
Abstract
This review summarizes direct and indirect analytical methods for the detection and quantification of the reactive oxygen species (ROS): 1O2, O ·−2 /HOO·, H2O2, HO·, and CO ·−3 in aqueous solution. Each section briefly describes the chemical properties of a specific ROS followed by a table (organized alphabetically by detection method, i.e., absorbance, chemiluminescence, etc.) summarizing the nature of the observable (associated analytical signal) for each method, limit of detection, application notes, and reaction of the probe molecule with the particular ROS.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
For the purpose of this review, reactive oxygen species (ROS) are defined as relatively short-lived molecules that contain oxygen atoms, with half-lives (t ½) in aquatic environments in the range of nanoseconds to hours (Bartosz 2006; Kearns 1971; Lu et al. 2006; Schmidt 2006; Zafiriou 1977; Zafiriou et al. 1984, 1990; Kieber et al. 2003; Waite et al. 1988). ROS are commonly found at picomolar to micromolar concentrations in environmental systems. This review focuses on the dominant ROS in surface waters and includes methods for the detection and quantification of singlet oxygen (1O2), superoxide (O ·−2 ) and its protonated form (hydroperoxyl radical; HOO·), hydrogen peroxide (H2O2), hydroxyl radical (HO·) and carbonate radical (CO ·−3 ).
In natural systems, 1O2, HOO·, HO·, H2O2, and CO ·−3 are capable of oxidizing a wide variety of molecules (including biomolecules) with relatively low selectivity and are involved in the attenuation of contaminants and the transformation of dissolved organic matter (DOM) in aquatic environments (Brezonik and Fulkerson-Brekken 1998; Canonica et al. 2005; Westerhoff et al. 1999, 2007). O ·−2 is more selective in its reactions with aqueous organic compounds than other ROS, but its reduction potential overlaps that of a range of biologically important metal ions (e.g., iron, copper, and manganese) that can themselves affect DOM oxidation (Goldstone and Voelker 2000). H2O2 is a thermodynamically powerful oxidant but its reaction rates with many compounds are typically slow compared to those of free radicals. Its conjugate base (HOO−) is also capable of acting as a reductant under some conditions, particularly for transition metal ions (Wood 1974; Koppenol and Butler 1985; Petlicki and van de Ven 1998).
ROS are usually generated by photolysis, electron transfer or energy transfer reactions (Bartosz 2006; Kearns 1971; Lu et al. 2006; Schmidt 2006; Zafiriou 1977; Zafiriou et al. 1984, 1990; Kieber et al. 2003). In the absence of other sinks, most free radical ROS undergo self-reaction (e.g., dimerization or disproportionation), while the electronically excited 1O2 rapidly decays through vibronic coupling with water (i.e., non-radiative decay). The steady-state concentration of 1O2 observed in natural waters is typically constrained by its interaction with water (resulting in a t ½ ~4 μs) with significant concentrations only observed in localized hydrophobic environments (Grandbois et al. 2008; Pogue et al. 2000). The bimolecular rate constants for the self-reactions of O ·−2 /HOO·, HO· and CO ·−3 are reasonably high (Czapski et al. 1994; Elliot et al. 1990; Zafiriou 1990; Scurlock and Ogilby 1996; Czapski and Dorfman 1964). However, in environmental systems, concentrations of these ROS are rarely high enough for self-reaction to be a significant sink for removal due to their large bimolecular rate constants with other sinks such as trace metal species and organic compounds. H2O2 does not react with itself but catalytically degrades through rapid reactions with trace metal ions and enzymes (Zepp et al. 1992; Rush and Bielski 1985; Duesterberg et al. 2005). Consequently, analyses of ROS have proven challenging because their lifetimes, with the exception of H2O2, are usually too short for ex-situ analysis.
Methods for ROS detection can be broadly classified as either direct or indirect. Due to the short lifetimes and typically low concentrations of ROS in aquatic systems, their direct observation is only possible on the sub-millisecond timescale, with the relatively stable H2O2 being an exception. Indirect methods involve the reaction of a particular ROS with a probe molecule to yield a more stable, long-lived analyte (Zafiriou et al. 1990). Such methods typically involve specific chemical derivatization (e.g., trapping a radical with a nitroxide or other spin trap) or are based on competitive kinetics. By virtue of introducing additional chemical reactions, all indirect techniques risk perturbing the observed system. In addition, both direct and indirect analyses suffer from the poor availability of standardized approaches to calibration (see Online Resource 1), particularly for use in the field.
Some important aspects to consider when choosing an ROS analysis method include: (1) the sensitivity of the method; (2) the selectivity and specificity of the method for the analyte of interest; and (3) the ability of the method to allow measurements with sufficiently fast time resolution. Specificity varies widely between methods, and should be carefully considered when choosing a method for ROS qualification and/or quantification. Additional analytical considerations are availability, robustness, portability (for field studies), the cost of the necessary instrumentation, and in some cases, the cost of the probe molecules. Largely due to these latter factors, much of the method development for aqueous ROS analysis has focused on ultraviolet (UV)/visible (Vis) light spectroscopic techniques and the use of relatively common and hence lower cost probe molecules. Spectroscopic detection strategies [including absorbance (UV/Vis), fluorescence (FL) and chemiluminescence (CL)] share a common approach with several other techniques for measuring rates of ROS formation and decay in laboratory experiments. These strategies are also compatible with methods such as steady-state kinetic analyses, stopped flow methods, time-resolved laser spectroscopy, flash photolysis and pulse radiolysis (Waite et al. 1988). However, applications of spectroscopic techniques for ROS analyses in natural waters are often limited by interference from DOM through background absorbance or FL, although the use of CL probes may circumvent these issues. All of these spectroscopic approaches can benefit, in certain circumstances, from the application of a preliminary “clean-up” technique such as the use of a concentrating resin, extraction, or chromatography [gas chromatography (GC) or high performance liquid chromatography (HPLC)] to remove interferences prior to analysis. Other analytical techniques for ROS detection, such as electron paramagnetic (spin) resonance (EPR), nuclear magnetic resonance (NMR), derivatization with attendant mass spectrometric (MS) analysis and liquid scintillation counting can also be quite useful but are less portable and often require considerable technical expertise to operate and can be expensive. For these reasons, when use of the aforementioned instrumentation is required, the observable species must be stable on timescales of days or more.
Earlier reviews have focused on the detection of specific ROS in specific media (e.g., cellular, aqueous, or organic solvents) (Bartosz 2006; Lu et al. 2006; Zafiriou et al. 1990; Gomes et al. 2005). This review is focused more broadly on comprehensively listing published methods that in the authors’ considered opinion are relevant for qualifying and quantifying ROS in environmental settings, particularly fresh and marine waters, groundwaters, and atmospheric waters. However, ‘relevance’ is defined by the needs of the researcher, and it is the authors’ hope that this review will also be useful for scientists working in engineered aquatic systems and biological systems. Since the probes reviewed in the compilation are by their nature reactive, it is important to note that the methods listed in the following tables require control studies in purified water to evaluate the possibility of probe degradation or competing reactions that may confuse the ROS signal. The low aqueous solubility of some probes may dictate the conditions of their use or result in their inadvertent partitioning from the aqueous phase to organic microenvironments (e.g., micellar or intra-DOM) (Grandbois et al. 2008; Lissi et al. 1993; Latch and McNeill 2006). The authors have personal experience with many of the methods but not all compiled in the tables. Because of this, when personal experience was lacking, additional pertinent information inserted into the tables was kept as faithful to the original cited text as possible.
The review is sectioned by ROS, with a brief introduction highlighting the fundamental chemical properties of the particular ROS followed by a critical evaluation and tabulation of relevant methods for detection of that ROS. The table entry for each method is arranged to display the identity of the probe molecule (using the nomenclature from the method’s literature citation), observable (analytical signal associated with the technique; e.g., absorbance, FL or CL emission, etc.), limit of detection (LOD) (as reported or calculated based on the original citation), application notes, approximate number of literature citations for the method (as of 09/2011), reaction schemes, and references for the method.
An Excel spreadsheet has also been prepared summarizing the information in the tables, enabling all the ROS analytical methods to be selected in terms of specific criteria such as the type of ROS to be analyzed while listing the relevant methods for that ROS, for example, in order of increasing LOD. This spreadsheet can be downloaded from the Web site http://neon.otago.ac.nz/research/bmp/data/ros_database.xlsx.
Singlet oxygen
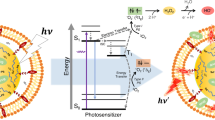
Ground state molecular oxygen exists as a triplet state with the lowest lying excited state of oxygen being a singlet state [O2(1Δg) or 1O2; Table 1] that lies 94 kJ/mol above the ground state (Table 1) (Khan et al. 1967; Wilkinson et al. 1995). The singlet state can be generated in solution by energy transfer from excited photosensitizers (S, e.g., humic substances or Rose Bengal; Eq. 1); or chemically, for example via the reaction between hypohalites and H2O2 (Eq. 2) (Zafiriou 1977; Khan and Kasha 1963; Schweitzer and Schmidt 2003; Schmidt 2006).
In sunlit waters, singlet oxygen concentrations have been measured in the range of ~10−12 to 10−13 M (Table 1) (Zepp et al. 1977; Haag and Hoigne 1986; Larson and Marley 1999; Egorov et al. 1989; Merkel and Kearns 1971; Wolff et al. 1981; Wick et al. 2000; Shao et al. 1994). The lifetime of singlet oxygen in aqueous solution is constrained through quenching by water with its lifetime in pure water being ~4 μs (Faust 1999; Egorov et al. 1989; Merkel and Kearns 1971). In natural waters its lifetime may be shorter due to the presence of additional quenchers, such as DOM (Table 1) (Faust 1999).
Direct measurement of the concentration of 1O2 is possible through observation of its emission at 1,268 nm (Hessler et al. 1994; Nonell and Braslavsky 2000). However, the intensity of this emission is too weak to be useful at low 1O2 concentrations. Consequently, use of this technique has been restricted to transient luminescence studies initiated by laser irradiation. The desire to measure environmentally relevant concentrations necessitated the development of molecular probes that can trap 1O2 or be used in competition kinetics (Table 2).
A suitable probe compound for photochemically generated 1O2 has to meet several requirements that are well summarized by Nardello et al. (1997): “The trap must be highly reactive towards 1O2, specific, compatible with aqueous media and must not perturb the system under study. Moreover, it must be transparent in the spectral range of the incident light in order to avoid photosensitization by the trap itself.” However, while many of the currently available probes meet some of these requirements, they do not meet all. Typical probes include anthracene- and pyrene-based compounds which are poorly soluble in water and absorb strongly in the UV-A and UV-B ranges appropriate for aquatic photochemistry (Evans and Upton 1985; Botsivali and Evans 1979; Wasserman et al. 1972; Corey and Taylor 1964). Therefore, the suite of probes matching all of the above mentioned requirements specified by Nardello et al. (1997) is small and includes only furfuryl alcohol (furan-2-ylmethanol; FFA) and 1,3-cyclohexadiene-1,4-diethanoate. FFA has the benefit of being commercially available and has been one of the most widely used probe compounds for singlet oxygen in aquatic photochemistry (Braun et al. 1986; Haag and Hoigne 1986; Haag et al. 1984a, b).
Furan derivatives, such as FFA, react with 1O2 to yield the corresponding molozonide (Table 2), which is unstable in water and rapidly hydrolyzes to other products, including the corresponding dicarbonyl. Steady-state concentrations of 1O2 are determined by measuring the rate constant of the loss of the furan probe and dividing by the second-order rate constant (Haag et al. 1984a; Latch et al. 2003). The rate constant is ~108 M−1 s−1 and hence two orders of magnitude below the diffusion-controlled limit (1010 M−1 s−1). Combined with the picomolar and sub-picomolar concentrations of 1O2 observed in natural waters, this results in long measurement times (tens of minutes) being required to observe a readily detectable decrease (>5%) in the concentration of FFA (Braun et al. 1986; Haag et al. 1984a, b).
The presence of 1O2 can also be tested through the addition of quenchers that promote non-radiative decay of this ROS back to the ground state. The addition of these materials result in an effective reduction in the rate of an observed process (k q), to a degree that is predictable (and therefore testable) based on the known rates of reaction between the added quencher and 1O2. The addition of quenchers can potentially disrupt the nature of the system and their suitability must be based on the system’s needs. For example, the azide ion (N3 −) is both a 1O2 quencher and a microbial poison, and is thus not suitable for microbiological studies examining the role of 1O2. Examples of frequently cited quenchers are N3 −, I−, and diazabicyclooctane (DABCO; Table 2) (Rubio et al. 1992; Hasty et al. 1972; Ouannes and Wilson 1968; Saito et al. 1975; Zepp et al. 1977). As with the furan derivatives, the interpretation of the results of these experiments is complicated by the additional reactivity of the quencher with HO· (Motohashi and Saito 1993).
An assay for 1O2 that avoids interference from HO· is the use of D2O as the reaction solvent (Table 2). Singlet oxygen has a longer lifetime in D2O solutions than H2O as a result of the relatively poor vibronic coupling between 1O2 and D2O (k d = 1.8 × 104 s−1) relative to H2O (k h = 2.4 × 105 s−1) (Wilkinson et al. 1995). Singlet oxygen’s reduced decay rate in D2O and/or D2O–water mixtures results in higher steady-state 1O2 concentrations leading to higher rate constants for oxidation of singlet oxygen acceptors (Merkel and Kearns 1972a, b; Merkel et al. 1972; Zepp et al. 1977). The use of the kinetic isotope effect in this instance allows the qualitative determination of 1O2 (through comparison of experimentally measured rates in the presence and absence of D2O) and its quantitative determination through application of the steady-state approximation to the rate of loss of a second probe (e.g., FFA). It should be noted that if the behavior of 1O2 is being monitored in a lipid membrane or other micro-heterogeneous phase (e.g., within a DOM micro-phase), many of these analytical measurements are rendered ineffective, since the reaction in this case is insensitive to the composition of the solvent and to the presence of quenchers that are present in the aqueous phase (Latch and McNeill 2006).
Superoxide and hydroperoxyl radical
Superoxide (Tables 3, 4) is the one-electron reduced form of triplet O2 and the conjugate base of the hydroperoxyl radical (Eq. 3; Table 3) (Hoigne 1975; Adams and Willson 1969; Bielski 1978). Because of its relatively low pK a (4.69), the O ·−2 anion dominates over HOO· in the majority of aqueous environments. O ·−2 can be generated during the redox cycling of transition metals (Eq. 4) and the photodegradation of DOM (Eq. 5) (Richard and Canonica 2005).
Superoxide concentrations have been reported over the range of 10−9 to 10−12 M in natural waters (Faust 1999; Fujiwara et al. 2006; Petasne and Zika 1987; Rose et al. 2008a, b; Voelker et al. 2000; Hansard et al. 2010; Heller and Croot 2010b; Shaked et al. 2010). The O ·−2 anion is relatively unreactive due to resonance stabilization, as reflected by the low rate constant for self-reaction of the anion (Eq. 6), but readily undergoes disproportionation through reaction with HOO· (Eq. 7). HOO· also reacts relatively rapidly with itself (Eq. 8) (Zafiriou 1977; Bielski 1978; Cooper and Zika 1983a).
The apparent second-order rate constant for disproportionation in terms of \({\text{T}}_{\text{O}_{2}^{\cdot - }}\), k obs, is thus highly pH dependent (Zafiriou 1977; Cooper and Zika 1983b; Bielski 1978) (Eqs. 9, 10), with a maximum value of ~107 M−1 s−1 at pH 4.69 (the pK a of O ·−2 /HOO·) that decreases by an order of magnitude for each unit increase in pH where the pH > pK a (Bielski 1978).
At the typical pH and superoxide concentrations found in seawater (8.1, 10−9 to 10−12 M), the uncatalyzed second-order disproportionation rate predicts a decay half-life of hours to hundreds of days. Measured half-lives of superoxide in seawater are much faster, ranging from 10 to 300 s, presumably due to catalysis by enzymes or transition metal ions (Hansard et al. 2010; Heller and Croot 2010b; Rose et al. 2010; Shaked et al. 2010; Rusak et al. 2011; Saragosti et al. 2010). This accelerated reactivity of O ·−2 thus presents significant analytical challenges, because standards (Online Resource 1) and samples at natural pH are stable for only seconds to minutes.
Superoxide absorbs strongly in the 230–350 nm region of the UV/Vis spectrum and can be quantified directly at micromolar concentrations by measuring its absorbance (Tables 3, 4). However, such a method is of limited value for measuring naturally occurring concentrations of superoxide due to the strong absorbance exhibited by other components of natural waters in this wavelength range. Many of the superoxide decay rate measurements in pure water were measured by millisecond ultraviolet spectroscopy (Bielski 1978). Superoxide can also be determined spectrophotometrically by measuring the rate of loss of compounds such as ferricytochrome c (FC), nitrobluetetrazolium (NBT), and 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) with which it readily reacts (Heller and Croot 2010a). These techniques have largely been used for measurements of superoxide production rates in biological systems due to their limited sensitivity (LOD ~ 1 μM–0.1 μM) and their lack of specificity (Olojo et al. 2005).
Due to superoxide’s brief lifetime and low steady-state concentrations (<1 nM) in natural waters, it is typically measured using highly sensitive CL probe molecules. Successful probes for decay or steady-state measurements must react at rates of at least ten times greater than that of natural superoxide disproportionation. Luminol is the most widely used CL probe for natural water analysis. This reagent has been used for the analysis of iron (Rose and Waite 2001; Xiao et al. 2002), chromium (Xiao et al. 2000), hydrogen peroxide (Yuan and Shiller 1999), and superoxide (Fujiwara et al. 2006), among other species, where these analytes are the rate-limiting species in the oxidation of luminol by superoxide. Unfortunately, because so many species can promote the CL of luminol in the presence of dissolved oxygen and hydrogen peroxide, this reagent is problematic for the selective analysis of superoxide in complex matrices.
In contrast, the selectivity, CL intensity, and commercial availability of MCLA (2-methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazo[l,2-a]pyrazin-3(7H)-one) and red-CLA([2-[4-[4-[3,7-dihydro-2-methyl-3-oxoimidazo[1,2-a]pyrazin-6-yl]phenoxy]butyramido]ethylamino]sulforhodamine101) make these probes particularly suitable for superoxide analysis in natural waters (Godrant et al. 2009; Hansard et al. 2010; Heller and Croot 2010b; Rose et al. 2008a, b; Shaked et al. 2010; Zheng et al. 2003). The entire class of CLA molecules react with superoxide through an expoxitane intermediate, with red-CLA and FCLA (3,7-dihydro-6-[4-[2-[N′-(5-fluoresceinyl)thioureido]ethoxy]phenyl]-2-methylimidazo[1,2-a]pyrazin-3-one) involved in a CL resonance energy transfer to shift the emission to longer wavelengths (Teranishi 2007). The CLA probes are also reactive with singlet oxygen (Suzuki et al. 1990), but selective analysis of superoxide is possible by first waiting ~100 μs for the singlet oxygen to decay. More significantly, the CLA reagents are not reactive to hydrogen peroxide, which is often present at concentrations in 100-fold excess to that of superoxide in natural waters. The selectivity of these probes is driven by their specific and relatively rapid second-order reaction rate with superoxide (~5 × 105 M−1 s−1). These rates of reaction are then 100 times that of the rate of disproportionation (Bielski 1978) and the rate of first order superoxide decay in natural samples (Hansard et al. 2010; Heller and Croot 2010b; Rose et al. 2008a, 2010; Shaked et al. 2010). The CLA reagents will react with superoxide and oxygen at pH and temperature dependent rates, thus requiring background CL measurements and standard additions of superoxide at ambient conditions with a constant oxygen concentration (creation of standards and correction for background CL described in Online Resource 1) (Godrant et al. 2009; Hansard et al. 2010). For superoxide flux measurements, a CLA probe is added several minutes before making chemiluminescence measurements to ensure complete reaction of the steady-state superoxide concentration in the samples. The CL signal is then assumed to arise solely from superoxide production in the sample with the chemiluminescence photon flux proportional to superoxide production rates (Godrant et al. 2009).
Of the analytical methods discussed above, MCLA and red-CLA have both been successfully used to measure steady-state concentrations and production/decay rates of superoxide in seawater. Detection limits of ~30 pM are reported for superoxide concentrations using MCLA (Hansard et al. 2010) while production/decay rates as low as ~1 pM/s can be determined using red-CLA (Godrant et al. 2009). Recently, there has been a substantial increase in the number of superoxide measurements being made in a wide range of oceanographic water types, most of which have utilized CL probes (Hansard et al. 2010; Heller and Croot 2010a, b; Rose et al. 2010; Shaked et al. 2010; Rusak SA 2011).
Hydrogen peroxide
Hydrogen peroxide (Table 5) is a weak acid (pK a 11.62) that is a ubiquitous component of surface and atmospheric waters (Eqs. 11–13). It is an important component of natural waters due to its impact on redox chemistry and biological processes, its role as an indicator of photochemical oxidation of DOM, as a photic zone tracer in the ocean, and its potential utility for in situ degradation of pollutants. Its presence in natural waters typically arises from the disproportionation of superoxide and the hydroperoxyl radical (von Sonntag and Schuchmann 1991). H2O2 production often occurs in association with the photoexcitation of DOM or the thermal oxidation of reduced transition metal ions (Eqs. 12–13), along with production from biological sources (Petasne and Zika 1987; Cooper et al. 1987; Thompson and Zafiriou 1983; Cooper and Zika 1983a).
The lifetime of H2O2 in the environment is dependent not only on pH, but also on the presence of transition metal ions, biological enzymatic decay, and some organic species that can catalyze its decomposition (Eq. 5) (Petasne and Zika 1997; Moffett and Zafiriou 1990, 1993; Moffett and Zika 1987). In ocean waters, its lifetime is on the order of days but in coastal waters it is much shorter (Hakkinen et al. 2004; Shaked et al. 2010). Hydrogen peroxide can be detected directly using spectrophotometric techniques, although its molar absorption coefficient (189 M−1 cm−1; Table 5) is low, thus limiting this analytical method to relatively pure solutions where the H2O2 concentration is high or the analytical pathlengths are long (e.g., atmospheric measurements) (Hochanadel 1952; Morgan et al. 1988).
Hydrogen peroxide has been measured at concentrations between 10−7 and 10−11 M (Table 5) in natural surface waters (Peake and Mosley 2004; Moore et al. 1993; Szymczak and Waite 1988, 1991; Cooper and Zika 1983a) and is normally in the micromolar range in atmospheric water (Kok et al. 1978; Zika et al. 1982). Concentrations in surface waters are generally highest in the photic zone and diminish toward the detection limit of most methods in the dark waters below the mixed surface layer, reflecting its dominant formation process by photochemical reactions.
Of the more than 30 analytical methods listed in Table 6, only 10 have a LOD that is useful for the H2O2 concentrations found in surface waters (LOD ≤ 50 nM). Of these, six employ a peroxidase enzyme to achieve the specificity required for H2O2. Because H2O2 is ubiquitous in water due to equilibrium with gas-phase H2O2 in the atmosphere (even in laboratory water), most methods require addition of catalase (an enzyme that decomposes H2O2 into water and O2) to the sample to eliminate H2O2 for analytical blanks prior to analysis.
Use of horseradish peroxidase (HRP) to provide specificity for the peroxide functional group can result in measurement of not only H2O2, but also other peroxy species such as peroxyacetic acid, methyl hydroperoxide, hydroxymethylperoxide, ethylhydroperoxide, and several propylperoxides formed through HRP-catlayzed reactions. These have all been shown to activate HRP and allow subsequent reaction with electron donors used to either develop (e.g., p-hydroxyphenylacetic acid or POHPAA) or diminish (e.g., scopoletin) FL. Using a post column chromatographic method based on the POHPAA technique, Miller et al. (2005) showed that any interference from organic peroxides is likely to be insignificant in the open ocean (Miller et al. 2005; Lee 1995). However, users of any peroxide method employing HRP should be aware of the potential contribution of organic peroxides in coastal and fresh waters.
While the 2-electron oxidation of HRP provides specificity for the peroxide functionality, it subsequently requires an electron donor to return HRP to its ground state. This second set of redox reactions is much less specific. This property of the enzyme is exploited in HRP-based methods, whereby the oxidized HRP subsequently reacts with a probe molecule to yield a product that is easily quantified, typically using spectroscopic methods such as absorbance or FL. This allows flexibility in terms of choosing the most suitable substrate for detection of H2O2 under particular measurement conditions. Phenolic compounds have fast reaction rates with the activated enzyme and all three FL methods discussed here take advantage of this fact. However, Miller and Kester (1988) have demonstrated that DOM in natural waters can also act as electron donors, likely via phenolic moieties which may need to be considered when using HRP-based methods under some conditions (Miller and Kester 1988).
While much research has been done with absorbance methods, the most commonly used and highly cited methods for determination of H2O2 in natural waters involve the HRP-catalyzed oxidation of probe compounds to yield products that either exhibit FL (e.g., p-hydroxyphenylacetic acid) or whose FL is diminished (e.g., scopoletin) after oxidation (Table 6). These fluorometric methods make use of readily available fluorophores, do not require specialized equipment other than a reliable fluorometer, and generally afford greater specificity, sensitivity and lower limits of detection compared to absorbance-based methods.
In many ways, current interest in the role of ROS in marine chemistry was inspired by early studies that used the HRP-catalyzed oxidation of scopoletin to analyze H2O2 in seawater (Perschke and Boda 1961; Zika et al. 1985a, b). While no longer the most commonly used method for the quantitative determination of peroxide, it is the seminal method from which many current methods evolved and so a presentation of some methodological detail is appropriate for any review. Specifically, when HRP, phenol, scopoletin, and H2O2 are together in a sample, the activated HRP enzyme catalyzes the production of a phenolic radical that then oxidizes scopoletin to a non-fluorescent product. This results in a stoichiometric decrease in scopoletin FL proportional to the concentration of H2O2 in the sample. Some studies have omitted phenol and still observed a decrease in scopoletin FL in the presence of HRP and H2O2. For example, Holm et al. (1987) substituted NaN3 for phenol to act as a more effective bactericide and H2O2 was still effectively measured using changes in scopoletin FL. It was noted, however, that the stoichiometry of the reaction varied without phenol. This is consistent with the greatly enhanced ability of naturally occurring phenolic compounds to compete as electron donors for activated HRP. In the absence of the fast-reacting phenol, a variable and wide variety of oxidized compounds having different reaction rates with scopoletin would generate variability in the observed decrease in FL. Additional details for the scopoletin method include the use of narrow-band optical filters and an excitation shutter (Donahue 1998), placing the sample in a dark fluorometer cell compartment for several minutes before measuring FL to minimize erroneous readings due to excitation from ambient light, storing reacted samples in the dark to reduce photobleaching, and carefully controlling pH. In fact, scopoletin FL is highly pH dependent and a buffer (usually phosphate buffer at pH 7) is required to ensure a meaningful and consistent relationship between the decrease in scopoletin FL emission intensity and the concentration of H2O2.
Hydrogen peroxide detection through the HRP-catalyzed dimerization of POHPAA to create a FL compound has been used extensively in rain, oceanic, and fresh water studies. In this method, two POHPAA radicals created via electron exchange with activated HRP dimerize to form a product that is FL at high pH using excitation and emission wavelengths of 313 and 400 nm, respectively (Miller and Kester 1988). The background FL of DOM in natural waters is variable and often pronounced at these wavelengths, thus requiring careful measurement of the natural FL in the sample prior to adding analytical reagents. This blank subtraction allows peroxide analysis in solutions with varying levels of naturally occurring fluorophores. As for all HRP-based methods, in samples with varying DOM content, standard additions are required to account for changes in stoichiometry due to competition between POHPAA and DOM for H2O2.
CL methods, while historically somewhat less useful in natural waters due to interferences from other ROS, have an inherent capacity for great sensitivity and have been used successfully in natural waters for measurement of H2O2. Both the luminol (Rose and Waite 2001) and acridinium ester methods (Cooper et al. 2000), when paired with portable and stable CL systems, have been demonstrated to be robust methods for analysis of H2O2 in oceanographic systems (Miller et al. 2005).
Luminol CL has been used most often for analysis of trace metals in natural water samples. However, in the presence of a metal catalyst, luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) is oxidized, yielding the luminol radical, and subsequently a diazaquinone species. The diazaquinone reacts stochiometrically with H2O2, forming an intermediate that decays spontaneously to 3-aminophthalate while emitting luminescence that can be detected at ~425 nm (Rose and Waite 2001). In solutions with an excess of metal catalyst and carefully controlled pH, H2O2 becomes the limiting reagent in the luminescence reaction, and the photon signal can be quantitatively related to the concentration of H2O2 (Yuan and Shiller 1999). Luminol-based determinations of H2O2 are generally performed using flow injection analysis, and offer subnanomolar detection limits in natural waters. Using a cobalt catalyst, Yuan and Shiller (1999) achieved a LOD of <1 nM, with a precision of 17 pM. The method is linear up to 300 nM H2O2; above 300 nM, the slope of the calibration curve decreases. As an excess of Co(II) must be added to the sample prior to analysis, the method does not exhibit interference from most other oxidants present in natural waters. Iron species, however, do compete with H2O2 in a secondary oxidation reaction that leads to a positive interference (Yuan and Shiller 1999; Rose and Waite 2001).
Similar to the use of luminol, acridinium ester reacts stochiometrically with H2O2 to form an intermediate structure that upon addition of base, forms a second intermediate which rapidly decays and emits luminescence that can be detected at ~470 nm (Cooper et al. 2000). Acridinium ester method advantages are that it minimizes interference from colored and FL organic compounds and metals naturally present in natural waters and it does not require the addition of metal complexes or other catalysts for the method to work. Miller et al. (2005) demonstrated that the use of the acridinium ester CL method and the POHPAA FL method in the open ocean gives indistinguishable and accurate results.
Hydroxyl radical
The hydroxyl radical (Table 7) is a non-selective oxidant that can be generated from a variety of sources, including photolysis of nitrate and nitrite ions (Eq. 14), transition metal complexes, and DOM, as well as Fenton-type reactions involving peroxides, hypohalites and several transition metal ions (Eq. 15), on photo-excited transition metal oxide surfaces and as a result of the aqueous decomposition of ozone (Zafiriou 1977; Zafiriou and True 1979a, b; Alfassi 1999; Weissler 1953; Makino et al. 1983; Dixon and Norman 1963; Zepp et al. 1992).
HO· reacts at near-diffusion-controlled rates with many substrates, resulting in low steady-state concentrations of HO· in sunlit natural waters (10−15 to 10−18 M) (Zepp et al. 1987; Brezonik and Fulkerson-Brekken 1998; Haag and Hoigne 1985). The corresponding low concentrations and brief lifetimes (~μs) for HO· pose significant challenges in quantifying this ROS. While HO· absorbs light in the UV region (Table 7), direct observation is not typically possible because of its limited lifetime and the presence of other chromophores absorbing in a similar wavelength region. Therefore, HO· is quantified either through the loss of a reagent or accumulation of a product. The key challenge is obtaining a compound that will react selectively and unambiguously with HO·, which either does not unduly influence the other aspects of the chemistry of the system under study, or alters the system in a predictable and well-defined manner.
The EPR technique is often used to measure the formation of stable paramagnetic aminoxyl radicals (spin adducts) by reaction of HO· with diamagnetic nitrone or nitroso (spin trap) compounds. The use of these diamagnetic compounds can also be beneficial in that they may allow for simultaneous quantification of several ROS through formation of different spin adducts, each with a characteristic EPR spectrum. However, the stability of the spin adduct in a given system needs to be carefully evaluated and controls established to confirm that radical trapping is the source of aminoxyl formation (Finkelstein et al. 1980c). For example, the adduct formed from the reaction of HO· with dimethylpyrrolidine oxide (DMPO, Table 8) can be readily transformed to a diamagnetic species by both Fe(III) and superoxide (Mizuta et al. 1997; Samuni et al. 1988); also, the superoxide DMPO-adduct decays to form either the HO· adduct or HO· itself (Finkelstein et al. 1979, 1982). Similarly, under acidic conditions the alternative spin trap (E)-2-methyl-N-((1-oxidopyridin-4-yl)methylene)propan-2-amine oxide (4-POBN, Table 8) hydrolyzes to the hydroxylamine, which can be readily oxidized to form the HO· adduct (Brezonik and Fulkerson-Brekken 1998; Janzen et al. 1978). pH can also significantly impact the stability of the hydroxyl spin adducts, with dramatic increases in stability observed for phenyl/pyridlyl-butylnitrone hydroxyl adducts when the pH is reduced from 8 to 6 (Janzen et al. 1992b). Spin trapping-type compounds have also been employed using alternative methods of quantitation to EPR. For example, 19F-NMR has been employed with a fluorinated-DMPO derivative, albeit with similar drawbacks to those already described for DMPO (Khramtsov et al. 2001). Although employed extensively in the biomedical literature, spin trapping with EPR detection appears to have had minimal use in natural systems. Of course, apart from these chemical limitations of the method, one of the biggest draw backs is the cost of EPR instrumentation, which is much greater than typical absorbance or fluorescence spectrometers.
An alternative to direct spin trapping of HO· is the inclusion of an additional reagent to convert HO· to a carbon-centered radical (such as the use of DMSO to form methyl radicals), which is then trapped and quantified. 3-aminomethyl-2,2,5,5,-tetramethyl-1-pyrrolidinyloxy (3-AMP, Table 8) and 3-amino-2,2,5,5,-tetramethyl-1-pyrrolidinyloxy (3-AP, Table 8) trap methyl radicals formed from the reaction of DMSO and HO·. The resultant complex can then be derivatized with fluorescamine and fluorometrically quantified after HPLC separation (with detection limits on the order of 10 nM) (Alaghmand and Blough 2007; Li et al. 1997a, 1999; Vaughan and Blough 1998). It is also possible to use a pre-fluorescamine derivatized aminoxyl probe in a similar fashion (Pou et al. 1993). This method can be employed under both oxic and anoxic conditions and has also been used to examine photochemical processes with minimal background production of the HO· derived product (Vaughan and Blough 1998).
The ability of HO· to undergo H-abstraction reactions has been employed for detection, typically using aliphatic alcohols/acids or halogenated alkanes as probe compounds (see Blough and Zepp 1995 for discusison of further probe compounds). Typical probe compounds employed in early studies involved quantitatively monitoring the loss of 1-chlorobutane (Haag and Hoigne 1985) or the reaction of 2-propanol with HO· yielding acetone, which can be quantified using HPLC after derivatization with 2,4-dinitrophenylhdrazine (Warneck and Wurzinger 1988).
Aromatic hydroxylation is another technique that is often employed for HO· quantification, particularly in natural aqueous environments. A wide variety of compounds have been reported for use in this type of HO· assay, including terephthalate, benzoic acid, p-chlorobenzoic acid, benzene and phthalhydrazide (Table 8) (Backa et al. 1997; Haag and Yao 1993; Miller et al. 2011; Page et al. 2010; Qian et al. 2001; Saran and Summer 1999; Vione et al. 2006; Zhou and Mopper 1990). These methods utilize the ability of HO· to add to an aromatic ring to initially form a hydroxycyclohexadienyl radical which can be further oxidized to a phenolic moiety by a range of oxidants. Dissolved oxygen is a suitable oxidant for this process, presumably proceeding via the mechanism suggested by Dorfman et al. (1962) involving HO2·/O ·−2 elimination to yield the diamagnetic hydroxylated species (Dorfman et al. 1962). The presence of other oxidants can alter the product distribution, and, as such, care must be taken to ensure consistent conditions for such a procedure to be analytically useful. In the hydroxylation of terephthalate, the yield of 2-hydroxyterephthalate increases by fivefold when IrCl6 2− is employed as the oxidant instead of the more abundant O2 (Fang et al. 1996). When benzoic acid is used as the probe compound, the more benign Fe(III)-EDTA (Ethylenediaminetetraacetic acid) oxidant is at least an order of magnitude less efficient than O2, and should not interfere with this procedure under oxic conditions (Maskos et al. 1990). Other routes to achieve aromatic hydroxylation are also possible, e.g., the hydroxylation of benzene to phenol by cytochrome P450 is a key process in the metabolism of benzene (Medinsky et al. 1995) with such processes representing potential interferences to aromatic hydroxylation probes. As O2 is necessary to oxidize the intermediate hydroxycyclohexadienyl radical, such probe systems are not suitable for investigating anoxic systems, where the absence of O2 would result in altered product distributions.
With aromatic hydroxylation probes, quantification can be undertaken by monitoring either the loss of the probe compound or the formation of one of the hydroxylated products, with the latter typically considered to be more selective for HO· than other strong oxidants and to offer sensitivity advantages. Since the hydroxylated product of analytical interest is only a fraction of the total amount of probe that has reacted with HO·, the yield of the hydroxylated product must be known in order to determine the quantity of HO· trapped. For benzoic acid, three hydroxybenzoic acid isomers are formed (Zhou and Mopper 1990). The proportion of ortho-, meta-, and para-substituted hydroxybenzoic acids has been found to be 36, 34, and 30%, respectively, under marine conditions (Zhou and Mopper 1990). When decarboxylation and ring fission products are accounted for, the fraction of HO· that reacts with benzoic acid to yield the para-isomer is determined to be 17%. Likewise, for phthalhydrazide, 20% of the reacted HO· yields the desired 5-hydroxy product (Miller et al. 2011) and 35% of the reacted terephthalate yields hydroxyterephthalate (Page et al. 2010). The higher yield from terephthalate is due to the symmetry of the probe molecule which yields only one possible hydroxy-isomer, thereby offering clear advantages in using this method.
For photochemical studies, it is essential to establish the stability of both the probe compound and the quantified product species with respect to direct photolysis by the light source. For the benzoic acid method, o-hydroxybenzoic acid has been shown to be stable with respect to direct photolysis using irradiation with λ > 313 nm and pH < 12 (Yang et al. 2004). The base (but not acid) form of m-hydroxybenzoic acid, however, is known to degrade in the presence of solar irradiation (Anastasio and McGregor 2001), whereas the para isomer is reportedly photo-stable (Zhou and Mopper 1990). With terephthalate, the probe compound itself is stable to solar irradiation, however, the hydroxylated product is directly photolyzed by 365 nm light with quantum yield (Φ) of (6.3 ± 0.1) × 10−3 (Page et al. 2010). In contrast, phthalhydrazide as a probe compound is directly photolyzed to the 5-hydroxy analyte, which is itself then seemingly stable to solar radiation (Miller et al. 2011). Although aromatic hydroxylation assays are advantageous with regards to sensitivity and selectivity, it is clear that caution must be used when applied to photochemical systems.
Further complications can arise when other oxidants are present in the system that are able to degrade the probe compound rendering it unavailable for reaction with any HO· that may be formed. For example, in studies of ozonation chemistry, precautions need to be taken to ensure the probe is unreactive towards ozone. In such cases, 4-chlorobenzoic acid has been found to be a suitable probe that is readily degraded by HO· yet is relatively unreactive towards O3. As such, HO· can be determined in the presence of O3 by monitoring the loss of 4-chlorobenzoic acid using HPLC (Haag and Yao 1993; Jans and Hoigné 1998).
There are significant challenges to determining HO· with all the methodologies described to date. In general, these all have some flaws or drawbacks that must be carefully considered and controlled as much as possible in any given system. Regardless of these potential problems, rigorous application of an appropriate method or indeed, potentially, several different methods (Table 8), enables quantitative insights to be made in the analysis of many systems. A discussion on the generation of standards to quantitatively determine HO· can be found in Online Resource 1.
Carbonate radical
The carbonate radical (Table 9) is produced in natural aqueous systems primarily through the oxidation of carbonate or bicarbonate ions by a one-electron oxidant such as HO· (Eq. 16) or the photoreactions of metal–carbonato complexes.
This radical has not been as widely measured in the aqueous environment as other transient oxidants. This is unfortunate, as its reduction potential overlaps that of I− and Br− and subsequent reactions of CO ·−3 could be a major source of reactive halogens in seawater. Its concentration in sunlit waters has been estimated at 10−13 to 10−15 M (Table 9) (Czapski et al. 1999; Faust 1999; Huang and Mabury 2000; Sulzberger et al. 1997; Larson and Zepp 1988). CO ·−3 reacts relatively slowly with itself, likely due to Coulombic repulsion (Behar et al. 1970a). It is much more selective than HO· with respect to reactions with organic species. For example, it reacts rapidly with phenols, anilines, and some amino acids (Busset et al. 2007; Chen and Hoffman 1973; Chen et al. 1975; Elango et al. 1985; Larson and Zepp 1988; Mak et al. 2007; Moore et al. 1977), but relatively slowly with saturated alkanes, aromatic hydrocarbons, etc. It primarily oxidizes organics through electron transfer as opposed to addition or atom transfer, and its role in environmental systems is underestimated because it has been so rarely measured. However, its impact may be greater than previously estimated, and it is interesting that the functional groups that CO ·−3 radicals are kinetically apt to react with are also those depleted most rapidly in the terrestrial DOM signature during mixing of fresh and marine waters.
CO ·−3 is strongly absorbing in the visible region (λ = 600 nm) and this is the primary method used for its detection in pump and probe experiments of its kinetics and reactivity (Weeks and Rabani 1966; Zuo et al. 1999). Its lifetime (~8 ms) is typically too brief to enable direct measurement in natural waters (Table 9) (Bonini et al. 1999; Canonica et al. 2005) and instead indirect quantitation methods are employed for this ROS. The presence of this radical is usually inferred through observation of the effects of the carbonate/bicarbonate ion on the oxidation of organic compounds by more oxidizing radicals such as HO· (Glaze et al. 1995; Glaze and Kang 1989). When the rate of oxidation of an HO· probe is reduced as a result of addition of carbonate ionic species, it is assumed that HO· has been scavenged by the carbonate anion resulting in the generation of CO ·−3 (Table 10). Although it is possible to trap this radical using nitrones with subsequent EPR detection (Table 10), the resulting adducts are subject to hydrolysis and so are difficult to characterize or quantify directly (Villamena et al. 2006, 2007; Wolcott et al. 1994; Yoon et al. 2002).
References
Abrams R, Altschul AM, Hogness TR (1942) Cytochrome c peroxidase II. The peroxidase–hydrogen peroxide complex. J Biol Chem 142(1):303–316
Acero JL, Von Gunten U (2001) Characterization of oxidation processes: ozonation and the AOP O3/H2O2. J Am Water Works Assoc 93(10):90–100
Adam W, Kazakov DV, Kazakov VP (2005) Singlet-oxygen chemiluminescence in peroxide reactions. Chem Rev 105(9):3371–3387. doi:10.1021/Cr0300035
Adams GE, Willson RL (1969) Pulse radiolysis studies on oxidation of organic radicals in aqueous solution. T Faraday Soc 65(563P):2981–2982
Adams GE, Boag JW, Michael BD (1964) Spectroscopic studies of reactions of OH radical in aqueous solutions. P Chem Soc London 4:492–505
Afanas’ev IB, Ostrakhovitch EA, Mikhal’chik EV, Korkina LG (2001) Direct enzymatic reduction of lucigenin decreases lucigenin-amplified chemiluminescence produced by superoxide ion. Luminescence 16(5):305–307
Alaghmand M, Blough NV (2007) Source-dependent variation in hydroxyl radical production by airborne particulate matter. Environ Sci Technol 41(7):2364–2370. doi:10.1021/Es061902o
Alfassi ZB (ed) (1999) General aspects of the chemistry of radicals. The chemistry of free radicals. Wiley, New York
Allouch A, Roubaud V, Lauricella R, Bouteiller JC, Tuccio A (2003) Preparation and use as spin trapping agents of new ester-nitrones. Org Biomol Chem 1(3):593–598
Allouch A, Roubaud V, Lauricella R, Bouteiller JC, Tuccio E (2005) Spin trapping of superoxide by diester-nitrones. Org Biomol Chem 3(13):2458–2462
Altschul AM, Abrams R, Hogness TR (1940) Cytochrome c peroxidase. J Biol Chem 136(3):777–794
Anastasio C, Matthew BM (2006) A chemical probe technique for the determination of reactive halogen species in aqueous solution: part 2—chloride solutions and mixed bromide/chloride solutions. Atmos Chem Phys 6(9):2439–2451
Anastasio C, McGregor KG (2001) Chemistry of fog waters in California’s Central Valley: 1. In situ photoformation of hydroxyl radical and singlet molecular oxygen. Atmos Environ 35(6):1079–1089. doi:10.1016/s1352-2310(00)00281-8
Andersen LK, Ogilby PR (2002) Absorption spectrum of singlet oxygen a1Δg → b1Σg+ in D2O: enabling the test of a model for the effect of solvent on oxygen’s radiative transitions. J Phys Chem A 106(46):11064–11069
Andreae WA (1955) Sensitive method for the estimation of hydrogen peroxide in biological materials. Nature 175(4463):859–860
Armstrong WA, Grant DW (1958) Highly sensitive chemical dosimeter for ionizing radiation. Nature 182(4637):747
Armstrong WA, Humphreys WG (1965) A L.E.T. independent dosimeter based on chemiluminescent determination of H2O2. Can J Chem 43(9):2576–2584
Armstrong WA, Black BA, Grant DW (1960) The radiolysis of aqueous calcium benzoate and benzoic acid solutions. J Phys Chem 64(10):1415–1419
Armstrong DA, Waltz WL, Rauk A (2006) Carbonate radical anion—thermochemistry. Can J Chem 84(12):1614–1619
Aubry JM, Rigaudy J, Cuong NK (1981a) A water-soluble rubrene derivative—synthesis, properties and trapping of 1O2 in aqueous-solution. Photochem Photobiol 33(2):149–153
Aubry JM, Rigaudy J, Ferradini C, Pucheault J (1981b) A search for singlet oxygen in the disproportionation of superoxide anion. J Am Chem Soc 103(16):4965–4966
Aurich HG (1982) Nitroxides. In: Patai S (ed) The chemistry of functional groups, supplemental F: the chemistry of amino, nitroso, and nitro compounds and their derivatives, part 1. Wiley, Chichester
Backa S, Jansbo K, Reitberger T (1997) Detection of hydroxyl radicals by a chemiluminescence method. A critical review. Holzforschung 51(6):557–564
Bader H, Sturzenegger V, Hoigne J (1988) Photometric-method for the determination of low concentrations of hydrogen-peroxide by the peroxidase catalyzed oxidation of N, N-Diethyl-P-Phenylenediamine (Dpd). Water Res 22(9):1109–1115
Baga AN, Johnson GRA, Nazhat NB, Saadallanazhat RA (1988) A simple spectrophotometric determination of hydrogen-peroxide at low concentrations in aqueous-solution. Anal Chim Acta 204(1–2):349–353
Baier A, Maier M, Engl R, Landthaler M, Baumler W (2005) Time-resolved investigations of singlet oxygen luminescence in water, in phosphatidylcholine, and in aqueous suspensions of phosphatidylcholine or HT29 cells. J Phys Chem B 109(7):3041–3046
Baier J, Fuss T, Pollmann C, Wiesmann C, Pindl K, Engl R, Baumer D, Maier M, Landthaler M, Baumler W (2007) Theoretical and experimental analysis of the luminescence signal of singlet oxygen for different photosensitizers. J Photochem Photobiol B 87(3):163–173
Barja G (2002) The quantitative measurement of H2O2 generation in isolated mitochondria. J Bioenerg Biomembr 34(3):227–233. doi:10.1023/A:1016039604958
Bartosz G (2006) Use of spectroscopic probes for detection of reactive oxygen species. Clin Chim Acta 368(1–2):53–76. doi:10.1016/j.cca.2005.12.039
Baxter RM, Carey JH (1983) Evidence for photochemical generation of superoxide ion in humic waters. Nature 306(5943):575–576
Behar D, Czapski G, Duchovny I (1970a) Carbonate radical in flash photolysis and pulse radiolysis of aqueous carbonate solutions. J Phys Chem 74(10):2206–2210
Behar D, Czapski G, Rabani J, Dorfman LM, Schwarz HA (1970b) Acid dissociation constant and decay kinetics of perhydroxyl radical. J Phys Chem 74(17):3209–3213
Benov L, Sztejnberg L, Fridovich I (1998) Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 25(7):826–831
Bielski BHJ (1978) Reevaluation of spectral and kinetic-properties of HO2 and O2 − free radicals. Photochem Photobiol 28(4–5):645–649
Bielski BHJ, Allen AO (1967) Radiation chemistry of aqueous tetranitromethane solutions in presence of air. J Phys Chem 71(13):4544–4549
Bielski BHJ, Shiue GG, Bajuk S (1980) Reduction of nitro blue tetrazolium by CO2 − and O ·−2 radicals. J Phys Chem 84(8):830–833
Blough NV, Zepp RG (1995) Reactive oxygen species in natural waters. In: Foote CS, Valentine JS, Greenburg A, Liebman JF (eds) Active oxygen in chemistry, vol 2. Blackie Academic and Professional, Glasgow, pp 280–333
Bonini MG, Radi R, Ferrer-Sueta G, Ferreira AMD, Augusto O (1999) Direct EPR detection of the carbonate radical anion produced from peroxynitrite and carbon dioxide. J Biol Chem 274(16):10802–10806
Bors W, Michel C, Saran M (1979a) Superoxide anions do not react with hydroperoxides. FEBS Lett 107(2):403–406
Bors W, Saran M, Michel C (1979b) Pulse-radiolytic investigations of catechols and catecholamines. 3. Adrenalone. J Phys Chem 83(19):2447–2452
Bors W, Saran M, Michel C (1982) Radical intermediates involved in the bleaching of the carotenoid crocin—hydroxyl radicals, superoxide anions and hydrated electrons. Int J Radiat Biol 41(5):493–501
Botsivali M, Evans DF (1979) New trap for singlet oxygen in aqueous-solution. J Chem Soc Chem Commun 24:1114–1116
Bottle SE, Micallef AS (2003) Synthesis and EPR spin trapping properties of a new isoindole-based nitrone: 1,1,3-trimethylisoindole N-oxide (TMINO). Org Biomol Chem 1(14):2581–2584
Bottle SE, Hanson GR, Micallef AS (2003) Application of the new EPR spin trap 1,1,3-trimethylisoindole N-oxide (TMINO) in trapping HO· and related biologically important radicals. Org Biomol Chem 1(14):2585–2589
Boveris A, Martino E, Stoppani AOM (1977) Evaluation of horseradish peroxidase-scopoletin method for measurement of hydrogen-peroxide formation in biological-systems. Anal Biochem 80(1):145–158
Braun AM, Frimmel FH, Hoigne J (1986) Singlet oxygen analysis in irradiated surface waters. Int J Environ Anal Chem 27:137–149
Brezonik PL, Fulkerson-Brekken J (1998) Nitrate-induced photolysis in natural waters: controls on concentrations of hydroxyl radical photo-intermediates by natural scavenging agents. Environ Sci Technol 32(19):3004–3010
Britigan B, Coffman T, Buettner G (1990) Spin trapping evidence for the lack of significant hydroxyl radical production during the respiration burst of human phagocytes using a spin adduct resistant to superoxide-mediated destruction. J Biol Chem 265(5):2650–2656
Buettner GR (1985) Spin trapping of hydroxyl radical. In: Greenwald RA (ed) CRC handbook of methods for oxygen radical research. CRC Press, Boca Raton, pp 151–155
Busset C, Mazellier P, Sarakha M, De Laat J (2007) Photochemical generation of carbonate radicals and their reactivity with phenol. J Photochem Photobiol A 185(2–3):127–132
Butler J, Jayson GG, Swallow AJ (1975) Reaction between superoxide anion radical and cytochrome-C. Biochim Biophys Acta 408(3):215–222
Buxton GV, Greenstock CL, Helman WP, Ross AB (1988) Critical-review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−) in aqueous solution. J Phys Chem Ref Data 17(2):513–886
Canonica S, Kohn T, Mac M, Real FJ, Wirz J, Von Gunten U (2005) Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds. Environ Sci Technol 39(23):9182–9188
Chance B (1943) The kinetics of the enzyme-substrate compound of peroxidase. J Biol Chem 151(2):553–577
Chen SR, Gee KR (2000) Redox-dependent trafficking of 2,3,4,5,6-pentafluorodihydrotetramethylrosaimine, a novel fluorogenic indicator of cellular oxidative activity. Free Radic Biol Med 28(8):1266–1278
Chen SN, Hoffman MZ (1973) Rate constants for reaction of carbonate radical with compounds of biochemical interest in neutral aqueous-solution. Radiat Res 56(1):40–47
Chen S, Cope VW, Hoffman MZ (1973) Behavior of CO3 − radicals generated in flash-photolysis of carbonatoamine complexes of Cobalt(III) in aqueous solution. J Phys Chem 77(9):1111–1116
Chen SN, Hoffman MZ, Parsons GH (1975) Reactivity of carbonate radical toward aromatic-compounds in aqueous-solution. J Phys Chem 79(18):1911–1912
Childs RE, Bardsley WG (1975) Steady-state kinetics of peroxidase with 2,2′-azino-di-(3-ethylbenzthiazoline-6-sulphonic acid) as chromogen. Biochem J 145(1):93–103
Cooper WJ, Zika RG (1983) Photochemical formation of hydrogen-peroxide in surface and ground waters exposed to sunlight. Science 220(4598):711–712
Cooper WJ, Saltzman ES, Zika RG (1987) The contribution of rainwater to variability in surface hydrogen-peroxide. J Geophys Res Ocean 92((C3)):2970–2980
Cooper WJ, Moegling JK, Kieber RJ, Kiddle JJ (2000) A chemiluminescence method for the analysis of H2O2 in natural waters. Mar Chem 70(1–3):191–200
Corey EJ, Taylor WC (1964) Study of peroxidation of organic compounds by externally generated singlet oxygen molecules. J Am Chem Soc 86(18):3881–3882
Crow JP (1997) Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide Biol Chem 1(2):145–157
Czapski G, Bielski BHJ (1963) Formation and decay of H2O3 and HO2 in electron-irradiated aqueous solutions. J Phys Chem 67(10):2180–2184
Czapski G, Dorfman LM (1964) Pulse radiolysis studies. V. Transient spectra and rate constants in oxygenated aqueous solutions. J Phys Chem 68(5):1169–1177
Czapski G, Holcman J, Bielski BHJ (1994) Reactivity of nitric-oxide with simple short-lived radicals in aqueous-solutions. J Am Chem Soc 116(25):11465–11469
Czapski G, Lymar SV, Schwarz HA (1999) Acidity of the carbonate radical. J Phys Chem A 103(18):3447–3450
Dasgupta PK, Hwang H (1985) Application of a nested loop system for the flow-injection analysis of trace aqueous peroxides. Anal Chem 57(6):1009–1012
Denham K, Milofsky RE (1998) Photooxidation of 3-substituted pyrroles: a postcolumn reaction detection system for singlet molecular oxygen in HPLC. Anal Chem 70(19):4081–4085
Dickson J, Odom M, Ducheneaux F, Murray J, Milofsky RE (2000) Coupling photochemical reaction detection based on singlet oxygen sensitization to capillary electrochromatography. Anal Chem 72(14):3038–3042
Dixon WT, Norman ROC (1963) Electron spin resonance studies of oxidation. 1. Alcohols. J Chem Soc 5:3119–3120
Donahue WF (1998) Interference in fluorometric hydrogen peroxide determination using scopoletin horseradish peroxidase. Environ Toxicol Chem 17(5):783–787
Dorfman LM, Taub IA, Buhler RE (1962) Pulse radiolysis studies. I. Transient spectra and reaction-rate constants in irradiated aqueous solutions of benzene. J Chem Phys 36(11):3051–3061
Draper WM, Crosby DG (1983a) The photochemical generation of hydrogen-peroxide in natural-waters. Arch Environ Con Tox 12(1):121–126
Draper WM, Crosby DG (1983b) Photochemical generation of superoxide radical-anion in water. J Agric Food Chem 31(4):734–737
Duesterberg CK, Cooper WJ, Waite TD (2005) Fenton-mediated oxidation in the presence and absence of oxygen. Environ Sci Technol 39(13):5052–5058. doi:10.1021/es048378a
Dukes EK, Hyder ML (1964) Determination of peroxide by automatic colorimetry. Anal Chem 36(8):1689–1690
Edman L, Rigler R (2000) Memory landscapes of single-enzyme molecules. Proc Natl Acad Sci USA 97(15):8266–8271
Egorov SY, Kamalov VF, Koroteev NI, Krasnovskii AA Jr, Toleutaev BN, Zinukov SV (1989) Rise and decay kinetics of photosensitized singlet oxygen luminescence in water. Measurements with nanosecond time-correlated single photon counting technique. Chem Phys Lett 163(4-5):421–424
Elango TP, Ramakrishnan V, Vancheesan S, Kuriacose JC (1985) Reactions of the carbonate radical with aliphatic-amines. Tetrahedron 41(18):3837–3843
Elliot AJ, Mccracken DR, Buxton GV, Wood ND (1990) Estimation of rate constants for near-diffusion-controlled reactions in water at high-temperatures. J Chem Soc Faraday Trans 86(9):1539–1547
Elovitz MS, von Gunten U (1999) Hydroxyl radical ozone ratios during ozonation processes. Ozone Sci Eng 21(3):239–260
Ernstbrunner EE, Girling RB, Grossman WEL, Hester RE (1978) Free-radical studies by resonance raman-spectroscopy. 2. Diazabicyclo-octane radical cation. J Chem Soc Faraday Trans 2(74):501–508
Evans DF, Upton MW (1985) Studies on singlet oxygen in aqueous-solution. 1. Formation of singlet oxygen from hydrogen-peroxide with 2-electron oxidants. J Chem Soc Dalton 6:1141–1145
Fang XW, Mark G, vonSonntag C (1996) OH radical formation by ultrasound in aqueous solutions. 1. The chemistry underlying the terephthalate dosimeter. Ultrason Sonochem 3(1):57–63
Farmilo A, Wilkinson F (1973) Mechanism of quenching of singlet oxygen in solution. Photochem Photobiol 18(6):447–450
Faust BC (1999) Aquatic photochemical reactions in atmospheric, surface, and marine waters. Influences on oxidant formation and pollutant degradation. In: Boule P (ed) Handbook of environmental chemistry, vol 2(Pt. L). Springer, Berlin, pp 101–122
Ferry JL, Fox MA (1999) Temperature effects on the kinetics of carbonate radical reactions in near-critical and supercritical water. J Phys Chem A 103(18):3438–3441
Finkelstein E, Rosen GM, Rauckman EJ, Paxton J (1979) Spin trapping of superoxide. Mol Pharmacol 16(2):676–685
Finkelstein E, Rosen GM, Rauckman EJ (1980a) Spin trapping—kinetics of the reaction of superoxide and hydroxyl radicals with nitrones. J Am Chem Soc 102(15):4994–4999
Finkelstein E, Rosen GM, Rauckman EJ (1980b) Spin trapping of superoxide and hydroxyl radical—practical aspects. Arch Biochem Biophys 200(1):1–16
Finkelstein E, Rosen GM, Rauckman EJ (1982) Production of hydroxyl radical by decomposition of superoxide spin-trapped adducts. Mol Pharmacol 21(2):262–265
Flors C, Fryer MJ, Waring J, Reeder B, Bechtold U, Mullineaux PM, Nonell S, Wilson MT, Baker NR (2006) Imaging the production of singlet oxygen in vivo using a new fluorescent sensor, singlet oxygen sensor green. J Exp Bot 57(8):1725–1734
Foote CS, Denny RW (1968) Chemistry of singlet oxygen VII. Quenching by β-carotene. J Am Chem Soc 90(22):6233–6235
Foote CS, Wuesthof MT, Wexler S, Burstain IG, Denny R, Schenck GO, Schulte-Elte K-H (1967) Photosensitized oxygenation of alkyl-substituted furans. Tetrahedron 23(6):2583–2584
Foote CS, Chang YC, Denny RW (1970a) Chemistry of singlet oxygen X. Carotenoid quenching parallels biological protection. J Am Chem Soc 92(17):5216–5218
Foote CS, Chang YC, Denny RW (1970b) Chemistry of singlet oxygen XI. Cis-trans isomerization of carotenoids by singlet oxygen and a probable quenching mechanism. J Am Chem Soc 92(17):5218–5219
Foote CS, Denny RW, Weaver L, Chang Y, Peters J (1970c) Quenching of singlet oxygen. Ann N Y Acad Sci 171(1):139–145
Frejaville C, Karoui H, Tuccio B, Lemoigne F, Culcasi M, Pietri S, Lauricella R, Tordo P (1994) 5-Diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO)—a new phosphorylated nitrone for the efficient in vitro and in vivo spin-trapping of oxygen-centered radicals. J Chem Soc Chem Commun 15:1793–1794
Frejaville C, Karoui H, Tuccio B, Lemoigne F, Culcasi M, Pietri S, Lauricella R, Tordo P (1995) 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide—a new efficient phosphorylated nitrone for the in vitro and in vivo spin-trapping of oxygen-centered radicals. J Med Chem 38(2):258–265
Fujimori K, Komiyama T, Tabata H, Nojima T, Ishiguro K, Sawaki Y, Tatsuzawa H, Nakano M (1998) Chemiluminescence of Cypridina luciferin analogs. Part 3. MCLA chemiluminescence with singlet oxygen generated by the retro-Diels–Alder reaction of a naphthalene endoperoxide. Photochem Photobiol 68(2):143–149
Fujiwara K, Kumata H, Kando N, Sakuma E, Aihara M, Morita Y, Miyakawa T (2006) Flow injection analysis to measure the production ability of superoxide with chemiluminescence detection in natural waters. Int J Environ Anal Chem 86(5):337–346
Giraud M, Valla A, Bazin M, Santus R, Momzikoff A (1982) A new water-soluble singlet oxygen probe. J Chem Soc Chem Commun 20:1147–1148
Glaze WH, Kang JW (1989) Advanced oxidation processes—test of a kinetic-model for the oxidation of organic-compounds with ozone and hydrogen-peroxide in a semibatch reactor. Ind Eng Chem Res 28(11):1580–1587
Glaze WH, Lay Y, Kang JW (1995) Advanced oxidation processes—a kinetic-model for the oxidation of 1,2-dibromo-3-chloropropane in water by the combination of hydrogen-peroxide and UV-radiation. Ind Eng Chem Res 34(7):2314–2323
Glover DJ, Landsman SG (1964) Spectrophotometric method for determination of tetranitromethane in solution and in air. Anal Chem 36(8):1690–1691
Godrant A, Rose AL, Sarthou G, Waite TD (2009) New method for the determination of extracellular production of superoxide by marine phytoplankton using the chemiluminescence probes MCLA and red-CLA. Limnol Oceanogr Methods 7:682–692
Goldstein S, Rosen GM, Russo A, Samuni A (2004) Kinetics of spin trapping superoxide, hydroxyl, and aliphatic radicals by cyclic nitrones. J Phys Chem A 108(32):6679–6685
Goldstone JV, Voelker BM (2000) Chemistry of superoxide radical in seawater: CDOM associated sink of superoxide in coastal waters. Environ Sci Technol 34(6):1043–1048
Gomes A, Fernandes E, Lima JLFC (2005) Fluorescence probes used for detection of reactive oxygen species. J Biochem Biophys Methods 65(2–3):45–80. doi:10.1016/j.jbbm.2005.10.003
Goto T, Takagi T (1980) Chemiluminescence of a Cypridina luciferin analogue, 2-methyl-6-phenol-3,7-dihyrdoimadizo[1,2a]-pyrazin-3-one, in the presence of the xanthine–xanthine oxidase system. Chem Soc Jpn 53:833–834
Grandbois M, Latch DE, McNeill K (2008) Microheterogeneous concentrations of singlet oxygen in natural organic matter isolate solutions. Environ Sci Technol 42(24):9184–9190
Greenstock CL, Ruddock GW (1976) Determination of superoxide (O2 −) radical-anion reaction-rates using pulse-radiolysis. Int J Radiat Phys Chem 8(3):367–369
Greenwood NN, Earnshaw A (1997) Chemistry of the elements, 2nd edn. Butterworth-Heinemann, Oxford
Guilbault GG, Brignac PJ Jr, Juneau M (1968) New substrates for the fluorometric determination of oxidative enzymes. Anal Chem 40(8):1256–1263
Gupta BL (1973) Microdetermination techniques for H2O2 in irradiated solutions. Microchem J 18(4):363–374
Gutteridge JMC, Maidt L, Poyer L (1990) Superoxide-dismutase and fenton chemistry—reaction of ferric EDTA complex and ferric-bipyridyl complex with hydrogen-peroxide without the apparent formation of iron(II). Biochem J 269(1):169–174
Haag WR, Hoigne J (1985) Photo-sensitized oxidation in natural-water via.OH radicals. Chemosphere 14(11–12):1659–1671
Haag WR, Hoigne J (1986) Singlet oxygen in surface waters. 3. Photochemical formation and steady-state concentrations in various types of waters. Environ Sci Technol 20(4):341–348
Haag WR, Yao CCD (1993) Ozonation of US drinking water sources: HO· concentration and oxidation-competition values. In: Ozone in water and wastewater treatment, Proceedings of the eleventh ozone world congress, August 29–September 13. pp S-17-119-126
Haag WR, Hoigne J, Gassman E, Braun AM (1984a) Singlet oxygen in surface waters—part I: furfuryl alcohol as a trapping agent. Chemosphere 113(5/6):631–640
Haag WR, Hoigne J, Gassman E, Braun AM (1984b) Singlet oxygen in surface waters–part II: quantum yields of its production by some natural humic materials as a function of wavelength. Chemosphere 113(5/6):641–650
Hakkinen PJ, Anesio AM, Graneli W (2004) Hydrogen peroxide distribution, production, and decay in boreal lakes. Can J Fish Aquat Sci 61(8):1520–1527
Halliwell B, Gutteridge JMC (1985) Hydroxyl radicals assayed by aromatic hydroxylation and deoxyribose degradation. In: Greenwald RA (ed) CRC handbook of methods for oxygen radical research. CRC Press, Boca Raton, pp 157–163
Hansard SP, Vermilyea WA, Voelker MB (2010) Measurements of superoxide radical concentration and decay kinetics in the Gulf of Alaska. Deep Sea Res I 57(9):1111–1119
Harbour JR, Chow V, Bolton JR (1974) Electron-spin resonance study of spin adducts of OH and HO2 radicals with nitrones in ultraviolet photolysis of aqueous hydrogen-peroxide solutions. Can J Chem 52(20):3549–3553
Hasty N, Merkel PB, Radlick P, Kearns DR (1972) Role of azide in singlet oxygen reactions: reaction of azide with singlet oxygen. Tetrahedron Lett 1:49–52
Hauser TR, Kolar MA (1968) Spectrophotometric determination of hydrogen peroxide in aqueous media with 1,2-di-(4-pyridyl)ethylene. Anal Chem 40:231–232
Heller MI, Croot PL (2010a) Application of a superoxide (O2 −) thermal source (SOTS-1) for the determination and calibration of O2 − fluxes in seawater. Anal Chim Acta 667(1–2):1–13. doi:10.1016/j.aca.2010.03.054
Heller MI, Croot PL (2010b) Superoxide decay kinetics in the southern ocean. Environ Sci Technol 44(1):191–196. doi:10.1021/Es901766r
Hessler DP, Frimmel FH, Oliveros E, Braun AM (1994) Solvent isotope effect on the rate constants of singlet-oxygen quenching by EDTA and its metal-complexes. Helv Chim Acta 77(3):859–868
Hideg E, Spetea C, Vass I (1994a) Singlet oxygen and free-radical production during acceptor-induced and donor-side-induced photoinhibition—studies with spin-trapping EPR spectroscopy. BBA Bioenerg 1186(3):143–152
Hideg E, Spetea C, Vass I (1994b) Singlet oxygen production in thylakoid membranes during photoinhibition as detected by EPR spectroscopy. Photosynth Res 39(2):191–199
Hochanadel CJ (1952) Effects of cobalt gamma-radiation on water and aqueous solutions. J Phys Chem 56(5):587–594
Hoigne J (1975) Aqueous radiation chemistry in relation to waste treatment. In: Radiation for a clean environment. International Atomic Energy Agency, Vienna, pp 219–232
Hoigne J (1997) Inter-calibration of OH radical sources and water quality parameters. Water Sci Technol 35(4):1–8
Holm TR, George GK et al (1987) Fluorometric determination of hydrogen peroxide in groundwater. Anal Chem 59:582–586
Hosaka S, Obuki M, Nakajima J, Suzuki M (2005) Comparative study of antioxidants as quenchers or scavengers of reactive oxygen species based on quenching of MCLA-dependent chemiluminescence. Luminescence 20(6):419–427. doi:10.1002/Bio.867
Huang J, Mabury S (2000) Steady state reactions of carbonate radicals in field waters. Environ Chem Toxicol 19(9):2181–2188
Huie RE, Clifton CL, Neta P (1991) Electron-transfer reaction-rates and equilibria of the carbonate and sulfate radical-anions. Radiat Phys Chem 38(5):477–481
Hurst JR, Mcdonald JD, Schuster GB (1982) Lifetime of singlet oxygen in solution directly determined by laser spectroscopy. J Am Chem Soc 104(7):2065–2067
Hwang H, Dasgupta PK (1985) Fluorimetric determination of trace hydrogen-peroxide in water with a flow-injection system. Anal Chim Acta 170:347–352
Hwang H, Dasgupta PK (1986) Fluorometric flow injection determination of aqueous peroxides at nanomolar level using membrane reactors. Anal Chem 58(7):1521–1524
Jankowski JJ, Kieber DJ, Mopper K (1999) Nitrate and nitrite ultraviolet actinometers. Photochem Photobiol 70(3):319–328
Jankowski JJ, Kieber DJ, Mopper K, Neale PJ (2000) Development and intercalibration of ultraviolet solar actinometers. Photochem Photobiol 71(4):431–440
Jans U, Hoigné J (1998) Activated carbon and carbon black catalyzed transformation of aqueous ozone into OH-radicals. Ozone Sci Eng J Int Ozone Assoc 20(1):67–90
Janzen EG, Wang YY, Shetty RV (1978) Spin trapping with alpha-pyridyl 1-oxide N-tert-butyl nitrones in aqueous-solutions—unique electron-spin resonance-spectrum for hydroxyl radical adduct. J Am Chem Soc 100(9):2923–2925
Janzen EG, Hinton RD, Kotake Y (1992a) Substituent effect on the stability of the hydroxyl radical adduct of alpha-phenyl N-tert-butyl nitrone (PBN). Tetrahedron Lett 33(10):1257–1260
Janzen EG, Kotake Y, Hinton RD (1992b) Stabilities of hydroxyl radical spin adducts of PBN-type spin traps. Free Radic Biol Med 12(2):169–173
Johnson RM, Siddiqi IW (1970) The determination of organic peroxides, vol 4. Monographs in organic functional group analysis. Pergamon Press, New York
Kambayashi Y, Ogino K (2003) Reestimation of Cypridina luciferin analogs (MCLA) as a chemiluminescence probe to detect active oxygen species: cautionary note for use of MCLA. J Toxicol Sci 28(3):139–148
Karoui H, Clement JL, Rockenbauer A, Siri D, Tordo P (2004) Synthesis and structure of 5,5-diethoxycarbonyl-l-pyrroline N-oxide (DECPO). Application to superoxide radical trapping. Tetrahedron Lett 45(1):149–152
Kearns DR (1971) Physical and chemical properties of singlet molecular oxygen. Chem Rev 71(4):395–427
Keston AS, Brandt R (1965) Fluorometric analysis of ultramicro quantities of hydrogen peroxide. Anal Biochem 11(1):1–5
Khan AU, Kasha M (1963) Red chemiluminescence of molecular oxygen in aqueous solution. J Chem Phys 39(8):2105–2106
Khan AU, Pitts JN, Smith EB (1967) Singlet oxygen in the environmental sciences: the role of singlet molecular oxygen in the production of photochemical air pollution. Environ Sci Technol 1(8):656–657
Khramtsov VV, Reznikov VA, Berliner LJ, Litkin AK, Grigor’ev IA, Clanton TL (2001) NMR spin trapping: detection of free radical reactions with a new fluorinated DMPO analog. Free Radic Biol Med 30(10):1099–1107. doi:10.1016/s0891-5849(01)00505-6
Kieber RJ, Helz GR (1986) Two-method verification of hydrogen peroxide determinations in natural waters. Anal Chem 58:2312–2315
Kieber DJ, Peake BM, Scully NM (2003) Reactive oxygen species in aquatic systems. In: Hebling EW, Zagrese H (eds) UV effects in aquatic organisms and ecosystems. Royal Society of Chemistry, Cambridge, pp 251–288
King DW, Cooper WJ, Rusak SA, Peake BM, Kiddle JJ, O’Sullivan DW, Melamed ML, Morgan CR, Theberge SM (2007) Flow injection analysis of H2O2 in natural waters using acridinium ester chemiluminescence: method development and optimization using a kinetic model. Anal Chem 79(11):4169–4176
Kishore K, Moorthy PN, Rao KN (1982) Riboflavin as a new versatile solute for the determination of OH radical rate constants by the competition kinetic technique. Radiat Phys chem 20(4):241–245
Klaning UK, Sehested K, Holcman J (1985) Standard gibbs energy of formation of the hydroxyl radical in aqueous-solution—rate constants for the reaction ClO2− and O3 reversible O3 − and ClO2. J Phys Chem 89(5):760–763
Klauschenz E, Haseloff RF, Volodarskii LB, Blasig IE (1994) Spin-trapping using 2,2-dimethyl-2H-imidazole-1-oxides. Free Radic Res 20(2):103–111
Klein GW, Bhatia K, Madhavan V, Schuler RH (1975) Reaction of OH with benzoic acid—isomer distribution in radical intermediates. J Phys Chem 79(17):1767–1774
Kok GL (1980) Measurements of hydrogen-peroxide in rainwater. Atmos Environ 14(6):653–656
Kok GL, Holler TP, Lopez MB, Nachtrieb HA, Yuan M (1978) Chemiluminescent method for determination of hydrogen-peroxide in ambient atmosphere. Environ Sci Technol 12(9):1072–1076
Kok GL, Thompson K, Lazrus AL, Mclaren SE (1986) Derivatization technique for the determination of peroxides in precipitation. Anal Chem 58(6):1192–1194
Koppenol WH (1976) Reactions involving singlet oxygen and superoxide anion. Nature 262(5567):420–421
Koppenol WH, Butler J (1985) Energetics of interconversion reactions of oxyradicals. Adv Free Radic Biol Med 1:91–131
Koppenol WH, Liebman JF (1984) The oxidizing nature of the hydroxyl radical—a comparison with the ferryl ion (FeO2+). J Phys Chem 88(1):99–101
Koppenol WH, Vanbuuren KJH, Butler J, Braams R (1976) Kinetics of reduction of cytochrome-C by superoxide anion radical. Biochim Biophys Acta 449(2):157–168
Kosaka H, Katsuki Y, Shiga T (1992) Spin trapping study on the kinetics of Fe2+ autoxidation: formation of spin adducts and their destruction by superoxide. Arch Biochem Biophys 293(2):401–408. doi:10.1016/0003-9861(92)90412-p
Kosaka K, Yamada H, Matsui S, Echigo S, Shishida K (1998) Comparison among the methods for hydrogen peroxide measurements to evaluate advanced oxidation processes: application of a spectrophotometric method using copper(II) ion and 2,9 dimethyl-1,10-phenanthroline. Environ Sci Technol 32(23):3821–3824
Kotake Y, Janzen EG (1991) Decay and fate of the hydroxyl radical adduct of alpha-phenyl-N-tert-butylnitrone in aqueous-media. J Am Chem Soc 113(25):9503–9506
Kraljic I, Mohsni SE (1978) New method for detection of singlet oxygen in aqueous-solutions. Photochem Photobiol 28(4–5):577–581
Kraljic I, Trumbore CN (1965) P-Nitrosodimethylaniline as an OH radical scavenger in radiation chemistry. J Am Chem Soc 87(12):2547–2550
Kwan WP, Voelker BM (2002) Decomposition of hydrogen peroxide and organic compounds in the presence of dissolved iron and ferrihydrite. Environ Sci Technol 36(7):1467–1476. doi:10.1021/es011109p
Larson RA, Marley KA (1999) Singlet oxygen in the environment. In: Boule P (ed) Handbook of environmental chemistry, vol 2(Pt. L). Springer, Berlin, pp 123–137
Larson RA, Zepp RG (1988) Reactivity of the carbonate radical with aniline derivatives. Environ Toxicol Chem 7(4):265–274
Latch E, McNeill K (2006) Microheterogeneity of singlet oxygen distributions in irradiated humic acid solutions. Science 311:1743–1747
Latch DE, Stender BL, Packer JL, Arnold WA, McNeill K (2003) Photochemical fate of pharmaceuticals in the environment: cimetidine and ranitidine. Environ Sci Technol 37(15):3342–3350. doi:10.1021/Es0340782
Lawrence GD (1985) Ethylene formation from methionine and its analogs. In: Greenwald RA (ed) CRC handbook of methods for oxygen radical research. CRC Press, Boca Raton, pp 157–163
Lazrus AL, Kok GL, Gitlin SN, Lind JA, Mclaren SE (1985) Automated fluorometric method for hydrogen-peroxide in atmospheric precipitation. Anal Chem 57(4):917–922
Lee M (1995) Hydrogen peroxide, methyl hydroperoxide, and formaldehyde in air impacted by biomass burning. University of Rhode Island, Kingston
Li JC, Cheng SM (1965) Spectrophotometric determination of titanium with 4-(2-pyridyl azo) resorcinol-hydrogen peroxide. Sci Sin 14(1):144–145
Li B, Gutierrez PL, Blough NV (1997) Trace determination of hydroxyl radical in biological systems. Anal Chem 69(21):4295–4302
Li Y, Zhu H, Kuppusamy P, Roubaud V, Zweier JL, Trush MA (1998) Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J Biol Chem 273(4):2015–2023
Li B, Gutierrez PL, Blough NV, Lester P (1999) Trace determination of hydroxyl radical using fluorescence detection. In: Methods of enzymology. Academic Press, London, pp 202–216
Li X, Zhang G, Ma H, Zhang D, Li J, Zhu D (2004) 4,5-Dimethylthio-4′-[2-(9-anthryloxy)ethylthio]tetrathiafulvalene, a highly selective and sensitive chemiluminescence probe for singlet oxygen. J Am Chem Soc 126:11543–11548
Lin CL, Rohatgi NK, Demore WB (1978) Ultraviolet-absorption cross-sections of hydrogen-peroxide. Geophys Res Lett 5(2):113–115
Lindig BA, Rodgers MAJ, Schaap AP (1980) Determination of the lifetime of singlet oxygen in D2O using 9,10-anthracenedipropionic acid, a water-soluble probe. J Am Chem Soc 102(17):5590–5593
Lion Y, Delmelle M, Vandevorst A (1976) New method of detecting singlet oxygen production. Nature 263(5576):442–443
Lissi EA, Encinas MV, Lemp E, Rubio MA (1993) Singlet oxygen O2(1Δg) bimolecular processes. Solvent and compartmentalization effects. Chem Rev 93:699–723
Loeff I, Swallow AJ (1964) On radiation chemistry of concentrated aqueous solutions of sodium benzoate. J Phys Chem 68(9):2470–2475
Lu C, Song G, Lin JM (2006) Reactive oxygen species and their chemiluminescence-detection methods. Trac Trends Anal Chem 25(10):985–995. doi:10.1016/j.trac.2006.07.007
Lymar SV, Schwarz HA, Czapski G (2000) Medium effects on reactions of the carbonate radical with thiocyanate, iodide, and ferrocyanide ions. Radiat Phys chem 59(4):387–392
MacManus-Spencer LA, McNeill K (2005) Quantification of singlet oxygen production in the reaction of superoxide with hydrogen peroxide using a selective chemiluminescent probe. J Am Chem Soc 127(25):8954–8955
MacManus-Spencer LA, Latch DE, Kroncke KM, McNeill K (2005) Stable dioxetane precursors as selective trap-and-trigger chemiluminescent probes for singlet oxygen. Anal Chem 77:1200–1205
MacManus-Spencer LA, Edhlund BL, McNeill K (2006) Singlet oxygen production in the reaction of superoxide with organic peroxides. J Org Chem 71:796–799
Macpherson AN, Telfer A, Barber J, Truscott TG (1993) Direct-detection of singlet oxygen from isolated photosystem-II reaction centers. Biochim Biophys Acta 1143(3):301–309
Madsen BC, Kromis MS (1984) Flow-injection and photometric-determination of hydrogen-peroxide in rainwater with N-ethyl-N-(sulfopropyl)aniline sodium-salt. Anal Chem 56(14):2849–2850
Mak AM, Whiteman M, Wong MW (2007) Reaction of the radical pair NO2 − and CO ·−3 with 2-[6-(4′-amino)phenoxy-3H-xanthen-3-on-9-yl]benzoic acid (APF). J Phys Chem A 111(33):8202–8210
Makino K, Mossoba MM, Riesz P (1983) Chemical effects of ultrasound on aqueous-solutions—formation of hydroxyl radicals and hydrogen-atoms. J Phys Chem 87(8):1369–1377
Malavolti NL, Pilosof D, Nieman TA (1984) Optimization of experimental-variables for the chemi-luminescent determination of glucose in microporous membrane flow cells. Anal Chem 56(12):2191–2195
Malehorn CL, Riehl TE, Hinze WL (1986) Improved determination of hydrogen-peroxide or lucigenin by measurement of lucigenin chemiluminescence in organized assemblies. Analyst 111(8):941–947
Mashiko S, Suzuki N, Koga S, Nakano M, Goto T, Ashino T, Mizumoto I, Inaba H (1991) Measurement of rate constants for quenching singlet oxygen with a Cypridina luciferin analog (2-methyl-6-[para-methoxyphenyl]-3,7-dihydroimidazo[1,2-a]pyrazin-3-one) and sodium-azide. J Biolumin Chemilumin 6(2):69–72
Maskiewicz R, Sogah D, Bruice TC (1979) Chemiluminescent reactions of lucigenin. 1. Reactions of lucigenin with hydrogen-peroxide. J Am Chem Soc 101(18):5347–5354
Maskos Z, Rush JD, Koppenol WH (1990) The hydroxylation of the salicylate anion by a fenton reaction and [Gamma]-radiolysis: a consideration of the respective mechanisms. Free Radic Biol Med 8(2):153–162
Matheson MS, Mulac WA, Weeks JL, Rabani J (1966) Pulse radiolysis of deaerated aqueous bromide solutions. J Phys Chem 70(7):2092–2099
Matheson IB, Lee J, Yamanash Bs, Wolbarsh Ml (1974) Measurement of absolute rate constants for singlet molecular oxygen(1ΔG) reaction with 1,3-diphenylisobenzofuran and physical quenching by ground-state molecular-oxygen. J Am Chem Soc 96(11):3343–3348
Matsubara C, Iwamoto T, Nishikawa Y, Takamura K, Yano S, Yoshikawa S (1985a) Colored species formed from the titanium(Iv)-4-(2′-pyridylazo)resorcinol reagent in the spectrophotometric determination of trace amounts of hydrogen-peroxide. J Chem Soc Dalton 1:81–84
Matsubara C, Kudo K, Kawashita T, Takamura K (1985b) Spectrophotometric determination of hydrogen-peroxide with titanium 2-((5-bromopyridyl)azo)-5-(N-propyl-N-sulfopropylamino)phenol reagent and its application to the determination of serum glucose using glucose-oxidase. Anal Chem 57(6):1107–1109
Matthews RW, Sangster DF (1965) Measurement by benzoate radiolytic decarboxylation of relative rate constants for hydroxyl radical reactions. J Phys Chem 69(6):1938–1946
Maurette MT, Oliveros E, Infelta PP, Ramsteiner K, Braun AM (1983) Singlet oxygen and superoxide—experimental differentiation and analysis. Helv Chim Acta 66(2):722–733
Mayneord WV, Anderson W, Evans HD, Rosen D (1955) Hydrogen peroxide yields in X-irradiated aqueous solutions—a sensitive method based on hydrazide chemiluminescence. Radiat Res 3(4):379–392
Mccord JM, Fridovich I (1968) Reduction of cytochrome C by milk xanthine oxidase. J Biol Chem 243(21):5753–5760
Medinsky MA, Kenyon EM, Schlosser PM (1995) Benzene: a case study in parent chemical and metabolite interactions. Toxicology 105(2–3):225–233
Merkel PB, Kearns DR (1971) Direct measurement of the lifetime of singlet oxygen in solution. Chem Phys Lett 12(1):120–122
Merkel PB, Kearns DR (1972a) Radiationless decay of singlet molecular oxygen in solution. An experimental and theoretical study of electronic-to-vibrational energy transfer. J Am Chem Soc 94(21):7244–7253
Merkel PB, Kearns DR (1972b) Remarkable solvent effects on lifetime of 1ΔG oxygen1. J Am Chem Soc 94(3):1029–1030
Merkel PB, Kearns DR (1975) Comment regarding the rate constant for the 1,3-diphenylisobenzofuran and singlet oxygen. J Am Chem Soc 97(2):462–463
Merkel PB, Nilsson R, Kearns DR (1972) Deuterium effects on singlet oxygen lifetimes in solutions. A new test of singlet oxygen reactions. J Am Chem Soc 94(3):1030–1031
Miller WL, Kester DR (1988) Hydrogen-peroxide measurement in seawater by (para-hydroxyphenyl)acetic acid dimerization. Anal Chem 60(24):2711–2715
Miller GW, Morgan CA, Kieber DJ, King DW, Snow JA, Heikes BG, Mopper K, Kiddle JJ (2005) Hydrogen peroxide method intercomparision study in seawater. Mar Chem 97(1–2):4–13
Miller CJ, Rose AL, Waite TD (2011) Phthalhydrazide chemiluminescence method for determination of hydroxyl radical production: modifications and adaptations for use in natural systems. Anal Chem 83(1):261–268. doi:10.1021/ac1022748
Mizuta Y, Masumizu T, Kohno M, Mori A, Packer L (1997) Kinetic analysis of the Fenton reaction by ESR-spin trapping. Biochem Mol Biol Int 43(5):1107–1120
Moffett JW, Zafiriou OC (1990) An investigation of hydrogen peroxide chemistry in surface waters of vineyard sound with H, 18O, and l8O2. Limnol Oceanogr 35(6):1221–1229
Moffett JW, Zafiriou OC (1993) The photochemical decomposition of hydrogen-peroxide in surface waters of the eastern Caribbean and Orinoco River. J Geophys Res Ocean 98(C2):2307–2313
Moffett JW, Zika RG (1987) Reaction-kinetics of hydrogen-peroxide with copper and iron in seawater. Environ Sci Technol 21(8):804–810
Moore JS, Phillips GO, Sosnowski A (1977) Reaction of carbonate radical-anion with substituted phenols. Int J Radiat Biol 31(6):603–605
Moore CA, Farmer CT, Zika RG (1993) Influence of the Orinoco River on hydrogen-peroxide distribution and production in the eastern Caribbean. J Geophys Res Ocean 98(C2):2289–2298
Morgan MS, Vantrieste PF, Garlick SM, Mahon MJ, Smith AL (1988) Ultraviolet molar absorptivities of aqueous hydrogen-peroxide and hydroperoxyl ion. Anal Chim Acta 215(1–2):325–329
Motohashi N, Saito Y (1993) Competitive measurement of rate constants for hydroxyl radical reactions using radiolytic hydroxylation of benzoate. Chem Pharm Bull 41(10):1842–1845
Mottola HA, Simpson BE, Gorin G (1970) Absorptiometric determination of hydrogen peroxide in submicrogram amounts with leuco crystal violet and peroxidase as catalyst. Anal Chem 42(3):410–411
Muller K, Ziereis K (1993) Dimethyl 3,3′-(4-methyl-1,3-naphthylene)dipropionate as a singlet oxygen trap in biological-systems. Arch Pharm 326(6):369–371
Nakano M (1990) Determination of superoxide radical and singlet oxygen based on chemiluminescence of luciferin analogs. Methods Enzymol 186:585–591
Nardello V, Azaroual N, Cervoise I, Vermeersch G, Aubry JM (1996) Synthesis and photooxidation of sodium 1,3-cyclohexadiene-1,4-diethanoate: a new colorless and water-soluble trap of singlet oxygen. Tetrahedron 52(6):2031–2046
Nardello V, Brault D, Chavalle P, Aubry JM (1997) Measurement of photogenerated singlet oxygen (1O2(1Δg) in aqueous solution by specific chemical trapping with sodium 1,3-cyclohexadiene-1,4-diethanoate. J Photochem Photobiol B 39(2):146–155
Neftel A, Jacob P, Klockow D (1984) Measurements of hydrogen-peroxide in polar ice samples. Nature 311(5981):43–45
Nelsen SF, Buschek JM (1974) Charge delocalization in saturated systems—radical cation of 1,3,6,8-tetraazatricyclo[4.4.1.1-3,8]dodecane. J Am Chem Soc 96(20):6424–6428
Niederlander HAG, Dejong MM, Gooijer C, Velthorst NH (1994a) Flow-injection system for determination of singlet oxygen quenching efficiencies utilizing online dioxetane chemiluminescence detection. Anal Chim Acta 290(1–2):201–214
Niederlander HAG, Nuijens MJ, Dozy EM, Gooijer C, Velthorst NH (1994b) Dioxetane chemiluminescence detection in liquid-chromatography based on photosensitized online generation of singlet molecular-oxygen—a thorough examination of experimental parameters and application to polychlorinated-biphenyls. Anal Chim Acta 297(3):349–368
Nonell S, Braslavsky SE (2000) Time-resolved singlet oxygen detection. Methods Enzymol 319:37–49
Ogilby PR, Foote CS (1982) Chemistry of singlet oxygen. 36. Singlet molecular-oxygen (1Δg) luminescence in solution following pulsed laser excitation—solvent deuterium-isotope effects on the lifetime of singlet oxygen. J Am Chem Soc 104(7):2069–2070
Oh BS, Song SJ, Lee ET, Oh HJ, Kang JW (2004) Catalyzed ozonation process with GAC and metal doped-GAC for removing organic pollutants. Water Sci Technol 49(4):45–49
Ohyashiki T, Nunomura M, Katoh T (1999) Detection of superoxide anion radical in phospholipid liposomal membrane by fluorescence quenching method using 1,3-diphenylisobenzofuran. BBA Biomembr 1421(1):131–139
Olive G, Mercier A, Le Moigne F, Rockenbauer A, Tordo P (2000) 2-Ethoxycarbonyl-2-methyl-3,4-dihydro-2H-pyrrole-1-oxide: evaluation of the spin trapping properties. Free Radic Biol Med 28(3):403–408
Olojo RO, Xia RH, Abramson JJ (2005) Spectrophotometric and fluorometric assay of superoxide ion using 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole. Anal Biochem 339(2):338–344. doi:10.1016/J.Ab.2005.01.032
Osman AM, Laane C, Hilhorst R (2001) Enhanced sensitivity of Cypridina luciferin analogue (CLA) chemiluminescence for the detection of O·− (2)− with non-ionic detergents. Luminescence 16(1):45–50
Oturan MA, Pinson J (1995) Hydroxylation by electrochemically generated oh radicals—monohydroxylation and polyhydroxylation of benzoic-acid—products and isomers distribution. J Phys Chem 99(38):13948–13954
Ouannes C, Wilson T (1968) Quenching of singlet oxygen by tertiary aliphatic amines. Effect of DABCO. J Am Chem Soc 90(23):6527–6528
Page SE, Arnold WA, McNeill K (2010) Terephthalate as a probe for photochemically generated hydroxyl radical. J Environ Monit 12(9):1658–1665
Patsoukis N, Papapostolou I, Georgiou CD (2005) Interference of non-specific peroxidases in the fluorescence detection of superoxide radical by hydroethidine oxidation: a new assay for H2O2. Anal Bioanal Chem 381(5):1065–1072. doi:10.1007/s00216-004-2999-x
Peake BM, Mosley LM (2004) Hydrogen peroxide concentrations in relation to optical properties in a fiord (Doubtful Sound, New Zealand). N Z J Mar Fresh 38(4):729–741
Perschke H, Boda E (1961) Determination of very small amounts of hydrogen peroxide. Nature 190(4772):257–258
Petasne RG, Zika RG (1987) Fate of superoxide in coastal sea water. Nature 325(6104):516–518
Petasne RG, Zika RG (1997) Hydrogen peroxide lifetimes in south Florida coastal and offshore waters. Mar Chem 56:215–225
Petlicki J, van de Ven TGM (1998) The equilibrium between the oxidation of hydrogen peroxide by oxygen and the dismutation of peroxyl or superoxide radicals in aqueous solutions in contact with oxygen. J Chem Soc Faraday Trans 94(18):2763–2767
Pi YZ, Schumacher J, Jekel M (2005) The use of para-chlorobenzoic acid (pCBA) as an ozone/hydroxyl radical probe compound. Ozone Sci Eng 27(6):431–436. doi:10.1080/01919510500349309
Pines DS, Reckhow DA (2002) Effect of dissolved cobalt(II) on the ozonation of oxalic acid. Environ Sci Technol 36(19):4046–4051. doi:10.1021/Es011230w
Pines DS, Reckhow DA (2003) Solid phase catalytic ozonation process for the destruction of a model pollutant. Ozone Sci Eng 25(1):25–39
Pogue BW, Paulsen KD, O’Hara JA, Hoopes PJDVM, Swartz H (2000) Modeling the oxygen microheterogeneity of tumors for photodynamic therapy dosimetry. In: Proceedings of SPIE-the International Society for Optical Engineering, vol 3909, pp 104–112
Posner GH, Lever JR, Miura K, Lisek C, Seliger HH, Thompson A (1984) A chemi-luminescent probe specific for singlet oxygen. Biochem Biophys Res Commun 123(2):869–873
Pou S, Huang YI, Bhan A, Bhadti VS, Hosmane RS, Wu SY, Cao GL, Rosen GM (1993) A fluorophore-containing nitroxide as a probe to detect superoxide and hydroxyl radical generated by stimulated neutrophils. Anal Biochem 212(1):85–90
Pou S, Ramos CL, Gladwell T, Renks E, Centra M, Young D, Cohen MS, Rosen GM (1994) A kinetic approach to the selection of a sensitive spin-trapping system for the detection of hydroxyl radical. Anal Biochem 217(1):76–83
Qian J, Mopper K, Kieber DJ (2001) Photochemical production of the hydroxyl radical in Antarctic waters. Deep Sea Res Part I Oceanogr Res Pap 48(3):741–759
Racine P, Auffray B (2005) Quenching of singlet molecular oxygen by Commiphora myrrha extracts and menthofuran. Fitoterapia 76(3–4):316–323
Reitberger T, Gierer J (1988) Chemiluminescence as a means to study the role of hydroxyl radicals in oxidative processes. Holzforschung 42(6):351–356
Richard C, Canonica S (2005) Aquatic phototransformation of organic contaminants induced by coloured dissolved natural organic matter. In: Handbook of environmental chemistry, vol 2 (Pt. M). Springer, Berlin, pp 299–323
Rizzi C, Marque S, Belin F, Bouteiller JC, Lauricella R, Tuccio B, Cerri V, Tordo P (1997) PPN-type nitrones: preparation and use of a new series of beta-phosphorylated spin-trapping agents. J Chem Soc Perkin Trans 2(12):2513–2518
Rodgers MAJ, Snowden PT (1982) Lifetime of O2(1ΔG) in liquid water as determined by time-resolved infrared luminescence measurements. J Am Chem Soc 104(20):5541–5543
Rose AL, Waite TD (2001) Chemiluminescence of luminol in the presence of iron(II) and oxygen: oxidation mechanism and implications for its analytical use. Anal Chem 73(24):5909–5920. doi:10.1021/Ac015547q
Rose AL, Moffet JW, Waite TD (2008a) Determination of superoxide in seawater using 2-methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazo[1,2-a]pyrazin-3(7H)-one chemiluminescence. Anal Chem 80(4):1215–1227
Rose AL, Webb EA, Waite TD, Moffett JW (2008b) Measurement and implications of nonphotochemically generated superoxide in the equatorial Pacific Ocean. Environ Sci Technol 42(7):2387–2393
Rose AL, Godrant A, Furnas M, Waite TD (2010) Dynamics of superoxide production in the Great Barrier Reef lagoon. Limnol Oceanogr 55(4):1521–1536
Rosen GM, Tsai P, Barth ED, Dorey G, Casara P, Spedding M, Halpern HJ (2000) A one-step synthesis of 2-(2-pyridyl)-3H-indol-3-one N-oxide: is it an efficient spin trap for hydroxyl radical? J Org Chem 65(14):4460–4463
Roubaud V, Lauricella R, Tuccio B, Bouteiller JC, Tordo P (1996) Decay of superoxide spin adducts of new PBN-type phosphorylated nitrones. Res Chem Intermediat 22(4):405–416
Roubaud V, Lauricella R, Bouteiller JC, Tuccio B (2002) N-2-(2-ethoxycarbonyl-propyl) alpha-phenylnitrone: an efficacious lipophilic spin trap for superoxide detection. Arch Biochem Biophys 397(1):51–56
Rubio MA, Martire DO, Braslavsky SE, Lissi EA (1992) Influence of the ionic-strength on O2(1Δg) quenching by azide. J Photochem Photobiol A 66(2):153–157
Rusak SA, Peake BM, Richard LE, Nodder SD, Cooper WJ (2011) Distributions of hydrogen peroxide and superoxide in seawater east of New Zealand. Mar Chem 127(1–4):155–169
Rush JD, Bielski BHJ (1985) Pulse radiolytic studies of the reactions of HO2/O2 − with Fe(II)/Fe(III) ions—the reactivity of HO2/O2 − with ferric ions and its implication on the occurrence of the Haber–Weiss reaction. J Phys Chem 89(23):5062–5066
Saito I, Inoue K, Matsuura T (1975) Occurrence of singlet-oxygen mechanism in photodynamic oxidations of guanosine. Photochem Photobiol 21(1):27–30
Samuni A, Black CD, Krishna CM, Malech HL, Bernstein EF, Russo A (1988) Hydroxyl radical production by stimulated neutrophils reappraised. J Biol Chem 263(27):13797–13801
Saragosti E, Tchernov D, Kastir A, Shaked Y (2010) Extracellular production and degradation of superoxide in the coral Stylophora pistillata and cultured Symbiodinium. PLoS ONE 5(9):e12508
Saran M, Summer KH (1999) Assaying for hydroxyl radicals: hydroxylated terephthalate is a superior fluorescence marker than hydroxylated benzoate. Free Radic Res 31(5):429–436. doi:10.1080/10715769900300991
Schmidt R (2006) Photosensitized generation of singlet oxygen. Photochem Photobiol 82(5):1161–1177
Schuler RH, Patterson LK, Janata E (1980) Yield for the scavenging of OH radicals in the radiolysis of N2O-saturated aqueous-solutions. J Phys Chem 84(16):2088–2089
Schuler RH, Hartzell AL, Behar B (1981) Track effects in radiation-chemistry—concentration-dependence for the scavenging of OH by ferrocyanide in N2O-saturated aqueous-solutions. J Phys Chem 85(2):192–199
Schwarz HA, Dodson RW (1984) Equilibrium between hydroxyl radicals and thallium(Ii) and the oxidation potential of OH(Aq). J Phys Chem 88(16):3643–3647
Schweitzer C, Schmidt R (2003) Physical mechanisms of generation and deactivation of singlet oxygen. Chem Rev 103(5):1685–1757
Scully NM, Tranvik LJ, Cooper WJ (2003) Photochemical effects on the interaction of enzymes and dissolved organic matter in natural waters. Limnol Oceanogr 48(5):1818–1824
Scurlock RD, Ogilby PR (1996) Quenching of O2 (a1Δg) by O2 (a1Δg) in solution. J Phys Chem 100(43):17226–17231
Shaked Y, Harris R, Klein-Kedem N (2010) Hydrogen peroxide photocycling in the Gulf of Aqaba, Red Sea. Environ Sci Technol 44(9):3238–3244. doi:10.1021/Es902343y
Shao C, Cooper WJ, Lean DRS (1994) Singlet oxygen formation in lake waters from mid-latitudes. In: Helz GR, Zepp RG, Crosby DG (eds) Aquatic and surface photochemistry. Lewis, Boca Raton, pp 215–221
Shimomura O, Wu C, Murai A, Nakamura H (1998) Evaluation of five imidazopyrazinone-type chemiluminescent superoxide probes and their application to the measurement of superoxide anion generated by Listeria monocytogenes. Anal Biochem 258(2):230–235
Smith GF, Mccurdy WH (1952) 2,9-Dimethyl-1,10-phenanthroline—new specific in spectrophotometric determination of copper. Anal Chem 24(2):371–373
Snyrychova I, Hideg E (2007) The first application of terephthalate fluorescence for highly selective detection of hydroxyl radicals in thylakoid membranes. Funct Plant Biol 34(12):1105–1111. doi:10.1071/Fp07150
Soh N (2006) Recent advances in fluorescent probes for the detection of reactive oxygen species. Anal Bioanal Chem 386(3):532–543
Song B, Wang GL, Tan MQ, Yuan JL (2006) A europium(III) complex as an efficient singlet oxygen luminescence probe. J Am Chem Soc 128(41):13442–13450
Southworth BA, Voelker BM (2003) Hydroxyl radical production via the photo-Fenton reaction in the presence of fulvic acid. Environ Sci Technol 37(6):1130–1136. doi:10.1021/Es020757l
Stolze K, Udilova N, Nohl H (2002) Spin adducts of superoxide, alkoxyl, and lipid-derived radicals with EMPO and its derivatives. Biol Chem 383(5):813–820
Stolze K, Udilova N, Rosenau T, Hofinger A, Nohl H (2003) Synthesis and characterization of EMPO-derived 5,5-disubstituted 1-pyrroline N-oxides as spin traps forming exceptionally stable superoxide spin adducts. Biol Chem 384(3):493–500
Stolze K, Udilova N, Rosenau T, Hofinger A, Nohl H (2004) Spin adducts of several N-2-(2-alkoxycarbonyl-propyl)-alpha-pyridylnitrone derivatives with superoxide, alkyl and lipid-derived radicals. Biochem Pharmacol 68(1):185–193
Stolze K, Udilova N, Rosenau T, Hofinger A, Nohl H (2005) Spin adduct formation from lipophilic EMPO-derived spin traps with various oxygen- and carbon-centered radicals. Biochem Pharmacol 69(2):297–305
Sugioka K, Nakano M, Kurashige S, Akuzawa Y, Goto T (1986) A chemiluminescent probe with a Cypridina luciferin analog, 2-methyl-6-phenyl-3,7-dihydroimidazo[1,2-a]pyrazin-3-one, specific and sensitive for O-2-production in phagocytizing macrophages. FEBS Lett 197(1–2):27–30
Sulzberger B, Canonica S, Egli T, Giger W, Klausen J, von Gunten U (1997) Oxidative transformations of contaminants in natural and in technical systems. Chimia 51(12):900–907
Sutton HC, Downes MT (1972) Reactions of HO2 radical in aqueous-solution with bromine and related compounds. J Chem Soc Faraday Trans 1 68(8):1498–1507
Suzuki N, Mizumoto I, Toya Y, Nomoto T, Mashiko S, Inaba H (1990) Steady-state near-infrared detection of singlet molecular-oxygen—a Stern–Volmer quenching experiment with luminol, superoxide-dismutase, and Cypridina luciferin analogs. Agric Biol Chem Tokyo 54(11):2783–2787
Szymczak R, Waite TD (1988) Generation and decay of hydrogen peroxide in estuarine waters. Mar Freshw Res 39:289–299
Szymczak R, Waite TD (1991) Photochemical activity in waters of the Great Barrier Reef. Estuar Coast Shelf Sci 33:605–622
Tamaoku K, Murao Y, Akiura K, Ohkura Y (1982) New water-soluble hydrogen donors for the enzymatic spectrophotometric determination of hydrogen-peroxide. Anal Chim Acta 136:121–127
Tanaka K, Miura T, Umezaqa N, Urano Y, Kikuchi K, Higuchi T, Nagano T (2001) Rational design of fluorescein-based fluorescence probes. Mechanism-based design of a maximum fluorescence probe for singlet oxygen. J Am Chem Soc 123:2530–2536
Telfer A, Bishop SM, Phillips D, Barber J (1994) Isolated photosynthetic reaction-center of photosystem-II as a sensitizer for the formation of singlet oxygen—detection and quantum yield determination using a chemical trapping technique. J Biol Chem 269(18):13244–13253
Teranishi K (2007) Development of imidazopyrazinone red-chemiluminescent probes for detecting superoxide anions via a chemiluminescence resonance energy transfer method. Luminescence 22(2):147–156. doi:10.1002/Bio.939
Thomas JK, Rabani J, Matheson MS, Hart EJ, Gordon S (1966) Absorption spectrum of hydroxyl radical. J Phys Chem 70(7):2409–2410
Thompson AM, Zafiriou OC (1983) Air-sea fluxes of transient atmospheric species. J Geophys Res Ocean Atmos 88(Nc11):6696–6708
Tuccio B, Lauricella R, Frejaville C, Bouteiller JC, Tordo P (1995) Decay of the hydroperoxyl spin adduct of 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide—an EPR kinetic-study. J Chem Soc Perkin Trans 2(2):295–298
Tuccio B, Zeghdaoui A, Finet JP, Cerri V, Tordo P (1996) Use of new beta-phosphorylated nitrones for the spin trapping of free radicals. Res Chem Intermediat 22(4):393–404
Umezawa N, Tanaka K, Urano Y, Kikuchi K, Higuchi T, Nagano T (1999) Novel fluorescent probes for singlet oxygen. Angew Chem Int Ed Engl 38(19):2899–2901
Vaughan PP, Blough NV (1998) Photochemical formation of hydroxyl radical by constituents of natural waters. Environ Sci Technol 32(19):2947–2953
Villamena FA, Zweier JL (2002) Superoxide radical trapping and spin adduct decay of 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BocMPO): kinetics and theoretical analysis. J Chem Soc Perkin Trans 2(7):1340–1344
Villamena FA, Locigno EJ, Rockenbauer A, Hadad CM, Zweier JL (2006) Theoretical and experimental studies of the spin trapping of inorganic radicals by 5,5-dimethyl-1-pyrroline N-oxide (DMPO). 1. Carbon dioxide radical anion. J Phys Chem A 110(49):13253–13258. doi:10.1021/Jp064892m
Villamena FA, Locigno EJ, Rockenbauer A, Hadad CM, Zweier JL (2007) Theoretical and experimental studies of the spin trapping of inorganic radicals by 5,5-dimethyl-1-pyrroline N-oxide (DMPO). 2. Carbonate radical anion. J Phys Chem A 111(2):384–391
Vione D, Falletti G, Maurino V, Minero C, Pelizzetti E, Malandrino M, Ajassa R, Olariu R-I, Arsene C (2006) Sources and sinks of hydroxyl radicals upon irradiation of natural water samples. Environ Sci Technol 40(12):3775–3781
Voelker BM, Sedlak DL, Zafiriou OC (2000) Chemistry of superoxide radical in seawater: reactions with organic Cu complexes. Environ Sci Technol 34(6):1036–1042
von Sonntag C, Schuchmann H-P (1991) The elucidation of peroxyl radical reactions in aqueous solution with the help of radiation-chemical means. Angew Chem Int Ed Engl 38:1229–12536
Waite TD, Sawyer DT, Zafiriou OC (1988) Panel 1: oceanic reactive chemical transients. Appl Geochem 3(1):9–17
Warneck P, Wurzinger C (1988) Product quantum yields for the 305-nm photodecomposition of nitrate in aqueous solution. J Phys Chem 92(22):6278–6283. doi:10.1021/j100333a022
Wasserman HH, Scheffer JR, Cooper JL (1972) Singlet oxygen reactions with 9,10-diphenylanthracene peroxide. J Am Chem Soc 94(14):4991–4996
Wasserman HH, Xia M, Wang J, Petersen AK, Jorgensen M, Power P, Parr J (2004) Singlet oxygen reactions of 3-methoxy-2-pyrrole carboxylic acid tert-butyl esters. A route to 5-substituted pyrrole precursors of prodigiosin and analogs. Tetrahedron 60:7419–7425
Weeks JL, Rabani J (1966) Pulse radiolysis of deaerated aqueous carbonate solutions. I. Transient optical spectrum and mechanism. 2. pK for OH radicals. J Phys Chem 70(7):2100–2106
Weissler A (1953) Sonochemistry—the production of chemical changes with sound waves. J Acoust Soc Am 25(4):651–657
Westerhoff P, Aiken G, Amy G, Debroux J (1999) Relationships between the structure of natural organic matter and its reactivity towards molecular ozone and hydroxyl radicals. Water Res 33(10):2265–2276
Westerhoff P, Mezyk SP, Cooper WJ, Minakata D (2007) Electron pulse radiolysis determination of hydroxyl radical rate constants with Suwannee river fulvic acid and other dissolved organic matter isolates. Environ Sci Technol 41(13):4640–4646
Wick LY, McNeill K, Rojo M, Medilanski E, Gschwend PM (2000) Fate of benzene in a stratified lake receiving contaminated groundwater discharges from a superfund site. Environ Sci Technol 34(20):4354–4362. doi:10.1021/Es000908p
Wilkinson F, Helman WP, Ross AB (1995) Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution an expanded and revised compilation. J Phys Chem Ref Data 24(2):663–1021
Williams DC, Huff GF, Seitz WR (1976) Evaluation of peroxyoxalate chemiluminescence for determination of enzyme generated peroxide. Anal Chem 48(7):1003–1006
Winston GW, Cederbaum AI (1985) Decarboxylation of 7-14C-benzoic acid. In: Greenwald RA (ed) CRC handbook of methods for oxygen radical research. CRC Press, Boca Raton, pp 169–175
Wolcott RG, Franks BS, Hannum DM, Hurst JK (1994) Bactericidal potency of hydroxyl radical in physiological environments. J Biol Chem 269(13):9721–9728
Wolff CJM, Halmans MTH, Vanderheijde HB (1981) The formation of singlet oxygen in surface waters. Chemosphere 10(1):59–62
Wood PM (1974) The redox potential of the system oxygen-superoxide. Fed Eur Biochem Soc Lett 44:22–23
Xiao CB, King DW, Palmer DA, Wesolowski DJ (2000) Study of enhancement effects in the chemiluminescence method for Cr(III) in the ng l−1 range. Anal Chim Acta 415(1–2):209–219
Xiao CB, Palmer DA, Wesolowski DJ, Lovitz SB, King DW (2002) Carbon dioxide effects on luminol and 1,10-phenanthroline chemiluminescence. Anal Chem 74(9):2210–2216. doi:10.1021/Ac015714m
Yamaguchi S, Kishikawa N, Ohyama K, Ohba Y, Kohno M, Masuda T, Takadate A, Nakashima K, Kuroda N (2010) Evaluation of chemiluminescence reagents for selective detection of reactive oxygen species. Anal Chim Acta 665(1):74–78. doi:10.1016/j.aca.2010.03.025
Yang XF, Guo XQ (2001) Study of nitroxide-linked naphthalene as a fluorescence probe for hydroxyl radicals. Anal Chim Acta 434(2):169–177
Yang X, Zhan M-J, Kong L-R, Wang L-S (2004) Determination of hydroxyl radicals with salicylic acid in aqueous nitrate and nitrite solutions. J Environ Sci (IOS Press) 16(4):687–689
Yao CCD, Haag WR (1991) Rate constants for direct reactions of ozone with several drinking-water contaminants. Water Res 25(7):761–773
Yoon JH, Jung J, Chung HH, Lee MJ (2002) EPR characterization of carbonate ion effect on TCE and PCE decomposition by gamma-rays. J Radioanal Nucl Chem 253(2):217–219
Young RH, Brewer D, Keller RA (1973) Determination of rate constants of reaction and lifetimes of singlet oxygen in solution by a flash-photolysis technique. J Am Chem Soc 95(2):375–379
Youngman RJ, Elstner EF (1985) Ethylene formation from methionine in the presence of pyridoxal phosphate. In: Greenwald RA (ed) CRC handbook of methods for oxygen radical research. CRC Press, Boca Raton, pp 165–168
Yu F, Xu D, Lei R, Li N, Li K (2008) Free-radical scavenging capacity using the Fenton reaction with rhodamine B as the spectrophotometric indicator. J Agric Food Chem 56(3):730–735. doi:10.1021/Jf072383r
Yuan JC, Shiller AM (1999) Determination of subnanomolar levels of hydrogen peroxide in seawater by reagent-injection chemiluminescence detection. Anal Chem 71(10):1975–1980
Zafiriou OC (1974) Sources and reactions of OH and daughter radicals in seawater. J Geophys Res 79(30):4491–4497
Zafiriou OC (1977) Marine organic photochemistry previewed. Mar Chem 5:497–522
Zafiriou OC (1990) Chemistry of superoxide ion-radical (O2 −) in seawater. 1. pK asw* (HOO) and uncatalyzed dismutation kinetics studied by pulse-radiolysis. Mar Chem 30(1–3):31–43
Zafiriou OC, True MB (1979a) Nitrate photolysis in seawater by sunlight. Mar Chem 8(1):33–42
Zafiriou OC, True MB (1979b) Nitrite photolysis in seawater by sunlight. Mar Chem 8:9–32
Zafiriou OC, Joussotdubien J, Zepp RG, Zika RG (1984) Photochemistry of natural-waters. Environ Sci Technol 18(12):A358–A371
Zafiriou OC, Blough NV, Micinski E, Dister B, Kieber D, Moffett J (1990) Molecular probe systems for reactive transients in natural-waters. Mar Chem 30(1–3):45–70
Zeghdaoui A, Tuccio B, Finet JP, Cerri V, Tordo P (1995) Beta-phosphorylated alpha-phenyl-N-tert-butylnitrone (PBN) analogs—a new series of spin traps for oxyl radicals. J Chem Soc Perkin Trans 2(12):2087–2089
Zehavi D, Rabani J (1972a) The oxidation of aqueous bromide ions by hydroxyl radicals a pulse radiolytic investigation. J Phys Chem 76(3):312–319
Zehavi D, Rabani J (1972b) Pulse radiolysis of aqueous ferro-ferricyanide system. 1. Reactions of OH, HO2, and O2 − radicals. J Phys Chem 76(25):3703–3709
Zepp RG, Wolfe NL, Baughman GL, Hollis RC (1977) Singlet oxygen in natural waters. Nature 267:421–423
Zepp RG, Baughman GL, Schlotzhauer PF (1981) Comparison of photochemical behavior of various humic substances in water: II. Photosensitized oxygenations. Chemosphere 10:119–126
Zepp RG, Hoigne J, Bader H (1987) Nitrate-induced photooxidation of trace organic-chemicals in water. Environ Sci Technol 21(5):443–450
Zepp RG, Skurlatov YI, Ritmiller LF (1988) Effects of aquatic humic substances on analysis for hydrogen-peroxide using peroxidase-catalyzed oxidations of triarylmethanes or para-hydroxyphenylacetic acid. Environ Technol Lett 9(4):287–298
Zepp RG, Faust BC, Hoigne J (1992) Hydroxyl radical formation in aqueous reactions (pH 3–8) of iron (II) with hydrogen peroxide: the photo-fenton reaction. Environ Sci Technol 26(2):313–319
Zhao HT, Joseph J, Zhang H, Karoui H, Kalyanaraman B (2001) Synthesis and biochemical applications of a solid cyclic nitrone spin trap: a relatively superior trap for detecting superoxide anions and glutathiyl radicals. Free Radic Bio Med 31(5):599–606
Zheng J, Springston SR, Weinstein-Lloyd J (2003) Quantitative analysis of hydroperoxyl radical using flow injection analysis with chemiluminescence detection. Anal Chem 75(17):4696–4700. doi:10.1021/Ac034429v
Zhou X, Mopper K (1990) Determination of photochemically produced hydroxyl radicals in seawater and freshwater. Mar Chem 30:71–88
Zhou MJ, Diwu ZJ, PanchukVoloshina N, Haugland RP (1997) A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem 253(2):162–168
Zika RG, Saltzman ES (1982) Interaction of ozone and hydrogen-peroxide in water—implications for analysis of H2O2 in air. Geophys Res Lett 9(3):231–234
Zika R, Saltzman E, Chameides WL, Davis DD (1982) H2O2 levels in rainwater collected in south Florida and the Bahama islands. J Geophys Res Ocean Atmos 87(Nc7):5015–5017
Zika RG, Moffett JW, Petasne RG, Cooper WJ, Saltzman ES (1985a) Spatial and temporal variations of hydrogen-peroxide in Gulf of Mexico waters. Geochim Cosmochim Acta 49(5):1173–1184
Zika RG, Saltzman ES, Cooper WJ (1985b) Hydrogen-peroxide concentrations in the Peru upwelling area. Mar Chem 17(3):265–275
Zuo Z, Cai Z, Katsumura Y, Chitose N, Muroya Y (1999) Reinvestigation of the acid-base equilibrium of the (bi)carbonate radical and pH dependence of its reactivity with inorganic reactants. Radiat Phys Chem 55:15–23
Acknowledgments
The authors would like to acknowledge valuable comments of the anonymous reviewers and the associate editor which significantly improved the manuscript. This work was supported in part by the US National Science Foundation, Grant no. OCE 0752473 and the College of Arts and Sciences at the University of South Carolina. This is publication 72 of the University of California Irvine, Urban Water Research Center.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Burns, J.M., Cooper, W.J., Ferry, J.L. et al. Methods for reactive oxygen species (ROS) detection in aqueous environments. Aquat Sci 74, 683–734 (2012). https://doi.org/10.1007/s00027-012-0251-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00027-012-0251-x