
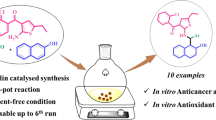
Abstract
A facile, green, and efficient method has been developed for the synthesis of pyrano[4, 3-b]pyrans via a one-pot reaction of aromatic aldehydes, malononitrile, and 4-hydroxy-6-methyl-pyran-2-one in the presence of ammonium acetate under solvent-free conditions using grinding method. The experimental simplicity, short reaction time, easy work-up, avoidance of organic solvents, and utilization of an inexpensive and readily available catalyst make this new methodology practical and economically attractive. The antitumor activity of the compounds was tested against human breast cancer cell line MCF-7. Compounds 4l, 4o, and 4p showed good cytotoxic activity comparable with standard drug Doxorubicin. On the other hand, compounds 4q and 4r exhibited potent growth inhibitory activity. Furthermore, the synthesized compounds were screened for in vitro antioxidant activity by DPPH assay. All the compounds assayed showed moderate to good free radical scavenging activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the past several years, chemists have been aware of the environmental implications of their chemistry. Nowadays, they are trying to develop new synthetic methods, reaction conditions, and uses of chemicals that reduce the risks to humans and the environment. Organic solvents are high on the list of hazardous chemicals because they are used in large amounts and are usually volatile liquids. Therefore, in recent years, solventless organic reactions have attracted great interest. They have many advantages such as high efficiency and selectivity, operational simplicity, low costs, mild reaction conditions, and reduced pollution (Tanaka and Toda, 2000; Shirini et al., 2007; Thirunarayanan and Vanangamudi, 2006). The grinding method for the solid-state reactions has been reported for Reformatsky reaction (Tanaka et al., 1991), Aldol condensation (Toda et al., 1990), Dieckmann condensation (Toda et al., 1998), Knoevenagel condensation (Ren et al., 2002), and other reactions (Ren et al., 2004, 2005; Schmeyers et al., 1998).
It is well known that fused pyran derivatives possess a variety of a pharmacological and biological properties, such as fungicidal, insecticidal, and acaricidal activity (Uher et al., 1994), antiviral and antileishmanial activity (Perez–Perez et al., 1995; Fan et al., 2010), and anticonvulsant activity (Aytemir et al., 2004). Moreover, they have been introduced as non-peptide human immunodeficiency virus (HIV) protease inhibitors (Wang et al., 1996; Pochet et al., 1996; Mazumder et al., 1996). Despite their wide range of pharmacological, industrial, and synthetic applications, the synthesis of pyrano[4, 3-b]pyrans has received little attention. Recently, a one-pot, three-component reaction of 4-hydroxy-6-methyl-pyran-2-one with malononitrile and aromatic aldehydes has enjoyed wider utilization in the synthesis of these compounds. A variety of reagents, such as piperidine (Piao and Imafuku, 1997; Stoyanov et al., 2000), TEBAC (Da-Qing et al., 2008), KF/Al2O3 (Wang et al., 2006), magnesium oxide (Seifi, and Sheibani 2008), DBU (Khurana et al., 2010), and ionic liquids (Shaabani et al., 2005) have been employed to accomplish this transformation. However, inspite of their potential utility, all of these methods suffer from one or more disadvantages, such as harsh reaction conditions, unsatisfactory yields, prolonged reaction times, tedious work-up, and use of organic solvents. As part of our current studies on the development of new routes to the synthesis of biologically active heterocycles, we wish to report, herein, a rapid and clean procedure for the preparation of pyrano[4, 3-b]pyrans. The present study also describes pyrano[4, 3-b]pyrans as potential cytotoxic and antioxidant agents.
Results and discussion
In an initial endeavor, equivalent amounts of benzaldehyde 1a, malononitrile 2, and 4-hydroxy-6-methyl-pyran-2-one 3, and ammonium acetate (10 mol %) were reacted in ethanol under reflux. After 2 h, only 64 % of the desired product 4a was obtained. In an attempt to improve the reaction yield and acknowledging the benefits of grinding, the same reaction was carried out by grinding under solvent-free conditions. Grinding for 10 min led to a yellow solid mass (the completion of the reaction was checked by TLC). The solid mass was washed with water, dried, and recrystallized from ethanol to give 2-Amino-7-methyl-5-oxo-4-phenyl-4H, 5H-pyrano[4, 3-b]pyran-3-carbonitrile 4a in 94 % yield. Other catalysts, such as basic alumina, Na2CO3, NaHCO3, K2CO3, and NH4Cl were not found as effective as ammonium acetate. The results are listed in Table 1. The effect of catalyst amount on the yield and rate was also investigated by using different amounts of ammonium acetate. Reaction with 5 mol % of the catalyst required longer reaction time and only 40 % yield of the product 4a was obtained. Excessive amount of catalyst (20 mol %) did not increase the yield remarkably.
The above optimized reaction condition was extended to a variety of aromatic aldehydes with different substituents. The results are summarized in Table 2. It is evident that all the aldehydes used, either bearing electron-donating or electron-withdrawing groups, reacted smoothly with malononitrile and 4-hydroxy-6-methyl-pyran-2-one to produce the corresponding pyrano[4, 3-b]pyran derivatives (4a–r) in excellent yields. It should be noted that the methodology worked well for heteroaromatic aldehydes (Table 2, entries 11–18).


Plausible reaction pathway for the formation of pyrano[4, 3-b]pyrans
The possible mechanism of the reaction is depicted in Scheme 1.
Cytotoxicity
In vitro cytotoxic activity of the synthesized compounds and Doxorubicin (Standard) was tested using the MTT assay against human breast cancer cell line MCF‐7. The results are presented in Table 3 as IC50 (μM) values. The data of Table 3 indicate that all the compounds tested exhibited moderate to remarkable cytotoxicity. The structure–activity relationship (SAR) suggested that insertion of halogens like Cl or Br into the aryl group of pyrano[4, 3-b]pyrans, as in 4e and 4f, induces an increase of the cytotoxicity. Introduction of the electron-donating substituents, such as Me and OMe, caused a decrease of the cytotoxic activity. This is why compounds 4b and 4c showed minimum inhibition with IC50 values of 54 and 57 μM, respectively. No significant effect on the antitumor activity was observed for the insertion of NO2 group into the aromatic ring of pyrano[4, 3-b]pyran, as in 4d, compared to that of its H substituted analog 4a. It is noteworthy to mention that replacement of aryl group of pyrano[4, 3-b]pyrans by heteroaromatic ring increased the cytotoxic activity to a great extent. Comparison of the data of the compounds 4l, 4m, and 4o indicates that insertion of Cl into the thiophene ring produces an increase in the cytotoxicity, whereas insertion of electron-donor Me group induces a decrease of the biological activity. Surprisingly, compounds 4q (IC50 = 7 μM) and 4r (IC50 = 5 μM), containing CN and Br substituted indole rings, respectively, showed potent cytotoxic effects.
Antioxidant activity
The synthesized compounds were screened for in vitro antioxidant activity by DPPH assay. The results obtained are depicted in Table 4 as IC50 (μM) values and compared with those of ascorbic acid as standard. Increased absorbance of the compounds with concentration (data not shown) reveals that compounds possess radical scavenging activity. Analysis of the data in Table 4 indicates that insertion of electron-donating Me and OMe groups, as in 4b and 4c, and halogens like Cl and Br, as in 4e and 4f, in the aryl group of pyrano[4, 3-b]pyrans induces an increase in the antioxidant property. On the contrary, introduction of Me in the thiophene ring and insertion of Br in the indole ring of pyrano[4, 3-b]pyrans, as in 4m and 4r, respectively, decreased the radical scavenging activity. The data of Table 4 indicate that insertion of electron-withdrawing substituents, such as NO2 and CN, as in 4d and 4q, produces a decrease in the antioxidant activity. Among the compounds tested, 4k and 4p exhibited potent free radical scavenging activity with IC50 values of 227.56 and 338.96 μM, respectively.
The overview of the results presented in Table 3 and Table 4 revealed that, in general, pyrano[4, 3-b]pyrans linked to heteroaromatic rings were more active than those containing aromatic rings. More studies are needed to be carried out to find the correlation between IC50 of the evaluated pyrano[4, 3-b]pyrans and their molecular descriptors, such as electronic, lipophilic, and steric parameters.
Conclusion
In summary, we have described a green and efficient method for the synthesis of pyrano[4, 3-b]pyran derivatives under solvent-free grinding conditions. The experimental simplicity, short reaction times, high yields, easy work-up procedure, avoidance of organic solvents, and utilization of an inexpensive and readily available catalyst make this novel protocol practical and economically attractive to prepare these derivatives. The synthesized compounds showed moderate to good in vitro cytotoxic activity against human breast cancer cell line MCF-7 when compared with standard drug Doxorubicin. Compounds 4q and 4r exhibited potent growth inhibitory activity. This study also describes pyrano[4, 3-b]pyrans as potential antioxidants.
Experimental section
General
All the chemicals were purchased from Merck and Sigma-Aldrich and used without further purification. Melting points were determined in open glass capillaries and are uncorrected. IR spectra were recorded on Perkin Elmer-1430 spectrophotometer using potassium bromide pellets. 1H and 13C NMR spectra were obtained at 400 MHz with a Bruker (AVANCE) spectrometer using DMSO-d6 as solvent and TMS as an internal standard. MS spectra were measured at Micromass ZMD ESI (70 eV) system.
General procedure for the synthesis of 2-Amino-4-aryl-5-oxo-4H, 5H-pyrano[4, 3-b]pyran-3-carbonitriles (4a–r)
Aromatic aldehyde 1 (2 mmol), malononitrile 2 (2.5 mmol), 4-hydroxy-6-methyl-pyran-2-one 3 (2 mmol), and ammonium acetate (10 mol %) were taken in a mortar and mixed thoroughly. The mixture was ground with a pestle at room temperature. The mixture becomes a sticky paste during the course of reaction which finally solidifies on completion of the reaction (monitored by TLC). The solid mass was washed with water, dried, and recrystallized from ethanol to obtain the pure product.
Spectroscopic and analytical data of selected compounds
2-Amino-4-(furan-2-yl)-7-methyl-5-oxo-4H, 5H-pyrano[4, 3-b]pyran-3-carbonitrile (4k, C14H10N2O4)
Pink crystals (from EtOH); m.p. 223–224 °C. IR (KBr):  = 3458, 3260, 3088, 2293, 1649, 1555, 1342, 1253, 1091, 790 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.25 (s, 3H, CH3), 4.47 (s, 1H, CH), 6.10 (s, 1H, =CH), 6.16 (d, 1H, J = 3.1 Hz, furan), 6.30 (dd, 1H, J
a
= 1.8, J
b
= 1.2 Hz, furan), 6.97 (s, 2H, NH2), 7.37 (d, 1H, J = 1.0 Hz, furan) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 19.43, 29.88, 55.36, 97.95, 98.40, 105.90, 110.17, 118.89, 141.55, 154.12, 158.58, 158.75, 161.13, 162.36 ppm. MS (70 eV): m/z = 270.0 (M+).
= 3458, 3260, 3088, 2293, 1649, 1555, 1342, 1253, 1091, 790 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.25 (s, 3H, CH3), 4.47 (s, 1H, CH), 6.10 (s, 1H, =CH), 6.16 (d, 1H, J = 3.1 Hz, furan), 6.30 (dd, 1H, J
a
= 1.8, J
b
= 1.2 Hz, furan), 6.97 (s, 2H, NH2), 7.37 (d, 1H, J = 1.0 Hz, furan) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 19.43, 29.88, 55.36, 97.95, 98.40, 105.90, 110.17, 118.89, 141.55, 154.12, 158.58, 158.75, 161.13, 162.36 ppm. MS (70 eV): m/z = 270.0 (M+).
2-Amino-7-methyl-5-oxo-4-(thiophen-2-yl)-4H, 5H-pyrano[4, 3-b]pyran-3-carbonitrile (4l, C14H10N2O3S)
Colorless crystals (from EtOH); m.p. 242–244 °C. IR (KBr): = 3475, 3376, 2926, 2212, 1654, 1540, 1301, 1032, 862, 754 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.23 (s, 3H, CH3), 4.66 (s, 1H, CH), 6.13 (s, 1H, =CH), 6.91 (dd, 1H, J
a
= 3.5 Hz, J
b
= 1.5 Hz, thiophene); 6.97 (d, 1H, J = 2.9 Hz, thiophene); 7.13 (s, 2H, NH2), 7.25 (d, 1H, J = 3.9 Hz, thiophene) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 19.40, 31.26, 57.81, 97.94, 100.89, 119.03, 124.44, 124.63, 126.61, 147.78, 157.72, 158.44, 161.22, 162.57 ppm. MS (70 eV): m/z = 286.1 (M+).
= 3475, 3376, 2926, 2212, 1654, 1540, 1301, 1032, 862, 754 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.23 (s, 3H, CH3), 4.66 (s, 1H, CH), 6.13 (s, 1H, =CH), 6.91 (dd, 1H, J
a
= 3.5 Hz, J
b
= 1.5 Hz, thiophene); 6.97 (d, 1H, J = 2.9 Hz, thiophene); 7.13 (s, 2H, NH2), 7.25 (d, 1H, J = 3.9 Hz, thiophene) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 19.40, 31.26, 57.81, 97.94, 100.89, 119.03, 124.44, 124.63, 126.61, 147.78, 157.72, 158.44, 161.22, 162.57 ppm. MS (70 eV): m/z = 286.1 (M+).
2-Amino-7-methyl-4-(5-methylthiophen-2-yl)-5-oxo-4H, 5H-pyrano[4, 3-b]pyran-3-carbonitrile (4m, C15H12N2O3S)
Brown crystals (from EtOH); m.p. 175–177 °C. IR (KBr): = 3476, 3358, 2965, 2226, 1643, 1548, 1352, 1272, 1082, 768 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.24 (s, 3H, CH3), 2.39 (s, 3H, CH3), 4.57 (s, 1H, CH), 6.04 (s, 1H, =CH), 6.55 (d, 1H, J = 1.6 Hz, thiophene), 6.74 (d, 1H, J = 3.3 Hz, thiophene), 6.84 (s, 2H, NH2) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 14.97, 19.44, 31.30, 58.19, 97.89, 101.02, 118.97, 124.29, 124.47, 138.17, 140.64, 157.51, 158.28, 161.30, 162.10 ppm. MS (70 eV): m/z = 301.1 (M++1).
= 3476, 3358, 2965, 2226, 1643, 1548, 1352, 1272, 1082, 768 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.24 (s, 3H, CH3), 2.39 (s, 3H, CH3), 4.57 (s, 1H, CH), 6.04 (s, 1H, =CH), 6.55 (d, 1H, J = 1.6 Hz, thiophene), 6.74 (d, 1H, J = 3.3 Hz, thiophene), 6.84 (s, 2H, NH2) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 14.97, 19.44, 31.30, 58.19, 97.89, 101.02, 118.97, 124.29, 124.47, 138.17, 140.64, 157.51, 158.28, 161.30, 162.10 ppm. MS (70 eV): m/z = 301.1 (M++1).
2-Amino-7-methyl-4-(3-methylthiophen-2-yl)-5-oxo-4H, 5H-pyrano[4, 3-b]pyran-3-carbonitrile (4n, C15H12N2O3S)
Brown crystals (from EtOH); m.p. 174–175 °C. IR (KBr): = 3472, 3375, 2962, 2223, 1648, 1552, 1332, 1275, 1076, 763 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.24 (s, 3H, CH3), 2.28 (s, 3H, CH3), 4.68 (s, 1H, CH), 6.15 (s, 1H, =CH), 6.73 (d, 1H, J = 5.0 Hz, thiophene), 7.03 (s, 2H, NH2), 7.15 (d, 1H, J = 2.5 Hz, thiophene) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 13.37, 19.39, 29.40, 58.40, 97.82, 100.93, 119.07, 122.69, 131.27, 133.46, 136.52, 157.64, 157.75, 161.09, 162.39 ppm. MS (70 eV): m/z = 301.1 (M++1).
= 3472, 3375, 2962, 2223, 1648, 1552, 1332, 1275, 1076, 763 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.24 (s, 3H, CH3), 2.28 (s, 3H, CH3), 4.68 (s, 1H, CH), 6.15 (s, 1H, =CH), 6.73 (d, 1H, J = 5.0 Hz, thiophene), 7.03 (s, 2H, NH2), 7.15 (d, 1H, J = 2.5 Hz, thiophene) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 13.37, 19.39, 29.40, 58.40, 97.82, 100.93, 119.07, 122.69, 131.27, 133.46, 136.52, 157.64, 157.75, 161.09, 162.39 ppm. MS (70 eV): m/z = 301.1 (M++1).
2-Amino-4-(5-chlorothiophen-2-yl)-7-methyl-5-oxo-4H, 5H-pyrano[4, 3-b]pyran-3-carbonitrile (4o, C14H9ClN2O3S)
Yellow crystals (from EtOH); m.p. 228–229 °C. IR (KBr): = 3463, 3358, 2952, 2216, 1632, 1546, 1306, 1028, 838, 767 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.22 (s, 3H, CH3), 4.24 (s, 1H, CH), 6.13 (s, 1H, =CH), 6.84 (d, 1H, J = 2.7 Hz, thiophene), 6.93 (s, 2H, NH2), 7.11 (d, 1H, J = 1.5 Hz, thiophene) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 19.35, 35.47, 58.28, 97.89, 101.18, 119.19, 123.95, 128.44, 133.26, 135.34, 159.97, 161.28, 162.07, 164.28 ppm. MS (70 eV): m/z = 320.0 (M+).
= 3463, 3358, 2952, 2216, 1632, 1546, 1306, 1028, 838, 767 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 2.22 (s, 3H, CH3), 4.24 (s, 1H, CH), 6.13 (s, 1H, =CH), 6.84 (d, 1H, J = 2.7 Hz, thiophene), 6.93 (s, 2H, NH2), 7.11 (d, 1H, J = 1.5 Hz, thiophene) ppm. 13C NMR (400 MHz, DMSO-d6): δ = 19.35, 35.47, 58.28, 97.89, 101.18, 119.19, 123.95, 128.44, 133.26, 135.34, 159.97, 161.28, 162.07, 164.28 ppm. MS (70 eV): m/z = 320.0 (M+).
In vitro cytotoxic activity
The cytotoxicity of the synthesized compounds was tested by MTT assay. MTT [(3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrasodium bromide)] is a pale yellow substrate that is cleaved by living cells to yield a dark blue formazan product. This process requires active mitochondria, and even freshly dead cells do not cleave significant amount of MTT. Thus the amount of MTT cleaved is directly proportional to the number of viable cells present, which is quantified by colorimetric methods. This assay was performed at Deshpande Laboratories, Bhopal using the standard operating procedures. Briefly, the compounds were dissolved in DMSO and serially diluted with complete medium to get the concentrations a range of test concentration. DMSO concentration was kept <0.1 % in all the samples. Cell lines maintained in appropriate conditions were seeded in 96 well plates and treated with different concentrations of the test samples and incubated at 37 °C, 5 % CO2 for 96 h. MTT reagent was added to the wells and incubated for 4 h; the dark blue formazan product formed by the cells was dissolved in DMSO under a safety cabinet and read at 550 nm. Percentage inhibitions were calculated and plotted with the concentrations used to calculate the IC50 values.
Free radical scavenging activity
The free radical scavenging activity of the synthesized compounds was tested by DPPH assay. DPPH is a free radical and accepts one electron or hydrogen radical to become a stable diamagnetic molecule. DPPH in methanol shows a strong absorption band at 517 nm (independent of pH from 5.0 to 6.5) and the solution appears to be deep violet in color. As the DPPH radical is scavenged by the donated hydrogen from the antioxidant, the absorbance is diminished according to the stoichiometry. The degree of discoloration indicates the scavenging potential of the antioxidant compounds. Briefly, solutions of different concentrations (100, 200, 300, 400, 500 μg mL−1) of the test compounds and ascorbic acid (standard) were prepared in methanol and added (1.5 mL) to the methanolic solution of DPPH (1.5 mL, 200 μM). The mixture was shaken vigorously and allowed to stand for 30 min in dark. After this, the absorbance was measured at 517 nm. Methanol (1.5 mL) was mixed with DPPH solution (1.5 mL, 200 μM) and used as a control.
The percentage of scavenging activity was calculated by using the formula:
Where, Ac = absorbance of control (1.5 mL of each of methanol and 200 μM DPPH solution), At = absorbance of test compound/ascorbic acid.
The percentage (%) inhibition curves for ascorbic acid and samples were plotted against concentration, from which IC50 values of percentage inhibition of DPPH by ascorbic acid and samples were calculated using regression equation.
References
Aytemir MD, Calis U, Ozalp M (2004) Synthesis and evaluation of anticonvulsant and antimicrobial activities of 3-hydroxy-6-methyl-2-substituted 4H-pyran-4-one derivatives. Arch Pharm 337:281
Da-Qing S, Li-Hui N, Qi-Ya Z (2008) One-pot three-component synthesis of pyrano[3,2-c]pyran-5-one derivatives in aqueous medium. Chin J Org Chem 28:1633
Fan X, Feng D, Qu Y, Zhang X, Wang J, Loiseau PM, Andrei G, Snoeck R, De-Clercq E (2010) Practical and efficient synthesis of pyrano[3,2-c]pyridone, pyrano[4,3-b]pyran and their hybrids with nucleoside as potential antiviral and antileishmanial agents. Bioorg Med Chem Lett 20:809
Khurana JM, Nand B, Saluja P (2010) DBU: a highly efficient catalyst for one-pot synthesis of substituted 3,4-dihydropyrano[3,2-c]chromenes, dihydropyrano[4,3-b]pyranes, 2-amino-4Hbenzo[h]chromenes and 2-amino-4H benzo[g]chromenes in aqueous medium. Tetrahedron 66:5637
Mazumder A, Wang S, Neamati N, Niclaus M, Sunder S, Chen J, Milne GWA, Rice WG, Burke TR Jr, Pommier Y (1996) Antiretroviral agents as inhibitors of both human immunodeficiency virus type 1 integrase and protease. J Med Chem 39:2472
Perez–Perez M, Balzarini J, Rozenski J, De-Clercq E, Herdewijn P (1995) Synthesis and antiviral activity of phosphonate derivatives of enantiomeric dihydro-2H-pyranyl nucleosides. Bioorg Med Chem Lett 5:1115
Piao MZ, Imafuku K (1997) Convenient synthesis of amino-substituted pyranopyranones. Tetrahedron Lett 38:5301
Pochet L, Doucet C, Schynts M, Thierry N, Boggetto N, Pirotte B, Jiang KY, Masereel B, de Tullio P, Delarge J, Reboud-Ravaux M (1996) Esters and amides of 6-(chloromethyl)-2-oxo-2H-1-benzopyran-3-carboxylic acid as inhibitors of α-chymotrypsin: significance of the “aromatic” nature of the novel ester-type coumarin for strong inhibitory activity. J Med Chem 39:2579
Ren ZJ, Cao WG, Tong WQ (2002) The Knoevenagel condensation reaction of aromatic aldehydes with malononitrile by grinding in the absence of solvents and catalysts. Synth Commun 32:3475
Ren ZJ, Cao WG, Ding WY, Shi W (2004) Solvent‐free stereoselective synthesis of cis‐1‐carbomethoxy‐2‐Aryl‐3,3‐dicyanocyclopropanes by grinding. Synth Commun 34:4395
Ren ZJ, Cao W, Tong W, Jin Z (2005) Solvent‐free, one‐pot synthesis of pyrano[2,3‐c]pyrazole derivatives in the presence of KF·2H2O by grinding. Synth Commun 35:2509
Schmeyers T, Toda F, Boy J, Kaupp G (1998) Quantitative solid–solid synthesis of azomethines. J Chem Soc Perkin Trans 2:989
Seifi M, Sheibani H (2008) High surface area MgO as a highly effective heterogeneous base catalyst for three-component synthesis of tetrahydrobenzopyran and 3,4-dihydropyrano[c]chromene derivatives in aqueous media. Catal Lett 126:275
Shaabani A, Samadi S, Badri Z, Rahmati A (2005) Ionic liquid promoted efficient and rapid one-pot synthesis of pyran annulated heterocyclic systems. Catal Lett 104:39
Shirini F, Marjani K, Nahzomi HT (2007) Silica triflate as an efficient catalyst for the solvent-free synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Arkivoc 1:51
Stoyanov EV, Ivanova IC, Heber D (2000) General method for the preparation of substituted 2-amino-4H,5H-pyrano[4,3-b]pyran-5-ones and 2-amino-4H-pyrano[3,2-c]pyridine-5-ones. Molecules 5:19
Tanaka K, Toda F (2000) Solvent-free organic synthesis. Chem Rev 100:1025
Tanaka K, Kishigami S, Toda F (1991) Reformatsky and Luche reaction in the absence of solvent. J Org Chem 56:4333
Thirunarayanan G, Vanangamudi G (2006) Synthesis of some 4-bromo-1-naphthyl chalcones using silica-sulfuric acid reagent under solvent free conditions. Arkivoc 12:58
Toda F, Tanaka K, Hamai K (1990) Aldol condensations in the absence of solvent: acceleration of the reaction and enhancement of the stereoselectivity. J Chem Soc Perkin Trans 1:3207
Toda F, Suzuki T, Higa S (1998) Solvent-free Dieckmann condensation reactions of diethyl adipate and pimelate. J Chem Soc Perkin Trans 1:3521
Uher M, Konecny V, Rajniakove O (1994) Synthesis of 5-hydroxy-2-hydroxymethyl-4H-pyran-4-one derivatives with pesticide activity. Chem Pap 48:282
Wang S, Milne GWA, Yang X, Posey IJ, Nicklaus MC, Graham L, Rice WG (1996) Discovery of novel, non-peptide HIV-1 protease inhibitors by pharmacophore searching. J Med Chem 39:2047
Wang XS, Zhou JX, Zeng ZS, Li YL, Da-Qing S, Tu SJ (2006) One-pot synthesis of pyrano[3,2-c]pyran derivatives catalyzed by KF/Al2O3. Arkivoc 11:107
Acknowledgments
The authors are thankful to the authorities of Deshpande Laboratories Pvt. Ltd., Bhopal (M.P.), India and Pinnacle Biomedical Research Institute, Bhopal (M.P.), India for the studies of biological activities. The authors also thank The Director, SAIF, Panjab University, Chandigarh for NMR and MS spectral data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rajguru, D., Keshwal, B.S. & Jain, S. Solvent-free, green and efficient synthesis of pyrano[4, 3-b]pyrans by grinding and their biological evaluation as antitumor and antioxidant agents. Med Chem Res 22, 5934–5939 (2013). https://doi.org/10.1007/s00044-013-0586-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0586-4