
Abstract
Neurons are highly specialised cells with a large bioenergetic demand, and so require a healthy mitochondrial network to function effectively. This network is compromised in many neurological disorders, in which damaged mitochondria accumulate. Dysfunctional mitochondria can be removed via an organelle-specific autophagic pathway, a process known as mitophagy. The canonical mitophagy pathway is dependent on the actions of PINK1 (PTEN-induced putative kinase 1) and Parkin and has been well studied in immortalised cells and cultured neurons. However, evidence for a role of this mitophagy pathway in the brain is still limited, and studies suggest that there may be important differences in how neurons respond to mitochondrial damage in vitro and in vivo. Here, we first describe the evidence for a functional PINK1/Parkin mitophagy pathway in neurons, and review how this pathway is affected in disease models. We then critically evaluate the literature by comparing findings from in vitro models and more recent in vivo studies in flies and mice. The emerging picture implicates that alternative mitophagy pathways operate in neurons in vivo. New mouse models that employ fluorescent biosensors to monitor mitophagy in vivo will be instrumental to understand the relative role of the different clearance pathways in the brain under physiological and pathological conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuronal function is energetically costly and this energy demand is almost exclusively met by mitochondria. Apart from producing energy in the form of adenosine triphosphate (ATP) via oxidative phosphorylation, mitochondria also play a number of other important roles, such as buffering cytosolic Ca2+ and synthesising lipids. To carry out these functions effectively, mitochondria undergo morphological changes and localise to specific cellular compartments as required, brought about by fission and fusion [1]. Mitochondria are prone to damage as they are the main production sites of cellular reactive oxygen species (ROS), and this damage can lead to the release of toxic levels of ROS and/or the initiation of apoptosis. To prevent this, cells have evolved a number of quality control systems to recognise and repair damaged mitochondria, including mitochondrial proteases and the mitochondrial unfolded protein response [2]. However, if the damage is too severe for repair, mitochondria can even be entirely eliminated via a combination of the ubiquitin/proteasome and autophagic pathways [3]. This process of mitochondrial degradation is termed mitophagy.
The importance of mitochondrial homeostasis, including balanced fission and fusion, in neurons is highlighted by various mitochondrial diseases, which although systemic, often manifest in neurological symptoms, such as dominant optic atrophy and Charcot–Marie–Tooth disease [4, 5]. Further, mitochondrial dysfunction, including increased ROS production, altered transport and morphology, and changes in mitochondrial protein levels and activity, is a dominant feature of most neurodegenerative conditions, including Parkinson’s disease (PD), Alzheimer’s disease (AD), and Huntington’s disease (HD) [6,7,8,9,10,11]. Mutations in the two mitophagy-linked genes PARK2 (Parkin) and PARK6 (PTEN-induced putative kinase 1, PINK1) have been found to cause familial PD (but not AD or HD) [12, 13]. This intriguing connection between disturbed mitochondrial quality control and neurodegeneration in PD has spurred research in this area more generally, and altered mitophagy is starting to become implicated in a number of neurodegenerative diseases other than PD, such as AD [14], HD [15,16,17] and amyotrophic lateral sclerosis [18, 19].
Mechanisms of PINK1/Parkin mitophagy
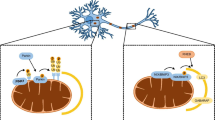
During oxidative phosphorylation, mitochondria shuttle electrons along a series of four protein complexes that form the electron transport chain located in the inner membrane. This process generates a proton gradient, and therefore, a voltage difference across the inner membrane. This membrane potential provides the energy to drive ATP synthase and generate ATP. Various mitochondrial insults, such as high ROS levels, mutations in mitochondrial genes, or chemical and environmental stress (e.g. respiratory chain inhibitors such as antimycin) can lead to a decrease in the mitochondrial membrane potential, impairing the ability of the electron transport chain to generate ATP. Such depolarisation signifies a dysfunctional mitochondrion and can trigger the mitophagy cascade [20]. Initially, the compromised membrane potential blocks mitochondrial import of the serine/threonine kinase PINK1, leading to its accumulation on the outer membrane (Fig. 1a) where it acts as a tag for damaged mitochondria [21]. In contrast, under physiological conditions, PINK1 cannot function as a tag, as it is continuously imported into mitochondria, where it is cleaved and degraded [22, 23]. However, when PINK1 accumulates, the kinase phosphorylates both basal ubiquitin on the outer membrane and the cytosolic ubiquitin ligase Parkin at serine 65 [24,25,26]. Phosphorylation of Parkin and its binding to phospho-ubiquitin constitute part of a multi-step activation process, whereby Parkin undergoes a conformational change, exposing its binding site for ubiquitin-conjugating enzymes [27, 28]. Parkin is thus both recruited onto mitochondria and activated. Activated Parkin then ubiquitinates a number of outer membrane proteins, including the voltage-dependent anion channel (VDAC) and the fusion proteins mitofusin (Mfn) 1 and 2. This action can be opposed by the deubiquitinase USP30, which is also located at the outer surface of mitochondria and there removes ubiquitin [29]. During mitophagy, however, the additional ubiquitin becomes available for phosphorylation by PINK1, thereby creating a positive feedback loop [30,31,32]. Importantly, these mitochondrial ubiquitin chains can interact with autophagy receptors, such as optineurin and nuclear dot protein 52 (NDP52), which recruit autophagosomal membranes around the mitochondria by binding to autophagosomal GABAA receptor-associated proteins (GABARAPs) [19, 33, 34]. Finally, these mito-autophagosomes are trafficked to the lysosome for elimination.

Pathways to mitophagy in neurons. a The PINK/Parkin pathway. 1. Normally, healthy mitochondria contain some basal ubiquitin moieties present on their outer membrane. 2. Following a mitochondrial insult, which decreases the inner membrane potential (ψ), import of PINK1 into mitochondria is stopped and the protein instead accumulates on the outer mitochondrial membrane, where it phosphorylates ubiquitin. 3. PINK1 phosphorylates Parkin and phospho-ubiquitin binds to Parkin, simultaneously recruiting it from the cytosol and activating it. Parkin ubiquitinates outer membrane proteins, generating a positive feedback loop. 4. Ubiquitin chains bind to autophagy receptors, such as optineurin, which in turn bind LC3 on autophagosomal membranes. Autophagosomes engulf mitochondria and are then trafficked to the lysosome for elimination. b Cardiolipin-mediated mitophagy. 1. Under normal conditions, the phospholipid cardiolipin resides in the inner membrane of mitochondria. 2. Following electron transport chain inhibition (e.g., rotenone) or treatment with other Parkinsonian toxins (e.g. 6-OHDA), cardiolipin relocates to the mitochondrial outer membrane. 3. Cardiolipin interacts with LC3, leading to the engulfment of mitochondria by autophagosomes. c GADH-mediated mitophagy. 1. Under normal conditions, GAPDH is mostly located in the cytosol. 2. Oxidative stress or mitochondrial damage caused by mutant Huntingtin expression, can lead to the association of catalytically inactive GAPDH with mitochondria. 3. Through an as yet unknown mechanism, GAPDH-decorated mitochondria can be directly taken up by lysosomes/late endosomes
Interestingly, there exists another PINK/Parkin-dependent pathway that, distinct from mitophagy, is involved in generating small mitochondria-derived vesicles. These are enriched for Syntaxin 17, allowing them to be targeted to lysosomes without requiring autophagy [35,36,37]. These vesicles form in response to mitochondrial stressors (e.g. antimycin) and contain oxidised proteins, but they are also generated under physiological conditions in cell lines [38]. Although such mitochondria-derived vesicles have not yet been described in neurons, their existence suggests an intriguing complement to mitophagy, which could improve the health of the mitochondrial network without eliminating mitochondria entirely.
Finally, a third pathway exists, as revealed in a recent study of Parkin-dependent degradation of mitochondria via the early-endosome system, in mouse embryonic fibroblasts that were unable to undergo autophagy due to the knock-out of autophagy-related proteins [39]. Instead, in response to depolarisation with the uncoupling agent FCCP, mitochondria became associated with Rab5-positive early endosomes, which then matured into Rab7-positive late endosomes, before fusing with lysosomes [39]. Again, whether this pathway is active in neurons still needs to be investigated.
PINK1/Parkin mitophagy in cultured neurons
Although the evidence regarding PINK1/Parkin mitophagy in cell lines is robust, there have been controversies about whether this pathway exists in neurons, and its physiological relevance also remains the subject of debate. Mitochondrial depolarisation induced with ionophores such as CCCP/FCCP consistently results in Parkin relocalisation in cell lines, but early studies were unable to observe translocation of both endogenous and overexpressed Parkin in primary neurons or in neurons derived from induced pluripotent stem cells (iPSCs) [40, 41]. After failing to observe Parkin translocation in primary neurons, one of these studies then forced HeLa cells to rely on mitochondrial respiration rather than glycolysis, thereby mimicking the way in which neurons generate energy, which severely compromised the cells’ ability to undergo Parkin translocation [40]. This suggested that differences in Parkin translocation between non-neuronal cell lines and neurons might be in part due to their different bioenergetic profiles [40]. However, subsequent studies revealed that cultured neurons are able to undergo Parkin translocation in response to a variety of stimuli, including ionophores, respiratory chain inhibitors, increases in ROS levels and glutamate excitotoxicity, or when modelling neurodegenerative disease [14, 42,43,44,45,46,47,48]. The observation of mitophagy in neurons is not trivial, however, as neurons are highly vulnerable to mitochondrial inhibitors, which cause cell death, and mitophagy occurs at a much lower frequency than in immortalised cells. For example, Cai et al. [44] found that incubating cortical neurons with CCCP could indeed lead to Parkin translocation; but only after at least 18 h, which is approximately 14–16 h longer than is required for most cells lines. Further, to induce Parkin translocation, the neurons had to be co-cultured with glia, and apoptosis inhibitors had to be added to the cultures. Interestingly, it was also reported that the antioxidant B27, a routine neuronal culture medium supplement, prevented Parkin translocation, but once this was removed, Parkin translocation occurred in cortical neurons after CCCP treatment or respiratory chain inhibition with rotenone or MPP + (1-methyl-4-phenylpyridinium) [46]. Another question relates to the neuronal compartment in which mitophagy occurs. Multiple studies initially noted that Parkin translocation and mitophagy were primarily observed in the somatodendritic domain [14, 29, 44]; however, a more targeted method to damage mitochondria revealed that these processes also occur in axons. This method, employed by Ashrafi et al., used the fluorophore KillerRed, which causes ROS formation upon stimulation with green light, to increase ROS levels in individual mitochondria in the axon [43]. Using this technique, as well as microfluidic chambers to specifically treat axons with the respiratory chain inhibitor antimycin, the authors elegantly demonstrated axonal Parkin translocation and mitophagy, as represented by mitochondrial colocalisation with autophagosomes and lysosomes.
Mitophagy in cellular models of Alzheimer’s disease
More recently, several studies have also examined neuronal mitophagy in the context of neurodegenerative diseases, including AD (Table 1). AD is characterised by two protein abnormalities: extracellular aggregates of the amyloid-β peptide (Aβ), generated from cleavage of the membrane-bound amyloid precursor protein (APP), and intracellular neurofibrillary tangles consisting of aggregated tau protein. In both cases, it is not only the aggregates themselves that are toxic but also their soluble precursors, such as oligomeric Aβ and hyperphosphorylated tau. Their formation can be modelled in cell culture and in vivo by overexpression of mutant forms of APP and tau found in families with neurodegenerative disease.
To investigate changes in mitophagy due to AD pathology, Ye et al. assessed Parkin levels in mitochondrial and cytosolic fractions from patient brains [14]. Mitochondrial Parkin was increased in patients compared to healthy controls, as was the autophagosome marker lipidated microtubule-associated protein 1A/1B-light chain 3 (LC3-II). Concurrently, cytosolic Parkin was depleted, consistent with an increased translocation of Parkin from this cellular compartment to mitochondria. Electron microscopy revealed increased numbers of defective mitochondria, consistent with other reports [49, 50], suggesting that these abnormal mitochondria might initiate the mitophagy pathway. However, the cells appeared to be overloaded with mitochondrial damage, indicating that mitophagy was not effectively removing damaged mitochondria. Moreover, in mouse primary neurons expressing a mutant form of human APP, with increased Aβ production, Parkin was recruited to mitochondria over time in the absence of any additional stimuli, and direct application of Aβ oligomers to wild-type neurons mimicked this effect [14]. This observation was accompanied by a decreased mitochondrial membrane potential. Together, these findings identify Aβ as a pro-mitophagic agent, which corresponds well with previous results regarding its mitochondrial toxicity, particularly the ability to depolarise mitochondria [51,52,53,54].
A study probing the mitochondrial effects of an amino-terminal fragment of tau, which was shown to be enriched in synaptosomes of AD patient brains, found that it caused mitochondrial fragmentation, Parkin translocation and mitophagy in cultured neurons [42, 55]. This was associated with synaptic loss and, eventually, cell death. Amino-terminal tau co-immunoprecipitated with both Parkin and ubiquitin-C-terminal hydrolase L1 (UCHL-1) in mitochondrial fractions from AD tissue [45]. The observation of this complex suggests that amino-terminal tau may recruit Parkin and UCHL-1 to induce mitophagy [42, 45]. Interestingly, full-length human tau (hTau) did not have these effects, illustrating the complexity of tauopathies, with tau having six isoforms, multiple cleavage products and many different post-translational modifications. In contrast, a second study, which examined the effect of full-length hTau on mitophagy, found that it actually impaired mitophagy, both in cultured neurons and in mice [56]. When hTau was expressed in vitro or virally targeted to the hippocampus in mice, the levels of mitochondrial proteins and DNA increased compared with controls [56]. The authors interpreted this accumulation as evidence of a mitophagy defect; however, the possibility of increased mitochondrial biogenesis was not ruled out. Tau was detected in the mitochondrial fractions from AAV-hTau mice and hTau-N2a cells and was shown to increase the mitochondrial membrane potential [56, 57], which would render mitochondria more resistant to PINK1 accumulation and the initiation of mitophagy. However, it was previously demonstrated that hTau-SHSY5Y cells did not exhibit changes in mitochondrial mass, despite an increased mitochondrial membrane potential, indicating that mitophagy was unlikely to be inhibited [57]. Clearly, different tau species have different effects on mitophagy and more investigation is required to better understand this aspect of tau pathology.
Although Parkin translocation can be observed in cultured neurons in response to various stimuli, several issues remain to be addressed. All of the above studies relied on overexpression of human Parkin to visualise its translocation. However, neurons contain endogenous Parkin and it is more relevant to know to the extent to which this pool translocates onto mitochondria. Thus far, only a handful of studies have been able to detect endogenous Parkin translocation, using western blotting of mitochondrial fractions whereby spatial information is lost [42, 44, 45, 48]. Furthermore, it is unclear to what extent Parkin translocation proceeds to mitophagy. Immortalised cells have been shown to lose their entire mitochondrial pool, but for neurons, this would be fatal. To our knowledge, there is only one report of basal mitophagy occurring in cultured neurons in the absence of Parkin overexpression [29]. This study used the pH-sensitive fluorophore mKeima, which exhibits a shift in its excitation/emission spectrum when it enters an acidic compartment such as the lysosome [58]. Using mitochondrially targeted mKeima, which is resistant to lysosomal proteases, the authors elegantly revealed an accumulation of mitochondria in lysosomes in cultured hippocampal neurons over a number of days, in the absence of any insult. This finding suggests that there is indeed a physiological role for mitophagy in neurons, and this warrants further exploration.
In summary, cultured neurons are clearly able to undergo PINK1/Parkin-mediated mitophagy. However, the role that this pathway plays under both physiological and pathological conditions and how relevant it is when Parkin is not overexpressed needs to be clarified; the main questions being whether neurons undergo PINK1/Parkin mitophagy in vivo and how and to what extent this pathway maintains cellular health.
Studies of neuronal mitophagy in Drosophila
Loss-of-function mutations in the genes encoding PINK1 and Parkin lead to PD, with an earlier disease onset than for sporadic cases [13, 59]. Following these discoveries, knock-out (KO) models were generated to better understand the function of these proteins and to recapitulate aspects of the human disease. Studies in Drosophila have been seminal in building our current understanding of the role of PINK1/Parkin in vivo [60,61,62,63,64,65,66,67]. In this review, however, we will only focus on studies that have addressed whether these proteins are involved in mitochondrial turnover. Burman et al. investigated mitochondria in dopaminergic neurons obtained from Parkin KO flies and found that they displayed a reduced membrane potential and increased length [60]. These results are consistent with the idea that abrogating Parkin-dependent mitophagy leads to an accumulation of depolarised mitochondria, as they can no longer be eliminated, and that Parkin can inhibit the fusion of mitochondria via degradation of Mfn 1 and 2 [68, 69]. Importantly, overexpression of the autophagy protein Atg8 rescued the mitochondrial membrane potential and neuronal loss, suggesting that impaired mitophagy was indeed contributing to the defects seen in Parkin KO flies [60]. Two recent papers extended these data by carrying out detailed analyses of mitochondrial trafficking and morphology in vivo in motor neurons of PINK1 or Parkin KO flies [63, 66]. The PINK1 KO mitochondria showed a decreased membrane potential and flux (number of mitochondria transported per minute) in axons, without changes to mitochondrial numbers [63]. A hypothesis based on the current model of PINK/Parkin mitophagy would predict greater numbers of mitochondria, as their elimination is compromised, but this was not observed. Moreover, KO of Parkin actually decreased the mitochondrial density in motor neuron axons, whereas the mitochondrial membrane potential, velocity and length remained unchanged [66], again in contrast to the prediction. This lead the authors to suggest that there might be a quality control filter at the soma/axon interface, so that only healthy (and therefore fewer) mitochondria were let through. Importantly, this study also investigated mitophagy by assessing the colocalisation with mitochondria and Atg8 in the soma and axons. There was no evidence for basal, physiological mitophagy, and starvation, a known activator of autophagy, did not lead to mitophagy either. However, although starvation has been shown to induce general autophagy, it would be more relevant to expose the fly larvae to mitochondrial-specific stress (such as respiratory chain inhibition) and then assess mitophagy. When the same cell type (motor neurons) was examined in vitro, basal mitophagy could be detected, and was significantly reduced in Parkin KOs. This important study helps to underscore the notion that mitophagy is more common and more easily observed in vitro compared to in vivo for the same type of neuron. This is not surprising, given that cultured neurons are particularly vulnerable to stress. Further, basal mitochondrial elimination in fly brains is very low, with the half-life of some mitochondrial proteins exceeding 30 days, and non-mitochondrial proteins generally displaying a much faster turnover (mostly below 12 days) [67]. Additional considerations are the relatively short life span of flies and the neuronal cell type used in these studies, as dopaminergic neurons are more vulnerable to Parkin KO than, for example, cholinergic neurons [60]—in line with PINK1/Parkin mutations causing specific loss of dopaminergic substantia nigra neurons in PD only after decades.
Strong evidence for a role of both PINK1 and Parkin in mitochondrial turnover came from Vincow et al. [67], who measured the half-lives of mitochondrial proteins in the heads of PINK1 and Parkin KO flies and compared them to those of wild-type flies. Mitochondrial protein half-lives were significantly longer in Parkin KOs and there was a selective effect of Parkin on the prolonged turnover of respiratory chain proteins. Unexpectedly, PINK1 KO flies did not display shorter mitochondrial protein half-lives in general, but there was a specific reduction in the half-lives of respiratory chain proteins. This study indicated a role for PINK1 and Parkin in protein turnover separate from general mitophagy [67].
Taken together, the above studies in Drosophila have highlighted that the roles of PINK1 and Parkin in neurons are very complex. They have provided in vivo evidence that these proteins are involved in the quality control and homeostasis of mitochondria, although the specifics of this involvement still need to be clarified. In many instances, the experimental observations have deviated from predictions based on the canonical PINK1/Parkin model. Mitophagy has also proven to be significantly more difficult to demonstrate in vivo than in vitro. Not unsurprisingly, given that dopaminergic neurons are specifically affected in PD, these cells appear more vulnerable to PINK1/Parkin loss. Such cell-type specific differences are an important consideration for any neuronal studies of this pathway, particularly as mammalian neuronal cultures are typically hippocampal or cortical. PINK1/Parkin mitophagy may be less relevant in these cells than in dopaminergic populations.
PINK/Parkin mitophagy in the mammalian nervous system
In humans, loss-of-function mutations in either PINK1 or Parkin can lead to PD. Therefore, it was surprising when the first PINK1 and Parkin KO mice were generated and did not recapitulate PD to any meaningful extent. The KO mice generally presented with mild phenotypes, no neuronal loss and few motor deficits, even at advanced ages [70,71,72,73,74]; for a summary of the KO mice published, see [75]. Although these mice do not model PD, they do provide insight into the roles of PINK1 and Parkin in the nervous system. Mitochondria have been found to be dysregulated when either protein was absent, although not usually in the way that a straightforward interpretation of the canonical PINK1-Parkin pathway would predict. To illustrate, in the midbrain of Parkin KO mice, mass spectrometry revealed decreases, rather than increases, in mitochondrial proteins [76]. Striatal mitochondria have exhibited reduced respiratory capacity in multiple KO studies, consistent with the role of Parkin in maintaining healthy mitochondria [76, 77]. However, no accumulation of mitochondria was found in either Parkin or PINK1 KOs, despite the presence of respiratory defects, thereby highlighting the fact that the roles of these proteins are more complex than simply an elimination of damaged mitochondria [77, 78]. Of note, Parkin/PINK1 KO specifically in the midbrain of adult mice was shown to cause mitochondrial defects and dopaminergic neuron loss via increases in the zinc-finger protein Parkin interacting substrate (PARIS), which represses the transcription factor PGC-1α, leading to impaired mitochondrial biogenesis rather than altered mitophagy [79,80,81].
Two interesting studies have specifically assessed aspects of PINK1/Parkin mitophagy, using a “two-hit” model for PD, whereby loss of Parkin (first “hit”) is combined with enhanced mitochondrial stress (second “hit”) [82, 83]. In both studies, a Parkin KO mouse strain was crossed with a second mouse model of mitochondrial dysfunction which was either a dopamine neuron-specific KO of the mitochondrial transcription factor A (TFAM) [82] or the mutator mouse, which expresses a proofreading-deficient polymerase γ (POLG), the mitochondrial replicase. The KO of TFAM on its own leads to mitochondrial DNA loss and respiratory chain deficiency [84]. When the TFAM KO was compared to the TFAM/Parkin double-KO, there was no change in the number of the abnormally enlarged mitochondria found in the TFAM KO at 2 months of age, when the abnormalities were already quite severe. This suggests that Parkin may not play a critical role in mitochondrial quality control under these circumstances, but a more detailed morphological and stereological analysis of the mitochondria could elucidate subtler effects. It is also possible that the mice were too young to observe the effect, since a Parkin KO is quite a mild insult and may take more time to be truly detrimental. However, at 8 months Parkin KO did not exacerbate the cell loss caused by TFAM KO alone either. When mCherry-Parkin was virally delivered to the midbrain of the TFAM KO mice, the authors were unable to observe Parkin translocation. This implies that, contrary to what had been predicted, Parkin translocation is not necessarily a response to the loss of mitochondrial DNA and a compromised respiratory chain function [82]. The trigger for PINK1 accumulation on the mitochondrial outer membrane (and subsequent Parkin recruitment) is a decrease in the membrane potential, which blocks mitochondrial protein import [22]. However, when the authors tested the mitochondrial import of the reporter protein mito-dsRed, it was unaffected. It is, therefore, reasonable to assume that the reason why Parkin translocation was not observed despite mitochondrial dysfunction is because the mitochondria were still able to maintain PINK1 import capabilities, preventing it from accumulating and initiating mitophagy. It appears that PINK1/Parkin-mediated mitophagy is a much more nuanced and specific quality control pathway than cell culture studies have suggested. How mice deal with impaired mitochondria in the absence of PINK1/Parkin mitophagy remains to be determined.
The second study to elucidate the role of Parkin in mitochondrial quality control in vivo used the mutator mouse, which accumulates mitochondrial DNA mutations [85]. Here, the double KO presented with dopamine neuron loss and motor impairment, whereas other brain regions were spared [83]. This suggests that a second “hit” is needed in Parkin KO mice to produce a PD phenotype. Further, the activity of mitochondrial complexes I and III was significantly reduced in the double KO mice. Importantly, the double KO mice had three times as much phospho-ubiquitin in the cortex than wild-type mice. Given that ubiquitin is phosphorylated by PINK1 to induce mitophagy, this provides evidence that the PINK1/Parkin mitophagy pathway is indeed activated in the brain [83].
Beyond PINK1/Parkin: alternative pathways to mitophagy
The evaluation of disease models other than mitochondrial-specific insults has also provided important evidence for mitophagy as a quality control process in the mouse brain. A mouse model of human A53T α-synuclein found in familial PD revealed large mitochondrial inclusions in α-synuclein-expressing dopaminergic neurons, which colocalised with endogenous LC3 and ubiquitin [86]. A small percentage of these inclusions were decorated with the autophagy adaptor protein p62/SQSTM1, supporting the notion that they were indeed mito-autophagosomes. Interestingly, the appearance of mito-autophagosomes preceded other neuronal defects exhibited by these mice, such as dendritic degeneration and large axonal varicosities [86], thereby placing a mitochondrial or mitophagy defect early in the aetiology of this model. To examine the role of PINK1 and Parkin in the formation of these mitochondrial inclusions, the A53T-mutant mice were crossed onto either a PINK1 or Parkin KO background [86]. Both crossings exacerbated the mitochondrial loss and led to an increase in the size and number of mito-autophagosomes. This is a fascinating observation, as it implies that PINK1 and Parkin are not required for the formation of mito-autophagosomes in this model. However, it also means that both proteins are involved in mitochondrial maintenance in the presence of A53T α-synuclein, given that the mitochondrial phenotype worsened in the absence of either protein [86]. It would have been interesting to determine whether endogenous Parkin colocalised to mitochondria in the A53T α-synuclein mouse to determine whether Parkin translocation was still involved (despite not being required) in the observed mitophagy. The outstanding question we are left with is, what is the mechanism of mitophagy in this model?
Parkin-independent mechanisms of mitophagy exist, although research in this area is still sparse. α-Synuclein interacts with the mitochondrial phospholipid cardiolipin [87], which has been shown to act as a mitophagy signal in primary cortical neurons, cell lines, primary fibroblasts, and yeast [88,89,90,91]. Cardiolipin normally resides in the inner mitochondrial membrane, but in response to CCCP and the PD toxins rotenone and 6-OHDA, it can relocate to the outer membrane. This externalisation enables direct binding of cardiolipin to LC3 and the initiation of mitophagy and is mediated by the inter-membrane space protein NDPK-D [91] (Fig. 1b). Cardiolipin has also indirectly been shown to initiate mitophagy in mouse embryonic fibroblasts, since knockdown of tafazzin, which catalyses cardiolipin remodelling (a post-synthetic modification), resulted in mitophagy impairment [89]. Importantly, in neurons, cardiolipin-mediated mitophagy was observed in response to PD toxins, but this did not involve increased PINK1 levels or Parkin translocation. Further, cardiolipin externalisation was greater in primary neurons than in HeLa cells when treated with CCCP [88]. These data suggest the intriguing possibility that in neurons cardiolipin-mediated mitophagy may be more relevant than Parkin-dependent mitophagy. However, further research is needed to advance these findings, and in particular, to establish whether this pathway is also active in vivo.
Another mitophagy mechanism, which involves glyceraldehyde 3-phosphate dehydrogenase (GAPDH)-mediated mitochondrial clearance, has recently been observed in HD models (Fig. 1c), although more studies in other disease models are needed [16]. Previously described in oxidatively stressed cardiac cells, this pathway involves the association of GAPDH with mitochondria, and the targeting of these mitochondria to lysosomes by an as yet unknown mechanism [92]. The catalytic activity of GAPDH is not required for this process, but its phosphorylation by protein kinase C∂ was shown to be a positive regulator of mitochondrial association with lysosomes-like structures. The same research group studied mitochondrial GAPDH in cellular models and a mouse model of HD [16]. In both cells and mice with poly-glutamine expansions found in HD, mitochondria were impaired and increased amounts of GAPDH were found associated with mitochondria [16]. Despite this accumulation, there was no increased mitochondrial degradation, suggesting that there may be a downstream impairment of this pathway in these models. This idea is supported by the finding that overexpression of catalytically inactive GAPDH decreased mitochondrial content (indicating mitophagy), rescued some mitochondrial defects, and improved cell viability [16]. The study did not investigate whether there were any concurrent changes in the PINK1/Parkin pathway and it will be interesting to determine whether these pathways work together in neurons, or whether GAPDH-mediated mitophagy is independently activated.
Iron chelation was able to activate mitophagy independent of PINK1/Parkin in neuroblastoma and osteosarcoma cells, as well as primary human fibroblasts [93]. Iron loss strongly induced mitophagy in fibroblasts from a PD patient with mutant Parkin, highlighting a separate mitophagy pathway in these cells. The mechanism of mitophagy in this circumstance is still unknown, although the process may be regulated by Drp-1 [94]. However, it is not known if iron loss can lead to mitophagy in neurons; rather the opposite was recently reported in mice with neuronal iron deficiency (induced by conditional KO of transferrin receptor 1), wherein abnormal mitochondria accumulated, indicative of ineffective elimination by mitophagy [95].
Together, these reports add greater complexity to mitochondrial quality control in neurons. Importantly, there is now clear evidence for mitophagy in the brain, and in neurons specifically [83, 86]. However, the role of the PINK1/Parkin pathway in vivo remains to be clarified. Further, defunct mitochondria need not necessarily be dealt with fully in the cell that contains them, as mitochondria have been found to get expelled from retinal ganglion cell axons, and taken up by astrocytes for degradation [96]. Recently, it was discovered that mitochondria in axons treated with a very low dose of antimycin were releasing syntaphilin-positive vesicles, which were transported to the endo-lysosomal system [97]. Therefore, different cell types and stimuli could affect which mitophagy pathway is being engaged. Quality control mechanisms most likely work together to maintain mitochondrial and therefore cellular, health in the brain.
New mouse models to investigate mitophagy
One reason that in vivo studies of mitophagy in the mammalian brain are lacking is the limited availability of appropriate tools. Thus far, electron microscopy has been used successfully to visualise mitochondria in autophagosomes [15, 98]; however, this method is both very time consuming and restricted by limited sampling of tissue. Other ways to detect mitophagy include co-immunostaining for mitochondria and autophagosomal markers, such as LC3 (e.g. [86]). This is faster, requires less-specialised equipment and can still provide quantitative results. However, the associations between mitochondria and some of these markers are transient and can be difficult to detect. An improvement on co-labelling strategies has been the use of mitochondrially targeted pH-sensitive fluorophores, which take advantage of the low pH of the lysosome to determine the localisation of mitochondria to this compartment. These include the fluorophore mKeima and fusion of pH-sensitive GFP to pH-insensitive RFP [58, 99]. Recently, these fluorophores have been used to generate transgenic mice. For example, there is now the mito-mKeima (“mt-Keima”) mouse available, which uses the pH-dependent excitation of mKeima to determine if mitochondria are in the cytosol (neutral pH, excitation at 458 nm) or in the lysosome (low pH, excitation at 561 nm) [100]. The resistance of mKeima to lysosomal proteases also extends the time window during which mitophagy can be captured. With this approach, the authors were able to compare basal mitophagy in a range of tissues and examine the effect of different experimental conditions (such as ageing and mutant Huntingtin expression) in vivo. Basal mitophagy was found to be relatively high in the dentate gyrus, lateral ventricle and Purkinje cells, but low in the striatum, cortex and substantia nigra, reflecting regional and cell-type differences [101]. It was also decreased in aged mice, adding age as an additional factor in the study of neuronal mitophagy. A serious limitation of this mouse model is that fresh tissue must be used to image mKeima, which restricts the possibility of co-staining with other markers (such as antibodies, which require tissue fixation) and complicates the identification of anatomical structures and neuronal populations. The mito-QC mouse, recently generated by McWilliams and et al., overcomes this limitation [102]. It uses a fusion of GFP to mCherry, targeted to the outer mitochondrial membrane. When mitochondria enter the lysosome, GFP fluorescence decreases due to its pH-sensitive nature, whereas mCherry fluorescence remains stable. GFP is also more amenable to lysosomal degradation, further eliminating its fluorescent signal. The advantages of this model are that the tissue can be fixed and the ratio of GFP to mCherry fluorescence offers a quantitative read-out of mitophagy [102, 103]. As the use of these mouse models widens and researchers start crossing them with other transgenic mice, our understanding of the role of mitophagy in the brain in physiological and pathological conditions will expand.
Conclusions and outlook
It is now evident that neurons are able to undergo mitophagy not only in vitro, but also in vivo. However, whereas the critical involvement of the PINK1/Parkin pathway in this process has been well established in culture systems and Drosophila, the situation in the mammalian brain is far more complex. Recent mouse models have allowed for the definitive observation of basal, physiological mitophagy in the healthy mouse brain and these findings will be extended as such models become more widely used. Similarly, altered mitophagy has been shown to be a feature of various neurodegenerative disease models, a field of research that is quickly gaining traction. However, the involvement of the PINK1/Parkin pathway in these instances has not yet been convincingly demonstrated, and more studies addressing this, and the role of alternative mitophagy pathways, are needed. Moving forward, new mouse models, which allow for better visualisation of mitophagy both ex vivo and in vivo, will be instrumental in advancing our understanding of the role of mitophagy in the brain.
References
Sheng Z-H, Cai Q (2012) Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13:77–93. https://doi.org/10.1038/nrn3156
Rugarli EI, Langer T (2012) Mitochondrial quality control: a matter of life and death for neurons. EMBO J 31:1336–1349
Nguyen TN, Padman BS, Lazarou M (2016) Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 26:733–744. https://doi.org/10.1016/j.tcb.2016.05.008
Delettre C, Lenaers G, Griffoin J-M et al (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26:207–210. https://doi.org/10.1038/79936
Züchner S, Mersiyanova IV, Muglia M et al (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36:449–451. https://doi.org/10.1038/ng1341
Keeney PM, Xie J, Capaldi RA, Bennett JP (2006) Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26:5256–5264. https://doi.org/10.1523/JNEUROSCI.0984-06.2006
Shirendeb U, Reddy AP, Manczak M et al (2011) Abnormal mitochondrial dynamics, mitochondrial loss and mutant Huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet 20:1438–1455. https://doi.org/10.1093/hmg/ddr024
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795. https://doi.org/10.1038/nature05292
Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20:2495–2509. https://doi.org/10.1093/hmg/ddr139
Panov AV, Gutekunst C-A, Leavitt BR et al (2002) Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 5:731–736. https://doi.org/10.1038/nn884
Wang X, Su B, Lee H et al (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103. https://doi.org/10.1523/JNEUROSCI.1357-09.2009
Matsumine H, Saito M, Shimoda-Matsubayashi S et al (1997) Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am J Hum Genet 60:588–596
Valente EM, Bentivoglio AR, Dixon PH et al (2001) Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet 68:895–900. https://doi.org/10.1086/319522
Ye X, Sun X, Starovoytov V, Cai Q (2015) Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet. https://doi.org/10.1093/hmg/ddv056
Guo X, Sun X, Hu D et al (2016) VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat Commun 7:12646. https://doi.org/10.1038/ncomms12646
Hwang S, Disatnik M-H, Mochly-Rosen D (2015) Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol Med 7:1307–1326. https://doi.org/10.15252/emmm.201505256
Khalil B, El Fissi N, Aouane A et al (2015) PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis 6:e1617. https://doi.org/10.1038/cddis.2014.581
Moore AS, Holzbaur ELF (2016) Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. PNAS 113:E3349–E3358. https://doi.org/10.1073/pnas.1523810113
Wong YC, Holzbaur ELF (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. PNAS 111:E4439–E4448. https://doi.org/10.1073/pnas.1405752111
Twig G, Elorza A, Molina AJA et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446. https://doi.org/10.1038/sj.emboj.7601963
Vives-Bauza C, Zhou C, Huang Y et al (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci 107:378–383. https://doi.org/10.1073/pnas.0911187107
Jin SM, Lazarou M, Wang C et al (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942. https://doi.org/10.1083/jcb.201008084
Takatori S, Ito G, Iwatsubo T (2008) Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett 430:13–17. https://doi.org/10.1016/j.neulet.2007.10.019
Kazlauskaite A, Kondapalli C, Gourlay R et al (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460:127–141. https://doi.org/10.1042/BJ20140334
Kim Y, Park J, Kim S et al (2008) PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun 377:975–980. https://doi.org/10.1016/j.bbrc.2008.10.104
Shiba-Fukushima K, Imai Y, Yoshida S et al (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. https://doi.org/10.1038/srep01002
Koyano F, Okatsu K, Kosako H et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. https://doi.org/10.1038/nature13392
Trempe J-F, Sauvé V, Grenier K et al (2013) Structure of Parkin reveals mechanisms for ubiquitin ligase activation. Science 340:1451–1455. https://doi.org/10.1126/science.1237908
Bingol B, Tea JS, Phu L et al (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510:370–375. https://doi.org/10.1038/nature13418
Kane LA, Lazarou M, Fogel AI et al (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205:143–153. https://doi.org/10.1083/jcb.201402104
Ordureau A, Sarraf SA, Duda DM et al (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56:360–375. https://doi.org/10.1016/j.molcel.2014.09.007
Sarraf SA, Raman M, Guarani-Pereira V et al (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376. https://doi.org/10.1038/nature12043
Heo J-M, Ordureau A, Paulo JA et al (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60:7–20. https://doi.org/10.1016/j.molcel.2015.08.016
Lazarou M, Sliter DA, Kane LA et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. https://doi.org/10.1038/nature14893
McLelland G-L, Lee SA, McBride HM, Fon EA (2016) Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol 214:275–291. https://doi.org/10.1083/jcb.201603105
McLelland G-L, Soubannier V, Chen CX et al (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33:282–295. https://doi.org/10.1002/embj.201385902
Soubannier V, McLelland G-L, Zunino R et al (2012) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22:135–141. https://doi.org/10.1016/j.cub.2011.11.057
Soubannier V, Rippstein P, Kaufman BA et al (2012) Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7:e52830. https://doi.org/10.1371/journal.pone.0052830
Hammerling BC, Najor RH, Cortez MQ et al (2017) A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun 8:14050. https://doi.org/10.1038/ncomms14050
Laar VSV, Arnold B, Cassady SJ et al (2011) Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet 20:927–940. https://doi.org/10.1093/hmg/ddq531
Rakovic A, Shurkewitsch K, Seibler P et al (2013) Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem 288:2223–2237. https://doi.org/10.1074/jbc.M112.391680
Amadoro G, Corsetti V, Florenzano F et al (2014) AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis 62:489–507. https://doi.org/10.1016/j.nbd.2013.10.018
Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206:655–670. https://doi.org/10.1083/jcb.201401070
Cai Q, Zakaria HM, Simone A, Sheng Z-H (2012) Spatial Parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 22:545–552. https://doi.org/10.1016/j.cub.2012.02.005
Corsetti V, Florenzano F, Atlante A et al (2015) NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum Mol Genet. https://doi.org/10.1093/hmg/ddv059
Joselin AP, Hewitt SJ, Callaghan SM et al (2012) ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet 21:4888–4903. https://doi.org/10.1093/hmg/dds325
Koyano F, Okatsu K, Ishigaki S et al (2013) The principal PINK1 and Parkin cellular events triggered in response to dissipation of mitochondrial membrane potential occur in primary neurons. Genes Cells 18:672–681. https://doi.org/10.1111/gtc.12066
Van Laar VS, Roy N, Liu A et al (2015) Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and in the presence of N-acetyl cysteine, mitophagy. Neurobiol Dis 74:180–193. https://doi.org/10.1016/j.nbd.2014.11.015
Balietti M, Giorgetti B, Casoli T et al (2013) Early selective vulnerability of synapses and synaptic mitochondria in the hippocampal CA1 region of the Tg2576 mouse model of Alzheimer’s disease. J Alzheimer’s Dis 34:887–896
Baloyannis SJ (2006) Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis 9:119–126
Ferreiro E, Oliveira CR, Pereira CMF (2008) The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis 30:331–342. https://doi.org/10.1016/j.nbd.2008.02.003
Paula-Lima AC, Adasme T, SanMartín C et al (2010) Amyloid β-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid Redox Signal 14:1209–1223. https://doi.org/10.1089/ars.2010.3287
Qiao H, Koya RC, Nakagawa K et al (2005) Inhibition of Alzheimer’s amyloid-β peptide-induced reduction of mitochondrial membrane potential and neurotoxicity by gelsolin. Neurobiol Aging 26:849–855. https://doi.org/10.1016/j.neurobiolaging.2004.08.003
Rhein V, Baysang G, Rao S et al (2009) Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell Mol Neurobiol 29:1063–1071. https://doi.org/10.1007/s10571-009-9398-y
Amadoro G, Corsetti V, Stringaro A et al (2010) A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimer’s Dis 21:445–470. https://doi.org/10.3233/JAD-2010-100120
Hu Y, Li X-C, Wang Z et al (2016) Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 7:17356–17368
Schulz KL, Eckert A, Rhein V et al (2012) A new link to mitochondrial impairment in tauopathies. Mol Neurobiol 46:205–216. https://doi.org/10.1007/s12035-012-8308-3
Katayama H, Kogure T, Mizushima N et al (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18:1042–1052. https://doi.org/10.1016/j.chembiol.2011.05.013
Kitada T, Asakawa S, Hattori N et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. https://doi.org/10.1038/33416
Burman JL, Yu S, Poole AC et al (2012) Analysis of neural subtypes reveals selective mitochondrial dysfunction in dopaminergic neurons from parkin mutants. PNAS 109:10438–10443. https://doi.org/10.1073/pnas.1120688109
Clark IE, Dodson MW, Jiang C et al (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166. https://doi.org/10.1038/nature04779
Deng H, Dodson MW, Huang H, Guo M (2008) The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. PNAS 105:14503–14508. https://doi.org/10.1073/pnas.0803998105
Devireddy S, Liu A, Lampe T, Hollenbeck PJ (2015) The organization of mitochondrial quality control and life cycle in the nervous system in vivo in the absence of PINK1. J Neurosci 35:9391–9401. https://doi.org/10.1523/JNEUROSCI.1198-15.2015
Park J, Lee SB, Lee S et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by Parkin. Nature 441:1157–1161. https://doi.org/10.1038/nature04788
Pesah Y, Pham T, Burgess H et al (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131:2183–2194. https://doi.org/10.1242/dev.01095
Sung H, Tandarich LC, Nguyen K, Hollenbeck PJ (2016) Compartmentalized regulation of Parkin-mediated mitochondrial quality control in the Drosophila nervous system in vivo. J Neurosci 36:7375–7391. https://doi.org/10.1523/JNEUROSCI.0633-16.2016
Vincow ES, Merrihew G, Thomas RE et al (2013) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. PNAS 110:6400–6405. https://doi.org/10.1073/pnas.1221132110
Glauser L, Sonnay S, Stafa K, Moore DJ (2011) Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem 118:636–645. https://doi.org/10.1111/j.1471-4159.2011.07318.x
Tanaka A, Cleland MM, Xu S et al (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380. https://doi.org/10.1083/jcb.201007013
von Coelln R, Thomas B, Savitt JM et al (2004) Loss of locus coeruleus neurons and reduced startle in parkin null mice. PNAS 101:10744–10749. https://doi.org/10.1073/pnas.0401297101
Goldberg MS, Fleming SM, Palacino JJ et al (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635. https://doi.org/10.1074/jbc.M308947200
Kitada T, Tong Y, Gautier CA, Shen J (2009) Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem 111:696–702. https://doi.org/10.1111/j.1471-4159.2009.06350.x
Perez FA, Palmiter RD (2005) Parkin-deficient mice are not a robust model of parkinsonism. PNAS 102:2174–2179. https://doi.org/10.1073/pnas.0409598102
Zhou H, Falkenburger BH, Schulz JB, et al (2007) Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Department of Pathology, Anatomy and Cell Biology Faculty Papers, pp 242–250
Pickrell AM, Youle RJ (2015) The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273. https://doi.org/10.1016/j.neuron.2014.12.007
Palacino JJ, Sagi D, Goldberg MS et al (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279:18614–18622. https://doi.org/10.1074/jbc.M401135200
Damiano M, Gautier CA, Bulteau A-L et al (2014) Tissue- and cell-specific mitochondrial defect in Parkin-deficient mice. PLoS One 9:e99898. https://doi.org/10.1371/journal.pone.0099898
Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. PNAS 105:11364–11369. https://doi.org/10.1073/pnas.0802076105
Lee Y, Stevens DA, Kang S-U et al (2017) PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Reports 18:918–932. https://doi.org/10.1016/j.celrep.2016.12.090
Shin J-H, Ko HS, Kang H et al (2011) PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 144:689–702. https://doi.org/10.1016/j.cell.2011.02.010
Stevens DA, Lee Y, Kang HC et al (2015) Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. PNAS 112:11696–11701. https://doi.org/10.1073/pnas.1500624112
Sterky FH, Lee S, Wibom R et al (2011) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. PNAS 108:12937–12942. https://doi.org/10.1073/pnas.1103295108
Pickrell AM, Huang C-H, Kennedy SR et al (2015) Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87:371–381. https://doi.org/10.1016/j.neuron.2015.06.034
Larsson N-G, Wang J, Wilhelmsson H et al (1998) Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 18:231–236. https://doi.org/10.1038/ng0398-231
Kujoth GC, Hiona A, Pugh TD et al (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481–484. https://doi.org/10.1126/science.1112125
Chen L, Xie Z, Turkson S, Zhuang X (2015) A53T human α-Synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci 35:890–905. https://doi.org/10.1523/JNEUROSCI.0089-14.2015
Ghio S, Kamp F, Cauchi R et al (2016) Interaction of α-synuclein with biomembranes in Parkinson’s disease—role of cardiolipin. Prog Lipid Res 61:73–82. https://doi.org/10.1016/j.plipres.2015.10.005
Chu CT, Ji J, Dagda RK et al (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15:1197–1205. https://doi.org/10.1038/ncb2837
Hsu P, Liu X, Zhang J et al (2015) Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11:643–652. https://doi.org/10.1080/15548627.2015.1023984
Shen Z, Li Y, Gasparski AN et al (2017) Cardiolipin regulates mitophagy through the protein kinase C pathway. J Biol Chem 292:2916–2923. https://doi.org/10.1074/jbc.M116.753574
Kagan VE, Jiang J, Huang Z et al (2016) NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ 23:1140–1151. https://doi.org/10.1038/cdd.2015.160
Yogalingam G, Hwang S, Ferreira JCB, Mochly-Rosen D (2013) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cδ (PKCδ) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem 288:18947–18960. https://doi.org/10.1074/jbc.M113.466870
Allen GFG, Toth R, James J, Ganley IG (2013) Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep 14:1127–1135. https://doi.org/10.1038/embor.2013.168
Kageyama Y, Hoshijima M, Seo K et al (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33:2798–2813. https://doi.org/10.15252/embj.201488658
Matak P, Matak A, Moustafa S et al (2016) Disrupted iron homeostasis causes dopaminergic neurodegeneration in mice. PNAS 113:3428–3435. https://doi.org/10.1073/pnas.1519473113
Davis C-HO, Kim K-Y, Bushong EA et al (2014) Transcellular degradation of axonal mitochondria. Proceed Natl Acad Sci. https://doi.org/10.1073/pnas.1404651111
Lin M-Y, Cheng X-T, Tammineni P et al (2017) Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94(595–610):e6. https://doi.org/10.1016/j.neuron.2017.04.004
Cummins N, Bartlett CA, Archer M et al (2013) Changes to mitochondrial ultrastructure in optic nerve vulnerable to secondary degeneration in vivo are limited by irradiation at 670 nm. BMC Neurosci 14:98. https://doi.org/10.1186/1471-2202-14-98
Rosado C, Mijaljica D, Hatzinisiriou I et al (2008) Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy 4:205–213. https://doi.org/10.4161/auto.5331
Sun N, Malide D, Liu J et al (2017) A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat Protoc 12:1576–1587. https://doi.org/10.1038/nprot.2017.060
Sun N, Yun J, Liu J et al (2015) Measuring in vivo mitophagy. Mol Cell 60:685–696. https://doi.org/10.1016/j.molcel.2015.10.009
McWilliams TG, Prescott AR, Allen GFG et al (2016) mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214:333–345. https://doi.org/10.1083/jcb.201603039
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521:525–528. https://doi.org/10.1038/nature14300
Hsieh C-H, Shaltouki A, Gonzalez AE et al (2016) Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19:709–724. https://doi.org/10.1016/j.stem.2016.08.002
Seibler P, Graziotto J, Jeong H et al (2011) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31:5970–5976. https://doi.org/10.1523/JNEUROSCI.4441-10.2011
Geisler S, Holmström KM, Treis A et al (2010) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6:871–878. https://doi.org/10.4161/auto.6.7.13286
Hirai K, Aliev G, Nunomura A et al (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21:3017–3023
Martín-Maestro P, Gargini R, Perry G et al (2016) PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum Mol Genet 25:792–806. https://doi.org/10.1093/hmg/ddv616
Goiran T, Duplan E, Chami M et al (2017) β-Amyloid precursor protein intracellular domain controls mitochondrial function by modulating phosphatase and tensin homolog-induced kinase 1 transcription in cells and in Alzheimer mice models. Biol Psychiat. https://doi.org/10.1016/j.biopsych.2017.04.011
Acknowledgements
The authors’ work is supported by the Estate of Dr Clem Jones AO and by Grants from the Australian Research Council (DP130101932), and the National Health and Medical Research Council of Australia (GNT1037746, GNT1003150). N. C. is supported by an Australian Government Research Training Program Scholarship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cummins, N., Götz, J. Shedding light on mitophagy in neurons: what is the evidence for PINK1/Parkin mitophagy in vivo?. Cell. Mol. Life Sci. 75, 1151–1162 (2018). https://doi.org/10.1007/s00018-017-2692-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2692-9