
Abstract
Status epilepticus (SE) is associated with a significant risk of cognitive impairment, and the increase of nitric oxide (NO) releasing has been reported during SE. We investigated the effects of neuronal nitric oxide synthase (nNOS) inhibitor, 7-nitroindazole (7-NI) and inducible nitric oxide synthase (iNOS) inhibitor, aminoguanidine (AG), on spatial performance of rats in the Morris water maze. Treatment with 7-NI, but not with AG, improved the performance of rats after SE not only in acquisition of the task but also in probe test. Furthermore, the level of SE-induced malondialdehyde (MDA), end product of lipid peroxidation, was significantly decreased only in animals receiving 7-NI injection. Taken together, the results of the present study provided evidence that the NO pathway contributed to oxidative stress after SE, and nNOS/NO pathway may underlie one of the potential mechanisms contributing to SE-induced spatial memory deficits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Status epilepticus (SE) is associated with cognitive impairment, cell loss and synaptic reorganization in the hippocampus. Animals can be used to evaluate memory, learning, and behavioral changes after SE, because animal models display many of the same anatomical and physiological changes observed in human SE [1, 2].
Nitric oxide (NO) is formed from arginine by the action of three kinds of different nitric oxide synthase (NOS) isoforms, two calcium-dependent forms, neuronal (nNOS), and endothelial (eNOS), and one calcium-independent form (iNOS). The increase of NO releasing has been reported during SE [3, 4]. NO has been viewed as a possible mediator of glutamate-induced neurotoxicity [5, 6]. The released NO appears to originate mainly from neurons [7]. The role of NO in SE-induced excitotoxicity has been studied in different experimental models, though the reported results are far from unequivocal.
Oxidative stress means an alteration in the delicate balance between free radicals and the scavenging capacity of antioxidant enzymes in favor of free radicals in the body systems. Brain tissue is particularly vulnerable to oxidative damage, possibly because of its high consumption of oxygen and the consequent generation of high quantities of free radical [8]. Growing data from experimental models and human brain suggest that oxidative stress and injury may play an important role in pathophysiology following acute neurological insults such as stroke and seizures. Membrane lipid derangements, including malondialdehyde (MDA), have been reported to contribute significantly to paroxysmal membrane malfunction during epileptogenesis; and enhanced free radical production and oxidative lipid damage have been demonstrated during seizures and seizure-mediated neuronal injury [9–12]. Further support for a role of free radicals in seizures comes from the successful use of exogenously administered antioxidants in protecting the brain against seizure-induced brain damage [13, 14].
Nitric oxide reacts rapidly with superoxide anion and produce speroxynitrite, which is a highly reactive free radical that has been shown to mediate much of the toxicity of NO [15]. Peroxynitrite can induce lipid peroxidation then to result in oxide damage. The NO toxicity seems to be produced by the direct effect of peroxynitrite [16].
The role of NO in SE-induced neurotoxicity remains unclear and debatable. Pilocarpine-induced SE in rat is a well-established model of temporal lobe epilepsy reproducing main characteristics of the disease [17, 18] and sensitive to pharmacological and environmental interventions [19–21]. The current study was designed to elucidate the role of NO in SE-induced spatial memory deficits and if NO-related oxide damages were involved in pilocarpine-treated rats.
Materials and methods
Animals and experimental procedures
Male Wistar rats (2 months old) weighing between 230 and 250 g were used in this study (Grade II, Certificate No.0068860, Experimental Animal Center, Chinese Academy of Medical Sciences, China). The experiments were conducted in accordance with the guidelines of the Medical Experimental Animal Administrative Committee of Nation. All efforts were made to minimize animal suffering and to reduce the number of animals used. The rats were housed under controlled temperature and light conditions (12 h light:12 h dark cycle with lights on at 08:00 a.m.), with ad libitum access to food and water.
Induction of seizures
Rats were injected with lithium chloride (intraperitoneally, 3 mEq/kg, Sigma, USA). About 20 h later, methylatropine (i.p., 1 mg/kg) was administered to limit the peripheral effects of the convulsant. SE was induced by injecting pilocarpine hydrochloride (subcutaneously, 30 mg/kg, Sigma) 30 min after methylatropine administration. The injection of diazepam (DZP, intramuscularly, 4 mg/kg) 60 min after SE onset allowed to improve survival. Rats in control group received the lithium- methylatropine, DZP treatment and saline instead of pilocarpine. For each animal, clinical signs of seizure activity were recorded. Briefly, within 5 min after pilocarpine injection, rats developed diarrhea, piloerection and other signs of cholinergic stimulation. During the following 15–20 min, rats exhibited head bobbing, scratching, chewing and exploratory behavior. Recurrent seizures started around 20–25 min after pilocarpine administration. These seizures which associated episodes of head and bilateral forelimb myoclonus with rearing and falling progressed to SE at approximately 35–40 min after pilocarpine injection, as described previously [22–24]. Drugs were given i.p. in a volume of 1 ml/kg body weight.
Drug treatments
Two different drugs, 7-nitroindazole (7-NI, Sigma) and aminoguanidine (AG, Sigma), acting on NOS were used. 7-NI, an inhibitor of NOS, exhibits selectivity for the brain enzyme [25] and has been described as potent selectivity for the neuronal isozyme in vivo [26]. 7-NI was generally used as a nNOS inhibitor in much study [27–29]). AG is an inducible NOS inhibitor. 7-NI was injected i.p. 30 min prior to pilocarpine treatment with a dose of 25 mg/kg and the injection was repeated twice at 1-h intervals after SE. AG, 100 mg/ml dissolved in physiological saline, was administered at 100 mg/kg i.p. 0, 6 h after pilocarpine treatment and then 50 mg/kg i.p for seven consecutive days. 7-NI and AG were given in a volume of 1 ml/kg body weight. The doses and injection schemes of NOS inhibitors employed were close to those used in the previous similar studies [10, 30, 31]. Only the rats showing SE were used for the further studies.
Tissue sampling
Rats were killed at 6 h, 24 h, 3 days, 7 days, 14 days after the onset of SE. Rats in 7-NI, AG and control groups were killed on 7 days. Brains were quickly removed, ice-cooled. Since numerous studies have identified an essential role for the hippocampus in spatial learning [32] and hippocampus is one of the most important brain regions involved in pathological process of epilepsy [33], the hippocampus was isolated, and stored at −70°C until further processing. Protein in hippocampus was measured using Coomassie brilliant blue with bovine serum album as standard.
Assay for NO and NOS activity
Rat hippocampus was weighed, and homogenized. Homogenization was carried either in saline (10% wt/vol) or HEPES buffer (20% wt/vol). Saline homogenate was used for estimation of NO contents, and NOS activity was measured in the HEPES-buffered homogenate.
The activities of cNOS (constitutive nitric oxide synthase, including the isoforms of nNOS and eNOS), iNOS as well as the concentrations of NO in the supernatant were determined by using commercially available kits (Jiancheng Bioengineering). All procedures completely complied with the manufacture’s instructions. The activities of enzymes were expressed as units per milligram protein.
Nitric oxide is a kind of extremely active molecule and turns rapidly to NO −2 and NO −3 in vivo. The assay of NO level was based on nitrate reductase specifically hydrogenise NO −3 to NO −2 and then was measured at a wavelength of 550 nm. NO is formed from l-arginine by the action of NOS. The assay of NOS activity was based on that NO can produce compounds with nucleophilic materials, which can be measured at a wavelength of 530 nm. cNOS is calcium-dependent and iNOS is calcium-independent, on which the discrimination of two kind of isoforms was based.
Assay for antioxidant enzymes and MDA
Saline homogenate was used for estimation of MDA contents, glutathione peroxidase (GSH-Px) and superoxide dismutase (SOD) activities. The activities of SOD, GSH-Px and the concentrations of the MDA in the supernatant were determined by using commercially available kits (Jiancheng Bioengineering, NanJing, China). All procedures completely complied with the manufacture’s instructions. The activities of enzymes were expressed as units per milligram protein.
The assay of SOD activity was based on its ability of inhibition the oxidation of oxymine by superoxide anion produced from the Xanthine-Xanthineoxiase system. One unit of SOD activity was defined as the amount that reduced the absorbance at 550 nm by 50%. The assay for GSH-Px activity was assayed by quantifying the rate of oxidation of the reduced glutathione to the oxidized glutathione by H2O2 catalyzed by GSH-Px. One unit of GSH-Px was defined as the amount that reduced the level of GSH by 1 μmol l−1 in 1 min/mg protein. Lipid peroxidation (LPO) was assessed by measuring the concentration of malondialdehyde, which can be measured at a wavelength of 532 nm by reacting with thiobarbituric acid (TBA) to form a stable chromophoric production. The level of MDA was expressed as nmol per milligram protein.
Antibodies and chemicals
Anti-nNOS, polyclonal-anti-rabbit IgG, horseradish peroxidase (HRP) conjugate, molecular weight marker, 4-Chloro-1 Naphthol, leupeptin, aprotinin,benzamidine, ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) were purchased from Sigma (USA). All reagents were analytical grade or the highest grade available. Antibodies against nNOS was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Western blot analyses
The left hippocampi were homogenized (1/10, w/v) in ice-cold buffer (50 m MTris-HCl (pH 7.5), 0.15 M NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 25 μg/ml leupeptin, 25 μg/ml aprotinin and 10 μM benzamidine). Equal amounts of protein for each sample (20μg of protein per lane) were separated by SDS/PAGE on 7.5% gels, blotted electrophoretically to PVDF membrane (Immobilon P), and incubated in Tris-buffered saline with Tween 20 (TBST) (50 mM Tris HCl pH 7.5), 150 mM NaCl, and 0.1% Tween 20) containing 3% bovine serum albumin for 30 min. Blots were incubated overnight with anti-nNOS (1:500) in 1% BSA. Blots were then subjected to three additional 10-min washings in TBST. Blot were incubated with alkaline HRP-conjugated monoclonal anti-rabbit IgG (1:10 00) in 1% BSA for 1 h at room temperature and three additional 10-min washes carried out with TBST. The membrane was incubated in 20 ml of fresh 0.05% 4-Chloro-1 Naphthol with 0.015% hydrogen peroxide in 20% methanol in TBS until color development. Images of immunoblots were analyzed with a computerized image analysis soft (Quantity One V4.31). We examined the changes on the protein concentration of nNOS after SE at 6 h, 24 h, 3 days, 7 days and 14 days.
Water maze task
A period of 3 days after SE, behavioral testing was performed on rats by the Morris water maze test. The water maze was a circular pool (diameter 150 cm, height 60 cm) filled with water (temperature 24°C) to a depth of 30 cm. An escape platform, 10 cm in diameter was submerged 2 cm below the water surface and remained constant in the northeast quadrant of the tank. The water had been made opaque by adding ink.
Acquisition of the task
The rats were trained in the water maze to locate the hidden platform using reference memory procedure. The procedure for both experiments consisted of 4 days of testing, with four trials, in which each animal was released from each of four different start positions each day; and order was determined in pseudorandom fashion. All animals were allowed to swim until the escape platform was located or for a maximum of 120 s. If the rat failed to navigate to each the platform within 120 s, it was gently guided to the platform and allowed to remain there for 10–15 s before the start of next trial. Latency was recorded using automated video tracking system. The inter-trial interval between the four trials given each day was approximately 5 min. Subjects who learn the task should show a decrease in latency and distance swam to the platform. They were given two trials per day with an intertrial interval of 8 h.
Probe test
Following acquisition of water maze task, the platform was removed from the tank, and the animal was allowed to swim for 120 s. The amount of time spent in target quadrant was measured.
Statistical analysis
Data are expressed as mean ± SEM. Two-way or one-way analysis of variance (ANOVA) followed by Turkey’s test was used to analyze the data. P-value less than 0.05 was considered statistically significant.
Results
SE-induced alteration in NO and NOS levels in rat hippocampus
An ANOVA revealed a significant group effect (F (5,39) = 26.996, P < 0.001) in the time course of NO level. NO significantly increased in SE group compared to controls at 6 h and on days 3 and 7(P < 0.001, respectively) according to post-hoc analysis (Fig. 1A.). We assayed the activities of cNOS and also for calcium-independent, iNOS. The time course in enzyme activity of both NOS isoforms is shown in Fig. 1B. There was significant group effect (F (5,39) = 12.596, P < 0.001) of cNOS activity, which significantly increased compared to controls only at 6 h after SE (P = 0.003). An ANOVA revealed a significant group effect (F(5,39) = 73.500, P < 0.001) of iNOS activity, which significantly increased compared to controls on days 3(P < 0.001) and 7(P < 0.001) after SE. The activity of iNOS did not significantly differ from controls during a period of 24 h (Fig. 1B). NOS activity assay did not differentiate between contribution of nNOS and eNOS to the total cNOS activity. To assess if SE induced changes in the expression of specific NOS isoforms, brains samples were analyzed by immunoblotting using specific antibodies to nNOS. ANOVA revealed a significant group effect (F (5,30) = 8.835, P < 0.001) and a following post-hoc analysis revealed that density of corresponding band shows an increased expression of nNOS protein at 6 h after seizures (P < 0.001) (Figs. 2A, B).

Time course of nitric oxide (NO) level (A) and constitutive nitric oxide synthase (cNOS), inducible nitric oxide synthase (iNOS) activity (B) in hippocampus in different time after status epilepticus (SE). Columns represent mean ± SEM.✭Values significantly different from controls (P < 0.05) (one way ANOVA followed by Turkey’s multiple comparison test) (n = 6–9 rats in each group)

Western blot showing the time course changes in neuronal nitric oxide synthase (nNOS) protein expression; Columns represent mean ± SEM. ✭Values significantly different from controls (P < 0.05) (one way ANOVA followed by Turkey’s multiple comparison test) (n = 6–9 rats in each group)
SE-induced alteration in MDA levels in rat hippocampus and SOD and GSH-Px activity
MDA, which is a measure of lipid peroxidation, was measured spectrophotometrically. An ANOVA revealed a significant group effect (F(5,39) = 106.435, P < 0.001). Post-hoc analysis indicated that the level of MDA significantly increased compared to controls at 6 h (P = 0.009) and on days 1(P = 0.004), 3 (P < 0.001) and 7(P < 0.001), while later on 14 day the levels were not different from controls (Fig 3A).

Time course of malondialdehyde (MDA) content (A) and superoxide dismutase (SOD), glutathione peroxidase GSH-Px activities (B) (Mean ± SEM) in hippocampus in different time after SE. Columns represent mean ± SEM. ✭Values significantly different from controls (P < 0.05) (one way ANOVA followed by Turkey’s multiple comparison test) (n = 6–9 rats in each group)
As enzymatic defense mechanisms, SOD and GSH-Px play pivotal roles in preventing cellular damage. A low steady-state level of intracellular superoxide is maintained by SOD. As shown in Fig.3B, there was a significant group effect in SOD activity (F (5,39) = 34.137, P < 0.001). Post-hoc analysis of these data showed that SOD activity significantly increased at 6 h (P < 0.001) and decreased on days 3 (P = 0.01) compared to controls. On the other hand, ANOVA indicated significant group differences (F (5,39) = 26.446, P < 0.001) in GSH-Px activity. Post-hoc analysis of these data showed that the activity significantly decreased on day 3(P < 0.001), 7 (P = 0.001) and 14 (P < 0.001) compared to controls (Fig.3B).
Effects of 7-NI and AG on SOD and GSH-Px activity and MDA levels
As shown in Fig. 4B, neither treatment with 7-NI nor AG affected SOD activity according to ANOVA (F (3, 27) = 1.251, P = 0.311). However, ANOVA revealed a significant group effect (F (3,27) = 40.053, P < 0.001) in GSH-Px activity. Post-hoc analysis revealed that GSH-Px activity significantly increased on 7-NI group compared to SE group (P = 0.003).On the other hand, AG had no obvious effects (P = 0.123). In addition, ANOVA revealed a significant group effect (F (3,27) = 40.053, P < 0.001) in MDA level. Post-hoc analysis revealed that MDA significantly decreased on 7-NI group compared to SE group (P < 0.001). However, there were no obvious effects on AG groups (P = 0.216) (Fig. 4A).

Effects of treatment with 7-NI and AG on SE-induced changes in the content of malondialdehyde (MDA) (A) and the activity of superoxide dismutase (SOD), glutathione peroxidase (GSH-Px) (B) in hippocampus. Columns represent mean ± SEM. ✭Values significantly different from controls (P < 0.05) #Values significantly different from SE group (P < 0.05) (one way ANOVA followed by Turkey’s multiple comparison test) (n = 6–9 rats in each group)
Effects of 7-NI and AG on the acquisition of the task
Statistical analyses (two way ANOVA) revealed a significant main effect of treatment and a significant interaction of treatment × times for latency to reach the platform (F (3,33) = 47.110, P < 0.001; F(21,264) = 4.131, P < 0.001; see Fig. 5A).

Effect of treatment on spatial learning (A) and swim speed (B) in the rat. Mean ± SEM values are shown as the time required to find an invisible platform submerged in water (escape latency) (n = 10–11 rats in each group)
Post-hoc analysis revealed subjects in SE group had longer latencies to the platform, as compared to control group, on times 1 (P = 0.015), times 2(P = 0.005), times 4 (P < 0.001), times 5 (P < 0.001), times 6 (P = 0.001), times 7 (P < 0.001), times 8 (P = 0.02). Post-hoc analysis revealed animals receiving 7-NI injection had shorter latencies to the platform, as compared to SE, on times 2 (P = 0.024), 4 (P = 0.001), 5 (P = 0.038). However, the latencies in AG rats had no difference compared to SE group at all times. A significant group effect between SE and 7-NI + SE group was found (P = 0.011), but a non-significant effect between SE (P = 0.98) and AG + SE group. There were no speed differences among the different groups (F (3, 33) = 1.095, P = 0.352) and interaction of treatment × times (F (21,264) = 0.528, P = 0.958; Fig. 5B).
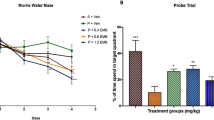
Effects of 7-NI and AG on the probe test
During the probe test trial, statistical analyses (one way ANOVA) revealed a significant main effect of treatment (F (3,33) = 17.092), P < 0.001). Post-hoc analysis revealed control rats spending a longer in the target quadrant than did SE (P < 0.001) (see Fig. 6). No differences were found for the time spent in the target quadrant between SE and AG groups (P = 0.994). However, animals receiving 7-NI injection spent longer than did SE rats (P = 0.009). There were no speed differences among the different groups ((F (3, 33) = 0.957, P = 0.425; Fig. 6B).

The changes of time spent in target quadrant (A) and swim speed (B) on retention of the spatial learning task (Mean ± SEM) ✭P < 0.05 compared to control group; # P < 0.05 compared with SE group (one way ANOVA followed by Turkey’s multiple comparison test) (n = 10–11 rats in each group)
Discussion
Oxide damages after Pilocarpine-induced SE
In brain, SOD and GSH-Px are considered to be the relatively more important antioxidant enzymes [34]. SOD removes superoxide anion, a kind of main reactive oxygen species, from the cell [35]. On the early state following SE, an increase SOD activity was observed. At the same time, the activity of GSH-Px was unchanged. The results of increased SOD activity following seizures are consistent with the report of Chavko et al. [36]. The increased SOD activity maybe an early compensatory reaction in response to increased MDA levels, since MDA increased in the same time. The activity of GSH-Px was unaltered until 3 days after SE, suggesting that the glutathione system may not be the major free radical scavenging system in the early state.
The level of MDA, which is a measure of lipid peroxidation, was found to be significantly raised after SE from 6 h in hippocampus, while it returned to control level on 14th day. The results implicated oxide damages were involved in SE induced neurotoxicity. The results of increased MDA level following SE are consistent with the report of Rajasekaran [10].
In the present study, the increase of MDA level at 6 h after SE corresponds to increases in NO levels. This observation suggests that NOS-mediated elevation of NO production could be potentially involved in the enhancement of MDA during the development of SE induced by polocarpine. NO reacts rapidly with superoxide anion and produces speroxynitrite, which is a highly reactive free radical that has been shown to mediate much of the toxicity of NO [15]. Chavko et al. [36] have demonstrated that seizures induce a significant increase in the production of protein nitrotyrosine, which is considered a footprint of peroxynitrite. The activity of iNOS did not change until 3 days while cNOS activity increased as early as at 6 h after SE, and further results from immunoblotting reveal nNOS maybe the mainly isoform resulting in the increased cNOS activity, which suggests that following acute increased NO is mainly produced through cNOS, especially nNOS.
Taken together, the results of the present study provide evidence for the participation of the NO pathway contributing to oxidative stress after SE. The pathway of nNOS/NO/peroxynitrite may underlie one of the potential mechanisms contributing to seizures-induced neurotoxicity.
Effects of 7-NI and AG on performance of SE rats in Morris water maze related oxidative damages
In agreement with previous work [37], SE induced deficits in spatial learning and memory. Experimentally induced seizures can lead to postictal impairment of learning [38]. Compared with SE animals, rats treatment with 7-NI improved the performance in a spatial maze task, which suggests 7-NI can prevent the SE-induced spatial memory impairment even though mechanisms still remain unclear.
In fact, several in vivo studies using 7-NI support a role of neuron-deirved NO in the excitotoxicity-related activity [39, 40].According to previous reports, 7-NI not only reduces NO generation but also prevents the delayed death of hippocampal neurons [30, 41], while the effects produced by 7-NI can be attributed to its ability to inhibit nNOS activity directly. According to Mackenzie et al. [42], 7-NI produced the maximal effects at 0.5 h post-infection and recovery at 24 h (i.p. administration). In this study, since the significant increase of nNOS appeared as early as at 6 h and recovered at 24 h after pilocarpin-induced SE, the rats was administered 7-NI treatment on the day of SE.
In addition, in our study, treatment with 7-NI not only prevented SE-induced increase in MDA level but decrease in GSH-Px activity in hippocampus, suggesting that MDA may be mainly derived from NO-related oxidative stress. NO alone is a poorly reactive species; however, it is able to undergo secondary reactions to form highly oxidizing and nitrating species, NO2, N2O3, and peroxynitrite, which, especial peroxynitrite, can lead to oxide damages, such as lipid peroxidation [43]. These secondary reactive nitrogen species are also capable of modifying a diversity of biomolecular structures in cell [44, 45], including antioxidant enzymes. On the other report, 7-NI can retard seizures accompanied by reduction of seizure induced increase in nitrotyrosine levels [36], a mark of peroxynitrite.
We have demonstrated that 7-NI treatment significantly prevented SE-induced decease of GSH-Px activity. Interestingly, some in vitro studies have suggested that GSH-Px alone offered greater protection from oxidative stress than SOD in the brain [46]. NO in vitro and in cultured macrophage cell line has been shown to inhibit GSH-Px activity by interfering with seleno-cysteine residues [47]. According to another report [48], 7-NI can inhibit formation of H2O2. Both may account for its ability to increase in hippocampal GSH-Px activity in SE rats.
Take together, the neuroprotective effects of 7-NI may primarily relate to the inhibition of nNOS activity. Reduction of peroxynitrite and then prevention oxide damages may be another mechanism by which 7-NI resulted in preservation of memory after SE.
However, there are conflicting reports on the effects of 7-NI in memory of seizure rats. In previous study [49], pretreatment with 7-NI at 50 and 100 mg/kg, did not produce significant changes in memory formation, but at 150 and 200 mg/kg resulted in memory impairment in picrotoxin-induced convulsions. Since formation of NO during long-term potentiation is suggestive of a significant involvement of NO in learning and memory processes [50], the difference may be based on dose of 7-NI and disparate animal models, and the protective dose of 7-NI may be narrow, which depend on further research.
In the nervous system, nNOS is largely responsible for NO production, while iNOS has been implicated in some important central processes [51]. Since the activity of iNOS increased after seizure, which is accordant to the previous reports [41, 52], it is valuable to examine the probable contribution of iNOS in SE-induced memory deficits. In this study, we used AG, which is an irreversible specific inhibitor of iNOS [53]. According to the results, the activity of iNOS had increased on 3d and till 7d after SE. However, it seems to be necessary to pretreat with AG to prevent the increasing iNOS, since iNOS is a high-output isoform [54] and plays a potent neurotoxic role. However, AG did not affect the performance of SE rats in Morris water maze in our study. iNOS produces large amounts of NO continuously for long periods, a feature that is responsible for the cytotoxicity of NO, while iNOS may play an important role during chronic stress in SE rats.
In conclusion, the present data provide evidence that NO from nNOS but not iNOS related oxidative damage that was involved in spatial memory deficits in the early state in pilocarpin-induced SE rats.
References
Holmes GL, Thompson JL, Marchi TA, Gabriel PS, Hogan MA, Carl FG, Feldman DS (1990) Effects of seizures on learning, memory, and behavior in the genetically epilepsyprone rat. Ann Neurol 27:24–32
Rice AC, Floyd CL, Lyeth BG, Hamm RJ, De Lorenzo RJ (1998) Status epilepticus causes long-term NMDA receptor-dependent behavioral changes and cognitive deficits. Epilepsia 39:1148–1157
Milatovic D, Zivin M, Gupta RC, Dettbarn WD (2001) Alterations in cytochrome c oxidase activity and energy metabolites in response to kainic acid-induced status epilepticus. Brain Res 912:67–78
Gupta RC, Dettbarn WD (2003) Prevention of kainic acid seizures-induced changes in levels of nitric oxide and high-energy phosphates by 7-nitroindazole in rat brain regions. Brain Res 981:184–192
Dawson VL, Dawson TM, London ED, Bredt DS (1991) Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA 88:6368–6371
Dawson VL, Kizushi VM, Huang PL, Snyder SH, Dawson TM (1996) Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J Neurosci 16:2479–2487
Mulsch A, Busse R, Mordvintcev PI, Vanin AF, Nielsen EO, Scheel-Kruger J, Olesen SP (1994) Nitric oxide promotes seizure activity in kainate-treated rats. NeuroReport 5:2325–2328
Castagne V, Gautschi M, Lefevre K, Posada A, Clarke PGH (1999) Relationships between neuronal death and the cellular redox status: focus on the developing nervous system. Prog Neurobiol 59:397–423
Iihan A, Aladag MA, Kocer A, Boluk A, Gurel A, Armutcu F (2005) Erdosteine ameliorates PTZ-induced oxidative stress in mice seizure model. Brain Res Bull 30:495–499
Rajasekaran K (2005) Seizure-induced oxidative stress in rat brain regions: blockade by nNOS inhibition. Pharmacol Biochem Behav 80:263–272
Frantseva MV, Velazquez JL, Hwang PA, Carlen PL (2000) Free radical production correlates with cell death in an in vitro model of epilepsy. Eur J Neurosci 12:1431–1439
Patel M, Liang LP, Roberts LJ II (2001) Enhanced hippocampal F2-isoprostane formation following kainate-induced seizures. J Neurochem 79:1065–1069
Rauca C, Wiswedel I, Zerbe R, Keilhoff G, Krug M (2004) The role of superoxide dismutase and alpha-tocopherol in the development of seizures and kindling induced by pentylenetetrazol-influence of the radical scavenger alpha-phenyl-N-tert-butyl nitrone. Brain Res 1009:203–212
Gupta RC, Milatovic D, Dettbarn WD (2001) Depletion of energy metabolites following cetylcholinesterase inhibitor-induced status epilepticus: protection by antioxidants. Neurotoxicology 22:271–282
Brown GC (1999) Nitric oxide and mitochondrial respiration. Biochim Biophys Acta 1411:351–369
Beckman JS, Koppenol WH (1996) Nitric oxide superoxide, and peroxynitrite: the good, the bad and the ugly. Am J Physiol 271:C1424–C1437
Holmes GL, Khazipov R, Ben-Ari Y (2002) Seizure-induced damage in the developing human: relevance of experimental models. Prog Brain Res 135:321–334
Löscher W (2002) Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res 50:105–123
Stewart LS, Persinger MA (2001) Ketamine prevents learning impairment when administered immediately after status epilepticus onset. Epilepsy Behav 2:585–591
Faverjon S, Silveira DC, Fu DD, Cha BH, Akman C, Hu Y, Holmes GL (2002) Beneficial effects of enriched environment following status epilepticus in immature rats. Neurology 59:1356–1364
Kubova H, Mares P, Suchomelova L, Brozek G, Druga R, Pitkanen A (2004) Status epilepticus in immature rats leads to behavioural and cognitive impairment and epileptogenesis. Eur J Neurosci 19:3255–3265
Dube´ C, Boyet S, Marescaux C, Nehlig A (2001) Relationship between neuronal loss and interictal glucose metabolism during the chronic phase of the lithiumpilocarpine model of epilepsy in the immature and adult rat. Exp Neurol 167:227–241
Roch C, Leroy C, Nehlig A, Namer IJ (2002) Magnetic resonance imaging in the study of the lithium-pilocarpine model of temporal lobe epilepsy in adult rats. Epilepsia 43:325–335
Voutsinos-Porche B, Koning E, Clement Y, Kaplan H, Ferrandan A, Motte J, Nehlig A (2006) EAAC1 glutamate transporter expression in the rat lithium-pilocarpine model of temporal lobe epilepsy. J Cereb Blood Flow Metab 26:1419–1430
Moore PK, Babbedge RC, Wallace P, Gaffen ZA, Hart SL (1993) 7-Nitro indazole, an inhibitor of nitric oxide synthase, exhibits anti-nociceptive activity in the mouse without increasing blood pressure. Br J Pharmacol 108(2):296–297 Feb
Southan GJ, Szabo C (1996) Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem Pharmacol 51(4):383–394 Feb 23
Komatsu T, Sakurada C, Sasaki M, Sanai K, Tsuzuki M, Bagetta G, Sakurada S, Sakurada T (2007) Extracellular signal-regulated kinase (ERK) and nitric oxide synthase mediate intrathecal morphine-induced nociceptive behavior. Neuropharmacology. 52(5):1237–1243. Apr. Epub 2007 Jan 14
Di Matteo V, Benigno A, Pierucci M, Giuliano DA, Crescimanno G, Esposito E, Di Giovanni G (2006) 7-nitroindazole protects striatal dopaminergic neurons against MPP+-induced degeneration: an in vivo microdialysis study. Ann N Y Acad Sci 1089:462–471 Nov
Manzanedo C, Aguilar MA, Rodriguez-Arias M, Navarro M, Minarro J (2004) 7-Nitroindazole blocks conditioned place preference but not hyperactivity induced by morphine. Behav Brain Res 150(1–2):73–82 Apr 2
Montecot C, Rondi-Reig L, Springhetti V, Seylaz J, Pinard E (1998) Inhibition of neuronal (type 1) nitric oxide synthase prevents hyperaemia and hippocampal lesions resulting from kainate-induced seizures. Neuroscience 84:791–800
Danielisova V, Nemethova M, Burda J (2004) The protective effect of aminoguanidine on cerebral ischemic damage in the rat brain. Physiol Res 53:533–540
Lynch MA (2004) Long-term potentiation and memory. Physiol Rev 84:87–136
Lee SK, Kim DW, Kim KK, Chung CK, Song IC, Chang KH (2005) Effect of seizure on hippocampus in mesial temporal lobe epilepsy and neocortical epilepsy: an MRS study. Neuroradiology 47:916–923
Reiter RJ (1998) Oxidative damage in the central nervous system: protection by melatonin. Prog Neurobiol 56:359–384
Fridovich I (1989) Superoxide dismutases: an adaptation to a paramagnetic gas. J Biol Chem 264:19328–19333
Chavko M, Auker CR, McCarron RM (2003) Relationship between protein nitration and oxidation and development of hyperoxic seizures. Nitric Oxide 9:18–23
Erdogan F, Golgeli A, Arman F, Ersoya AO (2004) The effects of pentylenetetrazole-induced status epilepticus on behavior, emotional memory, and learning in rats. Epilepsy Behav 5:388–393
Boukhezra O, Riviello P, Fu DD, Lui X, Zhao Q, Akman C, Holmes GL (2003) Effect of the postictal state on visual–spatial memory in immature rats. Epilepsy Res 55:165–175
O’Neill MJ, Hicks C, Ward M (1996) Neuroprotective effects of 7-nitroindazole in the gerbil model of global cerebral ischemia. Eur J Pharmacol 310:343–354
Schulz JB, Henshaw DR, Siwek D, Jenkins BG, Ferrante R.J., Cipolloni P.B., Kowall NW, Rosen BR, Beal MF (1995) Involvement of free radicals in excitotoxicity in vivo. J Neurochem 64:2239–2247
Murashima YL, Yoshii M, Suzuki J (2000) Role of nitric oxide in the epileptogenesis of EL mice. Epilepsia 41:S195–S199
MacKenzie GM, Rose S, Bland-Ward PA, Moore PK, Jenner P, Marsden CD, (1994) Time course of inhibition of brain nitric oxide synthase by 7-nitro indazole. Neuroreport 5:1851–1852
Ebadi M, Sharma S (2006) Metallothioneins 1 and 2 attenuate peroxynitrite-induced oxidative stress in Parkinson disease. Exp Biol Med 231:1576–1583
Reiter TA (2006) NO chemistry: a diversity of targets in the cell. Redox Rep 11:194–206
Chirino YI, Orozco-Lbarra M, Pedraza-Chaverri J (2006) Role of peroxynitrite anion in different diseases. Rev Invest Clin 58:350–358
Erakovic V, Zupan G, Varljen J, Laginja J, Simonic A (2000) Lithium plus pilocarpine induced status epilepticus—biochemical changes. Neurosci Res 36:157–166
Asahi M, Fujii J, Suzuki K, Seo HG, Kuzuya T, Hori M, Tada M, Fujii S, Taniguchi N (1995) Inactivation of glutathione peroxidase by NO: implication for cytotoxicity. J Biol Chem 270:21035–21039
Mayer B, Klatt P, Werner ER, Schmidt K (1994) Molecular mechanisms of porcine brain nitric oxide synthase by the ntinociceptive drug 7-nitroindazole. Neuropharmacology 33:1253–1259
Vanaja P, Ekambaram P (2004) Demonstrating the dose- and time-related effects of 7-nitroindazole on picrotoxin-induced convulsions, memory formation, brain nitric oxide synthase activity, and nitric oxide concentration in rats. Pharmacol Biochem Behav 77:1–8
Zhuo M, Laitinen JT, Li XC, Hawkins RD (1999) On the respective roles of nitric oxide and carbon monoxide in long-term potentiation in the hippocampus. Learn Mem 6:63–76
Licinio J, Prolo P, McCann SM, Wong ML (1999) Brain iNOS: current understanding and clinical implications. Mol Med Today 5:225–232
Yang R, Huang ZN, Cheng JS (2000) Anticonvulsion effect of acupuncture might be related to the decrease of neuronal and inducible nitric oxide synthases. Acupunct Electrother Res 25:137–143
Al-Shabanah OA, Alam K, Nagi MN, Al-Rikabi AC, Al-Bekairi AM (2000) Protective effect of aminoguanidine, a nitric oxide synthase inhibitor, against carbon tetrachloride induced hepatotoxicity in mice. Life Sci 66:265–270
De Alba J, Cardenas A, Moro MA, Leza JC, Lorenzo P, Bosca L, Lizasoain I (1999) Down-regulation of neuronal nitric oxide synthase by nitric oxide after oxygen-glucose deprivation in rat forebrain slices. J Neurochem 72:248–254
Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (30470453, 30640037), Municipal Science Foundation Research of TianJin (06YFJMJC09400).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Z.W., Zhang, T. & Yang, Z. Involvement of Nitric Oxide in Spatial Memory Deficits in Status Epilepticus Rats. Neurochem Res 32, 1875–1883 (2007). https://doi.org/10.1007/s11064-007-9374-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-007-9374-1