
Abstract
New functionalized derivatives were prepared using alkyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate as a building block. Newly prepared compounds were well characterized using 1H NMR, IR, and mass spectral data. All the synthesized products were screened for their antioxidant properties. Among the tested compounds, indazole derivatives exhibited noticeable DPPH radical scavenging activity and reducing power capacity in comparison with the standard Glutathione.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Michael addition reaction is widely recognized as one of the key reaction for carbon–carbon bond formation (Krause and Hoffmann-Röder, 2001; Leonard et al., 1998; Fan et al., 2002). Chalcone is used as a Michael acceptor in organic synthesis. Michael addition reaction of 1,3-dicarbonyl compounds such as acetoacetic esters to chalcones leads to the synthesis of cyclohexenone derivatives, which become interesting intermediates for the synthesis of a variety of heterocyclic systems with diverse biological activities (Yadav et al., 2011; Padmavathi et al., 2000; Senguttuvan and Nagarajan, 2010; Vyas et al., 2009).
The search for new molecules with anti‐oxidant properties is a very active domain of research, since they can protect the human body from free radicals and retard the progress of many chronic diseases, such as atherosclerosis, stroke, diabetes, Alzheimer’s disease, some forms of cancer, and oxidative stress responsible for DNA, protein, and membrane damage. Antioxidants are also believed to play a very important role in the body defense system against reactive oxygen species, which are the harmful byproducts generated during normal cell aerobic respiration. Supplementation with antioxidants may help to maintain an adequate antioxidant status and therefore, the normal physiological function of a living system (Van Acker et al., 1996). Hence, there is considerable interest in the discovery and development of efficient synthetic or natural antioxidants. Many synthetic antioxidants, which are characterized by a better antioxidant activity than natural antioxidants and are more easily available, have been used in a wide variety of food products. Butylated hydroxytoluene and butylated hydroxyanisole (BHA) were originally developed to protect petroleum from oxidative gumming. However, these compounds have been used as antioxidants in human foods since 1954 and are perhaps the most common antioxidants used in foods today (Sherwin, 1976). Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a strong novel free radical scavenger, is used for treatment of patients with acute brain infarction (Kokura et al., 2005). This has attracted a great deal of research interest in synthetic antioxidants.
A number of synthetic compounds such as, indazoles (Hagihara et al., 2011), terphenyls (Wang and Pan, 2012), benzodiazepines (García-Santos et al., 2004), quinazolines (Saravanan et al., 2010), and benzisoxazoles (Jain and Kwon, 2003) have been extremely exploited for antioxidant activity. Indazole derivatives have gained interest in medicinal chemistry in view of their promising pharmacological properties including antioxidant activity. It is noticed that by injecting 7-nitroindazole, malondialdehyde (MDA), a major end product of lipid peroxidation, significantly decreased in epilepticus rats (Liu et al., 2007; Thomas et al., 2008). The antioxidant study of some of the terphenyl derivatives isolated from three edible and delicious mushrooms (Thelephora ganbajun, T. aurantiotincta and Boletopis grisea) indigenous to China, expressed higher antioxidant activities than α-tocopherol or BHA which are generally used as efficient free radical scavengers (Liu et al., 2004; Yang et al., 2004). Moreover, the compounds having groups like –SH, –OH, or –NH2, which are able to provide free electron either in the form of a negative charge or in the form of a lone pair of electrons, may show higher antioxidant activity due to their redox properties (Flora, 2009). In view of above observations and in continuation of our ongoing efforts on the synthesis of large range of new compounds from a single precursor 4,4′-difluoro chalcone (Samshuddin et al., 2011a, b, c, d, 2012a, b, c, d; Jasinski et al., 2010a, b, 2012a, b; Fun et al., 2010a, 2012; Baktir et al., 2011a, b, 2012), we converted the chalcone into its cyclohexenone derivative and used that as a key intermediate for the synthesis of new functionalized derivatives. All these derivatives are characterized by spectral data and screened for their antioxidant properties.
Results and discussion
Chemistry
Syntheses of new functionalized derivatives were carried out by reacting the cyclohexenone derivative of 4,4′-difluoro chalcone 2a/2b with various reagents according to the reaction sequence depicted in Scheme 1, 2, 3, 4 5.

Synthesis of terphenyl esters

Synthesis of indazole derivatives

Synthesis of dibenzodiazepine derivative

Synthesis of oxime and substituted guanidine derivatives

Synthesis of aminated derivatives of cyclohexenones
Cyclohexenones 2a/2b were prepared by the condensation of ethyl acetoacetate/methyl acetoacetate to the 4,4′-difluoro chalcone 1 by means of an intermediate Michael adduct according to the method described in our previous works (Fun et al., 2010b; Dutkiewicz et al., 2011). The cyclocondensation of acetoacetic esters with chalcones led to the generation of two chiral centers in cyclohexenones which would result in a mixture of diastereomers. No attempt was undertaken to separate the diastereomeric cyclohexenones and used as such for further reaction.
Synthesis of terphenyl esters
Iodine in methanol is used as a good reagent for the conversion of 2-cyclohexen-1-ones into the corresponding anisole derivatives (Kotnis, 1990; Tamura and Yoshimoto, 1980). The method was successfully applied to the conversion of cyclohexenones 2a and 2b to anisole derivatives 3a and 3b. Hence, this reaction provides a simple and good method for the synthesis of terphenyl derivatives from chalcone via cyclohexenone intermediate (Scheme 1).
The structure of compounds 3a and 3b was confirmed by their spectral data. In the IR spectrum of 3a, stretching band due to ester carbonyl group was observed at 1,724 cm−1. The 1H NMR spectrum showed two singlets integrating for three protons each at δ 3.58 and 3.91 ppm due to the protons of methoxy group and methyl group of ester, respectively. Moreover, there was no signal other than multiplets in the range δ 7.20–7.86 ppm which were due to the ten aromatic protons, hence confirming the proposed structure of terphenyl derivative 3a. The mass spectrum showed a molecular ion peak at m/z 355.1 (M+ + 1) which further confirmed the proposed structure. Similarly, the spectral data of 3b was in good agreement with the proposed structure.
Synthesis of indazole derivatives
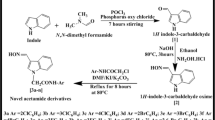
Both the cyclohexenones 2a and 2b containing 1,3-dicarbonyl system reacted with hydrazine hydrate in acid medium resulted in the formation of indazole derivative, 4,6-bis(4-fluorophenyl)-1,2,4,5-tetrahydro-3H-indazol-3-one 4a (Scheme 2 ). In the IR spectrum of 4a, the stretching band at 3,197 cm−1 was attributed to the presence of NH group of indazole moiety. An absorption band due to carbonyl group of indazole ring was seen at 1,718 cm−1. Two singlets at δ 9.57 and 11.59 ppm appeared in the 1H NMR spectrum were due to the two NH protons of indazole ring. Two doublet of doublets at δ 2.81 and 3.10 ppm were due to the two protons attached to C-5 carbon, adjacent to chiral center C-4. One more doublet of doublet seen at δ 4.17 ppm was due to a proton attached to chiral carbon C-4. Similarly a singlet observed at δ 6.72 ppm could be due to methylene proton of C-7. The mass spectrum showed a molecular ion peak at m/z 324.9 (M+ + 1) which confirmed the structure. The N-substituted indazole derivative 4b was obtained in similar way when cyclohexenone 2a or 2b was treated with 4-nitrophenyl hydrazine. The structure was confirmed by IR, 1H NMR, and mass spectral data.
In contrast to the above reaction, an indazole derivative 5 with aromatized ring-2 was obtained when cyclohexenone 2a or 2b was treated with phenyl hydrazine (Scheme 2). The formation of aromatized product could be due to the reaction carried out in acetic acid medium. In the IR spectrum of 5, absorption bands appeared at 3,120 and 1,641 cm−1 were due to NH group and carbonyl group of indazole ring, respectively. The 1H NMR spectrum supported the aromatization as there were no signals corresponding to aliphatic protons. A singlet appeared at δ 10.88 ppm was due to the NH proton of indazole ring. The mass spectrum showed a molecular ion peak at m/z 399.0 (M+ + 1) which confirmed the structure of 5.
Synthesis of dibenzodiazepine derivative
A dibenzodiazepine derivative 6 was obtained when cyclohexenone 2a or 2b was treated with o-phenylenediamine (Scheme 3). The absorption bands in IR spectrum showed stretching bands at 3,429 and 3,136 cm−1 due to NH group while strong stretching band at 1,712 cm−1 attributed to carbonyl of cyclic amide in dibenzodiazepine ring, supported the structure. Two singlets at δ 11.21 and 8.71 ppm appeared in the 1H NMR spectrum were due to the NH protons present in the dibenzodiazepine ring. Further, multiplets in the range δ 7.01–7.93 ppm appeared were due to the aromatic protons, confirming the proposed structure of dibenzodiazepine derivative 6. The mass spectrum showed a molecular ion peak at m/z 399.1 (M+ + 1) which confirmed the proposed structure.
Synthesis of oxime and substituted guanidine derivatives
It was reported that, the reaction of cyclohexenone derivatives with hydroxylamine hydrochloride yields benzisoxazole derivative (Rajanarendar et al., 2009). But, the cyclohexenone 2a or 2b with hydroxylamine hydrochloride in the presence of strong base afforded decarboxylated oxime derivative, (1E)-3,5-bis(4-fluorophenyl)-N-hydroxycyclohex-2-en-1-imine 7 (Scheme 4 ). The IR spectrum of compound 7 showed an absorption band at 3,255 cm−1 due to hydroxyl group of oxime. The 1H NMR spectrum showed a singlet at δ 10.96 ppm corresponding to the hydroxyl proton of oxime. A multiplet observed at δ 2.32 ppm was due to a proton attached to C-5 carbon of cyclohexene ring. Two mutiplets integrating for two protons each seen at δ 2.73 and 3.07 ppm were due to the protons attached to C-4 and C-6 carbon, respectively. Similarly, a singlet observed at δ 6.60 ppm was due to methylene proton of C-2. The mass spectrum showed a molecular ion peak at m/z 299.9 (M+ + 1) corresponding to the molecular formula C18H15F2NO.
It was aimed at the preparation of quinazoline derivative by reacting cyclohexenones 2a or 2b with amino guanidine hydrochloride (Senguttuvan and Nagarajan, 2009). But instead of cyclization, decarboxylated uncyclized product, N-[(1E)-3,5-bis(4-fluorophenyl)cyclohex-2-en-1-ylidene]carbonohydrazonic diamide 8 was obtained. The structure of the product 8 formed was confirmed by the spectral data. In the IR spectrum, the presence of absorption bands at 3,448, 3,313 cm−1 were due to amino groups present in the molecule. The absence of absorption band due to ester carbonyl group indicated the decarboxylation of the compound 8. Further, in the 1H NMR spectrum, the presence of two singlets at δ 5.44 and 5.76 ppm integrating for two protons each, revealed the presence of two different amino groups in the compound. Two doublet of doublets observed at δ 2.26 and 3.30 ppm were due to protons attached to C-4 carbon. Similarly, signals at δ 2.71 and 3.02 ppm were due to the protons attached to C-6 and C-5 carbon, respectively. The mass spectrum showed a molecular ion peak at m/z 340.9 (M+ + 1) corresponding to the molecular formula C19H18F2N4.
Synthesis of aminated derivatives of cyclohexenones
2-Aminated products 9a and 9b were obtained when cyclohexenones 2a and 2b treated with ammonium acetate in ethanol (Scheme 5). Their spectral data proved the formation of the products. In the IR spectrum of ethyl 2-amino-4,6-bis(4-fluorophenyl)cyclohexa-1,3-diene-1-carboxylate 9b, stretching bands appeared at 3,408, 3,311 cm−1 due to amino group and a strong stretching band at 1,649 cm−1 due to carbonyl group. The 1H NMR spectrum showed a triplet and a multiplet at δ 1.07 and 3.91 ppm integrating for three and two protons, respectively, were due to ethyl chain of ester. A doublet of doublet at δ 2.77 ppm, a multiplet at δ 3.04 ppm and also a doublet observed at δ 4.10 ppm integrating for one proton each, were due to the two protons attached to C-5 carbon and a proton attached to C-6 carbon, respectively. Two amino protons resonated at δ 7.85 ppm as a singlet. The mass spectrum showed a molecular ion peak at m/z 355.9 (M+ + 1) corresponding to the molecular formula C21H19F2NO2.
Similarly, uncyclized products were obtained when cyclohexenones 2a and 2b reacted with semicarbazide hydrochloride in the presence of a base (Scheme 5). The IR spectrum of ethyl (2E)-2-(2-carbamoylhydrazinylidene)-4,6-bis(4-fluorophenyl)cyclohex-3-ene-1-carboxylate 10b showed stretching bands at 3,460, 3,313, and 3,178 cm−1 due to NH group. A band at 1,683 cm−1 was due to amide carbonyl group, and another strong band at 1,734 cm−1 was due to ester carbonyl group. The presence of signals corresponding to ethyl chain of ester in the 1H NMR spectrum ruled out the formation of cyclized products. Furher, the 1H NMR spectrum of 10b showed a doublet at δ 2.73 ppm integrating for two protons was due to the protons attached to C-5 carbon. A multiplet at δ 3.36 ppm integrating for one proton was due to a proton attached to C-6 carbon, and a doublet at δ 3.82 ppm integrating for one proton was due to a proton attached to C-1 carbon. A broad singlet resonated at δ 6.07 ppm was observed due to the protons of NH2 group while a singlet at δ 10.15 ppm was observed due to NH proton. A molecular ion peak seen at m/z 413.8 (M+ + 1) in the mass spectrum confirmed the structure of 10b.
Antioxidant activity
DPPH radical scavenging assay
A rapid, simple and inexpensive method to measure antioxidant capacity of substances involves the use of the free radical, 2,2-diphenyl-1-picrylhydrazyl (DPPH). DPPH is widely used to test the ability of compounds to act as free radical scavengers or hydrogen donors. Antioxidants tested on DPPH were also found extremely effective in cell systems. This simple test further provides information on the ability of a compound to donate electrons during antioxidant action (Tiwari, 2004). The radical scavenging mechanism is based on the transfer of acidic H-atom from the compound to DPPH radical to form DPPH-H. The results are summarized in Table 1.
Among the tested compounds, compounds 4a, 4b, and 5 showed very high radical scavenging capacity with concentration of 1 mg/mL in comparison with the standard Glutathione. Such enhanced activity was due to the presence of acidic protons in the indazole moiety. Compounds 7 and 8 were also showing good activity due to the presence of oxime and guanidine functionalities in these molecules. Other compounds showed moderate activity.
Reducing power assay
Reducing power is associated with antioxidant activity and may serve as a significant reflection of the antioxidant activity (Oktay et al., 2003). Compounds with reducing power indicate that they are electron donors and can reduce the oxidized intermediates of lipid peroxidation processes, so that they can act as primary and secondary antioxidants (Chanda and Dave, 2009). Substances, which have reduction potential, react with potassium ferricyanide to form potassium ferrocyanide, which then reacts with ferric chloride to form ferrous complex that has an absorption maximum at 700 nm. Increased absorbance of the reaction mixture indicates the increased reducing power. The results are summarized in Table 1.
Reducing power assay is expressed in effective concentration (mg/mL) equivalent of 0.5 absorbance Glutathione. Among the tested compounds, many compounds showed significant reducing power capacity. Compounds 4a and 6 showed good reducing power capacity while compounds 4b, 5, 7, 8, and 9a exhibited moderate reducing power capacity in comparison with the standard Glutathione. The good reducing power capacity of these compounds was due to the presence of free NH group in the molecules.
Experimental section
Melting points were taken in open capillary tubes and are uncorrected. The purity of the compounds was confirmed by thin layer chromatography using Merck silica gel 60 F254-coated aluminum plates using ethyl acetate:n-hexane (3:7, v/v) as solvent system. IR spectra were recorded on Shimadzu-FTIR Infrared spectrometer in KBr (νmax in cm−1). 1H NMR (400 MHz) spectra were recorded on a Bruker AMX 400 spectrometer, with 5 mm PABBO BB-1H TUBES with TMS as internal standard. LCMS were obtained using Agilent 1200 series LC and Micromass zQ spectrometer. Elemental analysis was carried out using VARIO EL-III (Elementar Analysensysteme GmBH).
Synthesis of alkyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate (2a, b)
Cyclohexenone derivatives of 4,4′-difluoro chalcone viz. methyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2a and ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2b were prepared by the condensation of 4,4′-difluoro chalcone 1 with methyl acetoacetate and ethyl acetoacetate according to the method described in our previous works (Fun et al., 2010b; Dutkiewicz et al., 2011).
Synthesis of alkyl 4,4′′-difluoro-5′-methoxy-1,1′:3′,1′′-terphenyl-4′-carboxylate (3a, b)
A mixture of methyl/ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2a/2b (0.01 mol) and iodine (0.025 mol) in 20 mL methanol was refluxed for 24 h with stirring. The reaction mixture was cooled and poured into ice-cold water (50 mL). Organic contents were extracted with diethyl ether (25 mL) and washed with saturated sodium thiosulphate solution (2 × 25 mL) followed by water (25 mL). The precipitate obtained after evaporation of solvent was collected and recrystallized from methanol.
Methyl 4,4′′-difluoro-5′-methoxy-1,1′:3′,1′′-terphenyl-4′-carboxylate (3a)
Yield: 70 %; m.p. 126–128 °C. IR (KBr, νmax in cm−1): 3051 (Ar–H), 2983, 2943 (CH), 1724 (C=O), 1222 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 3.58 (s, 3H, OCH3), 3.91 (s, 3H, CO2CH3), 7.20–7.86 (m, 10H, Ar–H). LCMS: m/z 355.1 (M+ + 1). C H N Analysis; Calculated for C21H16F2O3: C, 71.18; H, 4.55. Found: C, 71.15; H, 4.57 %.
Ethyl 4,4′′-difluoro-5′-methoxy-1,1′:3′,1′′-terphenyl-4′-carboxylate (3b)
Yield: 63 %; m.p. 108–110 °C. IR (KBr, νmax in cm−1): 3062 (Ar–H), 2898 (CH), 1709 (C=O), 1220 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 0.98 (t, 3H, CH3), 3.62 (s, 3H, OCH3), 4.02 (q, 2H, CO2CH2), 7.18–7.81 (m, 10H, Ar–H). LCMS: m/z 369.1 (M+ + 1). C H N Analysis; Calculated for C21H16F2O3: C, 71.73; H, 4.93. Found: C, 71.70; H, 4.92 %.
Synthesis of 4,6-bis(4-fluorophenyl)-1,2,4,5-tetrahydro-3H-indazol-3-one (4a)
A mixture of ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2b (3.56 g, 0.01 mol) and hydrazine hydrate (0.75 g, 0.015 mol) in 20 mL ethanol in the presence of sulfuric acid (0.5 mL) was refluxed for 10 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate obtained after neutralization was collected by filtration and recrystallized in ethanol. Yield: 71 %; m.p. 211–214 °C. IR (KBr, νmax in cm−1): 3197 (N–H), 3043 (Ar–H), 2721 (C–H), 1718 (C=O), 1230 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.81 (dd, 1H, HA, J AB = 16.8 Hz, J AX = 3.6 Hz), 3.10 (dd, 1H, HB, J BA = 16.8 Hz, J BX = 8.02 Hz), 4.17 (dd, 1H, HB, J XB = 8.02 Hz, J XA = 3.8 Hz), 6.72 (s, 1H, CH), 7.00 -7.52 (m, 8H, Ar–H), 9.57 (s, 1H, NH), 11.59 (s, 1H, NH). LCMS: m/z 324.9 (M++1). C H N Analysis; Calculated for C19H14F2N2O: C, 70.36; H, 4.35; N, 8.64. Found: C, 70.33; H, 4.34; N, 8.62 %.
Synthesis of 4,6-bis(4-fluorophenyl)-2-(4-nitrophenyl)-1,2,4,5-tetrahydro-3H-indazol-3-one (4b)
A mixture of ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2b (3.56 g, 0.01 mol) and 4-nitrophenyl hydrazine (0.01 mol) in 20 mL ethanol in the presence of concentrated sulfuric acid (0.5 mL) was refluxed for 10 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate was collected by filtration and recrystallized in ethanol. Yield: 56 %; m.p. 204–206 °C. IR (KBr, νmax in cm−1): 3120 (NH), 3043 (Ar–H), 2924 (CH), 1724 (C=O), 1226 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.92 (broad d, 1H, CH2-HA, J AB = 16 Hz), 3.20 (dd, 1H, CH2–HB, J BA = 16.4 Hz), 4.40 (broad s, 1H, CH–HX), 6.91 (s, 1H, CH), 7.03–8.40 (m, 12H, Ar–H), 11.98 (s, 1H, NH). LCMS: m/z 445.8 (M+ + 1). C H N Analysis; Calculated for C25H17F2N3O3: C, 67.41; H, 3.85; N, 9.43. Found: C, 67.38; H, 3.84; N, 9.40 %.
Synthesis of 4,6-bis(4-fluorophenyl)-2-phenyl-1,2-dihydro-3H-indazol-3-one (5)
A mixture of ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2b (3.56 g, 0.01 mol) and phenyl hydrazine (1.08 g, 0.01 mol) in 20 mL glacial acetic acid in the presence of concentrated sulfuric acid (0.5 mL) was refluxed for 8 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate obtained after neutralization was collected by filtration and recrystallized in ethanol. Yield: 58 %; m.p. 271–273 °C. IR (KBr, νmax in cm−1): 3120 (NH), 3072 (Ar–H), 1641 (C=O), 1222 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 7.23–7.91 (m, 15H, Ar–H), 10.88 (s, 1H, NH). LCMS: m/z 399.0 (M+ + 1). C H N Analysis; Calculated for C25H16F2N2O: C, 75.37; H, 4.05; N, 7.03. Found: C, 75.35; H, 4.06; N, 7.00 %.
Synthesis of 1,3-bis(4-fluorophenyl)-5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-one (6)
A mixture of ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2b (3.56 g, 0.01 mol) and o-phenylenediamine (1.3 g, 0.012 mol) in 20 mL glacial acetic acid in the presence of concentrated sulfuric acid (0.5 mL) was refluxed for 10 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate obtained after neutralization was collected by filtration and recrystallized in ethanol. Yield: 47 %; m.p. 274–277 °C. IR (KBr, νmax in cm−1): 3429, 3136 (NH), 3066 (Ar–H), 1712 (C=O), 1226 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 7.01–7.93 (m, 14H, Ar–H), 8.71 (s, 1H, NH), 11.21 (s, 1H, NH). LCMS: m/z 399.1 (M+ + 1). C H N Analysis; Calculated for C25H16F2N2O: C, 75.37; H, 4.05; N, 7.03. Found: C, 75.36; H, 4.04; N, 7.01 %.
Synthesis of (1E)-3,5-bis(4-fluorophenyl)-N-hydroxycyclohex-2-en-1-imine (7)
A mixture of ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2b (3.56 g, 0.01 mol) and hydroxylamine hydrochloride (1.03 g, 0.015 mol) in 20 mL ethanol in the presence of 40 % NaOH (1 mL) was refluxed for 6 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate was collected by filtration and recrystallized in ethanol. Yield: 58 %; m.p. 164–166 °C. IR (KBr, νmax in cm−1): 3255 (OH), 3055 (Ar–H), 2900 (CH), 1600 (C=N), 1220 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.32 (m, 1H, C-1 H), 2.73 (m, 2H, C-2 CH2), 3.07 (m, 2H, C-6 CH2), 6.60 (s, 1H, CH), 7.13–7.64 (m, 8H, Ar–H), 10.96 (s, 1H, OH). LCMS: m/z 299.9 (M+ + 1). C H N Analysis; Calculated for C18H15F2NO: C, 72.23; H, 5.05; N, 4.68. Found: C, 72.20; H, 5.06; N, 4.66 %.
Synthesis of N-[(1E)-3,5-bis(4-fluorophenyl)cyclohex-2-en-1-ylidene]carbonohydrazonic diamide (8)
A mixture of ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2b (3.56 g, 0.01 mol) and aminoguanidine hydrochloride (0.012 mol) in 20 mL ethanol in the presence of 40 % NaOH (2 mL) was refluxed for 16 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate obtained after neutralization was collected by filtration and recrystallized from ethanol. Yield: 66 %; m.p. 186–188 °C. IR (KBr, νmax in cm−1): 3448, 3313 (NH), 3037 (Ar–H), 1581 (C=N), 1234 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.26 (dd, 1H, CH–HA, J AX = 12.4 Hz, J AB = 16.4 Hz), 2.71 (m, 2H, CH2), 3.02 (m, 1H, CH–HX), 3.30 (dd, 1H, CH–HB, JXB = 12.4 Hz, J BA = 16.4 Hz), 5.44 (s, 2H, NH2), 5.76 (s, 2H, NH2), 6.66 (s, 1H, CH), 7.11–7.58 (m, 8H, Ar–H). LCMS: m/z 340.9 (M+ + 1). C H N Analysis; Calculated for C19H18F2N4: C, 67.05; H, 5.33; N, 16.46. Found: C, 67.01; H, 5.35; N, 16.42 %.
Synthesis of alkyl 2-amino-4,6-bis(4-fluorophenyl)cyclohexa-1,3-diene-1-carboxylate (9a, b)
A mixture of methyl/ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2a/2b (0.01 mol) and ammonium acetate (0.02 mol) in 20 mL ethanol in the presence of acetic acid (1 mL) was refluxed for 14 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate obtained after neutralization was collected by filtration and recrystallized from ethanol.
Methyl 2-amino-4,6-bis(4-fluorophenyl)cyclohexa-1,3-diene-1-carboxylate (9a)
Yield: 68 %; m.p. 162–165 °C. IR (KBr, νmax in cm−1): 3298 (NH), 3068 (Ar–H), 2933 (CH), 1735 (C=O), 1228 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.68 (dd, 1H, CH), 2.97 (m, 1H, CH), 3.98 (s, 3H, CO2CH3), 4.07 (d, 1H, CH), 6.45 (d, 1H, CH), 6.92–7.39 (m, 8H, Ar–H), 7.91 (s, 2H, NH2). LCMS: m/z 341.9 (M+ + 1). C H N Analysis; Calculated for C20H17F2NO2: C, 70.37; H, 5.02; N, 4.10. Found: C, 70.34; H, 5.04; N, 4.06 %.
Ethyl 2-amino-4,6-bis(4-fluorophenyl)cyclohexa-1,3-diene-1-carboxylate (9b)
Yield: 70 %; m.p. 153–155 °C. IR (KBr, νmax in cm−1): 3408, 3311 (NH), 3049 (Ar–H), 2981 (CH), 1649 (C=O), 1222 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.07 (t, 3H, CH3), 2.77 (dd, 1H, CH), 3.04 (m, 1H, CH), 3.91 (m, 2H, CH2), 4.10 (d, 1H, CH), 6.50 (d, 1H, CH), 6.97–7.47 (m, 8H, Ar–H), 7.85 (s, 2H, NH2). LCMS: m/z 355.9 (M+ + 1). C H N Analysis; Calculated for C21H19F2NO2: C, 70.97; H, 5.39; N, 3.94. Found: C, 70.93; H, 5.38; N, 3.91 %.
Synthesis of alkyl (2E)-2-(2-carbamoylhydrazinylidene)-4,6-bis(4-fluorophenyl)cyclohex-3-ene-1-carboxylate (10a, b)
A mixture of methyl/ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate 2a/2b (0.01 mol) and semicarbazide hydrochloride (0.012 mol) in 20 mL ethanol in the presence of 10 % NaOAc (1 mL) was refluxed for 10 h. The reaction mixture was cooled and poured into ice-cold water (50 mL). The precipitate obtained after neutralization was collected by filtration and recrystallized from ethanol.
Methyl (2E)-2-(2-carbamoylhydrazinylidene)-4,6-bis(4-fluorophenyl)cyclohex-3-ene-1-carboxylate (10a)
Yield: 64 %; m.p. 192–194 °C. IR (KBr, νmax in cm−1): 3468, 3311, 3116 (NH), 3080 (Ar–H), 2904 (CH), 1734 (ester C=O), 1662 (amide C=O), 1228 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.68 (d, 2H, CH), 2.83 (m, 1H, CHA), 3.32 (m, 1H, CHB), 3.87 (d, 1H, CH), 4.04 (s, 3H, CO2CH3), 6.12 (s, 2H, NH2), 7.21–7.89 (m, 9H, Ar–H), 10.21 (s, 1H, NH). LCMS: m/z 399.8 (M+ + 1). C H N Analysis; Calculated for C21H19F2N3O3: C, 63.15; H, 4.80; N, 10.52. Found: C, 63.13; H, 4.78; N, 10.50 %.
Ethyl (2E)-2-(2-carbamoylhydrazinylidene)-4,6-bis(4-fluorophenyl)cyclohex-3-ene-1-carboxylate (10b)
Yield: 68 %; m.p. 214–217 °C. IR (KBr, νmax in cm−1): 3460, 3313, 3178 (NH), 3074 (Ar–H), 2931 (CH), 1683 (amide C=O), 1734 (ester C=O), 1220 (C–F). 1H NMR (400 MHz, DMSO-d6, δ ppm): 0.94 (t, 3H, CH3), 2.73 (d, 1H, CH), 2.84 (m, 1H, CHA), 3.36 (m, 1H, CHB), 3.82 (d, 1H, CH), 3.89 (q, 2H, CH2), 6.07 (s, 2H, NH2), 7.12–7.95 (m, 9H, Ar–H), 10.15 (s, 1H, NH). LCMS: m/z 413.8 (M+ + 1). C H N Analysis; Calculated for C22H21F2N3O3: C, 63.91; H, 5.12; N, 10.16. Found: C, 63.88; H, 5.14; N, 10.14 %.
Antioxidant activity
DPPH radical scavenging assay
The DPPH assay was based on the reported method (Blois, 1958). In brief, 1 mM solution of DPPH in ethanol was prepared, and this solution (1 mL) was added to sample solutions 1 mg/mL of DMSO. The mixture was shaken vigorously and allowed to stand at room temperature for 20 min. Then the absorbance was measured at 517 nm in a spectrophotometer. Lower absorbance of the reaction mixture indicated higher free radical scavenging activity. The capability to scavenge the DPPH radical was calculated using the following equation:
where A 0 is the absorbance of the control reaction and A 1 is the absorbance in the presence of the samples or standards. Each sample was assayed at 1 mg/mL and all experiments were carried out in triplicate.
Reducing power assay
The reducing power of the synthesized compounds was determined according to the method of Oyaizu (1986). Different concentrations of the samples (100–1,000 μg/mL) in DMSO (1 mL) were mixed with phosphate buffer (2.5 mL, 0.2 M, pH 6.6) and potassium ferricyanide (2.5 mL, 1 % solution). The mixture was incubated at 50 °C for 20 min. After which, 10 % trichloroacetic acid (2.5 mL) was added to the mixture, which was then centrifuged for 10 min. The upper layer of solution (2.5 mL) was mixed with distilled water (2.5 mL) and FeCl3 (0.5 mL, 0.1 %), and then, the absorbance at 700 nm was measured using a spectrophotometer. Higher absorbance of the reaction mixture indicated greater reducing power. All experiments were carried out in triplicate, and the reducing power assay was represented by effective concentration (mg/mL) equivalent of 0.5 absorbance Glutathione.
Conclusions
New functionalized derivatives were prepared using alkyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate as a building block. All the derivatives were characterized by 1H NMR, IR, and mass spectral data. All the synthesized products were screened for their antioxidant properties. Among the tested compounds, the indazole derivatives exhibited noticeable DPPH radical scavenging activity and reducing power capacity in comparison with the standard Glutathione.
References
Baktir Z, Akkurt M, Samshuddin S, Narayana B, Yathirajan HS (2011a) 2,4-Bis(4-fluorophenyl)-2,3-dihydro-1H-1,5-benzodiazepine. Acta Cryst E 67:1262–1263
Baktir Z, Akkurt M, Samshuddin S, Narayana B, Yathirajan HS (2011b) 3,5-Bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbaldehyde. Acta Cryst E 67:1292–1293
Betz R, Gerber T, Hosten E, Samshuddin S, Narayana B, Sarojini BK (2012) 2,2′-(Disulfanediyl)bis[4,6-(4-fluorophenyl)pyrimidine]. Acta Cryst E 68:476–477
Blois MS (1958) Antioxidant determinations by the use of a stable free radical. Nature 181:1199–1200
Chanda S, Dave R (2009) In vitro models for antioxidant activity evaluation and some medicinal plants possessing antioxidant properties: an overview. Afr J Microbiol Res 3:981–996
Dutkiewicz G, Narayana B, Veena K, Yathirajan HS, Kubicki M (2011) (1RS,6SR)-Ethyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate. Acta Cryst E 67:336
Fan QH, Li YM, Chan ASC (2002) Recoverable catalysts for asymmetric organic synthesis. Chem Rev 102:3385–3466
Flora SJS (2009) Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxidative Med Cell Longev 2:191–206
Fun HK, Hemamalini M, Samshuddin S, Narayana B, Yathirajan HS (2010a) 1-[3,5-Bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]ethanone. Acta Cryst E 66:582–583
Fun HK, Hemamalini M, Samshuddin S, Narayana B, Yathirajan HS (2010b) Methyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate. Acta Cryst E 66:864–865
Fun HK, Chia TS, Samshuddin S, Narayana B, Sarojini BK (2012) 2-[3,5-Bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4,6-bis(4-fluorophenyl)pyrimidine. Acta Cryst E 68:807–808
García-Santos G, Herrera F, Martín V, Rodriguez-Blanco J, Antolín I, Fernández-Marí F, Rodriguez C (2004) Antioxidant activity and neuroprotective effects of zolpidem and several synthesis intermediates. Free Radical Res 38:1289–1299
Hagihara M, Komori K, Sunamoto H, Nishida H, Tsuzaki Y, Takama A, Kido K, Fujimoto T, Matsugi T (2011) Novel indazole derivative or salt thereof, manufacturing intermediate therefor, antioxidant using same, and use for an indazole derivative or salt thereof. WO2011149011(A1)
Jain M, Kwon CH (2003) 1,2-Benzisoxazole phosphorodiamidates as novel anticancer prodrugs requiring bioreductive activation. J Med Chem 46:5428–5436
Jasinski JP, Guild CJ, Samshuddin S, Narayana B, Yathirajan HS (2010a) 2,3-Dibromo-1,3-bis(4-fluorophenyl)propan-1-one. Acta Cryst E 66:2018
Jasinski JP, Guild CJ, Samshuddin S, Narayana B, Yathirajan HS (2010b) 3,5-Bis(4-fluorophenyl)-1-phenyl-4,5-dihydro-1H-pyrazole. Acta Cryst E 66:1948–1949
Jasinski JP, Golen JA, Samshuddin S, Narayana B, Yathirajan HS (2012a) Synthesis, characterization and crystal structures of 3,5-bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carboxamide and 3,5-bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide. Crystals 2(3):1108–1115
Jasinski JP, Golen JA, Samshuddin S, Narayana B, Yathirajan HS (2012b) (6Z)-3,5-Bis(4-fluorophenyl)-6-(1-hydroxyethylidene)cyclohex-2-en-1-one. Acta Cryst E 68:638–639
Kokura S, Yoshida N, Sakamoto N, Ishikawa T, Takagi T, Higashihara H, Nakabe N, Handa O, Naito Y, Yoshikawa T (2005) The radical scavenger edaravone. Cancer Lett 229:223–233
Kotnis AS (1990) A rapid entry to highly functionalized p-methoxy benzoates. Tetrahedron Lett 31:481–484
Krause N, Hoffmann-Röder A (2001) Recent advances in catalytic enantioselective Michael additions. Synthesis 2001:171–196
Leonard J, Díez-Barra E, Merino S (1998) Control of asymmetry through conjugate addition reactions. Eur J Org Chem 1998:2051–2061
Liu JK, Hu L, Dong ZJ, Hu Q (2004) DPPH radical scavenging activity of ten natural p-terphenyl derivatives obtained from three edible mushrooms indigenous to China. Chem Biodiversity 1:601–605
Liu ZW, Zhang T, Yang Z (2007) Involvement of nitric oxide in spatial memory deficits in status epilepticus rats. Neurochem Res 32:1875–1883
Oktay M, Gulcin I, Kufrevioglu OI (2003) Determination of in vitro antioxidant activity of fennel (Foeniculum vulgare) seed extracts. LWT Food Sci Tech 36:263–271
Oyaizu M (1986) Studies on products of the browning reaction. Antioxidative activities of browning reaction products prepared from glucosamine. Jpn J Nutr 44:307–315
Padmavathi V, Reddy BJM, Balaiah A, Reddy KV, Reddy DB (2000) Synthesis of some fused pyrazoles and isoxazoles. Molecules 5:1281–1286
Rajanarendar E, Rao EK, Raju S (2009) Microwave assisted rapid and efficient synthesis of new 3-ethoxy-4,6-diaryl-4,5-dihydro-2,1-benzisoxazoles. Indian J Chem 48B:749–753
Samshuddin S, Narayana B, Sarojini BK (2011a) Ethyl 4,4′′-Difluoro-5′-hydroxy-1,1′:3′,1′′-terphenyl-4′-carboxylate. Molbank 2011:M745
Samshuddin S, Narayana B, Shetty DN, Raghavendra R (2011b) An efficient synthesis of 2,4,6-triaryl pyridines and their biological evaluation. Der Pharma Chemica 3(3):232–240
Samshuddin S, Narayana B, Baktir Z, Akkurt M, Yathirajan HS (2011c) Synthesis, characterization and crystal structure of 1-[3,5-bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]propan-1-one. Der Pharma Chemica 3(6):487–493
Samshuddin S, Butcher RJ, Akkurt M, Narayana B, Yathirajan HS, Sarojini BK (2011d) 1,3-Bis(4-fluorophenyl)-N, N′-(propane-1,3-diylidene)dihydroxylamine. Acta Cryst E 67:1954–1955
Samshuddin S, Narayana B, Sarojini BK, Yathirajan HS, Raghavendra R (2012a) Synthesis, characterization and biological evaluation of functionalized derivatives of versatile synthon 4,4′-difluoro chalcone. Der Pharma Chemica 4(4):1445–1457
Samshuddin S, Narayana B, Sarojini BK, Khan MTH, Yathirajan HS, Raj CGD, Raghavendra R (2012b) Antimicrobial, analgesic, DPPH scavenging activities and molecular docking study of some 1,3,5-triaryl-2-pyrazolines. Med Chem Res 21:2012–2022
Samshuddin S, Narayana B, Sarojini BK, Srinivasan R, Vinayachandra, Chandrashekar KR (2012c) Synthesis, characterization and biological evaluation of some pyrazoles derived from α, β-dibromo 4,4′-difluoro chalcone. Der Pharma Chemica 4(2):587–592
Samshuddin S, Narayana B, Shetty DN, Srinivasan R, Sarojini BK (2012d) (2E)-3-[4-(1H-Benzimidazol-2-ylmethoxy)-3-methoxyphenyl]-1-(4,4′′difluoro-5′-methoxy-1,1′:3′,1′′-terphenyl-4′-yl)prop-2-en-1-one. Molbank 2012:M764
Saravanan G, Alagarsamy V, Prakash CR (2010) Synthesis and evaluation of antioxidant activities of novel quinazolinone derivatives. Int J Pharm Pharm Sci 2:83–86
Senguttuvan S, Nagarajan S (2010) Synthesis of 2-amino-5-aryl-5,6-dihydro-7-(naphthalene-2-yl)quinazolin-4-ols. Int J Chem 2:108–112
Sherwin ER (1976) Antioxidants for vegetable oils. J Am Oil Chem Soc 53:430–436
Tamura Y, Yoshimoto Y (1980) An improved method for the conversion of cyclohexenones into anisoles. Chem Ind 1980:888–889
Thomas B, Saravanan KS, Kochupurackal P, Mohanakumar KP (2008) In vitro and in vivo evidences that antioxidant action contributes to the neuroprotective effects of the neuronal nitric oxide synthase and monoamine oxidase-B inhibitor, 7-nitroindazole. Neurochem Int 52:990–1001
Tiwari AK (2004) Antioxidants: new generation therapeutic base for treatment of polygenic disorders. Current Sci 86:1092–1102
Van Acker SABE, Van den Vijgh WJF, Bast F (1996) Structural aspects of antioxidant activity of flavonoids. Free Radic Biol Med 20:331–342
Vyas DH, Tala SD, Akbari JD, Dhaduk MF, Joshi HS (2009) Synthesis, antimicrobial and antitubercular activity of some cyclohexenone and indazole derivatives. Indian J Chem 48B:1405–1410
Wang CM, Pan XL (2012) DFT study on the antioxidant activity of a modeled p-terphenyl derivative. Chin J Struct Chem 31:894–902
Yadav JS, Rajora J, Srivastava YK (2011) Some transformations of benzimidazolyl chalcones using MAOS protocol—a green approach. Arch Appl Sci Res 3:192–198
Yang WM, Liu JK, Hu Z, Dong ZJ, Wu WL, Chen ZH (2004) Antioxidant properties of natural p-terphenyl derivatives from the mushroom Thelephora ganbajun. Naturforsch 59c:359–362
Acknowledgments
The authors are thankful to the Director, IISc, Bangalore for NMR data. BN thanks the UGC for financial assistance through SAP and BSR one time grant for the purchase of chemicals. SS thanks UGC for providing financial help for the research work through UGC-Research Fellowship in Sciences for Meritorious Students scheme.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Samshuddin, S., Narayana, B., Sarojini, B.K. et al. A study on the reactions of alkyl 4,6-bis(4-fluorophenyl)-2-oxocyclohex-3-ene-1-carboxylate and in vitro antioxidant activity of derivatives. Med Chem Res 22, 3002–3011 (2013). https://doi.org/10.1007/s00044-012-0304-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-012-0304-7