Hz, 2H), 7.83 (d, J = 15.5 Hz, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.64–7.66 (m, 2H), 7.47 (d, J = 15.5 Hz, 1H), 7.43–7.46 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 189.2, 146.6, 141.5, 134.4, 132.5, 131.2, 129.1, 128.9, 128.7, 121.2, 118.0, 116.0; IR (KBr) 3100 (arom. –CH str.), 2230 (arom. C≡N), 1662 (α,β-unsaturated keto group, –C=O str.), 1600, 1574 (C=C arom. str., C = C olefinic str.), 984 (oop –CH bend. vibration of alkene), 833 (1,4 disubstitution), 766 (arom. bend.) cm−1; Anal. calcd. for C16H11NO: C 82.38, H 4.75, N 6.00. Found: C 82.17, H 4.88, N 6.02; ESI MS (m/z, relative abundance) 234 [(M+H)+, 100].
2′-Aminochalcone 4c (Mannich and Dannehl, 1938)
Yellow flakes; yield: 52 %; R f = 0.68 (20 % EtOAc/cyclohexane); mp 50–54 °C; 1H NMR (CDCl3, 500 MHz) δ 7.85 (dd, J = 1.5, 8.5 Hz, 1H), 7.73 (d, J = 15.5 Hz, 1H), 7.62 (dd, J = 1.5, 8.5 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 15.5 Hz, 1H), 7.41 (ovlp. d, J = 8.5 Hz, 1H), 7.36–7.42 (m, 2H), 7.26–7.30 (m, 1H), 6.66–6.71 (m, 2H), 6.30 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 191.7, 151.0, 142.9, 135.3, 134.3, 131.0, 130.1, 128.9, 128.2, 123.2, 119.1, 117.3, 115.9; IR (KBr) 3444, 3325 (–NH str., primary amine), 1641 (α,β-unsaturated keto group, –C=O str.), 1605, 1573 (C=C arom. str., C=C olefinic str.), 1336 (arom. amino group, C–N str.), 738 (1,2 disubstitution) cm−1; ESI MS (m/z, relative abundance) 224 [(M+H)+, 100].
3′-Aminochalcone 4d (Karaman et al., 2010)
Yellow crystals obtained by column chromatography; yield: 33 %; R f = 0.35 (20 % EtOAc/cyclohexane); mp 90–94 °C; 1H NMR (CDCl3, 500 MHz) δ 7.79 (d, J = 16 Hz, 1H), 7.63 (d, J = 5.5 Hz, 1H), 7.62 (d, J = 5.5 Hz, 1H), 7.48 (d, J = 16 Hz, 1H), 7.41 (ovlp. d, J = 5.5 Hz, 1H), 7.40 (ovlp. d, J = 5.5 Hz, 1H), 7.38–7.42 (m, 2H), 7.26–7.32 (m, 2H), 6.88–6.90 (m, 1H), 3.85 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 190.7, 146.9, 144.5, 139.4, 135.0, 130.5, 129.5, 128.9, 128.4, 122.4, 119.4, 118.9, 114.4; IR (KBr) 3367, 3203 (–NH str., primary amine), 1660 (α,β-unsaturated keto group, –C=O str.), 1650, 1587 (C=C arom. str., C=C olefinic str.), 1336 (arom. amino group, C–N str.), 759 (1,3 disubstitution) cm−1; Anal. calcd. for C15H13NO: C 80.69, H 5.87, N 6.27. Found: C 80.90, H 5.79, N 6.41; ESI MS (m/z, relative abundance) 224 [(M+H)+,100].
4′-Aminochalcone 4e (Applequist and Gdanski, 1981)
Yellow crystalline needles; yield: 30 %; R f = 0.42 (20 % EtOAc/cyclohexane); mp 130–136 °C; 1H NMR (CDCl3, 500 MHz) δ 7.93 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 15.5 Hz, 1H), 7.62 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 15.5 Hz, 1H), 7.36–7.41 (m, 3H), 6.69 (d, J = 9 Hz, 2H), 4.22 (s, 2H); 13C NMR (CDCl3, 125 MHz) δ 188.1, 151.2, 143.1, 135.3, 131.0, 130.0, 128.8, 128.4, 128.2, 122.0, 113.9; IR (KBr) 3338, 3200 (–NH str., primary amine), 1628 (α,β-unsaturated keto group, –C=O str.), 1603, 1577 (C=C arom. str., C=C olefinic str.), 1341 (arom. amino group, C–N str.), 830 (1,4 disubstitution), 766 (arom. bend.) cm−1; Anal. calcd. for C15H13NO: C 80.69, H 5.87, N 6.27. Found: C 80.81, H 5.95, N 6.22; ESI MS (m/z, relative abundance) 224 [(M+H)+, 100], 249 [(M+Na)+, 10].
Synthesis of 3′-azidochalcone 4f
To a solution of 3-amino acetophenone (1.0 g, 7 mmol) in THF (10 mL), 20 mL of 10 % aqueous HCl was added. Sodium nitrite (1.0 g, 14.7 mmol) was added as a solid to this solution at 0 °C. After 45 min of stirring at 0–5 °C, sodium azide (4.81 g, 74 mmol) was added in small portions over 10 min, so that the temperature would not exceed 5 °C. The reaction was allowed to stir for a further 1–2 h at room temperature. The reaction mixture was extracted with Et2O (2 × 15 mL); the organic layers were washed with distilled water (2 × 20 mL) and saturated sodium chloride solution (20 mL). The washings were re-extracted with Et2O (15 mL), the combined Et2O layers were dried over Na2SO4 and evaporated to get the crude product 3f. To the solution of this crude 3-azido acetophenone in rectified spirit (30 mL), benzaldehyde (1.01 mL, 10 mmol) was added followed by an aqueous solution of 10 % KOH (10 mL). The mixture was stirred and kept overnight at room temperature. The contents of the reaction mixture were poured into crushed ice and acidified with dil. HCl (0.1–0.2 N). The precipitated 3′-azidochalcone was filtered off and recrystallised from rectified spirit. Further purification by column chromatography (20 % EtOAc/cyclohexane) afforded cream-coloured crystals. Yield: 14 % over two steps; R f = 0.68 (20 % EtOAc/cyclohexane); mp 46–50 °C; 1H NMR (CDCl3, 400 MHz) δ 7.75 (d, J = 15.6 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.56–7.59 (m, 3H), 7.41 (d, J = 7.6 Hz, 1H), 7.39 (d, J = 15.6 Hz, 1H), 7.34–7.43 (m, 3H), 7.14–7.17 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 188.5, 144.6, 139.9, 138.9, 133.7, 129.8, 129.0, 128.0, 127.5, 123.9, 122.1, 120.7, 117.8; IR (thin film) 3062 (arom. –CH str.), 2113 (asym. NNN str.), 1583 (α,β-unsaturated keto group, –C=O str.), 985 (oop –CH bend. vibration of alkene), 759 (1,3 disubstitution) cm−1; Anal. calcd. for C15H11N3O: C 72.28, H 4.45, N 16.86. Found: C 72.08, H 4.49, N 16.77; ESI MS (m/z, relative abundance) 222 [(M−N2)+, 100], 250 [(M+H)+, 30].
Synthesis of 4′-azidochalcone 4g (Zarghi et al., 2006)
Sodium nitrite (1.0 g, 14.7 mmol) was added as a solid to a 30 mL solution of 4-amino acetophenone (1.0 g, 7.4 mmol) in trifluoroacetic acid at 0 °C. After 45 min of stirring at 0–5 °C, sodium azide (4.81 g, 74 mmol) was added in small portions over 10 min, so that the temperature would not exceed 5 °C. An aliquot of Et2O (20 mL) was added, and the reaction was allowed to stir in the dark for 2 h. The reaction mixture was washed with distilled H2O (2 × 15 mL) and saturated sodium chloride solution (20 mL). The washings were re-extracted with Et2O (15 mL); the Et2O layers combined, dried over Na2SO4 and evaporated to get a dark brown residue from which the desired azido acetophenone 3g was extracted by boiling with hexane (3 × 20 mL). Pure 4-azido acetophenone (Kym et al., 1993) eventually precipitated as light yellow crystals after keeping the hexane extract in refrigerator overnight. Yield: 79 %; R f = 0.60 (20 % EtOAc/cyclohexane); mp 40–44 °C; IR (KBr) 3000 (arom. –CH str.), 2128 (asym. NNN str.), 1685 (C=O str.), 1654, 1596 (C=C arom. str.), 831 (1,4 disubstitution), 721 (arom. bend.) cm−1.
To the solution of 4-azido acetophenone (0.15 g, 0.9 mmol) in rectified spirit (30 mL), benzaldehyde (0.15 mL, 0.9 mmol) was added followed by an aqueous solution of 10 % KOH (5 mL). The mixture was stirred and kept overnight at room temperature. The contents of the reaction mixture were poured into crushed ice and acidified with dil. HCl (0.1–0.2 N). The precipitated 4′-azidochalcone was filtered off and recrystallised from rectified spirit to afford a light brown solid. Yield 79 %; R f = 0.72 (20 % EtOAc/cyclohexane); mp 80–84 °C; 1H NMR (CDCl3, 500 MHz) δ 8.05 (d, J = 9 Hz, 2H), 7.82 (d, J = 15.5 Hz, 1H), 7.64 (d, J = 9 Hz, 2H), 7.51 (d, J = 15.5 Hz, 1H), 7.42 (ovlp. d, J = 8.5 Hz, 1H),7.41–7.43 (m, 2H), 7.13 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3, 125 MHz) δ 188.6, 144.9, 144.7, 134.8, 134.7, 130.6, 130.4, 129.0, 128.4, 121.5, 119.0; IR (KBr) 3050 (arom. –CH str.), 2150 (asym. NNN str.), 1645 (α,β-unsaturated keto group, –C=O str.), 1600, 1570 (C=C arom. str., C=C olefinic str.), 985 (oop –CH bend. vibration of alkene), 830 (1,4 disubstitution), 775 (arom. bend.) cm−1; Anal. calcd. for C15H11N3O: C 72.28, H 4.45, N 16.86. Found: C 72.15, H 4.32, N 16.91; ESI MS (m/z, relative abundance) 222 [(M−N2)+, 100], 250 [(M+H)+, 40].
Synthesis of 4′-Methanesulphonamide chalcone 4h (Zarghi et al., 2006)
To a solution of 4-aminoacetophenone (0.5 g, 3.7 mmol) in dry CH2Cl2 (10 mL), Et3N (1.18 mL, 8 mmol) was added followed by methanesulphonyl chloride (0.32 mL, 4 mmol) at 0 °C. After maintaining the temperature between 0 and 5 °C for 3–4 h, the reaction mixture was gradually warmed to room temperature and the contents were stirred overnight. Once TLC indicated the completion of the reaction, the mixture was diluted with CH2Cl2 (20 mL) and washed with water (2 × 15 mL) followed by saturated sodium chloride solution (15 mL). Drying (Na2SO4) and then distillation of the organic extracts afforded the crude intermediate 3h as a light yellow solid. To the crude 4-methanesulphonamide acetophenone dissolved in rectified spirit (30 mL), benzaldehyde (1.01 mL, 10 mmol) was added followed by an aqueous solution of 10 % KOH (10 mL). The mixture was stirred and kept overnight at room temperature. The contents of the reaction mixture were poured into crushed ice and acidified with dilute HCl (0.1–0.2 N). The precipitated chalcone derivative was filtered off, recrystallised from rectified spirit and further purified by column chromatography (20 % EtOAc/cyclohexane) to get yellow crystals. Yield: 10 % over two steps; R f = 0.24 (20 % EtOAc/cyclohexane); mp 112–116 °C; 1H NMR (CDCl3, 500 MHz) δ 8.06 (d, J = 7.25 Hz, 1H), 8.05 (d, J = 7.25 Hz, 1H), 7.83 (d, J = 16 Hz, 1H), 7.65 (d, J = 7.25 Hz, 1H), 7.64 (d, J = 7.25 Hz, 1H), 7.51 (d, J = 16 Hz, 1H), 7.44 (ovlp. d, J = 6.25 Hz, 1H), 7.43 (ovlp. d, J = 6.25 Hz, 1H), 7.42–7.44 (m, 1H), 7.33 (d, J = 6.25 Hz, 1H), 7.32 (d, J = 6.25 Hz, 1H), 7.15 (s, 1H), 3.12 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 188.9, 145.1, 141.1, 134.8, 134.4, 131.1, 130.7, 130.6, 129.4, 129.0, 128.5, 128.3, 121.5, 118.4, 118.2, 40.1; IR (thin film) 3248 (–NH str.), 3035 (arom. –CH str.), 1653 (α,β-unsaturated keto group, –C=O str.), 1601, 1500 (C=C arom. str., C=C olefinic str.), 1338 (asym. SO2 str.), 1178 (sym. SO2 str.), 934 (SN str.), 837 (1,4 disubstitution) cm−1; Anal. calcd. for C16H15NO3S: C 63.77, H 5.02, N 4.65, S 10.64. Found: C 63.79, H 4.98, N 4.49, S 10.78; ESI MS (m/z, relative abundance) 300 [(M−H)+, 100].
Synthesis of 4′-benzenesulphonamide chalcone 4i (Moustafa and Ahmad, 2003)
To a solution of 4-aminoacetophenone (0.5 g, 3.7 mmol) in dry CH2Cl2 (10 mL), Et3N (1.18 mL, 8 mmol) was added followed by benzenesulphonyl chloride (0.51 mL, 4 mmol) at 0 °C. After maintaining the reaction mixture between 0 and 5 °C for 3–4 h, the contents were gradually brought up to room temperature and stirred overnight. After TLC indicated the completion of the reaction, the mixture was diluted with CH2Cl2 (20 mL) and washed with water (2 × 15 mL) followed by saturated sodium chloride solution (15 mL). Drying (Na2SO4) and then distillation of the organic layer afforded the crude acetophenone intermediate 3i as a yellow solid. This crude 4-benzenesulphonamide acetophenone was dissolved in rectified spirit (30 mL). Benzaldehyde (1.01 mL, 10 mmol) was added to it followed by an aqueous solution of 10 % KOH (10 mL). The mixture was stirred and kept overnight at room temperature. The contents of the reaction mixture were poured into crushed ice and acidified with dilute HCl (0.1–0.2 N). The precipitated product was filtered off and recrystallised from rectified spirit affording the title compound as light yellow crystals. Yield: 16 % over two steps; R f = 0.44 (20 % EtOAc/cyclohexane); mp 126–130 °C; 1H NMR (CDCl3, 500 MHz) δ 7.99 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.84 (d, J = 16 Hz, 1H) 7.70 (tt, J = 2, 8 Hz, 2H), 7.64–7.66 (m, 2H), 7.57 (s, 1H), 7.56–7.59 (ovlp. m, 2H), 7.49 (d, J = 16 Hz, 1H), 7.42–7.45 (m, 2H), 7.17 (dt, J = 2, 8 Hz, 2H); 13C NMR (CDCl3, 125 MHz) δ 189.4, 145.8, 139.5, 139.3, 137.9, 134.6, 134.2, 131.9, 130.9, 129.3, 129.2, 129.1, 128.6, 121.5; IR (thin film) 3344 (–NH str.), 3066 (arom. –CH str.), 1662 (α,β-unsaturated keto group, –C=O str.), 1595, 1489 (C=C arom. str., C=C olefinic str.), 1340 (asym. SO2 str.), 1166 (sym. SO2 str.), 920 (SN str.), 867 (1,4 disubstitution) cm−1; Anal. calcd. for C21H17NO3S: C 69.40, H 4.71, N 3.85, S 8.82. Found: C 69.29, H 4.64, N 3.78, S 8.99; ESI MS (m/z, relative abundance) 362 [(M−H)+, 100].
Synthesis of 2′-methanesulphonamide chalcone 5a (Batt et al., 1993)
To a solution of 2′-aminochalcone (0.18 g, 0.81 mmol) in dry CH2Cl2 (5 mL), pyridine (0.66 mL, 0.83 mmol) was added followed by methanesulphonyl chloride (0.07 mL, 0.9 mmol) at 0 °C under N2 atmosphere. The contents were stirred overnight at room temperature by gradually warming the reaction mixture from 0 °C. After completion of the reaction as indicated by TLC, the reaction mixture was diluted with CH2Cl2 (20 mL), washed with water (2 × 15 mL) and saturated sodium chloride solution (15 mL), dried over Na2SO4 and distilled off. The crude compound thus obtained was purified using column chromatography (10 % EtOAc/cyclohexane). Bright yellow crystals; yield: 19 %; R f = 0.35 (20 % EtOAc/cyclohexane); mp 74–76 °C; 1H NMR (CDCl3, 500 MHz) δ 11.23 (s, 1H), 8.04 (dd, J = 1.5, 8.5 Hz, 1H), 7.85 (d, J = 15.5 Hz, 1H), 7.80 (dd, J = 1.5, 8.5 Hz, 1H), 7.66 (d, J = 6.75 Hz, 1H), 7.65 (d, J = 6.75 Hz, 1H), 7.59 (d, J = 15.5 Hz, 1H), 7.58 (t, J = 1 Hz, 1H), 7.43–7.46 (m, 3H), 7.20–7.23 (m, 1H), 3.08 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 192.7, 146.3, 140.7, 134.9, 134.5, 131.1, 131.0, 129.1, 128.7, 123.4, 122.8, 121.8, 118.9, 40.2; IR (thin film) 3200 (–NH str.), 3053 (arom. –CH str.), 1643 (α,β-unsaturated keto group, –C=O str.), 1593, 1500 (C=C arom. str., C=C olefinic str.), 1342 (asym. SO2 str.), 1170 (sym. SO2 str.), 981 (SN str.), 767 (1,2 disubstitution) cm−1; Anal. calcd. for C16H15NO3S: C 63.77, H 5.02, N 4.65, S 10.64. Found: C 63.66, H 5.10, N 4.76, S 10.63; ESI MS (m/z, relative abundance) 300 [(M−H)+, 100].
Synthesis of 2′-benzenesulphonamide chalcone 5b (Donnelly and Farrell, 1989)
To a solution of 2′-aminochalcone (0.4 g, 3 mmol) in dry CH2Cl2 (10 mL), Et3N (0.7 mL, 5 mmol) was added followed by benzenesulphonyl chloride (0.51 mL, 4 mmol) at 0 °C. The contents were stirred overnight at room temperature by gradually warming the reaction mixture from 0 °C. After the completion of the reaction as indicated by TLC, the reaction mixture was diluted with CH2Cl2 (20 mL) and washed with water (2 × 15 mL) and saturated sodium chloride solution (15 mL). The organic extracts were then dried over Na2SO4 and evaporated under reduced pressure to give the crude compound which was subsequently purified by column chromatography (10 % EtOAc/cyclohexane) to afford yellow crystals of the title compound. Yield: 25 %; R f = 0.47 (20 % EtOAc/cyclohexane); mp 76–78 °C; 1H NMR (CDCl3, 500 MHz) δ 11.19 (s, 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.79–7.83 (m, 2H), 7.75 (d, J = 8.5 Hz, 1H), 7.66 (d, J = 15.5 Hz, 1H), 7.58–7.60 (m, 2H), 7.49 (t, J = 7.5 Hz, 1H), 7.38–7.44 (m, 6H), 7.34 (d, J = 15.5 Hz, 1H), 7.14 (t, J = 7.5 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 192.8, 146.1, 139.9, 139.4, 134.4, 134.3, 132.9, 131.0, 130.6, 129.1, 129.0, 128.6, 127.3, 124.9, 123.2, 122.1, 120.8; IR (thin film) 3144 (–NH str.), 3070 (arom. –CH str.), 1641 (α,β-unsaturated keto group, –C=O str.), 1593, 1494 (C=C arom. str., C=C olefinic str.), 1336 (asym. SO2 str.), 1163 (sym. SO2 str.), 931 (SN str.), 746 (1,2 disubstitution) cm−1; Anal. calcd. for C21H17NO3S: C 69.40, H 4.71, N 3.85, S 8.82. Found: C 69.43, H 4.88, N 3.97, S 8.81; ESI MS (m/z, relative abundance) 362 [(M−H)+, 100].
Determination of biological activity by in vitro methods
Anti-inflammatory activity
A well-established literature protocol that involved inhibition of protein denaturation was followed with minor modifications for the estimation of in vitro anti-inflammatory activity (Mizushima and Kobayashi, 1968). The standard drug ibuprofen and test compounds were dissolved in minimum amount of DMF and diluted with phosphate buffer saline (0.2 M, pH 7.4) in such a way that concentration of DMF in all solutions was less than 2.5 %. Each of the test solutions was mixed with bovine serum albumin (BSA) solution (2 mL, 2 mmol) in phosphate buffer saline and incubated at 27 ± 1 °C in an incubator for 15 min. Denaturation was induced by keeping the reaction mixture at 60 ± 1 °C in a water bath for 10 min. After cooling, the turbidity was measured at 660 nm with a UV visible spectrophotometer and the percentage of inhibition of denaturation calculated from control where no drug is added. The percentage of inhibition was calculated using the formula: percentage inhibition = A C − A S/A C × 100, where A C = absorbance of control and A S = absorbance of test sample.
Antioxidant activity
The method of Doble and co-workers was adopted with minor modifications for carrying out the DPPH assay (Sivakumar et al., 2011). Stock solutions of the compounds were prepared by solubilising 2 mg of the compound in 200 µL of DMF and then making up the volume to 10 ml with methanol. Corresponding dilutions of 10, 20, 30, 40 and 50 µg mL−1 were prepared with methanol. 0.1 mM DPPH solution and test samples were taken in a ratio of 1:2, and the mixture was vortexed and kept for equilibration by incubating in dark at 25 °C for 20 min. The optical density was measured at 517 nm in a UV spectrophotometer with ascorbic acid being used as the positive control. Pure methanol was used as the blank, and DPPH solution and methanol in a ratio of 1:2 were used as the control. The actual decrease in the absorption induced by the test compound was estimated by subtracting the absorbance of the test from that of the control. The percentage of scavenging of DPPH was calculated using the formula:
where A C = absorbance of control and AS = absorbance of test sample
Statistical analysis
All assays were carried out in triplicate, and results were expressed as mean ± SD. All statistical analysis was performed using Graph Pad (Version-6) Prism software. ANOVA was used to test the differences between the percentage inhibition values of the different derivatives in both the anti-inflammatory and antioxidant assays followed by Tukey’s multiple comparison tests, and p < 0.05 was considered statistically significant.
Results and discussion
Drug-likeness assessment
The drug-likeness of compounds was evaluated by calculating the Lipinski parameters using the high-speed molecular properties calculator, a free module in the Molsoft software package. All chalcone derivatives have less than or equal to two hydrogen bond donors and possess no more than three hydrogen bond acceptors, the total maximum permissible count being 10. Besides, none of their molecular weights exceeded 500 daltons. The octanol–water partition coefficient (logP) is not more than 5 for any of the ligands except compound 5b. These results that are shown in Table 1 strongly suggest drug-likeness, and the target compounds can be said to have properties that would make them amenable to oral administration in humans. Topological polar surface area (TPSA) of the target compounds and reference standard was below 70 Å2 implying easy permeability through cell membrane and ready penetrability across the blood brain barrier (BBB).
Chemistry
Out of the ten target compounds obtained, eight were synthesised by direct Claisen-Schmidt condensation of substituted acetophenones and benzaldehyde while sulphonamides 5a and 5b were prepared from 2′-aminochalcone (4c) as clearly illustrated in Scheme 1. The structures of all the synthesised derivatives were assigned on the basis of IR, 1H & 13C NMR, mass spectral data as well as elemental analysis and comparison with published data. It was observed that the results are in agreement with the proposed structures.
The IR spectra of 4a and 4b showed characteristic peaks at 1,943 cm−1 corresponding to C–F stretch of the –CF3 group and 2,230 cm−1 that is attributable to C≡N stretching of the –CN group, respectively. The 19F NMR of 3′-trifluoromethylchalcone gives a singlet at −63.00 ppm corresponding to the three equivalent fluorine atoms of the aromatic –CF3 group. With regard to the amino-substituted chalcones, it was difficult to purify the meta and para analogues due to the interference of side products. While 3′-aminochalcone (4d) was purified by repeated column chromatography of the crude product, precipitation of pure 4e occurred upon dissolving it in boiling ethanol and keeping overnight at room temperature. A broad singlet around 4 ppm in the 1H NMR represents the two protons of the –NH2 group. Deuterated water (D2O) exchange spectrum of 4d clearly demonstrates the exchange of –NH2 protons with deuterium atom(s). As seen in Fig. 2, the –NH2 peak disappears and a singlet corresponding to HOD appears around 4.7 ppm post equilibration of the sample.

D2O exchange observed in 3′-aminochalcone
The meta and para azidochalcones (4f and 4g, respectively) were synthesised in two steps involving the preparation of intermediate azido acetophenones and their subsequent condensation with benzaldehyde to afford the target compounds. The intermediate 4-azido acetophenone (3g) was isolated and characterised, whereas 3f could not be purified and was consequently coupled with benzaldehyde as such. The crude 4f was then purified by column chromatography. It is pertinent to note that 4f gradually darkens from off-white to brownish colour when kept at room temperature. The characteristic NNN stretching of azides was displayed by both the azidochalcones in their IR spectra at around 2,200–2,100 cm−1. Although the diagnostic molecular ion peaks were seen in the mass spectra of azidochalcones, the characteristic and most intense peak was at m/z 222 representing the fragment corresponding to the loss of nitrogen. The chalcone bearing the azide group at the 2′-(ortho) position was also targeted, but the efforts for its isolation failed. The formation of this product was confirmed by the mass spectrum showing an intense peak at 222 as well as deduced from IR spectrum which displayed the characteristic peak of azide group between 2,200 and 2,100 cm−1. The presence of a stubborn impurity was, however, seen in the compound’s 1H and 13C NMR spectra which led to its eventual disqualification from further in vitro study.
Two benzenesulphonamides and two methanesulphonamides, each appended to the 2′- or 4′- position of the unsubstituted chalcone, comprise the sulphonamides designed for the study. The 2′-sulphonamide chalcones 5a and 5b were synthesised by direct sulphonylation of 4c, whereas the 4′-sulphonamides 4h and 4i were accessed from 4-amino acetophenone (3e) via the intermediary 4-sulphonamide acetophenones. Pyridine or Et3N was employed as a base in the sulphonamide synthesis. The reaction, though slower when carried out using Et3N compared to pyridine, was, however, driven to completion under the former conditions. Three out of the four sulphonamides were purified by column chromatography which was difficult to be performed due to the similarity in the R f values of the impurities to that of the desired products. The remaining analogue, 4′-benzenesulphonamide chalcone (4i), was purified by stirring in boiling ethanol for 5–10 min and filtering the solution. The pure product being insoluble in hot ethanol is obtained as the filter cake and the impurities go along with the hot ethanol. Analogous to the aminochalcones, the presence of an exchangeable amide proton was tested by recording the D2O exchange spectra of all the sulphonamides (data not shown). As expected, the exchange phenomenon resulted in the disappearance of amide proton singlet in the range of 11–11.5 ppm and the concomitant appearance of a singlet corresponding to HOD in the range of 4.5–5 ppm.
Pharmacological evaluation
In our study, the in vitro anti-inflammatory potential of chalcone derivatives was evaluated against denaturation of BSA. The results indicated a concentration-dependent inhibition of protein denaturation by the entire chalcone library throughout the concentration range of 10–30 µg mL−1. Ibuprofen that was used as reference drug also exhibited a similar concentration-dependent profile of inhibition. It is clear from the data (Table 2) that the substitution pattern on the phenyl ring of the acetophenic group of chalcone moiety plays an important role in modulating inflammation. Inspection of the tabulated values easily sets apart the four sulphonamides as the better anti-inflammatory analogues with the 2′-benzenesulphonamide chalcone 5b emerging as the best compound within this subset. The para azido- and aminochalcones also fared better than ibuprofen, while the rest of the target compounds exhibited activity similar to that of the reference. Significantly, none of the library members were inactive in this assay.
From a SAR perspective, electron-withdrawing substituents in the form of a trifluoromethyl group (via inductive effect) in 4a and a cyano group (via mesomeric effect) in 4b had no effect on the anti-inflammatory activity. With respect to the azide group, the para regioisomer was preferred over the meta congener. This particular finding is in conformation with reports published by Knaus and co-workers revealing the structural as well as functional basis for the incorporation of an azide as a key bioisosteric pharmacophore in containing inflammation (Habeeb et al., 2001; Rao et al., 2004). A similar and subtler positional effect was mirrored in the case of the amino substituent as well. Previous studies in our group have led to the finding that electron-donating substituents, especially of the mesomeric type like hydroxy- and amino-, placed at the 2′-position potentiate the anti-inflammatory activity (Balasubramanian et al., 2013). Since free radicals have been definitively implicated in the progression of inflammatory damage, a regiochemical preference for the ortho position can be unambiguously explained by the superior ability of such a substituent to form an intramolecular hydrogen bond (iHB) and consequently, the relative ease of free radical formation. More specifically, the unique hydrogen bonding ability in 4c allows for the formation of an oxygen-centred radical rather than a nitrogen-centred radical, the former being a more favourable process owing to the lower bond dissociation energy (BDE) of O–H versus N–H bond (Bendary et al., 2013). Taken together with the results of this work, a useful activity rank order presents itself in the context of regioisomeric aminochalcones: ortho > para > meta.
A marked increase in the percent inhibition of the sulphonamides over the parent chalcone underscores the importance of this functional group as a ring A substituent. Sulphur-based functional groups have been successfully employed in blockbuster anti-inflammatory drugs. While sulphonamides are present in coxibs (celecoxib, valdecoxib and parecoxib) and oxicams (meloxicam, piroxicam and tenoxicam), reverse sulphonamides i.e. sulphonanilides are seen in drugs such as nimesulide and flosulide. The role of methylsulphonyl group in potentiating anti-inflammatory activity has been highlighted in depth (Gans et al., 1990; Talley, 1999). A detailed investigation has provided further valuable insight into the binding mode and consequently, the selectivity imparted by a p-NHSO2CH3 substituent in cyclooxygenase-2 (COX-2) inhibition (Garavito and DeWitt, 1999; Zarghi et al., 2006). Therefore, it is quite natural that the sulphonamides have emerged superior in this library. Even though target-specific in silico analysis was not a part of our study, it is not difficult to envision the benzenesulphonamide 5b as the most optimal compound because the phenyl group therein would likely participate in hydrophobic interactions with the relevant residues in the COX-2-binding pocket similar to those established in the case of a p-NHSO2CH3 group.
Recently, a report published by Menichini and co-workers (Conforti et al., 2009) as well as findings in our laboratory (manuscript accepted for publication in Free Rad and Antiox) have further corroborated the correlation between antioxidant and anti-inflammatory activity. Moreover, literature suggests that antioxidant and anti-inflammatory activities can be expected to co-exist owing to the involvement of reactive oxygen species (ROS) in cyclooxygenase and lipoxygenase-mediated inflammatory pathways from arachidonic acid (Chebil et al., 2007; Melagraki et al., 2009). The focused library under consideration in this study was, therefore, felt suitable for carrying out the antioxidant assay as it would help satisfy the twin objectives of SAR exploration and an extension of anti-inflammatory:antioxidant activity correlation. However, the results displayed in Table 2 indicate that the derivatives did not fare well in their antioxidant activity, with many showing negligible percentage inhibition compared to the reference compound. In fact, identification of the unsubstituted chalcone as one of the active compounds renders the members of this focused library ineffective for their ability to probe antioxidant SAR. Interestingly enough, one of the suboptimal anti-inflammatory compounds bearing the trifluoromethyl group was the only library member that exhibited better antioxidant potential than the parent. On a similar note, the potential anti-inflammatory sulphonamides had very poor antioxidant ability. These observations preclude the possibility of extending any meaningful correlation between the two activities. The azide-bearing chalcones 4f and 4g turned out to be the least active analogues in the DPPH radical scavenging assay.
Conclusions
The study reveals the design, synthesis and evaluation of anti-inflammatory and antioxidant activity of a series of monosubstituted chalcone derivatives. Most of the target compounds were found to have significant anti-inflammatory activity with many showing activity profile similar to that of the standard drug. Some key structural features that are beneficial for anti-inflammatory activity have been identified. The utility of sulphonamides in containing inflammation has been further corroborated in this work. We have shown that a shift of the azide group from the para to the meta position is detrimental to the activity. A clear regiochemical preference for primary amino groups being placed in the ortho position of ring A has emerged reinforcing our earlier observations. 2′-Benzenesulphonamide derivative (5b) possessed the best anti-inflammatory potential in this series. These results can act as an impetus for further research in designing potent anti-inflammatory synthetic chalcone derivatives. However, we realise that these compounds are not the right probes for studying antioxidant activity and consequently, a possible correlation between the two activities could not be extended.
References
Applequist DE, Gdanski RD (1981) Kinetic study of the hemolytic brominolysis of 1,2-diarylcyclopropanes. J Org Chem 46(12):2502–2510
Balasubramanian R, Vijayagopal R (2012) Design and in silico analysis of ring-A monosubstituted chalcones as potential anti-inflammatory agents. Bull Pharm Res 2(2):70–77
Balasubramanian R, Iqbal H, Vijayagopal R, Chandrika B (2013) Synthesis and preliminary evaluation of a focused chalcone library for anti-inflammatory activity. Ind J Pharm Educ Res 47(4):31–38
Bandgar BP, Gawande SS, Bodade RG, Totre JV, Khobragade CN (2010) Synthesis and biological evaluation of simple methoxylated chalcones as anticancer, anti-inflammatory and antioxidant agents. Bioorg Med Chem 18(3):1364–1370
Batt DG, Goodman R, Jones DG, Kerr JS, Mantegna LR, McAllister C, Newton RC, Nurnberg S, Welch PK, Covington MB (1993) 2-Substituted chalcone derivatives as inhibitors of interleukin-1 biosynthesis. J Med Chem 36(10):1434–1442
Bendary E, Francis RR, Ali HMG, Sarwat MI, El Hady S (2013) Antioxidant and structure-activity relationships (SARs) of some phenolic and aniline compounds. Ann Agric Sci 58(2):173–181
Chebil L, Anthoni J, Humeau C, Gerardin C, Engasser JM, Ghoul M (2007) Enzymatic acylation of flavonoids: effect of the nature of the substrate, origin of lipase, and operating conditions on conversion yield and regioselectivity. J Agric Food Chem 55(23):9496–9502
Cheng MS, Shili R, Kenyon GA (2000) Solid phase synthesis of chalcones by Claisen–Schmidt condensations. Chin Chem Lett 11(10):851–854
Conforti F, Sosa S, Marrelli M, Menichini F, Statti GA, Uzunov D, Tubarob A, Menichini F (2009) The protective ability of Mediterranean dietary plants against the oxidative damage: the role of radical oxygen species in inflammation and the polyphenol, flavonoid and sterol contents. Food Chem 112(3):587–594
Detsi A, Majdalani M, Kontogiorgis CA, Hadjipavlou-Litina D, Kefalas P (2009) Natural and synthetic 2′-hydroxy-chalcones and aurones: synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorg Med Chem 17(23):8073–8085
Donnelly JA, Farrell DF (1989) The chemistry of 2′-amino analogues of 2′-hydroxychalcone and its derivatives. J Org Chem 55(6):1757–1761
Gacche R, Khsirsagar M, Kamble S, Bandgar B, Dhole N, Shisode K, Chaudhari A (2008) Antioxidant and anti-inflammatory related activities of selected synthetic chalcones: structure–activity relationship studies using computational tools. Chem Pharm Bull 56(7):897–901
Gans KR, Galbraith W, Roman RJ, Haber SB, Kerr JS, Schmidt WK, Smith C, Hewes WE, Ackerman NR (1990) Anti-inflammatory and safety profile of DuP 697, a novel orally effective prostaglandin synthesis inhibitor. J Pharmacol Exp Ther 254(1):180–187
Garavito RM, DeWitt DL (1999) The cyclooxygenase isoforms: structural insights into the conversion of arachidonic acid to prostaglandins. Biochim Biophys Acta 1441(2–3):278–287
Habeeb AG, Rao PNP, Knaus EE (2001) Design and synthesis of celecoxib and rofecoxib analogues as selective cyclooxygenase-2 (COX-2) inhibitors: replacement of sulfonamide and methylsulfonyl pharmacophores by an azido bioisostere. J Med Chem 44(18):3039–3042
Isa NM, Abdelwahab SI, Mohan S, Abdul AB, Sukari MA, Taha MME, Syam S, Narrima P, Cheah S, Ahmad S, Mustafa MR (2012) In vitro anti-inflammatory, cytotoxic and antioxidant activities of boesenbergin A, a chalcone isolated from Boesenbergia rotunda (L.) (fingerroot). Braz J Med Biol Res 45(6):524–530
Karaman I, Gezegen H, Gürdere MB, Dingil A, Ceylan M (2010) Screening of biological activities of a series of chalcone derivatives against human pathogenic microorganisms. Chem Biodivers 7(2):400–408
Kumar CV, Ramaiah D, Das PK, George MV (1985) Photochemistry of aromatic α, β-epoxy ketones. Substituent effects on oxirane ring-opening and related ylide behavior. J Org Chem 50(16):2818–2825
Kumar V, Kumar S, Hassan M, Wu H, Thimmulappa RK, Kumar A, Sharma SK, Parmar VS, Biswal S, Malhotra SV (2011) Novel chalcone derivatives as potent nrf2 activators in mice and human lung epithelial cells. J Med Chem 54(12):4147–4159
Kym PR, Carlson KE, Katzenellenbogen JA (1993) Progestin 16α, l7α-dioxolane ketals as molecular probes for the progesterone receptor: synthesis, binding affinity, and photochemical evaluation. J Med Chem 36(9):1111–1119
Mannich C, Dannehl M (1938) Über die bildung eines chinolonderivatives aus o-amino-acetophenon und benzaldehyd. Ber Dtsch Chem Ges 71(9):1899–1901
Maydt D, De Spirt S, Muschelknautz C, Stahl W, Müller TJJ (2013) Chemical reactivity and biological activity of chalcones and other α, β-unsaturated carbonyl compounds. Xenobiotica 43(8):711–718
Melagraki G, Afantitis A, Igglessi-Markopoulou O, Detsi A, Koufaki M, Kontogiorgis C, Hadjipavlou-Litina DJ (2009) Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur J Med Chem 44(7):3020–3026
Meng CQ, Ni L, Worsencroft KJ, Ye Z, Weingarten MD, Simpson JE, Skudlarek JW, Marino EM, Suen KL, Kunsch C, Souder A, Howard RB, Sundell CL, Wasserman MA, Sikorski JA (2007) Carboxylated, heteroaryl-substituted chalcones as inhibitors of vascular cell adhesion molecule-1 expression for use in chronic inflammatory diseases. J Med Chem 50(6):1304–1315
Mizushima Y, Kobayashi M (1968) Interaction of anti-inflammatory drugs with serum proteins, especially with some biologically active proteins. J Pharm Pharm 20(3):169–173
Moustafa OS, Ahmad RA (2003) Synthesis and antimicrobial activity of some new cyanopyridines, isoxazoles, pyrazoles, and pyrimidines bearing sulfonamide moiety. Phosphorus Sulfur Silicon Relat Elem 178(3):475–484
Nowakowska Z (2007) A review of anti-infective and anti-inflammatory chalcones. Eur J Med Chem 42(2):125–137
Orlikova B, Tasdemir D, Golais F, Dicato M, Diederich M (2011) Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr 6(2):125–147
Rao PNP, Uddin MJ, Knaus EE (2004) Design, synthesis, and structure-activity relationship studies of 3,4,6-triphenylpyran-2-ones as selective cyclooxygenase-2 inhibitors. J Med Chem 47(16):3972–3990
Reddy MVB, Shen YC, Ohkoshi E, Bastowd KF, Qian K, Lee KH, Wu TS (2012) Bis-chalcone analogues as potent NO production inhibitors and as cytotoxic agents. Eur J Med Chem 47(1):97–103
Roome T, Dar A, Ali S, Naqvi S, Choudhary MI (2008) A study on antioxidant, free radical scavenging, anti-inflammatory and hepatoprotective actions of Aegiceras corniculatum (stem) extracts. J Ethnopharmacol 118(3):514–521
Saldanha LA, Elias G, Rao MN (1990) Oxygen radical scavenging activity of phenylbutenones and their correlation with anti-inflammatory activity. Arzneimittelforschung 40(1):89–91
Schinella GR, Tournier HA, Prieto JM, Mordujovich de Buschiazzo P, Ríos JL (2002) Antioxidant activity of anti-inflammatory plant extracts. Life Sci 70(9):1023–1033
Sing JP, Dulawat M, Jaitawat N, Chundawat SS, Devpura A, Dulawat SS (2012) Microwave enhanced Claisen–Schmidt condensation: a green route to chalcones. Indian J Chem B 51(11):1623–1627
Sivakumar PM, Prabhakar PK, Doble M (2011) Synthesis, antioxidant evaluation, and quantitative structure–activity relationship studies of chalcones. Med Chem Res 20(4):482–492
Solomon VR, Lee H (2012) Anti-breast cancer activity of heteroaryl chalcone derivatives. Biomed Pharmacother 66(3):213–220
Srinivasan B, Johnson TE, Lad R, Xing C (2009) Structure-activity relationship studies of chalcone leading to 3-hydroxy-4,3′,4′,5′-tetramethoxychalcone and its analogues as potent nuclear factor κB inhibitors and their anticancer activities. J Med Chem 52(22):7228–7235
Talley JJ (1999) Selective inhibitors of cyclooxygenase-2 (COX-2). Progr Med Chem 36:201–234
Tran TD, Nguyen TT, Do TH, Huynh TN, Tran CD, Thai KM (2012) Synthesis and antibacterial activity of some heterocyclic chalcone analogues alone and in combination with antibiotics. Molecules 17(6):6684–6696
Venkatachalam H, Nayak Y, Jayashree BS (2012) Evaluation of the antioxidant activity of novel synthetic chalcones and flavonols. Int J Chem Eng Appl 3(3):216–219
Vogel S, Barbic M, Jürgenliemk G, Heilmann J (2010) Synthesis, cytotoxicity, anti-oxidative and anti-inflammatory activity of chalcones and influence of A-ring modifications on the pharmacological effect. Eur J Med Chem 45(6):2206–2213
Wilhelm A, Lopez-Garcia LA, Busschots K, Fröhner W, Maurer F, Boettcher S, Zhang H, Schulze JO, Biondi RM, Engel M (2012) 2-(3-Oxo-1,3-diphenylpropyl)malonic acids as potent allosteric ligands of the PIF pocket of phosphoinositide-dependent kinase-1: development and prodrug concept. J Med Chem 55(22):9817–9830
Wu J, Li J, Cai Y, Pan Y, Ye F, Zhang Y, Zhao Y, Yang S, Li X, Liang G (2011) Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem 54(23):8110–8123
Yadav VR, Prasad S, Sung B, Aggarwal BB (2011) The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int Immunopharmacol 11(3):295–309
Zarghi A, Zebardast T, Hakimion F, Shirazi FH, Rao PNP, Knaus EE (2006) Synthesis and biological evaluation of 1,3-diphenylprop-2-en-1-ones possessing a methanesulfonamido or an azido pharmacophore as cyclooxygenase-1/-2 inhibitors. Bioorg Med Chem 14(20):7044–7050
Acknowledgments
This work was partially funded by a grant (No. 051/SPS/2012/CSTE) of Kerala State Council for Science, Technology and Environment (KSCSTE). The authors would like to thank the Department of Organic Chemistry, Cochin University of Science and Technology (CUSAT) for providing necessary laboratory facilities.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Iqbal, H., Prabhakar, V., Sangith, A. et al. Synthesis, anti-inflammatory and antioxidant activity of ring-A-monosubstituted chalcone derivatives. Med Chem Res 23, 4383–4394 (2014). https://doi.org/10.1007/s00044-014-1007-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-014-1007-z