
Abstract
Histone deacetylases have emerged as important drug target with a multitude of therapeutic potentials for their inhibitors. With the purpose of designing new chemical entities with enhanced inhibitory potencies against histone deacetylases, a two-dimensional (2D) quantitative structure-activity relationship (QSAR) study was carried out on aminophenylbenzamides and acrylamide derivatives as inhibitors of these deacetylases. The developed model was validated by standard QSAR parameters and through a detailed structural analysis of how it reproduces and explains the differences in the experimentally known activity data. The model showed a good correlative and predictive ability, with a cross-validated correlation coefficient of 0.594. The conventional and predictive correlation coefficients were found to be 0.725 and 0.577 for the antiproliferative activity of aminophenylbenzamides and acrylamide series, respectively. The study indicated that their antiproliferative activity is largely explained by the steric factors of the substituents, highlighting the role of the size and shape of the inhibitor in forming effective binding interactions with histone deacetylase. The models could be usefully employed to design inhibitors with greatly enhanced potency and selectivity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epigenetic modifications are increasingly being recognized as playing a major role in normal cellular biochemistry, physiology, and disease processes particularly in cancer, where improper gene expression has been shown to play a crucial part in the aetiology of the disease. Histone deacetylase (HDAC) enzymes, which regulate the level of histone acetylation, are one of the major groups mediating epigenetic control. HDAC are so named because histones were the first target substrates identified for these enzymes. However, it has become increasingly clear that this is a somewhat inappropriate designation given the increasing number of non-histone targets. HDAC enzymes act by regulating the level of biological acetylation and deacetylation reactions of their targets. Acetylation of the N-terminal region of histone proteins promotes gene expression (Garea et al., 2004; Somech et al., 2004). The aberrant recruitment of transcription corepressors results in hypoacetylation of the histone proteins and suppression of gene transcriptional activities, consequently leading to malignant cell proliferation. Thus, inhibition of HDACs, which induces histone hyperacetylation, provides a potential target for the development of synthetic anticancer drugs (Bouchain et al., 2003; Curtin, 2004; Weinmann et al. 2004 ; Monneret, 2005). In mammalian cells, HDACs are particularly responsible for the deacetylation of N-terminal lysine residues in the core histones such as H2A, H2B, H3, and H4. Deacetylation of these histones is associated with a relatively compact and inaccessible chromatin state, which generally correlates with lower gene transcription (McLaughlin et al., 2003). There has been substantial progress in the development of drugs that target epigenetic control processes as a new class of mechanism-based cancer therapeutics (McLaughlin et al., 2004), and one of the fastest moving areas is the development of histone deacetylase inhibitors (HDACIs). Cell-based studies have shown that HDACIs have a powerful antiproliferative property, causing cell-cycle arrest, apoptosis, and differentiation; these antiproliferative effects are far more pronounced in tumor cells than in normal cells. As a consequence, various HDACIs have entered, and in some cases completed, early clinical trials where, importantly, they have been found to exhibit a generally favorable toxicity profile (Inche et al., 2006). Given the importance of histone deacetylation as a major mechanism used by cells to regulate enzymes and other proteins and the association of many maladies with its aberrations (Carey, 2006), HDACs have increasingly become important targets and the hunt for HDAC inhibitors has been intensified and attracted great attention in drug discovery over the years (Pandolfi, 2001).
TSAR is an integrated analysis package for the interactive investigation of quantitative structure–activity relationships (Klocker et al., 2002; Kovatcheva et al., 2003). It provides the functions required to carry out any QSAR investigation whether in pharmaceutical or other areas of application. In common with all QSAR methods (Kubinyi, 1997; Dessalew et al., 2007a, 2007b, 2007c), it is based on a numerical description of molecular structure and employs statistics to obtain a correlation. Molecular structures are represented with a variety of two- (2D) and three-dimensional (3D) descriptors, the activity–descriptor relationship is computed by various standard statistical tools such as multiple regression, partial least-square regression, and neural-network analysis, and the output is displayed in the form of a model highlighting substituent points that are strongly correlated with the pharmacotoxicological properties under investigation. The TSAR methodology assumes that a suitable sampling of these structural descriptors provides all the information needed for understanding their biological properties. TSAR has been employed to investigate the effect of l-alanine ester variation on the anti-HIV activity and cytotoxicity of phosphoramidate derivatives (Knaggs et al., 2000). Recently, Chang and coworkers (Chang et al., 2007) have applied TSAR to study the neuroprotective activity of terpenoids and found that that the neuroprotection was mainly governed by the lipophilicity, shape index, and electrostatic property of the terpenoids studied. Similarly investigators (Lohray et al., 2006) have applied TSAR to analyze the structural requirement for the antibacterial properties of phenyloxazolidinone derivatives. These results all attest to the usefulness of such a methodology in understanding the structural requirements for the pharmacological properties of a given series.
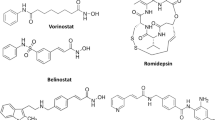
The intense research on small-molecule inhibitors of histone deacetylase has produced a diverse class of chemical scaffolds, which includes hydroxamic acids (Nishino et al., 2004; Jose et al., 2004), phthalimides (Shinji et al., 2005), mercaptoacetamides (Chen et al., 2005), and alkanoic acids (Marson et al., 2007). Figure 1 shows some inhibitors of these deacetylating enzymes. Although diverse in structure and large in number, none of them has progressed into a therapeutically useful agent. One of the effects hampering the development of an HDAC inhibitor drug is the difficulty in attaining selectivity and their weak binding affinity. TSAR, in common with other QSAR tools, is generally employed to enhance and optimize the binding affinity using a series of compounds acting on the same target with the same mechanism of action. As a quantitative pharmacophore mapping tool, such a methodology is valuable in pinpointing the structural requirements for the observed pharmacotoxicological properties by the series. Such insights are an aid to the design of a new entity having an acceptable level of potency and selectivity. In this paper, we report a 2D QSAR study carried out on HDACIs inhibitors in the anticipation of obtaining a model that could account for the quantitative differences in bioactivity seen in this series and to capitalize on these insights to design ligands with pronounced inhibitory potency and selectivity.

Examples of HDAC inhibitors
Computational details
Dataset for analysis
In vitro biological activity data reported as half maximum inhibitory concentration (IC50) values for the inhibition of histone deacetylase by series of aminophenylbenzamides and aminophenylacrylamide derivatives (Mordei et al., 2006) was used for the current study. As biological activities are generally skewed and are measures of the free energy of binding, the reported inhibitory constant values were converted into corresponding pIC50 values using the following formula:
Molecular modeling
The structures of 29 aminophenol derivatives were selected for QSAR studies (Table 3). The structures were sketched using ChemDraw Ultra 5.0 and were exported to TSAR 3.3 software. Three-dimensional structures of all the molecules were generated. Partial charges were derived using the Charge-2 CORINA 3D package in TSAR 3.3 and their geometries were optimized using the Cosmic module of TSAR. The calculations were terminated when the energy difference or the energy gradient were smaller than 1 × 10−5 and 1 × 10−10 kcal/mol, respectively.
Molecular descriptors were calculated with TSAR version 3.3. Descriptor values were obtained for substituents that varied at a common point on the generic structure. TSAR affords the calculation of the following descriptors: molecular surface area and volume, moments of inertia, ellipsoidal volume, Verloop parameters, dipole moments, lipole moments, molecular mass, Wiener index, molecular connectivity indices, molecular shape indices, electrotopological state indices, log P, number of defined atoms (carbon, nitrogen, etc.), rings (aromatic and aliphatic), and groups (methyl, hydroxyl, etc.). Vamp which is a semiempirical molecular orbital package in TSAR version 3.3, was used to calculate the electrostatic properties such as the total energy, electronic energy, nuclear repulsion energy, accessible surface area, atomic charge, mean polarizability, heat of formation, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) eigenvalues, ionization potential, total dipole, polarizability, and dipole components. Structure optimization was performed in vacuo using default parameters with the Austin model 1 (AM1) Hamiltonian. Pairwise correlation analysis of the calculated descriptors was performed. The model was obtained using descriptors that are strongly correlated with the antiproliferative activity. The descriptors and their characteristics are given in Table 2.
Statistical analysis
The relationship between the structural parameters (TSAR descriptors) and the biological activities was quantified by the multiple linear regressions implemented in TSAR version 3.3. Values for F-to-enter and F-to-leave were both 4. The cross-validation analysis was performed using the leave-one-out (LOO) method in which one compound is removed from the dataset and its activity is predicted using the model derived from the rest of the dataset. The cross-validated r 2 and conventional r 2 values that resulted in the lowest prediction error was taken. Unless otherwise stated, the default values for the other parameters were used.
Predictive correlation coefficient \( {\left( {r^{2}_{{{\text{pred}}}} } \right)} \)
The predictive capabilities of the 2D QSAR models was determined from a set of nine compounds that were excluded during model development. The optimization, charge derivation, and all other steps of the test sets were the same as that of the training set compounds as described above, and their activities were predicted using the model produced by the training set. The predictive correlation co-efficient \( {\left( {r^{2}_{{{\text{pred}}}} } \right)} \), based on the test set molecules, was computed using
where SD is the sum of the squared deviations between the biological activities of the test set and the mean activities of the training set molecules, and the predictive residual sum of squares (PRESS) is the sum of the squared deviations between the predicted and actual activity values for each molecule in the test set.
Results and discussion
The 2D QSAR TSAR study was carried out using aminophenol derivatives. Molecules, which lack biological inhibitory activity in numerical form, have been removed from the analysis. Following this, 29 molecules were left for the current study. These were partitioned into a training set of 22 and a test set of 7 compounds at random with a bias given to both chemical and biological diversity in both the training and test set molecules so as to form a 4:1 ratio of the training set to the test set for a standard QSAR study. Despite the ambiguity of the drug–receptor interaction in general, a statistically significant model was obtained from the TSAR study.
The TSAR multiple regression analysis is summarized in Table 1. The cross-validated correlation coefficient defines the goodness of the prediction whereas the non-cross-validated conventional correlation coefficient indicates the goodness of fit of the QSAR model. The F-test value indicates the degree of statistical confidence. As it is evident from Table 1, a cross-validated correlation coefficient of 0.594 was obtained using the leave-one-out cross-validation procedure. This indicates a good internal predictive capability of the developed model. The model also exhibited a non-cross-validated correlation coefficient of 0.725. The external predictive capability of a QSAR model is generally checked using test sets. All other procedures, including the geometry optimization, the charge computation, and the calculation of the structural descriptors of the nine test set molecules, were done in a manner analogous to the molecules in the training set. A predictive correlation coefficient of 0.577 was obtained from the study, indicating its usefulness in predicting the activities of external molecules. Yet another way to evaluate the usefulness of the developed model is to test for statistical stability. To this end, the standard error of the estimate and predictive residual sum of squares may be employed. The low values of the standard error of estimate (0.2985) and that of PRESS (0.0729 for the training set and 1.937 for the test sets) further testify to the statistical significance of the developed model. Table 2 shows the descriptors included in the final QSAR model and their statistical significance.
The structures of the inhibitors chosen and their actual and predicted activity are shown in Table 3. Figure 2 shows plots of the actual versus the predicted pIC50 values for the training and test molecules. Histograms of the residuals of the training set and test set molecules are presented in Fig. 3.

Plots of the actual versus the predicted pIC50 values for the training set (filled symbols) and test molecules (empty symbols)

Histograms of residuals for the molecules in the training set (empty bars) and the test set (filled bars)
The QSAR model with a high statistical significance is represented by Eq. 1:
where X 1 is Verloop L, X 2 is Verloop B2, and X 3 is Verloop B3.
The statistics for this equation are shown in Table 1. As the model shows, the antiproliferative activity increases with an increase in the Verloop B2 parameter while the activity was found to decrease with an increase in the values for the Verloop L and Verloop B3 parameters of the substituent. Verloop L alone correlates with the antiproliferative activity with r = 0.42 whereas Verloop B2 shows a correlation with the antiproliferative activity with r = 0.59. This is reflected in Eq. 1, in which the Verloop B2 parameter is more correlated with the activity than the Verloop L descriptor.
The Verloop parameters (Verloop, 1976a, 1976b, 1977) are a set of multidimensional steric descriptors that define a box that can be used to characterize the shape and volume of the substituent, which are very important in explaining the steric influence of substituents in the interaction of organic compounds with macromolecular drug receptors. The length parameter, Verloop L, is defined as the maximum length of the substituent along the axis of the bond between the first atom of the substituent and the parent molecule whereas the Verloop B1–B5 parameters describe the width of the substituent in the direction perpendicular to L. The higher activity of compound 26t as compared to compound 25 may be explained on the basis of this width parameter. The dimethoxyphenyl group in the former is bulkier than the monomethoxyphenyl of compound 25, with the former having a higher width and hence a higher antiproliferative activity. The decrease of the antiproliferative activity with the length of substituents is demonstrated by the results for compounds 2t, 3, 4, and 5, which differ only in a single substitution. Compound 3 is more active than compounds 2t, 4, and 5 as its F substitution is shorter than the dimethylamino of compound 2t, the methoxy of compound 5, and the trifluoromethoxy of compound 4. Compounds 5 and 6 have more or less the same length parameter. Compound 6 is more active than compound 5 as it is more bulky and hence has a higher value of the Verloop B2 parameter, which is directly correlated with the antiproliferative activity, as shown in Eq. 1. This impact of the B2 parameter is shown even more clearly by the higher activity of compound 14 (−0.301) as compared to that of the compounds 13t (−0.699) and 9t (−1.079). Compound 14 has a trimethoxy substitution compared to the dimethoxy substitution in compound 13t and the monomethoxy substitution compound 9t, and hence has a higher value of the B2 parameter. This study suggests that the antiproliferative activity exhibited by the series taken is largely explained by steric factors and that substituents with a given shape, i.e., low length parameter and higher Verloop B2 parameter, are expected to enhance antiproliferative activity. Considering the fact that the 2D QSAR model was able to reproduce the experimental facts and that it was validated by the appropriate statistical procedures, it could be useful in designing a more potent inhibitor. Currently we are using the developed model to design anticancer agents with a higher potency and selectivity in our laboratory.
Conclusion
A QSAR analysis using 29 aminophenol derivatives was successfully carried out to build a statistically significant model possessing a good correlative and predictive capability for the inhibition of HDAC. The 2D QSAR model was validated by standard statistical means and how it reproduces and explains the differences in the experimental activity data. The detailed structural investigation revealed that the antiproliferative activity exhibited by the series is predominantly explained by the steric factors of the substituent, and hence modulation of the steric bulk could be used to optimize activity. This study has provided useful clues about the structural requirement for effective inhibitor–histone deacetylase binding interaction and hence for the improvement of the observed biological activity. This analysis could be of help in the rational design of potential drug candidates with an enhanced inhibitory potency.
References
Anderson KV, Lengyel JA (1980) Changing rates of histone mRNA synthesis and turnover in Drosophila embryos. Cell 21:717–727
Bouchain G, Frechette S, Woo SH, Khalil EA, Leit S, Fournel M, Yan PT, Trachy-Bourget MC, Beaulieu C, Li Z, Besterman J, Delorme D (2001) Design and synthesis of a novel class of histone deacetylase inhibitors. Bioorg Med Chem Lett 11:2847–2850
Carey N, Thangue BN (2006) Histone Deacetylase Inhibitors: Gathering Pace. Curr opin Pharmacol 6:369–375
Chen B, Petukhov PA, Jung Ml, Valena A, Eliseeva A, Dritscilo A, Kozikowski P (2005) Chemistry and biology of mercaptoacetamides as novel histone reacetylase inhibitiors. Bioorg Med Chem Lett 15:1389–1392
Curtin M, Glaser K (2003) Histone deacetylase inhibitors: the Abbott experience. Curr Med Chem 10:2373–2392
Chang HJ, Kim HJ, Chun HS (2007) Quantitative structure–activity relationship (QSAR) for neuroprotective activity of terpenoids. Life Sci 80:835–841
Dessalew N, Bharatam PV (2007a) 3D-QSAR and molecular docking study on bisarylmaleimide series as glycogen synthase kinase 3, cyclin dependent kinase 2 and cyclin dependent kinase 4 inhibitors: An insight into the criteria for selectivity. Eur J Med Chem 42:1014–1027
Dessalew N, Patel DS, Bharatam PV (2007b) 3D-QSAR and molecular docking studies on pyrazolopyrimidine derivatives as glycogen synthase kinase-3b inhibitors. J Mol Graph Mod 25:885–895
Dessalew N, Bharatam PV, Singh SK (2007c) 3D-QSAR CoMFA Study on aminothiazole derivatives as cyclin-dependent kinase 2 inhibitors. QSAR Comb Sci 26:85–91
Garea VA, Esteller M (2004) Histone deacetylase inhibitors; understanding a new wave of anticancer agents. Int J Cancer 112:171–178
Jose B, Oniki Y, Kato T, Nishino N, Sumida Y, Yoshida M (2004) Novel histone deacetylase inhibitory activity of cyclic tetrapeptide with trifluoromethyl and pentafluoro ethyl ketones. Bioorg Med Chem Lett 14:5343–5346
Klocker J, Wailzer B, Buchbauer G, Wolschann P (2002) Bayesian neural networks for aroma classification. J Chem Inf Comput Sci 42:1443–1449
Knaggs MH, McGuigan C, Harris SA, Heshmati P, Cahard D, Gilberta IH, Balzarini J (2000) A QSAR study investigating the effect of L-alanine ester variation on the anti-HIV activity of some phosphoramidate derivatives of d4T Bioorg Med Chem Lett 10:2075–2078
Kovatcheva A, Buchbauer G, Golbraikh A, Wolschann P (2003) QSAR Modeling of alpha-campholenic derivatives with sandalwood odor (2003) J Chem Inf Comput Sci 43:259–266
Kubinyi H (1997) QSAR and 3D-QSAR in drug design Part 1: methodology. Drug Discovery Today 2:457–467
Lohray BB, Gandhi N, Srivastava KB, Lohray VB (2006) 3D QSAR studies of N-4-arylacryloylpiperazin-1-yl-phenyloxazolidinones: A novel class of antibacterial agentsq. Bioorg Med Chem Lett 16:3817–3823
Marson CM, Mahadevan T, Dines J, Sengmany S, Morrell JM, Alao JP, Joel SP, Vigushin DM, Coombes RC (2007) Structure activity relationships of aryloxy alkanoicacid hydroxamides as potent inhibitors of histone deacetylases. Bioorg Med Chem Lett 17:136–14
McLaughlin F, Finn P, La Thangue NB (2003) The cell cycle, chromatin and cancer: mechanism based therapeutics come of age. Drug Disc Today 8:793–802
McLaughlin F, La Thangue NB (2004) Histone deacetylase inhibitors open new doors in cancer therapy. Biochem Pharmacol 68:1139–1144
Mordei O, Leit S, Zhou N, Frechette F, Bouchain C, Woo HS, Paguin I, Raeppel S, Frechette S, Vaisburg A, Fournal M, Kalita A, Lu A, Claude M, Bourget T, Yan P, Liu J, Li Z, Rahil J, Macleod RA, Besterman MJ, Delorme D (2006) Substituted N-(2-aminophenlyl)-benzamides, (E)-N-(2-aminophenyl)-acryl amides and their analogues: Novel classes of histone deacetylase inhibitors. Bioorg Med Chem Lett 16:4048–4052
Monneret C (2005) Histone deacetylase inhibitors. Eur J Med Chem 40:1–13
Nishino N, Yoshikawa D, Blatanbe LA, Kato T, Jose BL, Komatsu Y, Shinta R, Yoshida M, Kato T (2004) Chlamydocin-hydroxamic acid analogues as histone deacetylase inhibitors. Bioorg Med Chem Lett 12:5777–5784
Pandolfi PP (2001) Transcription therapy for cancer. Oncogene 20:3116–3127
Inche AG, La Thangue NB (2006) Keynote review: Chromatin control and cancer-drug discovery: realizing the promise. Drug Discovery Today 11:97–109
Somech R, Izraeli S, Simon JA (2004) histone deacetylase inhibitors- a new tool to treat cancer. Cancer Treatm Rev 30:401–472
Shinji C, Nakamura T, Maeda S, Yoshida M, Hashimoto Y, Miyachi H (2005) Design and synthesis of phthalimide-type histone deacetylase inhibitors. Bioorg Med Chem Lett 15:4427–4431
Uesato S, Kitagawa M, Nagaoka Y, Maeda T, Kuwajima H, Yamori T (2002) Novel histone deacetylase inhibitors: N-hydroxycarboxamides possessing a terminal bicyclic aryl group. Bioorg Med Chem Lett 12:1347– 1349
Verloop A, Tipker HW (1976a) Development and application of new steric substituent parameters in drug design. In: Ariens EJ (ed.), Drug design 7, 165–207. New York: Academic
Verloop A, Tipker J (1976b) Use of linear free energy related and other parameters in the study of fungicidal selectivity. Pestic Sci 7:379–390
Verloop A, Tipker JA (1977) Comparative study of new parameters in drug design. In: Keverling BJA (ed.) Biological activity and chemical structure. Amsterdam: Elsevier pp. 63–81
Weinmann H, Ottow E (2004) recent advances in medicinal chemistry of histone deacetylase inhibitors. Ann Rep Med Chem 39:185–196
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dessalew, N. QSAR study on aminophenylbenzamides and acrylamides as histone deacetylase inhibitors: An insight into the structural basis of antiproliferative activity. Med Chem Res 16, 449–460 (2007). https://doi.org/10.1007/s00044-007-9085-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-007-9085-9