
Abstract
In the present study, a novel series of indolyl-pyrimidines (1–13) were synthesized starting from 4-hydrazinopyrimidine-5-carbonitrile 3. Elemental analysis, IR, 1H-NMR, 13C-NMR, and mass spectral data elucidated structure of newly synthesized compounds. All compounds were screened for their in vitro antibacterial, antifungal, and some for antioxidant activities. Compounds 5, 9g, 9i, and 9j showed pronounced antimicrobial activity against Staphylococcus aureus, Bacillus cereus, Escherichia coli, Candida albicans, and Aspergillus flavus compared to the reference drugs, while compounds 3 and 9g displayed promising free radical scavenging activity and found to be more potent than standard, ascorbic acid (vitamin C). Further, some compounds were evaluated for cytotoxic activity by SRB assay method against human colon carcinoma (CaCo-2) and showed that compounds 4 and 9g were found to be the highly active compared to the reference drug doxorubicin. Their structure and activity relationship were discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pyrimidine derivatives and related fused heterocycles are important classes of heterocyclic compounds that exhibit a broad spectrum of biological activities such as anticancer (Cocco et al., 2006; Ibrahim and El-Metwally, 2010; Le Brazidec et al., 2012), antiviral (Martinez-Montero et al., 2012), antibacterial (Kotaiah et al., 2012), antioxidant (Abu-Hashem et al., 2011), and anti-inflammatory (Hanna, 2012). Another important class of pyrimidine is 2-thiopyrimidine (2-TP) and its derivatives. Similarly, the related thiouracil derivatives are potential chemotherapeutics as antiviral, anticancer, and antimicrobial agents (Grigoryan et al., 2005; Al-Masoudi et al., 2011; Agbaje et al., 2011). In particular, 6-n-propyl-2-thiouracil (6-PTU) is antithyroid drug (Cooper, 2005) where its S-alkylation and N3-alkylation products have been recently reported as novel antibacterial and cytotoxic agents (Prachayasittikul et al., 2009). 6-Aryl-5-cyano-2-thiour-acils possess antimicrobial and antitumor activities. 1-Alkyl-2-(alkylthio)-4-aryl-6-oxo-1,6-dihydropyrimidine-5-carbonitriles are displayed promising anticancer activity against leukemia, non-small cell lung, melanoma, and renal cancer (Taher and Helwa, 2012). 6-(4-Bromophenyl)-5-cyano-4-oxo-3,4-dihydropyrimidin-2-yl 2-(4-bromophenyl)-N′-(4-hydroxyphenyl)-2-oxo-ethane-hydrazonothioate possessed superior antibacterial activity against the gram positive bacteria Staphylococcus aurus and Bacillus subtilis compared to amoxicillin (Taher and Abou-Seri, 2012). Moreover, 6-aryl-5-cyano-2-thiouracils possess inhibition of hepatitis C viral NS5B RNA dependent RNA polymerase (Ding et al., 2006). Reports from our laboratory revealed that several hydrazinopyrimidine derivatives show significant biological activities (Mohamed et al., 2011a, b, c, 2012a, b; Awad et al., 2013). In particular, hydrazinopyrimidine-5-carbonitriles were utilized as a pharmacophoric tool for the development of more efficacious antimicrobial and anticancer agents (Ram et al., 1987; Kadry et al., 2008; Fathalla et al., 2009; El-zahar et al., 2011). This study was undertaken in view of the fact that hyrazone (Onnis et al., 2009; Edrees et al., 2010), triazole (Atta, 2011; Singh et al., 2012), and pyrazole (Nitulescu et al., 2010; Chaudhari et al., 2011) moieties have been reported to possess significant chemotherapeutic activities. In the field of medicinal chemistry, azoles belong to a class of antimicrobial agents that are widely used and studied because of their safety profile and high therapeutic index. Ribavirin, rizatriptan, alprazolam, vorozole, letrozole, and anastrozole are the best examples of drugs containing 1,2,4-triazole moiety (Ashok et al., 2007; Hancu et al., 2007; Cai et al., 2007). Among azole-based drugs, conazoles, such as itraconazole, fluconazole, voriconazole, and ravuconazole, constitute a major class being used for the treatment of fungal infections (Gupta et al., 2007; Schiller and Fung, 2007). Moreover, indole and its derivatives play an important role as biologically active compounds. 2- and 3-aryl-indoles displayed noteworthy antimicrobial activity such as 3-phenylindole which is an inhibitor of brassinin glucosyltrans-ferase, a phytoalexin detoxifying enzyme from the fungus, Sclerotinia sclerotiorum (Leboho et al., 2009).
Indole-3-carbinol (I3C) has anti-proliferative and anti-estrogenic activities in human breast cancer cells (Souli et al., 2008). 1-Benzyl-I3C displayed enhanced potency in suppressing the growth of both estrogen responsive (MCF-7) and estrogen-independent (MDA-MB-231) human breast cancer cells (Nguyen et al., 2010). Also, 2-indolylmethanones (Mahboobi et al., 2005), camalexin (indolyl thiazole),4-(3-indolyl)oxazoles, and 5-(3-indolyl)-1,3,4-oxadiazoles were reported as potential anticancer agents against many types of human cancer cell lines (Kumar et al., 2010).Arbidol (ethyl 6-bromo-5-hydroxy-1H-indole-3-carboxylate derivative) is a broad-spectrum antiviral agent that inhibits acute and chronic HCV infection (Sellitto et al., 2010). Di-indolylmethane derivatives possess potential radical scavenging activity (Benabadji et al., 2004). In view of these observations, herein we report the synthesis of some new indolyl-pyrimidines incorporating different potent pharmacophores as trial to develop a novel antimicrobial, antioxidant, and anticancer agents. Their structure and activity relationship were also examined.
Experimental
All melting points were uncorrected and measured using Electro-thermal IA 9100 apparatus (Shimadzu, Japan). IR spectra were recorded as potassium bromide pellets on a Perkin-Elmer 1650 spectrophotometer (USA), Faculty of Science, Cairo University, Cairo, Egypt. 1H-NMR and 13C-NMR spectra were determined on a Varian Mercury (300 MHz) spectrometer (Varian, UK), and chemical shifts were expressed as ppm against TMS as internal reference (Faculty of Science, Cairo University, Cairo, Egypt). Mass spectra were recorded on 70 eV EI Ms-QP1000 EX (Shimadzu, Japan), Faculty of Science, Cairo University, Cairo, Egypt. Microanalyses were operated using a Vario, Elementar apparatus (Shimadzu, Japan), Organic Microanalysis Unit, Faculty of Science, Cairo University, Cairo, Egypt. Column chromatography was performed on (Merck) Silica gel 60 (particle size 0.06–0.20 mm) using chloroform: methanol (3:1) solvent system. All new compounds yielded spectral data consistent with the proposed structure and microanalysis within ±0.4 % of the theoretical values. Compound 1 was prepared as reported in the literature (Mohamed et al., 2011b). Target compounds were synthesized as outlined in Schemes 1 and 2.

Synthesis of compounds 1–9

Synthesis of compounds 10–13
Synthesis
6-(1H-Indol-3-yl)-4-oxo-2-thioxo-1,2,3,4-tetrahydro-pyrimidine-5-carbonitrile (1)
A mixture of ethylcyanoacetate (0.01 mol), thiourea (0.01 mol), indole-3-carboxaldehyde (0.01 mol), and 25 mL sodium ethoxide/ethanol was stirred for 1 h at room temperature. The reaction mixture poured onto ice and neutralized with 2 N HCl, the produced solid was filtered off, dried, and recrystallized from DMF/Water to give compound 1 as a yellow solid. Yield: 80 %; m.p.: 195–198 °C; IR (KBr) cm−1:3300, 2230, 1275; MS (EI) m/z: 268 (M+, 60.5 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 6.6–7.5 (m, 5H, Ar–H), 10.0 (s, 1H, NH, D2O exchangeable), 10.5 (s, 1H, NH, D2O exchangeable), 11.2 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 80 (C, C-5), 117 (CN), 105 (C, C-3′), 115 (CH, C-7′), 120 (CH, C-4′, C-6′), 122 (CH, C-5′), 124 (CH, C-2′), 128 (C, C-8′), 134 (C, C-9′), 160 (C=O), 168 (C, C-6), 180 (C=S); Anal. Calcd. for C13H8N4OS (268.29): C, 58.20; H, 3.01; N, 20.88; S, 11.95 %. Found: C, 58.32; H, 3.20; N, 20.73; S, 12.01 %.
4-Chloro-6-(1H-indol-3-yl)-2-thioxo-1,2-dihydropyrimidine-5-carbonitrile (2)
A mixture of indolyl-pyrimidine derivative 1 (0.01 mol) was heated under reflux in phosphorus oxychloride (25 mL) and phosphorus pentachloride (0.01 mol) for 8 h, cooled and poured onto ice to give precipitate, which was washed with petroleum ether, filtered off, and dried under vacuum to give compound 2 as a brown solid. Yield: 50 %;m.p.: 150–152 °C; IR (KBr) cm−1: 3200, 2220, 1275; MS (EI) m/z: 285 (M+, 60.5 %), 287 (M+2, 20.4 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 7.0–7.7 (m, 5H, Ar–H), 10.2 (s, 1H, NH, D 2 O exchangeable), 11.2 (s, 1H, NH, D 2 O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 88.5 (C, C-5), 102 (C, C-3′), 115.4 (CH, C-7′), 117 (CN), 122.2 (CH, C-4′, C-6′), 124 (CH, C-5′), 128 (CH, C-2′), 130.5 (C, C-8′), 136 (C, C-9′), 159.2 (C=N), 160.1 (C, C-6), 180 (C=S); Anal. Calcd. for C13H7ClN4S (286.73): C, 54.45; H, 2.46; N, 19.54; S, 11.18 %. Found: C, 54.60; H, 2.55; N, 19.56; S, 11.29 %.
4-Hydrazino-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (3)
A mixture of 4-chloropyrimidine 2 (0.01 mol) and hydrazine hydrate (20 mL, 99 %) was refluxed in methanol (30 mL) for 20 min, cooled, stirred for 24 h, and poured onto ice water. The solid obtained was filtered, dried, and recrystallized from DMF/water to yield compound 3 as a brown solid. Yield: 60 %; m.p.: 220–222 °C; IR (KBr) cm−1:3460, 3320, 2200, 1270; MS (EI) m/z: 282 (M+, 40 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 5.1–5.7 (s, 3H, NH, NH2, D2O exchangeable), 7.0–7.8 (m, 5H, Ar–H), 10.3 (s, 1H, NH, D2O exchangeable), 11.3 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 84.5 (C, C-5), 102 (C, C-3′), 111 (CH, C-7′), 117 (CN), 122.8 (CH, C-4′, C-6′), 124.5 (CH, C-5′), 127.2 (CH, C-2′), 128 (C, C-8′), 136 (C, C-9′), 164 (C=N), 167.1 (C, C-6), 180 (C=S); Anal. Calcd. for C13H10N6S (282.32): C, 55.30; H, 3.57; N, 29.77; S, 11.36 %. Found: C, 55.40; H, 3.66; N, 29.90; S, 11.45 %.
7-(1H-Indol-3-yl)-3-methyl-5-thioxo-5,6-dihydro[1,2,4]triazolo[4,3-c]pyrimidine-8-carbonitrile (4)
A mixture of hydrazine derivative 3 (0.01 mol) and acetic anhydride (30 mL) was heated under reflux for 4 h. The solid obtained was filtered, dried, and recrystallized from acetic acid to yield compound 4 as a black crystal. Yield: 55 %; m.p.: 265–267 °C; IR (KBr) cm−1: 3250, 2225, 1597, 1275; MS (EI) m/z: 306 (M+, 45 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.3 (s, 3H, CH3), 6.5–7.8 (m, 5H, Ar–H), 10.1 (s, 1H, NH, D2O exchangeable), 11.5 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 20.6 (CH3), 80.2 (C, C-5), 102.9 (C, C-3′), 115.4 (CH, C-7′), 117 (CN), 122.4 (CH, C-4′, C-6′), 124.5 (CH, C-5′), 127.1 (CH, C-2′), 130.5 (C, C-8′), 139.6 (C, C-9′), 151.1, 160 (2C=N), 162.6 (C, C-6), 175.2 (C=S); Anal. Calcd. for C15H10N6S (306.34): C, 58.81; H, 3.29; N, 27.43; S, 10.47 %. Found: C, 58.90; H, 3.33; N, 27.55; S, 10.56 %.
7-(1H-Indol-3-yl)-5-thioxo-5,6-dihydro[1,2,4]triazolo[4,3-c]pyrimidine-8-carbonitrile (5)
A mixture of hydrazine derivative 3 (0.01 mol) and triethyl orthoformate (30 mL) was heated under reflux for 5 h. The solid obtained was crystallized from acetic acid to yield compound 5 as dark brown crystals. Yield: 60 %; m.p.: 238–240 °C; IR (KBr) cm−1: 3237, 2225, 1582; MS (EI) m/z: 292 (M+, 42 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 7.0–7.9 (m, 5H, Ar–H), 8.3 (s, 1H, C5-H), 9.7 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 90 (C, C-5), 103 (C, C-3′), 116.6 (CH, C-7′), 117.9 (CN), 122.5 (CH, C-4′, C-6′), 124.2 (CH, C-5′), 127.2 (CH, C-2′), 129 (C, C-8′), 137 (C, C-9′), 152 (2C=N), 167 (C, C-6), 179 (C=S); Anal. Calcd. for C14H8N6S (292.31): C, 57.52; H, 2.76; N, 28.75; S, 10.97 %. Found: C, 57.67; H, 2.85;N, 28.90; S, 11.10 %.
7-(1H-Indol-3-yl)-5-thioxo-5,6-dihydro[1,2,4]triazolo[1,5-c]pyrimidine-8-carbonitrile (6) and 7-(1H-indol-3-yl)-2-methyl-5-thioxo-5,6-dihydro[1,2,4]triazolo[1,5-c]-pyrimidine-8-carbonitrile (7)
A mixture of hydrazine derivative 3 (0.01 mol) and formic or acetic acid (30 mL) was heated under reflux for 8–10 h, then cooled, and poured onto ice water. The solid obtained was crystallized from acetic acid to yield compounds 6 and 7 as black crystals, respectively.
6: Yield: 52 %; m.p.: 230–232 °C;IR (KBr) cm−1:3300, 2229, 1608, 1270; MS (EI) m/z: 292 (M+, 60 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 7.0–7.8 (m, 5H, Ar–H), 8.6 (s, 1H, C3-H), 10.3 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 88.2 (C, C-5), 102 (C, C-3′), 115.4 (CH, C-7′), 117.2 (CN), 122.2 (CH, C-4′, C-6′), 124.2 (CH, C-5′), 128 (CH, C-2′), 130.5 (C, C-8′), 136 (C, C-9′), 155.2 (2C=N), 160 (C, C-6), 180 (C=S); Anal.Calcd. for C14H8N6S (292.31): C, 57.52; H, 2.76; N, 28.75; S, 10.97 %. Found: C, 57.66; H, 2.78; N, 28.89; S, 11.17 %.
7: Yield: 55 %; m.p.: 243–245 °C; IR (KBr) cm−1: 3310, 2216, 1606, 1270; MS (EI) m/z: 306 (M+, 62 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 7.0–7.9 (m, 5H, Ar–H), 10.2 (s, 1H, NH, D2O exchangeable), 11.5 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 20.2 (CH3), 75 (C, C-5), 105 (C, C-3′), 112.1 (CH, C-7′), 117 (CN), 124.5 (CH, C-4′, C-6′), 127 (CH, C-5′), 128 (CH, C-2′), 130.5 (C, C-8′), 136 (C, C-9′), 148, 160 (2C=N), 169 (C, C-6), 178 (C=S); Anal.Calcd. for C15H10N6S (306.34): C, 58.81; H, 3.29; N, 27.43; S, 10.47 %. Found: C, 58.90; H, 3.34; N, 27.55; S, 11.01 %.
8-(1H-Indol-3-yl)-3,4-dioxo-6-thioxo-3,4,6,7-tetrahydro-2H-pyrimido[6,1-c][1,2,4]triazine-9-carbonitrile (8)
A solution of hydrazine derivative 3 (0.01 mol) and diethyloxalate (0.01 mol) in absolute ethanol (40 mL) was heated under reflux for 12 h. The solid obtained was crystallized from the benzene to yield compound 8 as reddish brown crystals. Yield: 65 %; m.p.: 240–242 °C; IR (KBr) cm−1: 3450, 3230, 2220, 1675, 1274; MS (EI) m/z: 336 (M+, 10 %), 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 7.0–7.9 (m, 5H, Ar–H), 8.2 (s, 1H, NH, D2O exchangeable), 9.8 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 88.2 (C, C-5), 106 (C, C-3′), 118.8 (CH, C-7′), 117.2 (CN), 122.5 (CH, C-4′, C-6′), 124.3 (CH, C-5′), 127.2 (CH, C-2′), 128 (C, C-8′), 136 (C, C-9′), 158.2 (C=N), 160 (C, C-6), 165.9 (2C=O), 180 (C=S); Anal. Calcd. for C15H8N6O2S (336.32): C, 53.57; H, 2.40; N, 24.99; S, 9.53 %. Found: C, 53.60; H, 2.49; N, 25.12; S, 10.14 %.
4-[(2E)-2-Substituted-(benzylidene)hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitriles (9a–9j)
A mixture of hydrazine derivative 3 (0.01 mol) and appropriate aldehyde (0.01 mol) in ethanol (30 mL) was heated under reflux for 4–7 h. The solid obtained was crystallized from benzene to yield compounds 9a–j, respectively.
4-[(2E)-2-Benzylidene-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9a)
Yield: 68 %; yellowish brown crystals; m.p.: 190–192 °C; IR (KBr) cm−1: 3220, 2225, 1606; MS (EI) m/z: 70 (M+, 15 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.3 (s, 1H, NH, D2O exchangeable), 6.4–7.8 (m, 10H, Ar–H), 8.6 (s, 1H, CH=N), 10.4 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 72 (C, C-5), 111 (C, C-3′), 117 (CN), 120.8 (CH, C-7′), 122.5 (CH, C-4′, C-6′), 124 (CH, C-5′), 127 (CH, C-2′), 128 (C, C-8′), 129 (CH, C-3″, C-5″), 130 (CH, C-2″, C-6″), 132 (CH, C-4″), 135 (C, C-1″), 136 (C, C-9′), 154.3 (N=CH), 163 (C=N), 168.5 (C, C-6), 180 (C=S); Anal. Calcd. for C20H14N6S (370.43): C, 64.85; H, 3.81; N, 22.69; S, 8.66 %. Found: C, 64.95; H, 3.95; N, 22.77; S, 8.80 %.
4-[(2E)-2-(4-Fluorobenzylidene)-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9b)
Yield: 62 %; yellow crystals; m.p.: 198–200 °C; IR (KBr) cm−1:3300, 2224, 1608; MS (EI) m/z: 388 (M+, 13 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.4 (s, 1H, NH, D2O exchangeable), 6.5–7.8 (m, 9H, Ar–H), 8.4 (s, 1H, CH=N), 10.4 (s, 1H, NH, D2O exchangeable), 11.2 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 79 (C, C-5), 105 (C, C-3′), 111 (CH, C-7′), 115 (CH, C-3″, C-5″), 117 (CN), 120 (CH, C-4′, C-6′), 122 (CH, C-5′), 124 (CH, C-2′), 126 (CH, C-2″, C-6″), 128 (C, C-8′), 128.7 (C, C-1″), 130 (C, C-4″), 138 (C, C-9′), 154.3 (N=CH), 164 (C=N), 166 (C, C-6), 180 (C=S); Anal. Calcd. for C20H13FN6S (388.42): C, 61.84; H, 3.37; N, 21.64; S, 8.26 %. Found: C, 61.96; H, 3.49; N, 21.69; S, 8.33 %.
4-[(2E)-2-(4-Bromobenzylidene)-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9c)
Yield: 60 %; light brown crystals; m.p.: 170–172 °C; IR (KBr) cm−1: 3230, 2225, 1608; MS (EI) m/z: 448 (M+, 18 %), 450 (M+2, 18.6 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.4 (s, 1H, NH, D2O exchangeable), 6.6–7.9 (m, 9H, Ar–H), 8.4 (s, 1H, CH=N), 10.5 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 79.5 (C, C-5), 106 (C, C-3′), 111 (CH, C-7′), 117 (CN), 120 (CH, C-3″, C-5″), 122 (CH, C-4′, C-6′), 124 (CH, C-5′), 125 (CH, C-2″, C-6″), 126 (CH, C-2′), 128 (C, C-8′), 130 (C, C-1″), 132 (C, C-4″), 136 (C, C-9′), 153 (N=CH), 164 (C=N), 167 (C, C-6), 182 (C=S); Anal.Calcd. for C20H13BrN6S (449.32): C, 53.46; H, 2.92; N, 18.70; S, 7.14 %. Found: C, 53.55; H, 3.02; N, 18.82; S, 7.28 %.
4-[(2E)-2-(4-Chlorobenzylidene)-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9d)
Yield: 60 %; yellow crystals; m.p.: 195–197 °C; IR (KBr) cm−1: 3360, 2222, 1605; MS (EI) m/z: 403 (M+, 25 %), 405 [M+2, 8.3 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.3 (s, 1H, NH, D2O exchangeable), 6.1–7.8 (m, 9H, Ar–H), 8.5 (s, 1H, CH=N), 10.5 (s, 1H, NH, D2O exchangeable), 11.5 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 73.6 (C, C-5), 102 (C, C-3′), 110 (CH, C-7′), 117 (CN), 120 (CH, C-4′, C-6′), 122 (CH, C-5′), 123 (CH, C-2′), 124 (CH, C-3″, C-5″), 126 (C, C-8′), 128 (CH, C-2″, C-6″), 130 (C, C-1″), 131.2 (C, C-4″), 136 (C, C-9′), 154.7 (N=CH), 164 (C=N), 171 (C, C-6), 180 (C=S); Anal.Calcd. for C20H13ClN6S (404.87): C, 59.33; H, 3.24; N, 20.76; S, 7.92 %. Found: C, 59.45; H, 3.37; N, 20.88; S, 8.08 %.
4-[(2E)-2-(2-Methoxybenzylidene)-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9e)
Yield: 62 %; brown crystals; m.p.: 184–186 °C; IR (KBr) cm−1: 3350, 2223, 1606; MS (EI) m/z: 400 (M+, 10 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.3 (s, 1H, NH, D2O exchangeable), 3.81 (s, 3H, OCH3), 6.5–7.8 (m, 9H, Ar–H), 8.5 (s, 1H, CH=N), 10.4 (s, 1H, NH, D2O exchangeable), 11.5 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 56 (O–CH3), 78.5 (C, C-5), 111 (C, C-3′), 114.9 (CH, C-3″), 116 (C, C-1″), 117 (CN), 120 (CH, C-7′), 122.3 (CH, C-4′, C-6′), 120.9 (C, C-5″), 124.5 (CH, C-5′), 127.7 (CH, C-2′), 128.5 (C, C-8′), 130 (CH, C-6″), 131 (CH, C-4″), 136 (C, C-9′), 152 (N=CH), 160 (C, C-2″), 163 (C=N), 169 (C, C-6), 182 (C=S); Anal. Calcd. for C21H16N6OS (400.45): C, 62.98; H, 4.03; N, 20.99; S, 8.01 %. Found: C, 63.11; H, 4.23; N, 21.15; S, 8.12 %.
4-[(2E)-2-(3,4-Dimethoxybenzylidene)-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-etrahydro-pyrimidine-5-carbonitrile (9f)
Yield: 63 %; brown crystals; m.p.: 178–180 °C; IR (KBr) cm−1: 3375, 2225, 1609; MS (EI) m/z: 430 (M+, 12 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.4 (s, 1H, NH, D2O exchangeable), 3.4, 3.81 (s, 6H, OCH3), 6.6–7.9 (m, 8H, Ar–H), 8.5 (s, 1H, CH=N), 10.5 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 56.3, 56.6 (O–2CH3), 73.6 (C, C-5), 105 (C, C-3′), 111.6 (CH, C-7′), 115 (CH, C-5″), 116 (CH, C-2″), 117 (CN), 120 (CH, C-6″), 122.6 (CH, C-4′, C-6′), 124.3 (CH, C-5′), 126 (C, C-1″), 127.8 (CH, C-2′), 128.4 (C, C-8′), 136.9 (C, C-9′), 138 (C, C-3″), 140 (CH, C-4″), 154.5 (N=CH), 162 (C=N), 167.2 (C, C-6), 184 (C=S); Anal. Calcd. for C22H18N6O2S (430.48): C, 61.38; H, 4.21; N, 19.52; S, 7.45 %. Found: C, 61.45; H, 4.39; N, 19.69; S, 7.56 %.
4-[(2E)-2-(3,4,5-Trimethoxybenzylidene)-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyimidi-ne-5-carbonitrile (9g)
Yield: 65 %; yellow crystals; m.p.: 194–196 °C; IR (KBr) cm−1: 3380, 2220, 1606; MS (EI) m/z: 460 (M+, 11 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.4 (s, 1H, NH, D2O exchangeable), 3.2–3.8 (s, 9H, 3OCH3), 6.5–7.7 (m, 7H, Ar–H), 8.5 (s, 1H, CH=N), 10.4 (s, 1H, NH, D2O exchangeable), 11.3 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 56.3 (O–2CH3), 56.5 (O–CH3), 79.7 (C, C-5), 104 (C, C-3′), 107 (CH, C-2″, C-6″), 111.6 (CH, C-7′), 117 (CN), 122.6 (CH, C-4′, C-6′), 124.2 (CH, C-5′), 126 (C, C-1″), 127.8 (CH, C-2′), 128.4 (C, C-8′), 130 (C, C-4″), 136.9 (C, C-9′), 142 (C, C-3″, C-5″), 154.2 (N=CH), 162 (C=N), 166 (C, C-6), 182 (C=S); Anal. Calcd. for C23H20N6O3S (460.50): C, 59.99; H, 4.38; N, 18.25; S, 6.96 %. Found: C, 60.30; H, 4.46; N, 18.35; S, 7.08 %.
4-[(2E)-2-(4-Dimethylamino)benzylidene)-hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9h)
Yield: 62 %; brown crystals; m.p.: 185–187 °C; IR (KBr) cm−1: 3300, 2222, 1608; MS (EI) m/z: 413 (M+, 18 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.4 (s, 1H, NH, D2O exchangeable), 3 (s, 6H, N(CH3)2), 6.8–8.1 (m, 9H, Ar–H), 8.4 (s, 1H, CH=N), 10.3 (s, 1H, NH, D2O exchangeable), 11.4 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 40.3 (N–2CH3), 80 (C, C-5), 102 (C, C-3′), 111 (CH, C-7′), 113 (CH, C-3″, C-5″), 117 (CN), 120 (CH, C-4′, C-6′), 122.4 (CH, C-5′), 125 (C, C-1″), 126.5 (CH, C-2′), 128.6 (C, C-8′), 130 (CH, C-2″, C-6″), 134.5 (C, C-9′), 140 (C, C-4″), 154.5 (N=CH), 162 (C=N), 169 (C, C-6), 184 (C=S); Anal. Calcd. for C22H19N7S (413.49): C, 63.90; H, 4.63; N, 23.71; S, 7.75 %. Found: C, 63.98; H, 4.78; N, 23.88; S, 7.88 %.
6-(1H-Indol-3-yl-4-[(2E)-2-(4-nitrobenzylidene)-hydrazino]-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9i)
Yield: 64 %; reddish brown crystals; m.p.: 200–202 °C; IR (KBr) cm−1: 3380, 2220, 1609; MS (EI) m/z: 415 (M+, 15 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.4 (s, 1H, NH, D2O exchangeable), 6.8–8.2 (m, 9H, Ar–H), 8.4 (s, 1H, CH=N), 10.4 (s, 1H, NH, D2O exchangeable), 11 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 82 (C, C-5), 104 (C, C-3′), 112 (CH, C-7′), 117 (CN), 119 (CH, C-4′, C-6′), 120 (CH, C-5′), 122.8 (CH, C-2′), 124.6 (C, C-8′), 128 (CH, C-3″, C-5″), 129 (CH, C-2″, C-6″), 132 (C, C-9′), 137 (C, C-1″), 145 (C, C-4″), 154.9 (N=CH), 164 (C=N), 169.2 (C, C-6), 180 (C=S); Anal. Calcd. for C20H13N7O2S (415.42): C, 57.82; H, 3.15; N, 23.60; S, 7.72 %. Found: C, 57.96; H, 3.27; N, 23.80; S, 7.82 %.
6-(1H-Indol-3-yl-4-[(2E)-2-(4-methylbenzylidene)-hydrazino]-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9j)
Yield: 66 %; brown crystals; m.p.: 196–198 °C; IR (KBr) cm−1: 3356, 2223, 1609; MS (EI) m/z: 384 (M+, 19 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.2 (s, 3H, CH3), 2.4 (s, 1H, NH, D2O exchangeable), 6.5–7.9 (m, 9H, Ar–H), 8.4 (s, 1H, CH=N), 9.5 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 12.5 (CH3), 82 (C, C-5), 105 (C, C-3′), 114 (CH, C-7′), 117 (CN), 120 (CH, C-4′, C-6′), 122 (CH, C-5′), 124.6 (CH, C-2′), 128.5 (C, C-8′), 129 (C, C-1″), 130 (CH, C-2″, C-6″), 131 (CH, C-3″, C-5″), 132 (C, C-4″), 134.5 (C, C-9′), 154.1 (N=CH), 164 (C=N), 168 (C, C-6), 182 (C=S); Anal. Calcd. for C21H16N6OS (384. 45): C, 65.61; H, 4.19; N, 21.86; S, 8.34 %. Found: C, 65.77; H, 4.28; N, 21.95; S, 8.45 %.
4-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(1H-indol-3-yl)-2-thioxo-1,2-dihydropyrimidine-5-carbonitrile (10)
A mixture of hydrazine derivative 3 (0.01 mol) and acetyl acetone (0.1 mL, 0.001 mol) in acetic acid (15 mL) was refluxed for 15 h. The reaction mixture was cooled and poured onto ice water. The solid obtained was crystallized from ethanol to yield compound 10 as brown crystals. Yield: 55 %; m.p.: 238–240 °C; IR (KBr) cm−1: 3348, 2227, 1610; MS (EI) m/z: 346 (M+, 27 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 2.3, 2.5 (s, 6H, 2CH3), 6.8 (s, 1H, C4–H), 7–7.9 (m, 5H, Ar–H), 10.0 (s, 1H, NH, D2O exchangeable), 11.0 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 10, 15.3 (2CH3), 92 (C, C-5), 102 (C, C-3′), 103 (C, C-4″), 117.7 (CH, C-7′), 117 (CN), 122.2 (CH, C-4′, C-6′), 124 (CH, C-5′), 128.2 (CH, C-2′), 134.4 (C, C-8′), 138.3 (C, C-9′), 146 (C, C-5″), 148 (C=N), 164 (C=N), 169 (C, C-6), 174 (C=S); Anal. Calcd. for C18H14N6S (346.40): C, 62.41; H, 4.07; N, 24.26; S, 9.26 %. Found: C, 62.55; H, 4.19; N, 24.38; S, 9.33 %.
7-(1H-Indol-3-yl)-3-oxo-5-thioxo-2,3,5,6-tetrahydro[1,2,4]-triazolo[4,3-c]pyrimidine-8-carbonitrile (11)
A mixture of hydrazine derivative 3 (0.01 mol) and ethylchloroformate (0.02 mol) in pyridine (30 mL) was heated under reflux for 12 h and poured on 2 N HCl. The solid obtained was crystallized from dimethyl formamide to yield compound 11 as black crystals. Yield: 60 %; m.p.: 230–232 °C; IR (KBr) cm−1: 3210, 2229, 1665; MS (EI) m/z: 308 (M+, 48 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 7.0–7.8 (m, 5H, Ar–H), 8.3 (s, 1H, NH, D2O exchangeable), 10.1 (s, 1H, NH, D2O exchangeable), 11.4 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 79.5 (C, C-5), 102.9 (C, C-3′), 112.2 (CH, C-7′), 115.7 (CN), 124.3 (CH, C-4′, C-6′), 128 (CH, C-5′), 130.5 (CH, C-2′), 136 (C, C-8′), 147.8 (C, C-9′), 158 (C=N), 164 (C=O), 170.9 (C, C-6), 175 (C=S); Anal. Calcd. for C14H8N6OS (308.31): C, 54.54; H, 2.62; N, 27.26; S, 10.40 %. Found: C, 54.61; H, 2.77; N, 27.36; S, 10.54 %.
7-(1H-Indol-3-yl)-3,5-dithioxo-2,3,5,6-tetrahydro[1,2,4]triazolo[4,3-c]pyrimidine-8-carbonitrile (12)
To an ice-cold solution of hydrazine derivative 3 (0.01 mol) and KOH (0.01 mol) in ethanol (20 mL) was added dropwise with stirring CS2 (10 mL), then the reaction mixture was refluxed on a water-bath for 5 h. The reaction mixture cooled, poured onto ice water, neutralized with 2 N HCl, filtered, and crystallized from ethanol to yield compound 12 as black crystals. Yield: 60 %; m.p.: 237–239 °C; IR (KBr) cm−1: 3290, 2222, 1650, 1580; MS (EI) m/z: 324 (M+, 30 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 6.5–7.9 (m, 5H, Ar–H), 9.5 (s, 1H, NH, D2O exchangeable), 10.3 (s, 1H, NH, D2O exchangeable), 11.5 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 75 (C, C-5), 104.2 (C, C-3′), 111.5 (CH, C-7′), 117 (CN), 124.2 (CH, C-4′, C-6′), 128 (CH, C-5′), 130.5 (CH, C-2′), 136.2 (C, C-8′), 147.8 (C, C-9′), 159.9 (C=N), 169 (C, C-6), 178, 188.2 (2C=S); Anal. Calcd. for C14H8N6S2 (324.38): C, 51.84; H, 2.49; N, 25.91; S, 19.77 %. Found: C, 51.95; H, 2.55; N, 26.10; S, 20. 09 %.
7-(1H-Indol-3-yl)-5-thioxo-5,6-dihydrotetrazolo[1,5-c]pyrimidine-8-carbonitriles (13)
A cold solution (0–5 °C) of sodium nitrite (1 g, 0.144 mol) in H2O (15 mL) was added gradually within 15 m to a cold stirred solution of hydrazine derivative 3 (0.01 mol) in 2 N HCl (15 mL). After addition, the reaction mixture was then further stirred for 4 h at the same temperature, and then it was diluted with cold H2O. The solid obtained was crystallized from ethanol to yield compound 13 as yellowish brown crystals. Yield: 62 %; m.p.: 210–212 °C; IR (KBr) cm−1: 3345, 2227, 1507; MS (EI) m/z: 293 (M+, 48 %); 1H-NMR (DMSO-d 6, 300 MHz) δ (ppm): 7.0–7.9 (m, 5H, Ar–H), 10, 11.2 (s, 2H, 2NH, D2O exchangeble); 13C-NMR (300 MHz, DMSO-d 6) δ (ppm): 79 (C, C-5), 102.3 (C, C-3′), 116.5 (CH, C-7′), 117.5 (CN), 120.3 (CH, C-4′, C-6′), 124.4 (CH, C-5′), 128.7 (CH, C-2′), 138.2 (C, C-8′), 140.2 (C, C-9′), 160 (C=N), 166 (C, C-6), 182 (C=S); Anal. Calcd. for C13H7N7S (293.30): C, 53.23; H, 2.41; N, 33.43; S, 10.93 %. Found: C, 53.38; H, 2.56; N, 33.57; S, 11.04 %.
Antimicrobial activity
All compounds were evaluated for antibacterial activity against several pathogenic representative Gram-positive bacteria (S.aureus ATCC12600, B.cereus ATCC14579), Gram-negative bacteria (Escherichia coli ATCC11775), and (Candida albicans ATCC26555, Aspergillus flavus ATCC 11495) as a representative for fungi using the disk diffusion method (Bauer et al., 1996). All microorganisms used were obtained from the culture collection of the Department of Microbiology, Micro Analytical Centre, Faculty of Science, Cairo University, Cairo, Egypt. Media for disk sensitivity tests were the nutrient agar and Muller-Hinton agar (MHA) purchased from Difco (USA). Non-sterile powder of tested compounds was dissolved in sterile DMSO to yield 10 mg/mL solution, and passed through a 0.2 μm membrane filter (MilliporeCorp, SA).
Penicillin (Bioanalyse, Turkey) and Fluconazole (Sigma-Aldrich, USA) were used as reference substances. Inhibition zones were measured in millimeters at the end of an incubation period (Table 1).
Antibacterial evaluation
All the newly synthesized compounds were screened for their antibacterial activity against Gram-positive bacteria viz. S. aureus (ATCC12600) and B. cereus (ATCC14579), and Gram-negative bacteria viz. E. coli ATCC11775 by disk diffusion method (Bauer et al., 1996).
For the antibacterial assay, standard inoculums (1–2 × 107c.f.u/Ml 0.5 McFarland standards) were introduced onto the surface of sterile agar plates, and a sterile glass spreader was used for even distribution of the inoculums. The disks measuring 6 mm in diameter were prepared from Whatman no. 1 filter paper and sterilized by dry heat at 140 °C for 1 h. The sterile disks previously soaked in a known concentration of the test compounds were placed in the nutrient agar medium. The plates were inverted and incubated for 24 h at 37 °C. The inhibition zones were measured and compared with the standard drug Penicillin. The data are presented in Table 1.
Minimum inhibitory concentration (MIC) was determined by the broth micro-dilution procedures (NCCLS, 1982; Rostom et al., 2009). Bacterial colonies of the test organisms were suspended directly into a small volume of 0.9 % saline and further diluted until turbidity matched the Mc Farland Standard no: 0.5 Petri dishes containing Mueller-Hinton agar were impregnated with these microbial suspensions. The stock solutions of the synthesized compounds were prepared in dimethyl sulfoxide (DMSO), which had no effect on the organisms in the concentrations studied. All of the dilutions were done with distillated water. The solution of the newly synthesized compounds and standard drug were prepared at 1024, 512, 256, 128, 64, 32, and 16 μg/mL. DMSO was used as negative control. Penicillin was used as reference drug for antibacterial activity. All the inoculated plates were incubated at 37 °C, and results were evaluated after 24 h for bacteria. The lowest concentration of the compounds that prevented visible growth was considered minimal inhibitor concentrations (MICs) (Table 2).
Antifungal evaluation
Preliminary in vitro anti-fungal testing using diffusion disk method
All compounds 1–13 were also evaluated for in vitro antifungal activity against two fungi viz. C. albicans (ATCC26555) and A. flavus (ATCC 11495), by agar diffusion method (NCCLS, 1982). For the antifungal assay, Sabourands agar media was prepared by dissolving peptone (1 g), d-glucose (4 g), and agar (2 g) in distilled water (100 mL) and adjusting the pH to 5.7. Normal saline was used to make a suspension of spore of fungal strain for lawning. A loopful of particular fungal strain was transferred to 3 mL saline to get a suspension of corresponding species. 20 mL of agar media was poured into each petri-dish, excess of suspension was decanted, and the plates were dried by placing in an incubator at 37 °C for 1 h. Using agar punch, wells were made, and each well was labeled. A control was also prepared and maintained at 37 °C for 3–4 days. The C. albicans was grown for 48 h at 28 °C in YPD broth (1 % yeast extract, 2 % peptone, and 2 % dextrose), harvested by centrifugation and then washed twice with sterile distilled water. A. flavus was plated in potato dextrose agar (PDA) (Difco) and incubated at 28 °C for 48 h. Spores were washed three times with sterile-distilled water and resuspended in distilled water to obtain an initial inoculums size of 105 spores/mL. Each plate contained three paper disks of the compound tested, standard drug, and solvent (DMSO). The plates were kept undisturbed for at least 2 h at room temperature. After incubation of the plates, the diameter of the inhibition zone was measured and compared with the standard drug Fluconazole (Table 1).
The in vitro antifungal activity of the compounds was tested in Sabouraud’s dextrose broth (Difco) by micro-dilution procedures (NCCLS, 1982; Rostom et al., 2009). Fungal colonies of the test organisms were suspended directly into a small volume of 0.9 % saline and further diluted until turbidity matched the Mc Farland Standard no: 0.5 Petri dishes containing Sabouraud Dextrose agar were impregnated with these microbial suspensions. The stock solutions of the synthesized compounds were prepared in DMSO, which had no effect on the organisms in the concentrations studied. All of the dilutions were done with distillated water. The solution of the newly synthesized compounds and standard drug were prepared at 1024, 512, 256, 128, 64, 32, and 16 μg/mL. DMSO was used as negative control. Fluconazole was used as reference drug for antifungal activity. All the inoculated plates were incubated at 28 °C, and results were evaluated after 48 h for fungi. The lowest concentration of the compounds that prevented visible growth was considered minimal inhibitor concentrations (MICs) (Table 2).
Antioxidant activity
Determination of radical scavenging activity using DPPH assay: indolyl-pyrimidine derivatives 3, 4, 8, 9g, and 11 were evaluated for their antioxidative potential through in vitro DPPH radical scavenging model. The DPPH (2,2-diphenyl-1-picrylhydrazyl) radical scavenging effect was carried out according to reported methods (Blois, 1958; Brand-Williams et al., 1995). Compounds of different concentrations were prepared in DMSO; 1 mL of each compound solutions having different concentrations (10, 25, 50, 100, 200, and 300 μg/mL) were taken in different test tubes; 4 mL of 0.1 mg/mL DMSO solution of DPPH was added and shaken vigorously. The tubes were then incubated in the dark room at RT for 20 min. A DPPH blank was prepared without compound, and DMSO was used for the baseline correction. Changes (decrease) in the absorbance at 517 nm were measured using a UV–Visible spectrophotometer (Shimadzu 160A). Scavenging of DPPH free radicals was calculated from the following equation:
where A c is absorbance of control, A t is absorbance of compound.
Results
The relation between DPPH scavenging percentage against compound concentrations is plotted to get compound concentration providing a 50 % decrease in absorbance of DPPH radical (IC50 %). The radical scavenging activities were expressed as IC50. Vitamin C served as reference compound. The results have been given in Table 3 and Fig. 3.
In vitro cytotoxicity activity
Indolyl-pyrimidines 3, 4, 8, 9g, and 11 were subjected to a screening system for evaluation of their anticancer activity against cell line of human cancer, namely colon (CaCo-2) cancer obtained from pharmacology screening unit of the National Cancer Institute (NCI), Cairo University, Egypt, following the Sulfo Rhod-amine-B-stain (SRB) assay method (Skehan et al., 1990) in comparison to the known anticancer drugs: Doxorubicin. The SRB assay, which was developed in 1990, is one of the most widely used methods where it relies on the ability of SRB to bind to protein components of the cells that have been fixed to tissue-culture plates by trichloroacetic acid (TCA). As the binding of SRB is stoichiometric, the amount of dye extracted from stained cells is directly proportional to the cell mass.
Materials, methods, and reagents
Fetal calf serum (FCS) was from Invitrogen Co. (Carlsbad, CA). DMEM medium was from Cambrex (New Jersey, USA). DMSO, doxorubicin, penicillin, streptomycin, and sulforhodamine B (SRB) were from Sigma Chemical Co. (St. Louis, USA). Samples: Stock solutions of compounds were prepared in DMSO and kept at 20 °C. Appropriate dilutions of the compounds were freshly prepared just prior to the assays. Final concentrations of DMSO did not interfere with the cell growth.
SRB cytotoxic assay
The cultured colon carcinoma cell CaCo-2 from the National Cancer Institute (NCI, Cairo, Egypt) is routinely maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal calf serum (FCS), antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin). Cells were plated in 96-multiwell plate (104 cells/well) for 24 h before treatment with the compounds to allow attachment of cells to the wall of the plate. Test compounds were dissolved in DMSO and diluted with saline to the appropriate volume. Different concentrations of the compound under test (0, 1, 2.5, 5, 10 μg/mL) were added to the cell monolayer. Triplicate wells were prepared for each individual dose. Monolayer cells were incubated with the compounds for 48 h at 37 °C and in an atmosphere of 5 % CO2. After 48 h, cells were fixed, washed, and stained with Sulfo-Rhodamine-B stain. Excess stain was washed with acetic acid, and attached stain was recovered with Tris–EDTA buffer. Color intensity is measured in an ELISA reader at a wavelength of 570 nm. Results are expressed as means of at least three independent experiments performed in duplicate. The results are expressed as growth inhibition of 50 % (IC50) of cells (Table 4).
Results and discussion
Chemistry
The synthetic strategy to synthesize the target indolyl-pyrimidine compounds 1–13 is depicted in Schemes 1 and 2. The synthesis of 4-chloro-2-thiopyrimidine 2 has been achieved via heating under reflux a mixture of 4-oxo-2-thiopyrimidine analog 1 with phosphorus oxychloride and phosphorus pentachloride in boiling water bath (Mohamed et al., 2011c), then converted the latter to 4-hydrazino derivative 3 by reaction with hydrazine hydrate (99 %) in methanol. This hydrazino derivative was the key compound for preparation of all the rest indolyl-pyrimidine derivatives. The condensation of 4-hydrazino derivative 3 with acetic anhydride, triethyl orthoformate (TEOF), and formic or acetic acid (Mohamed et al., 2005; Mohamed et al., 2011a; Taher and Helwa, 2012) afforded 7-(1H-indol-3-yl)-5-thioxo-[1,2,4]triazolo[4,3-c]pyrimidine-8-carbonitriles 4, 5, and 7-(1H-indol-3-yl)-5-thioxo-[1,2,4]triazolo[1,5-c]pyrimidine-8-carbonitriles 6, 7, while reaction of compound 3 with diethyl oxalate in refluxing ethanol yielded carbonitrile 8. Refluxing of 3 with appropriate aldehydes in absolute ethanol (Cocco et al., 2006) gave 4-[(2E)-2-benzylidene)hydrazino]-6-(1H-indol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitriles 9a–j as revealed in Scheme 1. The reflux of 3 with acetylacetone in acetic acid afforded compound 10 (El-zahar et al., 2011). Further, cyclocondensation of 3 with ethyl chloroformate or CS2/KOH afforded carbonitriles 11 and 12, respectively (Fathalla et al., 2012; Mohamed et al., 2012a). Finally, 7-(1H-indol-3-yl)-5-thioxo-5,6-dihydrotetrazolo[1,5-c]pyrimidine-8-carbonitrile 13 was produced by stirring with sodium nitrite in HCl (El-Sawy et al., 2010) as revealed in Scheme 2. The structures of new compounds were confirmed by MS, IR, 1H-NMR, 13C-NMR, as well as elemental analysis. The structure of 2 was established on the basis of IR, which showed CN band at 2220 cm−1 and absorbance band at 3200 cm−1 corresponding NH, while 1H-NMR spectrum revealed signals for two NH protons. IR spectrum of compound 3 showed two absorbance bands at 3460–3200 cm−1 corresponding NHNH2. 1H-NMR spectrum showed three singlets of NHNH2 around 5.1 and 5.7 ppm. The IR spectrum of 4 showed the absence of absorbance bands at 3460–3200 cm−1 for (NHNH2).Its 1H-NMR spectrum showed a singlet signal at δ 2.3 ppm (CH3) and two singlets at δ 10.1 and 11.5 ppm (2NH). The 1H-NMR for 5 showed singlet signals for C5–H and two NH. Also, MS spectra gave their molecular ion peaks. The 1H-NMR spectrum for 6 revealed singlet signals for C3–H and two NH, for 7 signals for CH3 and two NH. Compounds 8 and 9 were confirmed by spectral data, and the mass spectrum studies of these compounds gave additional evidence for the proposed structures. The 1H-NMR of 10 showed two singlet signals at 2.3, 2.5 ppm corresponding to two CH3 group and only two singlets corresponding to NH. The IR spectrum of product 10 was compatible with the proposed structure. The IR spectrum of compound 11 displayed absorbance band at 1665 cm−1 for C=O. The 1H-NMR and mass spectra supported the structure. The IR spectrum of 12 showed additional absorbance band corresponding to the new C=S, and 1H-NMR showed three singlet signals for three NH protons. The absence of absorbance bands at 3460–3200 cm−1 (NHNH2) in IR spectrum was characteristic for compound 13. All structures were assigned by their mass spectra, 13C-NMR and elemental analysis.
Antimicrobial studies
Antibacterial activity
The antibacterial data (Tables 1, 2) revealed that all tested compounds have moderate to high antibacterial activity, except compounds 4, 6, and 7 were either inactive or weakly active against the tested microorganism. As compared to the standard drug Penicillin, compounds 5 and 9g, i, and j showed very promising activity against S. aureus, B. cereus, E. coli (MIC = 16–32 μg/mL). Compound 3 shows potent antibacterial activity against S. aureus and E. coli (MIC = 16 and 32 μg/mL, respectively), compound 10 against B. cereus and E. coli (MIC = 32 μg/mL). Also, compound 13 shows potent activity against S. aureus (MIC = 16 μg/mL), B. cereus, and E. coli (MIC = 32 μg/mL).
Antifungal activity
The screening data of antifungal activity of these compounds show moderate to good activity against the tested fungi except compounds 4, 10, 11, and 13 were inactive against C. albicans and A. flavus. Compounds 5 and 9g, i, and j exhibited potent in vitro antifungal activity against C. albicans and A.flavus (MIC = 16 μg/mL) compared to the standard drug Fluconazole (MIC = 16 μg/mL). Also compound 2 showed pronounced antifungal activity against A. flavus (MIC = 16 μg/mL).
Structure–activity relationship (SAR)
The SAR of newly synthesized compounds based on the observed results explored the importance of the nature of the heterocycles nucleus and the nature of the substituent on position 4 of pyrimidine.
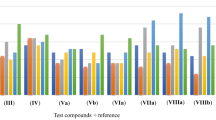
First, regarding the influence of the nature of the heterocycles nucleus, it is observed that triazolo[4,3-c]pyrimidine 5 acquired significant antimicrobial activity against Gram-positive (S. aureus, B. cereus), Gram-negative (E. coli) bacteria and fungi (C. albicans and A. flavus). On the other hand, increased antibacterial activity was achieved by cyclization to pyazolopyrimidine 10 or tetrazolopyrimidine 13 against S. aureus, B. cereus, and E. coli, while cyclization to pyrimido[6,1-c][1,2,4]triazine 8 or triazolo[4,3-c]pyrimidine 11 increased activity only against S. aureus or A. flavus. However, cyclization to triazolo[1,5-c]pyrimidines 6 and 7 or triazolo[4,3-c] pyrimidines as in 4 and 12 either decreased or diminished antimicrobial activity (Fig. 1).

Antimicrobial activity (Gram +ve, −ve, Fungi) of synthesized compounds
Regarding the nature of the substituent, 4-chloropyrimidine 2 shows significant activity against B. cereus, E. coli (MIC = 32 μg/mL), and A. flavus (MIC = 16 μg/mL) and moderate activity against S. aureus and C. albicans (MIC = 32 μg/mL). Compound 3, in which the chlorine in the 4-position is replaced by hydrazine hydrate, shows significant activity against S. aureus (MIC = 16 μg/mL), E. coli (MIC = 32 μg/mL), and moderate activity against B. cereus (MIC = 64 μg/mL) and A. flavus (MIC = 32 μg/mL).Conversion of 4-hydrazine derivative 3 to hydrazones (9a–j) caused a pronounced inhibition effect against S. aureus, B. cereus, E. coli, C. Albicans, and A. flavus (MIC = 16–32 μg/mL).
The in vitro antimicrobial activity of compounds 9a–j is shown to be increased when groups such as 3,4,5-(OCH3)3,-NO2, and -CH3 are present as in 9g, i and j, respectively (MIC = 16 μg/mL).
The SAR suggested that conversion of indolyl-pyrimidine to hydrazones 9g, i, and j or triazolo [4,3-c] pyrimidine 5 showed higher antibacterial and antifungal activities than other derivatives.
Antioxidant evaluation
DPPH free radical scavenging assay
Recent evidence suggests that free radicals, which are generated in any bioorganic redox processes, may induce oxidative damage in various components of human body (e.g., lipids, proteins, and nucleic acids). Antioxidants are very interesting, particularly in terms of prevention of the presumed deleterious effects of free radicals in the human body and in fats or other constituents of food stuffs. There is therefore a parallel increase in the use of methods for estimating the efficiency of such substances as antioxidants (Carocho and Ferreira, 2013). One such method that is currently popular is based on the use of the stable, free radical diphenyl picryl hydrazyl (DPPH). A freshly prepared DPPH solution exhibits a deep purple color with an absorption maximum at 517 nm. This purple color generally disappears when an antioxidant is present in the medium. Thus, antioxidant molecules can quench DPPH free radicals, by providing hydrogen atoms (Fig. 2) or by electron donation via a free radical attack on the DPPH molecule, and convert them to colorless product (Amarowicz et al., 2004; Siddhuraju and Becker, 2007).

The DPPH radical scavenging assay
Free radical scavenging activity of compounds 3, 4, 8, 9g, and 11 was evaluated by DPPH assay and compared to those of the well-known antioxidant vitamin C (Table 3). We can conclude that the studied compounds were able to reduce DPPH in a concentration-dependent manner. The tested samples were statistically different (P < 0.05, Kruskal–Wallis test) over the dose range used. Compounds 3 and 9g showed more potent-free radical scavenging activity (IC50 = 7.52 and 6.21 μg/mL, respectively) than the reference drug, vitamin C (IC50 = 10 μg/mL) (Fig. 3).

Screening of antioxidant activity by the DPPH assay shows that 3 and 9g have highest activity (more potent than the reference drug (RF). Each value represents a mean ± SEM (n = 3)
On critical overview of synthesized compounds, it has been found that compounds with electron donating group (–OCH3) on phenyl ring exhibited potent antioxidant activity.
Structure activity relationship (SAR)
Interpretation of the obtained results and considering the SAR of the tested compounds showed that, 4-hydrazinopyrimidine 3 and hydrazone derivative with substituent (R = 3,4,5-OCH3) 9g possess potent antioxidant activity. While, triazolo[4,3-c]pyrimidines as in 4 and 11 and pyrimido[6,1-c][1,2,4]triazine 8 showed moderate antioxidant activity.
Therefore, more active compounds (3 and 9g) because it quenches DPPH free radical by providing hydrogen atoms or by electron donation via a free radical attack on the DPPH molecule.
Cytotoxic activity
Five of the newly synthesized compounds (3, 4, 8, 9g, 11) were primary screened for their in vitro cytotoxicity against (CaCo-2) cell line (Table 4). Compounds 3 (with hydrazine hydrate group in molecule) and 8 (with triazine moiety) showed moderate cytotoxic activity against CaCo-2 cell line (IC50 = 28.59 and 38.55 μg/mL, respectively).Compound 11 (with triazole moiety) showed significant cytotoxic activity (IC50 = 15.97 μg/mL). Compounds 4 (with 3-methyl triazole moiety) and 9g (with phenyl hydrazono moiety) showed more potent activity (IC50 = 8.28 and 8.84 μg/mL, respectively) than the reference drug doxorubicin (IC50 = 12 μg/mL).
Compounds 4 and 9g that contain 3-methyl triazole and phenyl hydrazono moieties attached to indolyl-pyrimidine moiety are associated with remarkable cytotoxicity against CaCo-2 cancer cell line.
Structural–activity relationship (SAR)
From the above obtained results (Table 1), we can conclude that the anticancer activity is due to:
-
(i)
The presence of triazole, phenylhydrazone moieties.
-
(ii)
The presence of nitrile generally enhancing the activity.
-
(iii)
The presence of 2-thiouracil, indole moieties is essential for activity.
Summary
We have synthesized a series of novel indolyl-pyrimidine derivatives and have been screened as antimicrobial, antioxidant, and anticancer agents. Interpretation of the results with considering the SAR proved that the most promising antimicrobial compounds are 7-(1H-indol-3-yl)-5-thioxo-5,6-dihydro[1,2,4]triazolo[4,3-c]pyrimidine-8-carbonitrile (5), 6-(1H-indol-3-yl)-4-[(2E)-2-(3,4,5-trimethoxybenzylidene)hydrazino]-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9g), 6-(1H-indol-3-yl)-4-[(2E)-2-(4-nitrobenzylidene)hydrazino]-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9i) and 6-(1H-indol-3-yl)-4-[(2E)-2-(4-methylbenzylidene)hydrazino]-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (9j). Compounds 3 and 9g were found to possess promising antioxidant activity when compared with standard ascorbic acid (vitamin C). Compounds 4 and 9g were found to be the highly active compounds against CaCo-2 cell line compared to the reference drug doxorubicin. In conclusion, the preliminary biological studies lead to the identification of novel antimicrobials, antioxidant, and cytotoxic agents. The findings demonstrate indolyl-pyrimidines as novel leads for further development as medicinal agents.
References
Abu-Hashem AA, Youssef MM, Hussein HAR (2011) Synthesis, antioxidant, antitumor activities of some new thiazolopyrimidines, pyrrolothiazolopyrimidines and triazolopyrrolothiazolopyrimidines derivatives. J Chin Chem Soc 58:41–48
Agbaje OC, Fadeyi OO, Fadeyi SA, Myles LE, Okoro CO (2011) Synthesis and in vitro cytotoxicity evaluation of some fluorinated hexahydropyrimidine derivatives. Bioorg Med Chem Lett 21:989–992
Al-Masoudi NA, Saleh BA, Abdul Karim NA, Issa AY, Pannecouque C (2011) Synthesis and anti-HIV activity of new 2-thiolumazine and 2-thiouracil metal complexes. Heteroat Chem 22(1):44–50
Amarowicz R, Pegg RB, Rahimi-Moghaddam P, Barl B, Weil JA (2004) Free-radical scavenging capacity and antioxidant activity of selected plant species from the Canadian prairies. Food Chem 84:551–562
Ashok M, Holla BS, Poojary B (2007) Convenient one pot synthesis and antimicrobial evaluation of some new mannich bases carrying 4-methylthiobenzyl moiety. Eur J Med Chem 42:1095–1101
Atta KFM (2011) Synthesis and electrophilic substitutions of novel pyrazolo[1,5-c]-1,2,4-triazolo[4,3-a] pyrimidines. Molecules 16:7081–7096. doi:10.3390/molecules16087081
Awad SM, Fathalla OA, Wietrzyk J, Milczarek M, Soliman AM, Mohamed MS (2013) Synthesis of new pyrimidine derivatives and their antiproliferative activity against selected human cancer cell lines. Res Chem Intermed. doi:10.1007/s11164-013-1312-z
Bauer AW, Kirby MM, Sherris JC, Turck M (1996) Antibiotic susceptibility testing by a standardized single disc method. Am J Clin Pathol 45:493–496
Benabadji S, Wen R, Zheng JB, Dong X, Yaun S (2004) Anticarcinogenic and antioxidant activity of diindolylmethane derivatives. Acta Pharmacol Sin 25(5):666–671
Blois MS (1958) Antioxidant determinations by the use of a stable free radical. Nature 181:1199–1200
Brand-Williams W, Cuvelier ME, Berset C (1995) Use of a free-radical method to evaluate antioxidant activity. J Food Sci Technol 28:25–30
Cai S, Li QS, Borchardt RT, Kuczera K, Schowen RL (2007) The antiviral drug ribavirin is a selective inhibitor of S-adenosyl-lhomocysteine hydrolase from Trypanosoma cruzi. Bioorg Med Chem 15:7281–7287
Carocho M, Ferreira ICFR (2013) A review on antioxidants, prooxidants and related controversy: natural and synthetic compounds, screening and analysis methodologies and future perspectives. Food Chem Toxicol 51:15–25
Chaudhari PK, Sikotra KH, Shah VH (2011) Synthesis and biological studies of pyrazolo[3,4-d]pyrimidines. Orient J Chem 27(1):95–100
Cocco MT, Congiu C, Lilliu V, Onnis V (2006) Synthesis and in vitro antitumoral activity of new hydrazino pyrimidine-5-carbonitrile derivatives. Bioor Med Chem 14:366–372
Cooper DS (2005) Drug therapy—antithyroid drugs. N Engl J Med 352:905–917
Ding Y, Girardet J, Smith KL, Larson G, Prigaro B, Wu JZ, Yao N (2006) Parallel synthesis of 5-cyano-6-aryl-2-thiouracil derivatives as inhibitors for hepatitis C viral NS5B RNA-dependent RNA polymerase. Bioorg Chem 34:26–38
Edrees MM, Farghaly TA, El-Hag FAA, Abdalla MM (2010) Antimicrobial, antitumor and 5α-reductase inhibitor activities of some hydrazonoyl substituted pyrimidinones. Eur J Med Chem 45:5702–5707
El-Sawy ER, Bassyouni FA, Abu-bakr SH, Rady HM, Abdlla MM (2010) Synthesis and biological activity of some new 1-benzyland 1-benzoyl-3-heterocyclic indole derivatives. Acta Pharm 60:55–71
El-zahar MI, Abd el-karim SS, Haiba ME, Khedr MA (2011) Synthesis, antitumor activity and molecular docking study of novel benzofuran-2-ylpyrazole pyrimidine derivatives. Acta Pol Pharm 68(3):357–373
Fathalla OA, Zeid IF, Haiba ME, Soliman AM, Abd-Elmoez ShI, El-Serwy WS (2009) Synthesis, antibacterial and anticancer evaluation of some pyrimidine derivatives. World J Chem 4(2):127–132
Fathalla OAM, Ismail MAH, Anwar MM, Abouzid KAM, Ramadan AAK (2012) Novel 2-thiopyrimidine derivatives as CDK2 inhibitors: molecular modeling, synthesis, and anti-tumor activity evaluation. Med Chem Res. doi:10.1007/s00044-012-0051-9
Grigoryan LA, Kaldrikyan MA, Melik-Ogandzhanyan RG, Arsenyan FG, Stepanyan GM, Garibdzhanyan BG (2005) Synthesis and antitumor activity of 2-S-substituted pyrimidine derivatives. Pharm Chem J 39(9):468–472
Gupta A, Unadkat JD, Mao Q (2007) Interactions of azole antifungal agents with the human breast cancer resistance protein (BCRP). J Pharm Sci 96:3226–3235
Hancu G, Gaspar A, Gyeresi A (2007) Separation of 1,4-benzodiazepines by micellar electrokinetic capilary chromatography. J Biochem Biophys Methods 69:251–259
Hanna MM (2012) New pyrimido[5,4-e]pyrrolo[1,2-c]pyrimidines: synthesis, 2D-QSAR, anti-inflammatory, analgesic and ulcerogenicity studies. Eur J Med Chem 55:12–22
Ibrahim DA, El-Metwally AM (2010) Design, synthesis and biological evaluation of novel pyrimidine derivatives as CDK2 inhibitors. Eur J Med Chem 45:1158–1166
Kadry AM, Abdel Aal EH, Abdel-Fattah HA, Al-Mahmoudy AM (2008) Synthesis and antimicrobial activity of new triazolopyrimidine carbonitrile derivatives. Arkivoc 10:127–134
Kotaiah Y, Hari Krishna N, Naga Raju K, Rao CV, Jonnalagadda SB, Maddila S (2012) Synthesis and biological evaluation of novel isopropyl 2-thiazolopyrimidine-6-carboxylate derivatives. J Korean Chem Soc 56(1):68–73
Kumar D, Maruthi Kumar N, Chang K, Shah K (2010) Synthesis and anticancer activity of 5-(3-indolyl)-1,3,4-thiadiazoles. Eur J Med Chem 45:4664–4668
Le Brazidec J, Pasis A, Tam B, Boykin C, Black C, Wang D, Claassen G, Chong J, Chao J, Fan J, Nguyen K, Silvian L, Ling L, Zhang L, Choi M, Teng M, Pathan N, Zhao S, Li T, Taveras A (2012) Synthesis, SAR and biological evaluation of 1,6-disubstituted-1H-pyrazolo[3,4-d]pyrimidines as dual inhibitors of Aurora kinases and CDK1. Bioorg Med Chem Lett 22:2070–2074
Leboho TC, Michael JP, van Otterlo WAL, van Vuuren SF, de Koning CB (2009) The synthesis of 2- and 3-aryl indoles and 1,3,4,5-tetrahydropyrano[4,3-b]indoles and their antibacterial and antifungal activity. Bioorg Med Chem Lett 19:4948–4951
Mahboobi S, Sellmer A, Eichhorn E, Beckers T, Fiebig H, Kelter G (2005) Synthesis and cytotoxic activity of 2-acyl-1H-indole-4,7-diones on human cancer cell lines. Eur J Med Chem 40:85–92
Martinez-Montero S, Fernandez S, Sanghvi YS, Theodorakis EA, Detorio MA, Mcbrayer TR, Whitaker T, Schinazi RF, Gotor V, Ferrero M (2012) Synthesis, evaluation of anti-HIV-1 and anti-HCV activity of novel 2′,3′dideoxy-2′,2′-difluoro-4′-azanucleosides. Bioorg Med Chem 20:6885–6893
Mohamed MS, Rashad AE, Zaki MEA, Fathalla SS (2005) Synthesis and antimicrobial screening of some fused heterocyclic pyrroles. Acta Pharm 55:237–249
Mohamed MS, Hussein WM, McGeary RP, Vella P, Schenk G, Abd El-hameed RH (2011a) Synthesis and kinetic testing of new inhibitors for a metallo-β-lactamase from Klebsiella pneumonia and Pseudomonas aeruginosa. Eur J Med Chem 46:6075–6082
Mohamed MS, Awad SM, Ahmed NM (2011b) Synthesis and antimicrobial activities of new indolyl-pyrimidine derivatives. J Appl Pharm Sci 1(5):76–80
Mohamed MS, Awad SM, Ahmed NM (2011c) Synthesis and antimicrobial evaluation of some 6-aryl-5-cyano-2-thiouracil derivatives. Acta Pharm 61:171–185
Mohamed MS, Kamel R, Abd El-hameed RH (2012a) Evaluation of the anti-inflammatory activity of some pyrrolo[2,3-d]pyrimidine derivatives. Med Chem Res. doi:10.1007/s00044-012-0217-5
Mohamed MS, Awad SM, Ahmed NM (2012b) Anti-cancer activities of 6-aryl-5-cyano-2-thiouracil derivatives. Pharm Res 6(2):54–60
National Committee for Clinical Laboratory Standards (NCCLS) (1982) Standard methods for dilution antimicrobial susceptibility tests for bacteria, which grows aerobically. NCCLS, Villanova
Nguyen HH, Lavrenov SN, Sundar SN, Nguyen DHH, Senga MT, Marconett CN, Kung J, Staub RE, Preobrazhenskaya MN, Bjeldanes LF, Firestone GL (2010) 1-Benzyl-indole-3-carbinol is a novel indole-3-carbinol derivative with significantly enhanced potency of anti-proliferative and anti-estrogenic properties in human breast cancer cells. Chem Biol Interact 186:255–266
Nitulescu GM, Draghici C, Missir AV (2010) Synthesis of new pyrazole derivatives and their anticancer evaluation. Eur J Med Chem 45:4914–4919
Onnis V, Cocco MT, Fadda R, Congiu C (2009) Synthesis and evaluation of anticancer activity of 2-aryl amino-6-trifluoromethyl-3-(hydrazonocarbonyl)pyridines. Bioorg Med Chem 17:6158–6165
Prachayasittikul S, Sornsongkhram N, Pingaew R, Techatanachai S, Ruchirawat S, Prachayasittikul V (2009) Synthesis and novel bioactivities of substituted 6-propylthiouracils. Eur J Sci Res 36(2):236–245
Ram VJ, Vanden Berghe DA, Vlietinck AJ (1987) Syntheses and activities of novel pyrimidines derived from 5-cyano-6-aryl2-thiouracil. Liebigs Ann Chem 5:797–801
Rostom SAF, Ashour HMA, El Razik HAA (2009) Synthesis and biological evaluation of some novel poly substituted pyrimidine derivatives as potential anti microbial and anti cancer agents. Arch Pharm Chem Life Sci 342:299–310. doi:10.1002/ardp.200800223
Schiller SD, Fung HB (2007) Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther 29:1862–1886
Sellitto G, Faruolo A, Caprariis P, Altamura S, Paonessa G, Ciliberto G (2010) Synthesis and anti-hepatitis C virus activity of novel ethyl-1H-indole-3-carboxylates in vitro. Bioorg Med Chem 18:6143–6148
Skehan P, Storenge R, Scudiero D, Monks S, McMahon J, Vistica D, Warren J, Boesch H, Kenny S, Boyed MR (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112
Siddhuraju P, Becker K (2007) The antioxidant and free radical scavenging activities of processed cowpea (Vigna unguiculata (L.) Walp.) seed extracts. Food Chem 101:10–19
Singh R, Pujar GV, Purohit MN, Chandrashekar VM (2012) Synthesis, in vitro cytotoxicity, and anti-bacterial studies of new asymmetric bis-1,2,4-triazoles. Med Chem Res. doi:10.1007/s00044-012-0209-5
Souli E, Machluf M, Morgenstern A, Sabo E, Yannai S (2008) Indole-3-carbinol (I3C) exhibits inhibitory and preventive effects on prostate tumors in mice. Food Chem Toxicol 46:863–870
Taher AT, Abou-Seri SM (2012) Synthesis and bioactivity evaluation of new 6-aryl-5-cyano thiouracils as potential antimicrobial and anticancer agents. Molecules 17:9868–9886
Taher AT, Helwa AA (2012) Novel pyrimidinone derivatives: synthesis, antitumor and antimicrobial evaluation. Chem Pharm Bull 60(4):521–530
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mohamed, M.S., Youns, M.M. & Ahmed, N.M. Novel indolyl-pyrimidine derivatives: synthesis, antimicrobial, and antioxidant evaluations. Med Chem Res 23, 3374–3388 (2014). https://doi.org/10.1007/s00044-014-0916-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-014-0916-1