
Abstract
Circular RNAs (circRNAs) are members of the non-coding transcriptome; however, some of them are translated into proteins. These transcripts have important roles in both physiological and pathological mechanisms due to their ability to directly influence cellular signaling pathways. Specifically, circRNAs are regulators of transcription, translation, protein interaction, and signal transduction. An increased knowledge within their area is observed over the last few years, concomitant with the development of next-generation sequencing techniques. circRNAs are mostly tissue and disease specific with the ability of specifically changing the biological behavior of cells. The altered expression profile is currently investigated as novel minimally invasive diagnosis/prognosis tool and also therapeutic target in human disease. The diagnosis approach is based on their level modification within pathological states, especially cancer, where circRNAs’ therapies are intensively explored in anti-aging strategies, diabetes, cardiovascular diseases, and malignant pathologies, and are relying on the restoration of homeostatic profiles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since their discovery, significant improvements have been made in confirming the consequential role of non-coding RNAs (ncRNAs) in the coordination of physiological and pathological processes [1,2,3,4,5,6,7]. Nowadays, the transcription of the human genome into ncRNAs is well characterized. The major types of ncRNAs are: microRNAs (miRNAs), small interfering RNAs (siRNAs) and long non-coding RNAs (lncRNAs) [4, 5]. Intensive efforts are made to study the role of ncRNAs, with a special focus on their interactions with various transcripts and proteins [3, 8].
Recently, the previous status of some ncRNAs called circular RNAs (circRNAs) has been revised [9], being highlighted new functional roles [4, 10]. These transcripts have multiple cellular functions and the alteration of their homeostatic expression is associated with the onset and development of various pathologies [3, 11, 12]. Deep understanding of these mechanisms will impact a wide range of clinical niches, particularly within the development of new diagnosis and prognosis tools and targeted therapies [4, 10].
The history of cirRNAs began around 45 years ago, with an article published by Sanger et al. which described the sequencing method for bacteriophage F1 genome, focused on the analysis of nucleotide polymers found in a close loop [13]. Later on, with the help of electron microscopy and electrophoresis in the hepatitis delta (δ) virus, circRNAs were discovered [14]. In 1991, it was assessed that the exons of the tumor suppressor gene DCC (deleted in colorectal cancer) are joined through their splice site in a different order [15]. Two years later, circRNAs containing exons were found in the cytoplasm of eukaryotic cells [16]. At that time, they were considered a result of defective splicing [17]. However, circRNAs are highly conserved sequences due to their stable circular shape [18] and after general reconsideration regarding the role of ncRNAs in organism homeostasis and progress to a pathological state, circRNAs’ previous status has been revised [19,20,21].
Our paper intends to summarize the main implication of circRNAs in cellular communication and the translation of these mechanisms in pathological scenarios. Specifically, we present the mechanism of circRNAs biogenesis, their roles in cellular processes and the various methods used for their detection and quantification, together with the databases developed for their analysis in human disease. The general overview of circRNAs intends to encourage the progression of clinical uses of these molecules as biomarker or therapeutic tools.
Circular RNAs: definition and biogenesis
Nowadays, circRNAs are considered an important bridge between the coding and the non-coding RNAs. Initially, circRNAs were thought to be only the effect of the transcriptional noise [17], but further studies challenged this idea [19, 21, 22] through the identification of thousands of genomic loci in mammals specific for circRNAs; moreover, these sequences have important implication in cellular state [23].
CircRNAs are generated from intronic or exonic sequences [24] found in thousands of genomic loci [23]. Some of them are transcribed from a single gene or multiple genes [25]. An example from multiple coding genes is the PML/RARα fusion gene in leukemias. The primary transcript can undergo the process of back-splicing and give rise to a fusogenic circRNA (fcircRNA) that contains exons from both translocated genes [26].
CircRNAs can be originated from the primary form of: messenger RNA (pre-mRNA), ribosomal RNA (pre-rRNA), or transport RNA (pre-tRNA) [27] (Fig. 1). The pre-mRNA can be spliced into circRNAs through two main mechanisms: lariat formation and direct back-splicing [28, 29].

The biogenesis of circRNAs. a Through the pre-mRNA canonical splicing, the mature mRNA is generated. The alternative splicing pathways are: exon skipping and back-splicing. b The circularized transcript can contain only the exon part and it is named exonic circular RNA (ecircRNA/circRNA). c, d The flanking introns can form direct base-pairing (c) or RNA-binding protein (RNP)-mediated intron–intron binding (d). The resulted circular transcript can be an exon–intron circular RNA (EIcircRNA) or the introns are further removed and the two- or multiple exons containing ecircRNA are generated. The EIcircRNA can regulate its own transcription by interacting with U1 snRNA and RNA Pol II. e The intron can form a lariat during canonical or alternative splicing. The lariat may become more stable and circular thus giving rise to the intronic circular RNA (circRNA). f In bacterial cells, as well as in eukaryotic cells, it was proven that the primary tRNA has introns that are spliced and circularized into tricRNA. Their function still remains to be studied
Lariat formation mechanism is divided into two types of strategies: direct circularization of intronic lariat and circularization through exon skipping [17]. For the direct circularization of an intronic lariat, the pre-mRNA is cleaved at the 5′ end with the help of small nuclear RNA (snRNA) U1 and ligated through a 5′–2′ bound between a guanidine and an adenosine [30]. The intronic lariat is processed and kept in a circular form in the nucleus [31]. In the case of exon skipping, a “hetero-lariat” is formed, containing both exons and introns (ElcircRNA) [32, 33]. This strategy can continue with the removal of all introns and the generation of circular RNAs containing only exons, which are named exonic circular RNAs (EcircRNAs) [28].
Back-splicing can generate all of the three types of circular RNA: circular intronic RNAs (ciRNAs), EcircRNAs, and intron–exon circular RNAs (EIcircRNAs) [22]. Through the process of direct back-splicing, the 5′ splice donor is ligated to a 3′ splice acceptor via simple or RNA-binding proteins (RNP) base-pairing [15, 34]. This is facilitated by the presence of Alu repeats in the intronic area [28, 35]. There are various cis- or trans-acting factors responsible for bringing the donor and acceptor sites in proximity to one another [20]. Although the canonical splicing shares common regulatory factors with the alternative splicing, the interaction pattern discriminates between the two [36]. Moreover, some of these factors, such as nuclear factor 110 (NF110) [37] or the Quaking (QKI) protein [38, 39], remain bound to the circRNAs also in the cytoplasm and enhance their stability.
A greater diversity of circRNAs is found in bacteria (derived from primary transcripts of tRNAs [40] or rRNAs contain introns [41]). The introns of pre-tRNA are cleaved and circularized. These RNA products are named tRNA intronic circular RNAs (tricRNAs) and were discovered both in bacteria and in eukaryotic cells [40, 41]. In bacteria, the intron containing pre-rRNAs is passing through a similar process, resulting in a circularized rRNA transcript [41].
CircRNAs’ biological function
Important roles are now attributed to circRNAs sequences, probably with many others to be discovered. These transcripts are key regulator molecules, being also highly conserved due to their lack of 3′ poly-A tail, 5′ cap, and consequential circular shape [18]. circRNAs fulfill a variety of functions inside the cell [42] through their interaction with DNA, other coding or non-coding RNAs, and proteins [27].
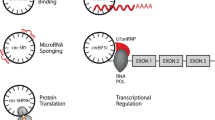
CircRNAs are involved in the regulation of transcription, post-transcription control (particularly splicing), and translation and post-translational signaling [17, 43]. They act as competing endogenous RNAs (ceRNA) for miRNAs [44, 45], with important consequences in the regulation of gene expression. It is supposed that their miRNA sponging activity is also connected with lncRNAs function; however further experimental data are needed [46]. CircRNAs functions are summarized in Fig. 2.

Circular RNAs (noted circRNA—for all circular RNA types) fulfill multiple functions inside the cell. In the nucleus a–c circRNAs can a interact with the histone methylation pattern and silence a specific locus; b regulate the transcription of their gene of origin through direct (circRNAs) or snU1-mediated (EIcircRNA) interaction with the RNA polymerase II; c compete with the mRNA for the available splicing machinery and interfere in the alternative splicing process. In the cytoplasm d–i the circRNAs can: d entrap certain transcription factors in the cytoplasm; e act as miRNAs sponges, especially the circRNAs with exon-containing transcripts; f be translated into proteins. g circRNAs interact with RNA-binding proteins and regulate the translation of some mRNAs. h circRNAs can act as protein scaffolds. They can entrap some proteins and impair their signal transduction role. i circRNAs can induce apoptosis by interfering within the processing of pre-rRNA subunits
CircRNAs as gene expression regulators
CircRNAs are generally tissue-specific [24], where 20% of the expressed genes are able to generate circular transcripts [47]; therefore, they are correlated with the expression profile of the parent genes [19]. As stated, circRNAs are involved in transcription and post-transcriptional gene expression regulation, including interference with key RNA-binding proteins.
CircRNAs and transcriptional control
circRNAs are active elements at transcriptional level, acting as positive regulators of Pol II transcription through their accumulation at the transcription site [48]. The circRNA ci-ankrd52 is a cis-acting positive regulator of its own gene, ANKRD52, by affecting the Pol II-mediated elongation during transcription [49]. The exonic–intronic circRNAs also called EIciRNAs are proved to increase the expression level of the parental gene through a transcriptional control that implies the interaction with the parental genes as cis forms, highlighting an important transcription regulatory strategy [50]. These transcripts can also interfere with the histone methylation pattern: e.g., circANRIL interaction with the polycomb repressive complex 1 is necessary for an effective histone 3-methylation and silencing of INK4a locus [51]. Circ-Amotl1 promotes the nuclear localization of the c-Myc transcription factor [52].
The EIciRNAs, circEIF3J and circPAIP2, stimulate the transcription of their own genes by interacting with the transcriptions machinery. These EIciRNAs form a direct RNA–RNA interaction with the U1 snRNA. TheEIciRNA–U1snRNA complex interacts with the U1A and U1C proteins, U2 snRNA and RNA polymerase II. The EIciRNAs are cis-regulators of their gene of origin, but they can also act as trans-regulators of distant genes [50]. This is the case of ciANKRD52 that interacts with the transcription elongation factor Pol II [49]. CircRNAs transcripts can also interact with regulatory proteins, affecting the translation of targeted mRNAs. For instance, CircPABPN1 inhibits the HuR protein, a positive regulator of PABPN1 mRNA, thus downregulating the translation of its own gene of origin [53].
CircRNAs and post-transcription control
Alternative pre-mRNA splicing is a contributor of post-transcriptional regulation and sustains the proteomic diversity [46]; circRNAs interfere with this mechanism. In the case of circMbl, the introns contain putative-binding sites for the MBL protein; thus, the MBL gene creates a negative-feedback loop to regulate its own expression mediated by splicing competitiveness [54]. The RNA-binding proteins (RBPs) act as an ON/OFF switch for lariat formation/back-splicing by facilitating or hindering the complementary base-pairing of the flanking introns [55]. The EcircRNAs entrap the exons from pre-mRNA and disrupts their translation [56]. There are also RBPs, such as ADAR1, which oppose the interaction between the two flanking introns by melting the newly formed stems of dsRNA and impairing the circRNA formation [57]. These introns usually contain reverse complementary sequences, such as AG–CT or GGG–CCC repeats, which allow the interaction with each other [55].
CircRNAs are able to impair rRNA processing: circANRIL induce apoptosis by inhibiting the rRNA nuclease processing and ribosome biogenesis [58].
In general, circRNAs are derived from exonic regions of pre-mRNAs, the fact that anticipates that their synthesis is implicitly disrupting the splicing of the mRNA sequences [56]. The preference toward back-splicing/exon skipping and circularization versus canonical splicing is regulated by the introns flanking the skipped exons. These are long introns, difficult to cut during splicing, and contain reverse complement sequences, which allow the interaction with each other [48].
CircRNA as miRNA sponging
The initial circularized transcript which contains both introns and exons is further processed into a final transcript in which the exon is either kept or discarded. If, in the final circularized form, the exon is present, the circRNAs have the ability of miRNA sponging, as in the case of ciRS-7 from Mbl gene and miR-7 [59]. This mechanism is mediated by the binding to the miRNA-specific RBP, Argonaute 2 (Ago2) [46, 60, 61]. The sponging capacity has important clinical applications in tumoral pathologies for targeting oncogenic miRNAs.
CircRNAs can be translated into proteins
Until recently, there were a lot of controversies regarding the translation of circRNAs. Part of them has the necessary characteristics to be translated into proteins, but the actual proteins were only recently found [62]. Some circRNAs have the full set of characteristics to be translated in proteins, which renders them as protein-coding RNAs. N6-Methyladenosine (m6A), is the most frequent modification of RNA and this alteration sustains protein translation from circRNAs [25, 63]. Moreover, these circRNAs interact with the ribosome [25] through their internal ribosome entry sites (IRESs) located in the 5′ untranslated region (5′ UTR). These circRNAs possess start and stop codons, similar to their corresponding linear mRNA [64]. With the help of mass spectrometry PRIDE database, 46 translated circRNAs were found, originated in 37 genes [65]. circMbl encodes a protein found in Drosophila [38]; circ-ZNF609 controls myoblast proliferation and is translated into a protein [64]; circ-SHPRH encodes the SHPRH-146aa protein, which functions as a tumor suppressor in human glioblastoma [66]. The bioinformatics tools demonstrate clearly that some circRNAs can be translated, but the probability that circRNAs are endogenously translated has been only indirectly tested [25]. In addition, the protein-coding capacity in the majority of circRNAs in vivo is still a matter of debate.
CircRNAs can entrap proteins and impair signal transduction
CircRNAs act as scaffold for some transcription factor at cytosolic level or nuclear level, as regulatory mechanisms or as transport systems [62]. CircRNAs perform vital functions in regulating a variety of signaling pathways like mitogen-activated protein kinase (MAPK), cell division kinases, phosphatidylinositol 3-kinase (PI3K), or Wnt/β-catenin pathway [67]. CircITCH is an important regulator of cell cycle and, at the same time, affects malignant transformation via Wnt signaling pathway [68].
CDK2 helps in the progression of cell cycle, while the p21 protein opposes its action. The circRNA fork head transcription factor, circ-Foxo3, entraps CDK2 and p21, and hence, it impairs cell cycle progression [69]. In the cytoplasm, circ-Foxo3 is able to bind the stress-activated transcription regulators: ID1, E2F1, FAK, and HIF1α, and block their nuclear import [70]. Therefore, circ-Foxo3 does not affect the expression level of these transcription factors, but it affects their localization. The ID1 [71], E2F1 [72], FAk [73], and HIF1α [74] are cancer-promoting transcription factors, which may explain why the circFoxo3 is underexpressed in cancer [45].
Foxo3 protein and circ-Foxo3 function as positively regulated tumor suppressors. In this case, the circ-Foxo3 has two functions: one is the binding, ubiquitination, and induced degradation of p53, and the other one consists in the interaction with MDM2 protein, thus hindering the MDM2 capability of ubiquitination and degradation of Foxo3 protein [25, 45, 75]. The exact mechanism by which circ-Foxo3 is capable of binding and entrapping these proteins needs to be elucidated [69].
Another important mechanism of gene expression regulation mediated by circRNAs consists in the HuR-mediated translational repression. The CircPABPN1 (hsa_circ_0031288) competes with PABPN1 mRNA for the binding of HuR protein. The HuR protein is a RNA-binding protein (RBP), indispensable for the ribosomal translation of the mRNA; thus, by entrapping the HuR protein, the circRNA suppresses the expression of the linear form. As the level of circRNAs increases, the level of available HuR is decreased [53].
CircRNAs in human disease
Non-communicable diseases, such as cardiovascular disease, diabetes, or cancer, have a major impact on the mortality rates worldwide. In parallel, efforts are made to implement the concept of healthy aging or regenerative medicine [76]. CircRNAs, due to their wide spread function, have impact on all the reminded sectors.
circRNAs in cardiovascular disease
circRNAs are an important effector of the cardiovascular system and are involved in the both physiological and pathological states (Table 1). Circ-Foxo3 is overexpressed in the cytoplasm of aged mice and involved in the regulation of cardiac senescence and myocardial protection [1, 70]; meanwhile, cANRIL is correlated with atherosclerosis risk [77]. CircANRIL binds a 60S assembly factor of ribosomal RNA and it stops rRNA processing, which activates the p53 pathway. In this way, atheroma cells enter apoptosis and are also subjected to a slower proliferation rate [58]. Another important transcript is represented by ciRNA HRCR (heart-related circRNA) that has a protective role in the case of pathological hypertrophy and heart failure [78].
In mice, mmu_circRNA_006636 is significantly downregulated after infarct injury. This circRNA binds to the Dnmt3B and impairs the methylation of Pink1 promoter, leading to its overexpression. Pink1 leads to FAM65B phosphorylation and blocking of autophagy in cardiomyocytes [79].
circRNAs in diabetes
With fast worldwide aging, the incidence of diabetes and its complications, such as diabetic retinopathy [80], are growing. In this context, circRNA molecular mechanisms offer valuable information (Table 1). In an animal models for type I and type II diabetes, it was demonstrated that circRNAs are important regulators β-cell dysfunction. ciRS-7/CDR1 acts as a miR-7 sponge [81]. CircHIPK3 functions as a ceRNA for miR-124 and miR-338, and it targets the expression of relevant pancreatic β-cell genes, such as Slc2a2, AKT1, and MTPN [82]. Diabetic retinopathy is an important issue in the most of the diabetes patient; there are different circRNAs with altered expression in this condition. Within all, circ_0005015 is a potential biomarker of this disease [83].
CircRNAs in aging and regenerative medicine
CircRNAs accumulation is considered as a hallmark of aging [87]. Although the process of aging is not considered a pathological state, it is connected with a wide range of diseases. The regenerative medicine sits at the opposite side by being more active in a young organism. CircRNAs have an ontogenic shift in their expression profile [88], which is why they are of extreme importance for aging and regenerative studies [89].
The accumulation of circRNAs in the neurons of aging animals is a common event. This is correlated with a decrease in the translational level of genes necessary for physiological function of the cell. The underlying reason for this probably consists in a reduced level of normal splicing and overgeneration of circRNAs [90].
In Caenorhabditis elegans, circRNAs accumulate all over the organisms. The deregulated circRNAs are composed of genes related to the life span of the animal. In addition, from 1166 circRNAs identified, 575 consist in de novo sequences. The accumulation of the circular form of transcripts was not done at the expense of linear forms [91]. Other study observed the accumulation of circRNAs in aging neurons accountable for decreased cellular proliferation rate, lower exosome generation potential, and a higher capacity of RBP stabilization of circRNAs [87].
The aging of ovaries show a change in the circRNA profiles, with a total of 194 upregulated circRNAs and 207 downregulated circRNAs. CircDDX-10 sponges either miR-1301 or miR-466 and it leads to the overexpression of SIRT3, a protein involved in epigenetic gene silencing [85].
CircRNAs are important regulators of cell senescence [70, 84]. For instance, circRNA100783 has a key role in chronic CD28-associated CD8(+)T cell aging [84]. Circ-Foxo3 promotes cardiac senescence via cytoplasmic entrapment of transcription factors related to stress and senescence responses [70].
In the brain of aging mice, circRNAs cargo increases, while the linear form has an approximately constant expression. Some of the upregulated circRNAs are: circAnkib1, circZfp609, Circ-Rims2, Circ-dac4, Circ-NFATc3, Circ-Top1, and Circ-Mtf2 [92]. In Caenorhabditis elegans, it was again found a global accumulation of circRNAs in older organisms. A total of 797 circRNAs are upregulated, with some of them presenting over a 40-fold increase in the expression level [91].
CircRNAs have a role in the regenerative medicine, especially for stem cell differentiation. The induced pluripotent stem cells (iPSC) have a lower expression of circALPK2, circCACNA1D, circSLC8A1, and circSPHKAP, when compared to differentiated cardiomyocytes [93]. Other study demonstrated that ciRNAs have the capacity to delay or impaired wound healing, particular for the case of patients with diabetes and atherosclerosis. This is the case of circ-Amotl1 that interacts with STAT3, which promotes the nuclear translocation and overexpression of Dnmt3a, with further impact on wound healing through the stimulated proliferation, migration, adhesion, and survival of epithelial cells at the wound site [86].
CircRNAs in cancer
CircRNAs are constantly found at deregulated levels in malignant pathologies with the potential of further use for diagnosis/prognosis tools or therapeutic target.
CircRNA-406483 and circRNA-404833 are overexpressed and the circRNA-001640, circRNA-006411, and circRNA-401977 are underexpressed in human lung cancer tissue versus normal one [94]. The CDR1-derived circRNA is a miR-7 sponge, with implication in multiple pathologies [1, 81, 95,96,97]. Inhibition of CDR1 is associated with impairment of colorectal cancer progression by increased expression level of miR-7 and positive regulation of epidermal growth factor receptor (EGFR) and insulin-like growth factor receptor 1 (IGF-1R) [98]. Overexpression of CDR1 is correlated with hepatic microvascular invasion in hepatic cancer [99]. Functional studies demonstrate that CDR1 has oncogenic role [100], by stimulating cell proliferation through the modulation of EGFR [101], CCNE1 and PIK3CD [100].
Endothelial circRNAs: cZNF292, cAFF1, and cDENND4C are involved in the regulation of hypoxia [102]. Another circRNA, circDENND4C, is upregulated in breast cancer. This circRNA interacts with HIF1α and promotes cancer cell proliferation [103].
The ANRIL locus in the genome is transcribed both as a linear long non-coding RNA and as a circRNA. It is observed that ANRIL, in linear form or circular form, regulates INK4/ARF gene expression. The change in form is correlated with disease risk via chromatin interaction at 9p21 locus [104], gastric cancer via interaction with TET2 gene [105], non-small cell lung cancer (NSCLC) by silencing KLF2 and p21 expression [106], and hepatocellular carcinoma via epigenetic silencing of KLF2 [107].
CircMTO1 suppresses hepatocellular carcinoma (HCC) progression acting as a sponge of oncogenic miR-9. It has a prognostic role in this disease, where a low expression level predicts a reduced survival rate [108]. In bladder cancer, circMYLK is upregulated, being involved in the regulation of VEGFA expression by acting as a ceRNA for miR-29a [109]. Further details regarding the role of circRNAs in various tumoral pathologies are found in Table 2.
Detection of circRNAs
The detection of circRNAs has reshaped the view of basic and translational research. The circRNAs do not have a poly-A tail and are not capped, which is why they cannot be quantified with the oligo dT enrichment method. Instead of this, scientists can use enzymatic digestion approaches using exonuclease (RNAase R), enzymes that destroy most of the linear transcripts.
The rapid progression of high-throughput RNA sequencing (RNAseq) approaches, sustained by novel bioinformatics tools and databases, allows the quantification of circRNAs in an accurate manner, leading to new insights into to the expression level and biological significance of these transcripts. These procedures for circRNAs analysis consist in the enrichment procedure using RNA exonucleases, enzymes that are capable to digest linear RNA species but not circular ones. These properties are exploited in the procedures of sample preparation or detection.
Most of the circRNAs’ next-generation sequencing analysis is based on in house pipelines and bioinformatics analysis. These methods allow the identification and characterization of thousands of different circRNAs [118].
Common RNAseq protocols introduce technical artefacts that can result in spurious identification of circRNAs isoforms. It is proven that technical artefacts can be introduced during the ligation and reverse transcription steps of RNAseq library preparation. In general, during most of the sequencing protocols, the libraries are prepared with or without exonucleases treatments (RNase R) followed by ribosomal RNA (rRNA) depletion and complex bioinformatics analysis for the evaluation of the expression level and interaction with coding or non-coding RNAs and proteins. CircRNAs are identified from the RNAseq data as reads having apparent splice junctions that couples the end of a split read fragment to the start of a downstream or an upstream fragment [119]. In both cases, the sequencing of the amplification products is mandatory to certify the accuracy of detection, and to prevent the false-positive detection or improper quantification [118, 120].
Arraystar is the most common circRNAs microarray platform, based on an adapted protocol that uses RNase R treatment and a specific circular junction probe design to achieve a high sensitivity and specificity through enriched circRNAs’ fraction [121]. Microarray can be considered as a robust tool for circRNAs profiling, being able to evaluate over 80,000 circRNAs in cervical tumors versus normal tissue and over 18,000 circRNAs in cell-free plasma samples using the Capital Bio Technology Human CircRNA Microarray (version v2.0) with two types of probes with different length (30 nt and 20 nt) [122]. Microarray is proved more efficient and accurate than RNAseq for circRNA profiling; for example, in the case of cervical cancer, around 80,000 circRNAs are identified and 25,000 of them are differently expressed in cervical tumors versus matched normal tissues [122].
Each of the novel cirRNAs identified by RNAseq needs to be validated through qRT-PCR and Northern blot [123]. qRT-PCR is a simple and informative technique. The cDNA synthesis is based on ‘outward-facing’ and ‘opposite-directed’ primers [124]. When they anneal to the circRNAs, their 3′ ends face away from each other. RT-PCR can give false-positive detection and improper quantification [118, 120]. To certify the accuracy of the scrambled junction and to avoid template switching or trans-splicing, the sequencing of the amplification products is mandatory [120] The qRT-PCR protocol using divergent primers has the advantage of targeting circRNA back-spliced junction sequence. In this case, only the circRNAs is amplified and not the equivalent linear RNA sequence [123].
Northern blot technique uses the migration of the linear RNAs, along with different circRNAs resulting from trans-splicing or tandem duplication. A specific hybridization protocol that can identify the back-spliced junction is required [47]. It is a very usefully approach used for the case of validation of primary structure predicted by bioinformatics tools, with a particular interest when is focused on the differentiation from the other transcripts [47].
Fluorescent in situ hybridization allows the evaluation of cell- and tissue-specific circRNAs expression based on the specific design of fluorescent probes complementary to the back-spliced junctions; these are further connected to high-resolution microscope [125]. Another technology is using the principle of targeting miRNAs using a fluorescent probe compatible for microscopic evaluation; this could be also applied to counteract linear transcripts expressed from the same gene locus as circRNAs [47, 125]. Table 3 presents the main techniques used for circRNAs detection and quantification together with their advantages and disadvantages.
Databases for circRNAs
Following the development of new high-throughput sequencing technologies, numerous databases were produced, allowing advancement of genomics, transcriptomics, proteomics, and metabolomics. In this way, a promising perspective was created for a more precise evaluation of the expression level of ncRNAs, including circRNAs [129]. The novel circRNAs need to be integrated in a database that allows the summarization of expression pattern for these sequences, integrating in the same time their main isomorphic forms with the exact sequence and the genomic annotation. One of the pioneers in this field is StarBase v2.0 [130, 131]. In Table 4, there is a summary of the key databases/online tools used for circRNA analysis, as well as their advantaged/disadvantages.
CircBase is a complex database that comprises the genomic localization and also the transcribed RNAs along with the information related to the in silico predicted spliced form, the overlapping coding transcripts; it is also connected with the literature data related to the expression level of the circRNAs [132]. Similar to CircBase, Circ2Traits attempts to bring together information about circRNAs that include data from experimental analysis of these RNAs, creating a comprehensive platform of circRNAs associated with human pathologies [133, 134]. CircNet is developed for continuously update with novel circRNAs, providing the genomic annotation together with different isoforms and interaction patterns with coding and non-coding transcripts [129].
There are several softwares used for circularization prediction of transcripts based on RNASeq data. The most important concern when it comes to choosing such a software package is the overestimation of circRNAs generated inside a cell. The CIRI software can generate up to 68% false-positive results, while the CIRCexplorer is the best predictor of circRNAs [135, 136]. This result may be reasoned by the fact that these software packages require the use of gene annotation [135, 137]. Other prediction tools consist in CircMarker [138], PcircRNA_finder [139], find_circ, circRNA_finder, MapSplice, and CirComPara, which focus on the evaluation of abundance and significance of circRNAs in particular cell types [135, 140]. The cancer-specific circRNA database (CSCD) is focused on malignant pathologies [141], while Tissue-Specific CircRNA Database (TSCD) is designed to bring light on circRNAs roles in organ development and tissue-specific disorders [142]. PcircRNA_finder is a software developed for circRNAs’ prediction in plants [139]. The most commonly encountered challenge is the overprediction of circRNAs from the RNASeq data.
CircInteractome is a web tool able to evaluate the interactions of circRNAs with RBPs or different coding and non-coding RNA transcripts. It also includes information regarding the presence of internal ribosomal entry sites (IRESs), primers for circular transcripts, and siRNA design tools for circRNAs targeting [44, 143].
In line with the latest studies regarding the protein-coding capacity of circRNAs, a new database, circRNADb, was developed. This database includes: an estimation of the circRNAs capacity to encode for proteins, the confirmed results from mass spectrometry analysis, and a tissue-specific pattern of circRNAs [65]. The exogenous circRNAs are presented as robust and stable protein expression in eukaryotic cells, as alternative to linear mRNA [144]. These are only some pioneers’ studies that need to be sustained by additional information.
The increase in sequencing depth allows the identification of novel circRNAs by RNAseq. This information, once integrated in public databases, can be used for the generation of specific probes for microarray or other specific determination methods [122]. However, the main problem of these databases is the restrain knowledge related the role of the altered circRNAs and the interconnection of circRNA-miRNAs/lncRNAs-mRNA [147] integrated in interconnected networks of several database.
Circulating circRNAs as disease biomarkers
Exosomes are exocytic microvesicles (30–100 nm) that are released by both healthy and pathological cells to facilitate the communication between them. These vehicles have a cell-type specific cargo, comprised of proteins, DNA, RNAs—mRNA, miRNAs, lncRNAs, lipids, and others [148]. Recently, it was demonstrated that exosomes also contain circRNAs [149]. Li et al. identified over 1000 circRNAs species in human serum exosomes (exo-circRNAs). Their length is mostly under 1000 nucleotides [148]. As follows, the circRNAs act as long-distance regulators, circulating in the body fluids more often in the exosomes than as a free form [150, 151].
CircRNAs are generally expressed at very low levels, but accumulate in the tissue to detectable level due to their high stability [152]. This allows them to be kept at room temperature for 24 h, with no observed modification in the expression level [149].
The exact mechanism regulating the loading of circRNAs into exosomes remains elusive [150]. The circRNAs are enriched in exosomes compared to the cell of origin. In addition, the sorting of circRNAs into exosomes may be regulated by changes in the associated miRNA levels [149].
Due to the high proliferation capacity and increased metabolism of tumor cells, the exosomal circRNAs are isolated in high concentrations. Still, the level of some circRNAs is more reduced in tumor tissue than in the normal one [42, 153]. For instance, circ_002059 is downregulated in gastric cancer tissues, when compared to normal tissue [154].
The ability of circRNAs to act as sponges for multiple targets, including miRNAs, proteins, and possibly lncRNAs, could be used in experimental strategies directed toward cancer inhibition. The synthesis of circular artificial sequences able to impair different oncogenic miRNAs in a simultaneous act could become a highly efficient therapeutic strategy used for the impairment of compensatory mechanisms in cancer.
If an artificial circRNA could sponge, at the same time, various oncogenic miRNAs associated with independent signaling profiles, the chance of resistance is significantly reduced. A superior level of cancer therapeutic modulation could be represented by hybrid circRNAs able to simultaneously bind oncogenic miRNAs and oncoproteins of the same pathway [155]. This strategy is especially important in the context of drug resistance in cancer, one of the main issues responsible for increased mortality rates [156,157,158,159].
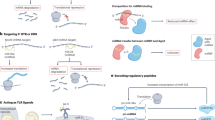
Engagement of these approaches in the preclinical research could result in a more extended modulation of non-coding and proteomic profiles, decreasing significantly the change of cancer cells to activate secondary survival pathways. The design of artificial circRNAs should target several upstream and downstream elements from a signaling pathway, thus preventing the development of secondary resistance (Fig. 3).

Therapeutic value of synthetic circRNAs. a The synthetic circRNAs can be design in such a way that it will be able to target multiple miRNAs with consequences toward impairment of multiple oncogenic pathways. Synthetic circRNA can be delivered as a single agent. Inside the cell, this circRNA will sponge multiple microRNAs; b a second option consists in synthetic circRNAs able to target multiple proteins leading to the impairment of multiple oncogenic pathways. Once delivered inside the cell, the oncogenic proteins will be sponged and their activity suppressed; c both types of circRNAs can be loaded in tumor cell exosomes which will enhance their targeting efficiency and amount that can be delivered
The therapeutic potential of circRNAs lays mainly in their ability to sponge microRNAs (Fig. 4). Some circRNAs could be subjected to inhibition, because they are overexpressed in cancerous cells and sponge the tumor suppressors’ miRNAs; as in the case of circGFRA1 and miR-34a [96] or circUBAP2 and miR-143 [160]. There are a number of in vitro studies based on this therapeutically strategy. Circ-SMARCA5 is an androgen-induced overexpressed circRNA in prostate cancer and its inhibition by siRNA targeting the back-splicing site leads to cell apoptosis and inhibited cell proliferation [161]. CircRNA-MYLK functions as a ceRNA by binding miR-29a and allowing the expression of VEGFA. The silencing of this circRNA was also proposed for the treatment of bladder cancer [109]. cZNF292 is a hypoxia-induced circRNA, whose expression silenced by siRNA leads to suppressed proliferation and angiogenesis in glioma [162].

The major strategies for circularRNA therapy. a A tumor suppressor circRNA inside the cell will sponge oncomiRs, resulting in the upregulation of tumor suppressor mRNAs. b In the intracellular environment, siRNA can target and repress the oncogenic circRNA and the upregulation of tumor suppressor mRNA. c Inside the cell, the number of tumor suppressor circRNAs is restored and these can directly inhibit the oncogenic mRNAs. d circRNAs can also function as miRNA delivery systems, where tumor suppressor miRNAs can further repress the oncogenic mRNA. All of the above-mentioned circRNA therapeutic strategies result in the mRNA-mediated tumor suppression
Other circRNAs sponge oncomiRs and their overexpression is also proposed as a therapeutic option. In hepatocellular carcinoma, circMTO1 sponges miR-9 and allows the expression of p21. siRNA targeted toward this circRNA promotes tumor growth, invasion, and proliferation [163].
Circular RNA-ITCH (circITCH) has also therapeutic potential by downregulating the Wnt/β-Catenin pathway [113] in: colorectal cancer [110], bladder cancer [116], or esophageal squamous cell carcinoma [112]. Circ-FOXO3 entraps the CDK2 and p21 proteins, thus, impairing cell cycle progression in cancer [69]. Circ-FOXO3, when transfected into breast cancer cells, causes a decrease in tumor size and it enhances cell death [75, 164].
On top of using circRNAs as therapeutic options, they can also be used as vectors, for microRNAs or proteins [155]. One important challenge that may come in the future development of circRNA-based therapies is the lately discovered fact where exogenous circRNAs are recognized by RIG-I as non-self-molecules through an unknown mechanism. The exogenous circRNAs, in contrast to the endogenous ones, do not possess the RNA associated proteins: U1, U2, and U4/U6/U5 tri-snRNP subunits, EIF4A3, MGN2, RNPS1, DDX39B, THOC4, and XPO5. Moreover, the intracellular delivery of exogenous circRNA gives rise to a rapid increase in the expression level of various inflammation-associated genes [155, 165]. Even if cancer-derived exosomes have an increased tropism for malignant cells, there is also the possibility of targeting healthy ones, resulting in phenotypic transformation or induced cell death. In these sense, hybrid strategies need to be taken in consideration, combining different delivery perspectives from the nanopharmacology domain [166].
Conclusions
CircRNAs are key regulatory elements in all the stages of cellular development. Some of them have the status of housekeeping transcripts, while others are tissue-specific. Moreover, during pathological states, the expression of circRNAs is shifting toward a differential one, the fact that is transforming these sequences in important diagnosis and prognosis tools, but also therapeutic targets. Moreover, the main known role of circRNAs, namely miRNAs sponging, highlights the idea of using these circular transcripts as inhibitory vectors for oncogenic overexpressed miRNAs. Moreover, similar techniques could be used for therapeutic strategies to eliminate the harmful activity of aberrantly overexpressed circRNAs. CRISPR/Cas9 has emerged as an important technique for the investigation of gene function and also therapeutic strategies [167, 168]. For the case of, circRNAs complete depletion via CRISPR/Cas9 of the genomic corresponding region could offer specific insights within the role of circRNAs in cellular homeostasis and also development toward pathological states.
The interference with important regulatory processes of mRNA metabolism on many levels: transcription, splicing, mRNA turnover, and translation, makes circRNAs key molecules within evaluation of pathological processes, with further implication in the possible novel clinical management tools. The utility of these circRNAs as circulating molecules in diagnosis procedures can be justified by the first clinical trials (NCT03334708) launched [169]. This study evaluates a panel of circRNAs, along with other molecular markers (proteins and proteases, functional DNA repair assays, exosomes, stromal elements, and circulating tumor DNA) for the diagnosis of the early stage pancreatic cancer.
The main problem remains the optimisation of the detection and quantification methods, where the research is still in the early phases. Even so, circRNAs hold great promises due to their complex regulatory capacity in different stages of development and also differential profiles in pathological states.
Abbreviations
- Ago2:
-
Argonaute 2
- circRNAs:
-
Circular RNAs
- EGFR:
-
Epidermal growth factor receptor
- ElcircRNA:
-
Exon–intron circular RNA
- EMT:
-
Epithelial-to-mesenchymal transition
- fcircRNA:
-
Fusogenic circRNA
- lncRNAs:
-
Long non-coding RNAs
- m6A:
-
N6-Methyladenosine
- miRNAs:
-
MicroRNAs
- ncRNAs:
-
Non-coding RNAs
- siRNAs:
-
Small interfering RNAs
- tricRNAs:
-
tRNA intronic circular RNAs
References
Geng HH, Li R, Su YM, Xiao J, Pan M, Cai XX, Ji XP (2016) The circular RNA Cdr1as promotes myocardial infarction by mediating the regulation of miR-7a on its target genes expression. PLoS One 11:e0151753
Karreth FA, Reschke M, Ruocco A, Ng C, Chapuy B, Leopold V, Sjoberg M, Keane TM, Verma A, Ala U, Tay Y, Wu D, Seitzer N, Velasco-Herrera Mdel C, Bothmer A, Fung J, Langellotto F, Rodig SJ, Elemento O, Shipp MA, Adams DJ, Chiarle R, Pandolfi PP (2015) The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell 161:319–332
Braicu C, Calin GA, Berindan-Neagoe I (2013) MicroRNAs and cancer therapy—from bystanders to major players. Curr Med Chem 20:3561–3573
Braicu C, Catana C, Calin GA, Berindan-Neagoe I (2014) NCRNA combined therapy as future treatment option for cancer. Curr Pharm Des 20:6565–6574
Seles M, Hutterer GC, Kiesslich T, Pummer K, Berindan-Neagoe I, Perakis S, Schwarzenbacher D, Stotz M, Gerger A, Pichler M (2016) Current Insights into Long Non-Coding RNAs in Renal Cell Carcinoma. Int J Mol Sci 17(4):573. https://doi.org/10.3390/ijms17040573
Redis RS, Berindan-Neagoe I, Pop VI, Calin GA (2012) Non-coding RNAs as theranostics in human cancers. J Cell Biochem 113:1451–1459
Gulei D, Magdo L, Jurj A, Raduly L, Cojocneanu-Petric R, Moldovan A, Moldovan C, Florea A, Pasca S, Pop LA, Moisoiu V, Budisan L, Pop-Bica C, Ciocan C, Buiga R, Muresan MS, Stiufiuc R, Ionescu C, Berindan-Neagoe I (2018) The silent healer: miR-205-5p up-regulation inhibits epithelial to mesenchymal transition in colon cancer cells by indirectly up-regulating E-cadherin expression. Cell Death Dis 9:66
Irimie AI, Braicu C, Sonea L, Zimta AA, Cojocneanu-Petric R, Tonchev K, Mehterov N, Diudea D, Buduru S, Berindan-Neagoe I (2017) A looking-glass of non-coding RNAs in oral cancer. Int J Mol Sci 18(12):2620. https://doi.org/10.3390/ijms18122620
Roberts TC, Morris KV (2013) Not so pseudo anymore: pseudogenes as therapeutic targets. Pharmacogenomics 14:2023–2034
Irimie AI, Braicu C, Pileczki V, Petrushev B, Soritau O, Campian RS, Berindan-Neagoe I (2016) Knocking down of p53 triggers apoptosis and autophagy, concomitantly with inhibition of migration on SSC-4 oral squamous carcinoma cells. Mol Cell Biochem 419:75–82
Berindan-Neagoe I, Braicu C, Gulei D, Tomuleasa C, Calin GA (2017) Noncoding RNAs in lung cancer angiogenesis. In: Simionescu D, Simionescu A (eds) Physiologic and pathologic angiogenesis—signaling mechanisms and targeted therapy. InTech, Rijeka
Lekka E, Hall J (2018) Noncoding RNAs in disease. FEBS Lett 592:2884–2900
Sanger F, Donelson JE, Coulson AR, Kossel H, Fischer D (1973) Use of DNA polymerase I primed by a synthetic oligonucleotide to determine a nucleotide sequence in phage fl DNA. Proc Natl Acad Sci USA 70:1209–1213
Kos A, Dijkema R, Arnberg AC, van der Meide PH, Schellekens H (1986) The hepatitis delta (delta) virus possesses a circular RNA. Nature 323:558–560
Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B (1991) Scrambled exons. Cell 64:607–613
Cocquerelle C, Mascrez B, Hetuin D, Bailleul B (1993) Mis-splicing yields circular RNA molecules. FASEB J Off Publ Feder Am Soc Exp Biol 7:155–160
Wang X, Zhang Y, Huang L, Zhang J, Pan F, Li B, Yan Y, Jia B, Liu H, Li S, Zheng W (2015) Decreased expression of hsa_circ_001988 in colorectal cancer and its clinical significances. Int J Clin Exp Pathol 8:16020–16025
Xie H, Ren X, Xin S, Lan X, Lu G, Lin Y, Yang S, Zeng Z, Liao W, Ding YQ, Liang L (2016) Emerging roles of circRNA_001569 targeting miR-145 in the proliferation and invasion of colorectal cancer. Oncotarget 7(18):26680–26691. https://doi.org/10.18632/oncotarget.8589
Huang C, Shan G (2015) What happens at or after transcription: insights into circRNA biogenesis and function. Transcription 6:61–64
Tung MC, Lin PL, Wang YC, He TY, Lee MC, Yeh SD, Chen CY, Lee H (2015) Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 6:41692–41705
Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, Chen LL, Yang L (2016) Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 26:1277–1287
Chen LL, Yang L (2015) Regulation of circRNA biogenesis. RNA Biol 12:381–388
Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, Chen LL (2016) The biogenesis of nascent circular RNAs. Cell Rep 15(3):611–624. https://doi.org/10.1016/j.celrep.2016.03.058
Xin Z, Ma Q, Ren S, Wang G, Li F (2016) The understanding of circular RNAs as special triggers in carcinogenesis. Brief Funct Genom 16(2):80–86. https://doi.org/10.1093/bfgp/elw001
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, Shenzis S, Samson M, Dittmar G, Landthaler M, Chekulaeva M, Rajewsky N, Kadener S (2017) Translation of circRNAs. Mol Cell 66:9–21
Guarnerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM, Lo-Coco F, Tay Y, Beck AH, Pandolfi PP (2016) Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell 165:289–302
Zhang Y, Liang W, Zhang P, Chen J, Qian H, Zhang X, Xu W (2017) Circular RNAs: emerging cancer biomarkers and targets. J Exp Clin Cancer Res 36:152
Yang Z, Xie L, Han L, Qu X, Yang Y, Zhang Y, He Z, Wang Y, Li J (2017) Circular RNAs: regulators of cancer-related signaling pathways and potential diagnostic biomarkers for human cancers. Theranostics 7:3106–3117
Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H (2015) Circular RNA: a new star of noncoding RNAs. Cancer Lett 365:141–148
Konarska MM, Grabowski PJ, Padgett RA, Sharp PA (1985) Characterization of the branch site in lariat RNAs produced by splicing of mRNA precursors. Nature 313:552–557
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495:384–388
Suzuki H, Aoki Y, Kameyama T, Saito T, Masuda S, Tanihata J, Nagata T, Mayeda A, Takeda S, Tsukahara T (2016) Endogenous multiple exon skipping and back-splicing at the DMD mutation hotspot. Int J Mol Sci 17(10):1722. https://doi.org/10.3390/ijms17101722
Zhu LP, He YJ, Hou JC, Chen X, Zhou SY, Yang SJ, Li J, Zhang HD, Hu JH, Zhong SL, Zhao JH, Tang JH (2017) The role of circRNAs in cancers. Biosci Rep 37:BSR20170750
Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell-Badge R (1993) Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73:1019–1030
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19:141–157
Chou MY, Rooke N, Turck CW, Black DL (1999) hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol Cell Biol 19:69–77
Li X, Liu CX, Xue W, Zhang Y, Jiang S, Yin QF, Wei J, Yao RW, Yang L, Chen LL (2017) Coordinated circRNA biogenesis and function with NF90/NF110 in viral infection. Mol Cell 67(214–227):e7
Wang Y, Wang Z (2015) Efficient backsplicing produces translatable circular mRNAs. RNA 21:172–179
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ (2015) The RNA binding protein quaking regulates formation of circRNAs. Cell 160:1125–1134
Noto JJ, Schmidt CA, Matera AG (2017) Engineering and expressing circular RNAs via tRNA splicing. RNA Biol 14:978–984
Danan M, Schwartz S, Edelheit S, Sorek R (2012) Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res 40:3131–3142
Abu N, Jamal R (2016) Circular RNAs as promising biomarkers: a mini-review. Front Physiol 7:355
Halbeisen RE, Galgano A, Scherrer T, Gerber AP (2008) Post-transcriptional gene regulation: from genome-wide studies to principles. Cell Mol Life Sci CMLS 65:798–813
Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M (2016) CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol 13:34–42
Lu WY (2017) Roles of the circular RNA circ-Foxo3 in breast cancer progression. Cell Cycle (Georgetown, Tex) 16:589–590
Lyu D, Huang S (2017) The emerging role and clinical implication of human exonic circular RNA. RNA Biol 14:1000–1006
Huang S, Yang B, Chen BJ, Bliim N, Ueberham U, Arendt T, Janitz M (2017) The emerging role of circular RNAs in transcriptome regulation. Genomics 109:401–407
Han YN, Xia SQ, Zhang YY, Zheng JH, Li W (2017) Circular RNAs: a novel type of biomarker and genetic tools in cancer. Oncotarget 8:64551–64563
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL (2013) Circular intronic long noncoding RNAs. Mol Cell 51:792–806
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, Zhu P, Chang Z, Wu Q, Zhao Y, Jia Y, Xu P, Liu H, Shan G (2015) Exon–intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22:256–264
Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM (2010) Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 38:662–674
Yang Q, Du WW, Wu N, Yang W, Awan FM, Fang L, Ma J, Li X, Zeng Y, Yang Z, Dong J, Khorshidi A, Yang BB (2017) A circular RNA promotes tumorigenesis by inducing c-myc nuclear translocation. Cell Death Differ 24:1609–1620
Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S, Kim J, Noh JH, Kim KM, Martindale JL, Gorospe M (2017) Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol 14:361–369
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S (2014) circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56:55–66
Ferracin M, Gautheret D, Hubé F, Mani S, Mattick J, Andersson Ørom U, Santulli G, Slotkin R, Szweykowska-Kulinska Z, Taube J, Vazquez F, Yang J-H (2015) The non-coding RNA Journal Club: highlights on recent papers. Non-coding RNA 1:87
Ebbesen KK, Hansen TB, Kjems J (2017) Insights into circular RNA biology. RNA Biol 14:1035–1045
Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, Rajewsky N (2015) Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10:170–177
Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, Kohlmaier A, Herbst A, Northoff BH, Nicolaou A, Gabel G, Beutner F, Scholz M, Thiery J, Musunuru K, Krohn K, Mann M, Teupser D (2016) Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun 7:12429
Zhao ZJ, Shen J (2015) Circular RNA participates in the carcinogenesis and the malignant behavior of cancer. RNA Biol 14(5):514–521. https://doi.org/10.1080/15476286.2015.1122162
Werfel S, Nothjunge S, Schwarzmayr T, Strom TM, Meitinger T, Engelhardt S (2016) Characterization of circular RNAs in human, mouse and rat hearts. J Mol Cell Cardiol 98:103–107
Zhang XQ, Yang JH (2018) Discovering circRNA-microRNA interactions from CLIP-Seq data. Methods Mol Biol (Clifton, NJ) 1724:193–207
Gomes CPC, Salgado-Somoza A, Creemers EE, Dieterich C, Lustrek M, Devaux Y (2018) Circular RNAs in the cardiovascular system. Non-coding RNA Res 3:1–11
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen L-L, Wang Y, Wong CCL, Xiao X, Wang Z (2017) Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res 27:626
Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, Laneve P, Rajewsky N, Bozzoni I (2017) Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell 66:22–37
Chen X, Han P, Zhou T, Guo X, Song X, Li Y (2016) circRNADb: a comprehensive database for human circular RNAs with protein-coding annotations. Sci Rep 6:34985
Zhang M, Huang N, Yang X, Luo J, Yan S, Xiao F, Chen W, Gao X, Zhao K, Zhou H, Li Z, Ming L, Xie B, Zhang N (2018) A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene 37:1805
Greene J, Baird A-M, Brady L, Lim M, Gray SG, McDermott R, Finn SP (2017) Circular RNAs: biogenesis, function and role in human diseases. Front Mol Biosci 4:38
Ding H-X, Lv Z, Yuan Y, Xu Q (2018) The expression of circRNAs as a promising biomarker in the diagnosis and prognosis of human cancers: a systematic review and meta-analysis. Oncotarget 9(14):11824–11836. https://doi.org/10.18632/oncotarget.23484
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB (2016) Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 44:2846–2858
Du WW, Yang W, Chen Y, Wu ZK, Foster FS, Yang Z, Li X, Yang BB (2017) Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur Heart J 38:1402–1412
Ling MT, Wang X, Zhang X, Wong YC (2006) The multiple roles of Id-1 in cancer progression. Differentiation 74:481–487
Engelmann D, Putzer BM (2012) The dark side of E2F1: in transit beyond apoptosis. Cancer Res 72:571–575
Sulzmaier FJ, Jean C, Schlaepfer DD (2014) FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14:598–610
Masoud GN, Li W (2015) HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 5:378–389
Du WW, Fang L, Yang W, Wu N, Awan FM, Yang Z, Yang BB (2017) Induction of tumor apoptosis through a circular RNA enhancing Foxo3 activity. Cell Death Differ 24:357–370
Hui L (2017) Assessment of the role of ageing and non-ageing factors in death from non-communicable diseases based on a cumulative frequency model. Sci Rep 7:8159
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE (2010) Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet 6:e1001233
Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, Zhou LY, Sun T, Wang M, Yu T, Gong Y, Liu J, Dong YH, Li N, Li PF (2016) A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J 37:2602–2611
Zhou LY, Zhai M, Huang Y, Xu S, An T, Wang YH, Zhang RC, Liu CY, Dong YH, Wang M, Qian LL, Ponnusamy M, Zhang YH, Zhang J, Wang K (2018) The circular RNA ACR attenuates myocardial ischemia/reperfusion injury by suppressing autophagy via modulation of the Pink1/FAM65B pathway. Cell Death Differ. https://doi.org/10.1038/s41418-018-0206-4
He X, Pu C, Quan Y, Ou C, Zhou S (2018) Circular RNA HIPK3: an emerging player in diabetes. Transl Cancer Res 1:S715–S717
Xu H, Guo S, Li W, Yu P (2015) The circular RNA Cdr1as, via miR-7 and its targets, regulates insulin transcription and secretion in islet cells. Sci Rep 5:12453
Stoll L, Sobel J, Rodriguez-Trejo A, Guay C, Lee K, Veno MT, Kjems J, Laybutt DR, Regazzi R (2018) Circular RNAs as novel regulators of beta-cell functions in normal and disease conditions. Mol Metab 9:69–83
Zhang SJ, Chen X, Li CP, Li XM, Liu C, Liu BH, Shan K, Jiang Q, Zhao C, Yan B (2017) Identification and characterization of circular RNAs as a new class of putative biomarkers in diabetes retinopathy. Investig Ophthalmol Vis Sci 58:6500–6509
Wang Y-H, Yu X-H, Luo S-S, Han H (2015) Comprehensive circular RNA profiling reveals that circular RNA100783 is involved in chronic CD28-associated CD8(+)T cell ageing. Immun Ageing I&A 12:17. https://doi.org/10.1186/s12979-015-0042-z
Cai H, Li Y, Li H, Niringiyumukiza JD, Zhang M, Chen L, Chen G, Xiang W (2018) Identification and characterization of human ovary-derived circular RNAs and their potential roles in ovarian aging. Aging 10:2511–2534
Yang ZG, Awan FM, Du WW, Zeng Y, Lyu J, Wu D, Gupta S, Yang W, Yang BB (2017) The circular RNA interacts with STAT3, increasing its nuclear translocation and wound repair by modulating Dnmt3a and miR-17 function. Mol Ther J Am Soc Gene Ther 25:2062–2074
Knupp D, Miura P (2018) CircRNA accumulation: a new hallmark of aging? Mech Ageing Dev 173:71–79
Veno MT, Hansen TB, Veno ST, Clausen BH, Grebing M, Finsen B, Holm IE, Kjems J (2015) Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol 16:245
Xu T, Wu J, Han P, Zhao Z, Song X (2017) Circular RNA expression profiles and features in human tissues: a study using RNA-seq data. BMC Genom 18:680
Hall H, Medina P, Cooper DA, Escobedo SE, Rounds J, Brennan KJ, Vincent C, Miura P, Doerge R, Weake VM (2017) Transcriptome profiling of aging Drosophila photoreceptors reveals gene expression trends that correlate with visual senescence. BMC Genom 18:894
Cortes-Lopez M, Gruner MR, Cooper DA, Gruner HN, Voda AI, van der Linden AM, Miura P (2018) Global accumulation of circRNAs during aging in Caenorhabditis elegans. BMC Genom 19:8
Gruner H, Cortes-Lopez M, Cooper DA, Bauer M, Miura P (2016) CircRNA accumulation in the aging mouse brain. Sci Rep 6:38907
Lei W, Feng T, Fang X, Yu Y, Yang J, Zhao ZA, Liu J, Shen Z, Deng W, Hu S (2018) Signature of circular RNAs in human induced pluripotent stem cells and derived cardiomyocytes. Stem Cell Res Ther 9:56
Zhao J, Li L, Wang Q, Han H, Zhan Q, Xu M (2017) CircRNA expression profile in early-stage lung adenocarcinoma patients. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 44:2138–2146
Zeng Y, Xu Y, Shu R, Sun L, Tian Y, Shi C, Zheng Z, Wang K, Luo H (2017) Altered expression profiles of circular RNA in colorectal cancer tissues from patients with lung metastasis. Int J Mol Med 40:1818–1828
Peng L, Yuan XQ, Li GC (2015) The emerging landscape of circular RNA ciRS-7 in cancer (Review). Oncol Rep 33:2669–2674
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338
Tang W, Ji M, He G, Yang L, Niu Z, Jian M, Wei Y, Ren L, Xu J (2017) Silencing CDR1as inhibits colorectal cancer progression through regulating microRNA-7. OncoTargets Ther 10:2045–2056
Xu L, Zhang M, Zheng X, Yi P, Lan C, Xu M (2017) The circular RNA ciRS-7 (Cdr1as) acts as a risk factor of hepatic microvascular invasion in hepatocellular carcinoma. J Cancer Res Clin Oncol 143:17–27
Yu L, Gong X, Sun L, Zhou Q, Lu B, Zhu L (2016) The circular RNA Cdr1as act as an oncogene in hepatocellular carcinoma through targeting miR-7 expression. PLoS One 11:e0158347
Yang X, Xiong Q, Wu Y, Li S, Ge F (2017) Quantitative proteomics reveals the regulatory networks of circular RNA CDR1as in hepatocellular carcinoma cells. J Proteome Res 16:3891–3902
Boeckel JN, Jae N, Heumuller AW, Chen W, Boon RA, Stellos K, Zeiher AM, John D, Uchida S, Dimmeler S (2015) Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res 117:884–890
Liang G, Liu Z, Tan L, Su AN, Jiang WG, Gong C (2017) HIF1alpha-associated circDENND4C promotes proliferation of breast cancer cells in hypoxic environment. Anticancer Res 37:4337–4343
Nakaoka H, Gurumurthy A, Hayano T, Ahmadloo S, Omer WH, Yoshihara K, Yamamoto A, Kurose K, Enomoto T, Akira S, Hosomichi K, Inoue I (2016) Allelic imbalance in regulation of ANRIL through chromatin interaction at 9p21 endometriosis risk locus. PLoS Genet 12:e1005893
Deng W, Wang J, Zhang J, Cai J, Bai Z, Zhang Z (2016) TET2 regulates LncRNA-ANRIL expression and inhibits the growth of human gastric cancer cells. IUBMB Life 68:355
Nie FQ, Sun M, Yang JS, Xie M, Xu TP, Xia R, Liu YW, Liu XH, Zhang EB, Lu KH, Shu YQ (2015) Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther 14:268–277
Huang MD, Chen WM, Qi FZ, Xia R, Sun M, Xu TP, Yin L, Zhang EB, De W, Shu YQ (2015) Long non-coding RNA ANRIL is upregulated in hepatocellular carcinoma and regulates cell apoptosis by epigenetic silencing of KLF2. J Hematol Oncol 8:50
Cui Y, Zhang F, Zhu C, Geng L, Tian T, Liu H (2017) Upregulated lncRNA SNHG1 contributes to progression of non-small cell lung cancer through inhibition of miR-101-3p and activation of Wnt/beta-catenin signaling pathway. Oncotarget 8:17785–17794
Zhong Z, Huang M, Lv M, He Y, Duan C, Zhang L, Chen J (2017) Circular RNA MYLK as a competing endogenous RNA promotes bladder cancer progression through modulating VEGFA/VEGFR2 signaling pathway. Cancer Lett 403:305–317. https://doi.org/10.1016/j.canlet.2017.06.027
Huang G, Zhu H, Shi Y, Wu W, Cai H, Chen X (2015) cir-ITCH plays an inhibitory role in colorectal cancer by regulating the Wnt/β-catenin pathway. PLoS One 10:e0131225
Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z, Yang J, Fan J, Liu L, Qin W (2016) Hsa_circ_0001649: a circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark Sect A Dis Mark 16:161–169
Paller CJ, Rudek MA, Zhou XC, Wagner WD, Hudson TS, Anders N, Hammers HJ, Dowling D, King S, Antonarakis ES, Drake CG, Eisenberger MA, Denmeade SR, Rosner GL, Carducci MA (2015) A phase I study of muscadine grape skin extract in men with biochemically recurrent prostate cancer: safety, tolerability, and dose determination. Prostate 75:1518–1525
Wan L, Zhang L, Fan K, Cheng ZX, Sun QC, Wang JJ (2016) Circular RNA-ITCH suppresses lung cancer proliferation via inhibiting the Wnt/beta-catenin pathway. Biomed Res Int 2016:1579490
Luo YH, Zhu XZ, Huang KW, Zhang Q, Fan YX, Yan PW, Wen J (2017) Emerging roles of circular RNA hsa_circ_0000064 in the proliferation and metastasis of lung cancer. Biomed Pharmacother Biomed Pharmacother 96:892–898
Li Y, Jiang F, Chen L, Yang Y, Cao S, Ye Y, Wang X, Mu J, Li Z, Li L (2015) Blockage of TGFβ-SMAD2 by demethylation-activated miR-148a is involved in caffeic acid-induced inhibition of cancer stem cell-like properties in vitro and in vivo. FEBS Open Biol 5:466–475
Yang C, Yuan W, Yang X, Li P, Wang J, Han J, Tao J, Li P, Yang H, Lv Q, Zhang W (2018) Circular RNA circ-ITCH inhibits bladder cancer progression by sponging miR-17/miR-224 and regulating p21, PTEN expression. Mol Cancer 17:19
Yang Y, Gao X, Zhang M, Yan S, Sun C, Xiao F, Huang N, Yang X, Zhao K, Zhou H, Huang S, Xie B, Zhang N (2018) Novel role of FBXW7 circular RNA in repressing glioma tumorigenesis. J Natl Cancer Inst 110(3). https://doi.org/10.1093/jnci/djx166
Jeck WR, Sharpless NE (2014) Detecting and characterizing circular RNAs. Nat Biotechnol 32:453–461
Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt LM, Teupser D, Hackermüller J, Stadler PF (2014) A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol 15:R34
Barrett SP, Salzman J (2016) Circular RNAs: analysis, expression and potential functions. Development (Cambridge, England) 110(3):1838–1847
Su H, Lin F, Deng X, Shen L, Fang Y, Fei Z, Zhao L, Zhang X, Pan H, Xie D, Jin X, Xie C (2016) Profiling and bioinformatics analyses reveal differential circular RNA expression in radioresistant esophageal cancer cells. J Transl Med 14:225
Li S, Teng S, Xu J, Su G, Zhang Y, Zhao J, Zhang S, Wang H, Qin W, Lu ZJ, Guo Y, Zhu Q, Wang D (2018) Microarray is an efficient tool for circRNA profiling. Brief Bioinform. https://doi.org/10.1093/bib/bby006
Panda AC, Gorospe M (2018) Detection and analysis of circular RNAs by RT-PCR. Bio-protocol 8(6):e2775. https://doi.org/10.21769/BioProtoc.2775
Li L, Guo J, Chen Y, Chang C, Xu C (2017) Comprehensive CircRNA expression profile and selection of key CircRNAs during priming phase of rat liver regeneration. BMC Genom 18:80
Urbanek MO, Nawrocka AU, Krzyzosiak WJ (2015) Small RNA detection by in situ hybridization methods. Int J Mol Sci 16:13259–13286
Schneider T, Schreiner S, Preusser C, Bindereif A, Rossbach O (2018) Northern blot analysis of circular RNAs. Methods Mol Biol (Clifton, NJ) 1724:119–133
Grozdanov PN, Macdonald CC (2014) High-throughput sequencing of RNA isolated by cross-linking and immunoprecipitation (HITS-CLIP) to determine sites of binding of CstF-64 on nascent RNAs. Methods Mol Biol (Clifton, NJ) 1125:187–208
Carbonell A (2017) Immunoprecipitation and high-throughput sequencing of ARGONAUTE-bound target RNAs from plants. Methods Mol Biol (Clifton, NJ) 1640:93–112
Liu YC, Li JR, Sun CH, Andrews E, Chao RF, Lin FM, Weng SL, Hsu SD, Huang CC, Cheng C, Liu CC, Huang HD (2016) CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res 44:D209–D215
Li JH, Liu S, Zhou H, Qu LH, Yang JH (2014) starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42:D92–D97
Xu, Y. (2017) An overview of the main circRNA databases. Non-coding RNA Investig 1:1–22. https://doi.org/10.21037/ncri.2017.11.05
Glazar P, Papavasileiou P, Rajewsky N (2014) circBase: a database for circular RNAs. RNA 20:1666–1670
Hancock JM (2014) Circles within circles: commentary on Ghosal et al. (2013) “Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits”. Front Genet 5:459
Ghosal S, Das S, Sen R, Basak P, Chakrabarti J (2013) Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet 4:283
Hansen TB, Veno MT, Damgaard CK, Kjems J (2016) Comparison of circular RNA prediction tools. Nucleic Acids Res 44:e58
Gao Y, Wang J, Zhao F (2015) CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16:4
Hansen TB (2018) Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol 6:20
Li X, Chu C, Pei J, Mandoiu I, Wu Y (2017) CircMarker: a fast and accurate algorithm for circular RNA detection. BMC Genomics19(6):572. https://doi.org/10.1186/s12864-018-4926-0
Chen L, Yu Y, Zhang X, Liu C, Ye C, Fan L (2016) PcircRNA_finder: a software for circRNA prediction in plants. Bioinformatics (Oxford, England) 32:3528–3529
Gaffo E, Bonizzato A, Kronnie G, Bortoluzzi S (2017) CirComPara: a multi-method comparative bioinformatics pipeline to detect and study circRNAs from RNA-seq data. Non-coding RNA 3:8
Xia S, Feng J, Chen K, Ma Y, Gong J, Cai F, Jin Y, Gao Y, Xia L, Chang H, Wei L, Han L, He C (2018) CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res 46:D925–D929
Xia S, Feng J, Lei L, Hu J, Xia L, Wang J, Xiang Y, Liu L, Zhong S, Han L, He C (2017) Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief Bioinform 18:984–992
Panda AC, Dudekula DB, Abdelmohsen K, Gorospe M (2018) Analysis of circular RNAs using the web tool CircInteractome. Methods Mol Biol (Clifton, NJ) 1724:43–56
Wesselhoeft RA, Kowalski PS, Anderson DG (2018) Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun 9:2629
Zeng X, Lin W, Guo M, Zou Q (2017) A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 13:e1005420
Li X, Chu C, Pei J, Măndoiu I, Wu Y (2018) CircMarker: a fast and accurate algorithm for circular RNA detection. BMC Genom 19:175–183
Rong D, Sun H, Li Z, Liu S, Dong C, Fu K, Tang W, Cao H (2017) An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget 8:73271–73281
Braicu C, Tomuleasa C, Monroig P, Cucuianu A, Berindan-Neagoe I, Calin GA (2015) Exosomes as divine messengers: are they the Hermes of modern molecular oncology? Cell Death Differ 22:34–45
Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, Chen D, Gu J, He X, Huang S (2015) Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res 25:981–984
Bao C, Lyu D, Huang S (2016) Circular RNA expands its territory. Mol Cell Oncol 3:e1084443
Lasda E, Parker R (2016) Circular RNAs co-precipitate with extracellular vesicles: a possible mechanism for circRNA clearance. PLoS One 11:e0148407
Lasda E, Parker R (2014) Circular RNAs: diversity of form and function. RNA 20:1829–1842
Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D (2015) Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 5:8057
Li P, Chen S, Chen H, Mo X, Li T, Shao Y, Xiao B, Guo J (2015) Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta Int J Clin Chem 444:132–136
Kristensen LS, Hansen TB, Veno MT, Kjems J (2017) Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 37:555
Aldea MD, Petrushev B, Soritau O, Tomuleasa CI, Berindan-Neagoe I, Filip AG, Chereches G, Cenariu M, Craciun L, Tatomir C, Florian IS, Crivii CB, Kacso G (2014) Metformin plus sorafenib highly impacts temozolomide resistant glioblastoma stem-like cells. J BUON Off J Balkan Union Oncol 19:502–511
Baguley BC (2010) Multiple drug resistance mechanisms in cancer. Mol Biotechnol 46:308–316
Wang J, Seebacher N, Shi H, Kan Q, Duan Z (2017) Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 8:84559–84571
Braicu C, Mehterov N, Vladimirov B, Sarafian V, Nabavi SM, Atanasov AG, Berindan-Neagoe I (2017) Nutrigenomics in cancer: revisiting the effects of natural compounds. Semin Cancer Biol 46:84–106
He R, Liu P, Xie X, Zhou Y, Liao Q, Xiong W, Li X, Li G, Zeng Z, Tang H (2017) circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J Exp Clin Cancer Res 36:145
Kong Z, Wan X, Zhang Y, Zhang P, Zhang Y, Zhang X, Qi X, Wu H, Huang J, Li Y (2017) Androgen-responsive circular RNA circSMARCA5 is up-regulated and promotes cell proliferation in prostate cancer. Biochem Biophys Res Commun 493:1217–1223
Yang P, Qiu Z, Jiang Y, Dong L, Yang W, Gu C, Li G, Zhu Y (2016) Silencing of cZNF292 circular RNA suppresses human glioma tube formation via the Wnt/beta-catenin signaling pathway. Oncotarget 7:63449–63455
Han D, Li J, Wang H, Su X, Hou J, Gu Y, Qian C, Lin Y, Liu X, Huang M, Li N, Zhou W, Yu Y, Cao X (2017) Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology 66:1151–1164
Yang W, Du WW, Li X, Yee AJ, Yang BB (2016) Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene 35:3919–3931
Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, Iwasaki A, Chang HY (2017) Sensing self and foreign circular RNAs by intron identity. Mol Cell 67(228–238):e5
Tomuleasa C, Braicu C, Irimie A, Craciun L, Berindan-Neagoe I (2014) Nanopharmacology in translational hematology and oncology. Int J Nanomed 9:3465–3479
Chira S, Gulei D, Hajitou A, Zimta AA, Cordelier P, Berindan-Neagoe I (2017) CRISPR/Cas9: transcending the reality of genome editing. Mol Ther Nucleic Acids 7:211–222
Gulei D, Berindan-Neagoe I (2017) CRISPR/Cas9: a potential life-saving tool. What’s next? Mol Ther Nucleic Acids 9:333–336
https://clinicaltrials.gov/ct2/show/NCT03334708?cond=NCT03334708&rank=1. Accessed 18 Jan 2018
Funding
This work was supported by a POC Grant, entitled “Clinical and economical impact of personalized targeted anti-microRNA therapies in reconverting lung cancer chemoresistance”-CANTEMIR (project no. 35/01.09.2016, Cod MySMIS 103375) and by PN-III-P1-1.2-PCCDI-2017-0737 (“Genomic mapping of population from polluted area with radioactivity and heavy metals to increase national security-ARTEMIS and PN-III-P2-2.1-PED-2016-0425 (project no 178 PED).
Author information
Authors and Affiliations
Contributions
CB wrote the paper; A-AZ participated for the introduction part and for figures concept; DG assisted for the table preparation and wrote the final part related to the circulating cirRNAs as biomarkers. AO was responsible for the figure design and for the part related to data-based and programs for circRNAs application. IBN was the design of the study, final correction of the manuscript. All the authors assisted in the preparation of the manuscript and editing and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Authors have no financial and non-financial competing interests to be declared.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Braicu, C., Zimta, AA., Gulei, D. et al. Comprehensive analysis of circular RNAs in pathological states: biogenesis, cellular regulation, and therapeutic relevance. Cell. Mol. Life Sci. 76, 1559–1577 (2019). https://doi.org/10.1007/s00018-019-03016-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03016-5