
Abstract
Neurons are specialized cells with complex and extended architecture and high energy requirements. Energy in the form of adenosine triphosphate, produced essentially by mitochondrial respiration, is necessary to preserve neuronal morphology, maintain resting potential, fire action potentials, and ensure neurotransmission. Pools of functional mitochondria are required in all neuronal compartments, including cell body and dendrites, nodes of Ranvier, growth cones, axons, and synapses. The mechanisms by which old or damaged mitochondria are removed and replaced in neurons remain to be fully understood. Mitophagy has gained considerable interest since the discovery of familial forms of Parkinson’s disease caused by dysfunction of PINK1 and Parkin, two multifunctional proteins cooperating in the regulation of this process. Over the past 10 years, the molecular mechanisms by which PINK1 and Parkin jointly promote the degradation of defective mitochondria by autophagy have been dissected. However, our understanding of the relevance of mitophagy to mitochondrial homeostasis in neurons remains poor. Insight has been recently gained thanks to the development of fluorescent reporter systems for tracking mitochondria in the acidic compartment of the lysosome. Using these tools, mitophagy events have been visualized in primary neurons in culture and in vivo, under basal conditions and in response to toxic insults. Despite these advances, whether PINK1 and Parkin play a major role in promoting neuronal mitophagy under physiological conditions in adult animals and during aging remains a matter of debate. Future studies will have to clarify in how far dysfunction of neuronal mitophagy is central to the pathophysiology of Parkinson’s disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mitophagy is a form of cargo-selective autophagy removing unnecessary mitochondria, or clearing aged or dysfunctional mitochondria to protect the cells from their deleterious effects. More than 50 years ago, electron microscopy images revealed the presence of altered mitochondrial profiles in atypically enlarged lysosomal vesicles in the liver and kidney, under pathological conditions (Ashford and Porter 1962; De Duve and Wattiaux 1966; Novikoff 1959; Novikoff and Essner 1962). This phenomenon was interpreted as progressive mitochondrial degeneration (Novikoff and Essner 1962). However, the term mitophagy was coined more recently with the emergence of evidence, initially from yeast, indicating that the autophagy of mitochondria does not occur at random, but rather reflects a selective program (Kissova et al. 2004; Lemasters 2005). The term was then used to describe the autophagy-dependent degradation of mitochondria in cultured hepatocytes, following nutrient deprivation or laser-induced photodamage (Kim et al. 2007; Rodriguez-Enriquez et al. 2006). An earlier precursor study in cultured primary sympathetic neurons reported complete removal of mitochondria under conditions of apoptosis induction in the presence of caspase inhibitors, presumably associated with loss of mitochondrial membrane potential and cytochrome c release (Tolkovsky et al. 2002). These studies highlighted a role for mitophagy in the regulation of mitochondrial degradation under conditions of metabolic remodeling or severe mitochondrial damage. Other types of programmed mitophagy were recognized to be central to specific developmental programs: during the maturation of erythrocytes, whereby the cells get rid of their mitochondria to acquire the ability to transport oxygen (Sandoval et al. 2008; Schweers et al. 2007); following fertilization of the oocyte, a stage marked by the removal of paternal mitochondria, underlying the universal principle of maternal mitochondrial inheritance in animals (Al Rawi et al. 2011; Sato and Sato 2011); during cardiomyocyte maturation in the neonatal period, whereby fetal mitochondria are replaced by adult mitochondria to sustain the transition from carbohydrates to fatty acid metabolism (Gong et al. 2015); or during the differentiation of myoblasts into mature myotubes, during which a distinct network of mitochondria replaces the old population to support the metabolic switch from glycolysis to oxidative phosphorylation (Sin et al. 2016).
It was not until 2008 that mitophagy emerged as a mitochondrial quality control mechanism of potential relevance to neuronal cells, with the groundbreaking discovery of the involvement of the RING-IBR-RING E3 ubiquitin ligase Parkin in the clearance of severely damaged mitochondria (Narendra et al. 2008). Parkin is encoded by the PARK2 gene, which carries loss-of-function mutations in autosomal recessive forms of Parkinson’s disease (PD), a common most often sporadic movement disorder caused by the progressive degeneration of the dopaminergic (DA) neurons in a region of the midbrain termed substantia nigra pars compacta (SNc). Why these neurons die in PD remains a mystery. However, mitochondrial dysfunction had been suspected to play a role since the 1980s, when the mitochondrial neurotoxin MPTP was identified as the cause of Parkinsonism in young people consuming drugs (Corti et al. 2011; Exner et al. 2012; Langston and Ballard 1983; Schapira and Gegg 2011). The discovery of Parkin-dependent mitophagy provided a cellular mechanism leading to mitochondrial dysfunction in PD. The role of this mechanism was strengthened a few years later, when work from several laboratories demonstrated that the product of another gene responsible of autosomal recessive PD, the mitochondrial serine/threonine kinase PINK1, was central to the activation of Parkin and its mitochondrial recruitment during mitophagy (Geisler et al. 2010; Matsuda et al. 2010; Narendra et al. 2010). Thanks to an unprecedented collective effort of the scientific community in the field during the past 10 years, enlightened by recent descriptions of the crystal structure of Parkin (Byrd and Weissman 2013; Caulfield et al. 2015; Gladkova et al. 2018; Kumar et al. 2017a; Riley et al. 2013; Sauve et al. 2018; Seirafi et al. 2015; Spratt et al. 2014; Trempe et al. 2013; Wauer and Komander 2013) and PINK1 (Kumar et al. 2017b; Okatsu et al. 2018; Schubert et al. 2017), we have gained a comprehensive view of the molecular mechanisms underlying this specific mitophagy program, which have been summarized in excellent recent reviews (McWilliams and Muqit 2017; Truban et al. 2017). Although other mitophagy programs have been studied at the molecular level (Chu 2018; McWilliams and Muqit 2017), PINK1/Parkin-dependent mitochondrial clearance is undoubtedly the best characterized. Comparatively, however, our understanding of the relevance of PINK1/Parkin-dependent mitophagy to mitochondrial homeostasis in neurons, particularly in those that degenerate in PD, remains poor (Jang et al. 2018; Palikaras et al. 2018).
PINK1/Parkin-Dependent Mitophagy in Brief
Our current understanding of PINK1/Parkin-dependent mitophagy is that it is a program activated by specific stress-related stimuli. Its activation depends on mitochondrial protein import arrest, caused either by toxins leading to mitochondrial depolarization, such as the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Geisler et al. 2010; Matsuda et al. 2010; Narendra et al. 2008; Narendra et al. 2010), by dysfunction of protein import components or proteases involved in PINK1 processing (Bertolin et al. 2013; Greene et al. 2012; Jin et al. 2010), or by unfolded protein stress in the mitochondrial matrix (Fiesel et al. 2017; Jin and Youle 2013). These conditions interfere with the translocation of PINK1 into the organelle; leading to its accumulation in proximity of the translocase of outer mitochondrial membrane (Bertolin et al. 2013; Hasson et al. 2013; Lazarou et al. 2012; Okatsu et al. 2012; Okatsu et al. 2013); its dimerization and autophosphorylation; the phosphorylation of ubiquitin moieties associated with the outer mitochondrial membrane (Koyano et al. 2014; Kane et al. 2014; Kazlauskaite et al. 2014; Matsuda 2016); the recruitment and activation of the otherwise inactive Parkin ligase (Riley et al. 2013 Trempe et al. 2013; Wauer and Komander 2013), by binding to phospho-ubiquitin; and its stabilization in an active state through the PINK1-dependent phosphorylation of its N-terminal ubiquitin-like domain (Byrd and Weissman 2013; Caulfield et al. 2015; Chaugule et al. 2011; Gladkova et al. 2018; Kazlauskaite and Muqit 2015; Kondapalli et al. 2012; Ordureau et al. 2015; Ordureau et al. 2014; Sauve et al. 2018; Shiba-Fukushima et al. 2012; Spratt et al. 2014; Yamano et al. 2015). As a consequence, Parkin ubiquitylates a number of proteins of the outer mitochondrial membrane by forming different types of ubiquitin chains, which are further phosphorylated by PINK1, recruiting additional Parkin molecules, and thereby feeding an amplification loop (Okatsu et al. 2015; Ordureau et al. 2015; Ordureau et al. 2014; Sarraf et al. 2013). This process is negatively regulated by specific deubiquitylases (Bingol et al. 2014; Cornelissen et al. 2014; Durcan et al. 2014). The ubiquitylation of mitochondrial proteins leads to the recruitment of specific ubiquitin-binding autophagy receptors, including NDP52 and optineurin, which in turn promotes association of upstream autophagy-related proteins, thereby priming mitochondria for autophagy (Heo et al. 2015; Itakura et al. 2012; Lazarou et al. 2015; Wei et al. 2017; Yamano et al. 2018). Evidence has been provided that mitophagy is initiated in proximity of contact sites between mitochondria and the endoplasmic reticulum (ER) (Gautier et al. 2016; Gelmetti et al. 2017; Yang and Yang 2013) and that it requires dissociation between the two organelles to proceed efficiently (McLelland et al. 2018). This is achieved by a rapid burst of ubiquitylation of the Parkin substrate Mitofusin 2 (Mfn2), localized on both outer mitochondrial and ER membranes, which in turn leads to disassembly of Mfn2 complexes and destruction of the mitochondria-ER contact (McLelland et al. 2018).
Neurons: Cells on a Tight Energy Budget
The human brain does not exceed 2% of the body’s weight but its energy budget represents 20% of that of the body (Harris et al. 2012; Mink et al. 1981). While neural activity drives ATP synthesis by both glycolysis and oxidative phosphorylation, most of the brain ATP is generated by oxidative phosphorylation (Hall et al. 2012; Rangaraju et al. 2014). This makes brain cells highly reliant on mitochondria. Neurons have a uniquely complex cellular architecture intimately linked to their role in information processing and transmission. They are composed of a cell body containing the nucleus and enriched in other essential organelles, including the Golgi apparatus, the endoplasmic reticulum, lysosomes, and mitochondria. A system of highly branched dendrites that emerge (or "emerging") from the cell body is adapted to receive information from other neurons, and a single extension of variable length, the axon, is responsible for transmitting the electrical signal. The axon ramifies into several terminals that pass the signal to the dendrites of other neurons through the specialized compartment of the synapse. In recent years, thanks to the development of techniques for single neurons tracing and 3-dimensional reconstruction of neuronal morphology, it has been calculated that individual neurons can generate axons of tens of centimeters in length in the rodent brain (Matsuda et al. 2009; Wu et al. 2014). Extrapolations based on these studies and on available morphometric data predict that, in the human brain, such axons reach lengths of up to tens of meters (Bolam and Pissadaki 2012; Matsuda et al. 2009). DA neurons of the SNc, in particular, have an unmyelinated, extremely large, and highly branched axonal arbor. In humans, the average total length of the axonal arbor of a single DA neuron of the SNc has been estimated to exceed 4.5 m (Bolam and Pissadaki 2012). Such an axon can generate up to 2.4 million synapses in the projection region of the striatum.
Neurons require a lot of energy to preserve their architecture, maintain the resting potential, fire action potentials, and ensure neurotransmission in the pre- and post-synaptic compartments (Attwell and Laughlin 2001). Based on a computational model for DA neurons, integrating their morphological complexity and electrophysiological properties, it has been calculated that the energy cost associated with axon potential propagation and recovery of resting membrane potential increases exponentially with the number of levels of branches of the axon, and according to a power law of the axonal surface area and number of branch points (Pissadaki and Bolam 2013). The disproportionately higher energy demand associated with the large and highly branched axonal arbor of the SNc DA neurons compared with that of smaller axons, such as those of the DA neurons in the less susceptible ventral tegmental area (VTA), may well be one of the determinants of the special vulnerability of these neurons in PD. In a recent study, it was indeed shown that cultured mouse SNc DA neuron displayed larger axonal arbors than cultured DA neurons from the VTA of the olfactory bulb (Pacelli et al. 2015). This property correlated with higher density of mitochondria specifically in the axonal compartment, higher oxygen consumption rates, higher levels of reactive oxygen species, and greater sensitivity to mitochondrial neurotoxins. Strikingly, treatment of the SNc DA neurons with Semaphorin 7A, a guidance molecule known to modulate axonal arborization in these neurons, reduced the size of the axonal arbor and at the same time diminished oxygen consumption rates and neuronal vulnerability to toxins, highlighting the intimate relationship between the complex morphological architecture of these neurons and their energy requirement and special sensitivity.
Mitochondrial Homeostasis in Neurons: How Are Mitochondria Rejuvenated?
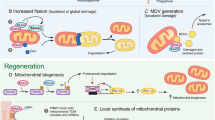
In contrast to other cell types in the body, neurons do not divide and are therefore destined to last a lifetime. Mitochondria are required in different neuronal compartments and have been shown to accumulate at nodes of Ranvier, in growth cones, axonal branches, and synapses (Hollenbeck and Saxton 2005). How do neurons manage to maintain appropriate pools of healthy mitochondria throughout their complex architecture and the lifespan of the organism? A combination of functionally intertwined mechanisms that are active in other cell types are also at play here (Misgeld and Schwarz 2017; Rugarli and Langer 2012; Shutt and McBride 2013). These include (1) transport from the cell soma to remote peripheral sites in the dendritic arbor, axon, and axonal terminals; (2) fusion and fission cycles that shape the morphology and size of the organelle to adapt them to metabolic demand, transport, and clearance; (3) pathways for the degradation of individual mitochondrial proteins, portions of mitochondria, or whole organelle; and (4) biogenesis of individual components and whole organelles. Studies in cultured neurons and in various in vivo models, including C. elegans, Drosophila, zebrafish, and mouse, have demonstrated that although most mitochondria are stationary, a fraction of about 10–40% moves along axons at any given time (Misgeld and Schwarz 2017). To enter the axon, a mitochondrion has to be separated from the network (Berthet et al. 2014; Verstreken et al. 2005) and in the axon, it can move in both anterograde and retrograde directions. However, the proportion of mitochondria moving from the soma to the peripheral arbor is higher than that transported back from the periphery to the cell body (Misgeld et al. 2007; Pilling et al. 2006; Plucinska et al. 2012; Wang et al. 2011), which lends support to the idea that that “old” stationary mitochondria are, at least in part, turned over at axonal terminals.
Calculations of protein turnover rates based on isotopic labeling at the organism level suggest that mitochondria have an average lifetime of several days, a duration that appears to be conserved during evolution and longer in the brain than in other organs (Price et al. 2010; Vincow et al. 2013). The “rejuvenation” of mitochondria through the replacement of individual components or whole organelles, particularly at distal sites, is probably operated by several parallel pathways. It is admitted that mitochondrial biogenesis is not restricted to the neuronal cell body (Harbauer 2017). Transcripts for nucleus-encoded mitochondrial proteins are present in axons (Aschrafi et al. 2016; Shigeoka et al. 2016), which are endowed with the protein synthesis machinery (Koenig and Giuditta 1999), and there is evidence for replication of mitochondrial DNA (mtDNA) in axons (Amiri and Hollenbeck 2008). Moreover, the smooth endoplasmic reticulum populates even the most remote neuronal compartments, including the thin branches of the axon terminals, often interacting closely with mitochondria, providing the capacity for local membrane lipid biosynthesis (Berridge 1998; Wu et al. 2017). In principle, mitochondrial protein synthesis and mitochondrial biogenesis may thus occur in the neuronal periphery, although the extent to which this is indeed the case has yet to be fully appreciated (Saxton and Hollenbeck 2012). Fusion with younger mitochondria is another key mechanism by which mitochondrial competence is preserved in neurons, through the redistribution of essential mitochondrial constituents and the dilution of damaged components. The relevance of this process to neurons is illustrated by the consequence of mutations in genes encoding components of the mitochondrial fusion machinery, which cause neurodegenerative diseases in humans (Chen and Chan 2010). Finally, new mitochondria may be supplied in neurons by non-canonical pathways, such as through the transfer of mitochondria from astrocytes (Hayakawa et al. 2016), although the extent to which this occurs awaits further experimental corroboration (Berridge et al. 2016).
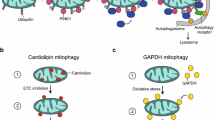
How are “old” or damaged mitochondrial components or whole organelles degraded in neurons? Several complementary pathways for the turnover of mitochondrial components have been identified, and again, their importance to neuronal homeostasis is underscored by the neurodegenerative diseases engendered by their dysfunction linked to genetic mutations (Misgeld and Schwarz 2017; Rugarli and Langer 2012). These involve the removal of individual proteins by mitochondrial proteases or by the cytosolic proteasome, the vesicular delivery of mitochondrial cargo to the lysosome through mitochondria-derived vesicles (MDVs), intraneuronal mitophagy, and transcellular mitophagy (Table 1). The relevance of each of these pathways, the physiological circumstances in which they are solicited and the specific subcellular compartments in which they take place, remains to be fully understood. Notably, in addition to its role in mitophagy, the PINK1/Parkin pathway has also been involved in the regulation of the degradation of outer mitochondrial membrane proteins by the ubiquitin-proteasome pathway (Tanaka et al. 2010, Karbowski and Youle 2011) and in the delivery of selected damaged mitochondrial components to the lysosome by MDVs (McLelland et al. 2014; McLelland et al. 2016; Sugiura et al. 2014) (Table 1).
Mitophagy in Cultured Neurons: Evidence for or Against
Most studies investigated mitophagy in immortalized cell lines, following treatment with chemical uncouplers or inhibitors of the mitochondrial respiratory chain. The question of whether this process is relevant to primary cells, particularly to neurons, under physiological conditions, has been highly debated (Grenier et al. 2013). Relatively, quickly after the discovery of Parkin-dependent mitophagy, studies in different primary neuronal models, including DA neurons differentiated from induced pluripotent stem cells (iPSCs), provided evidence for mitochondrial translocation of exogenously expressed Parkin (Cai et al. 2012; Joselin et al. 2012; Seibler et al. 2011; Wang et al. 2011) in cells treated with various mitochondrial toxins. As in other cell types, recruitment of Parkin to mitochondria was dependent on the presence of PINK1 (Seibler et al. 2011). Very recently, it was also shown that exposure to mitochondrial uncouplers stabilizes endogenous PINK1 (Oh et al. 2017; Soutar et al. 2018) and activates endogenous Parkin in primary rodent and human neurons, according to the same molecular mechanisms identified in cell lines overexpressing Parkin (Barini et al. 2018; Oh et al. 2017; Ordureau et al. 2018; McWilliams et al. 2018a).
Some studies provided direct evidence for mitophagic events in neurons. By co-expressing fluorescent proteins targeted to autophagic vesicles (GFP-LC3) or lysosomes (GFP-LAMP1), it was shown that autophagosomes are co-recruited to Parkin-positive mitochondria and that mitochondria are engulfed by lysosomal vesicles in primary mouse cortical neurons treated with the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Cai et al. 2012). These authors also showed accumulation of depolarized mitochondria in cells silenced for endogenous Parkin, strongly supporting the involvement of mitophagy in mitochondrial maintenance in neurons subjected to acute mitochondrial stress. At the same time, however, other researchers reported absence of Parkin translocation to mitochondria or recruitment of autophagic vesicles in primary rat cortical neurons exposed to a mitochondrial uncoupler, using similar approaches (Van Laar et al. 2011). These authors proposed that the bioenergetic properties of neurons, their strong reliance on oxidative phosphorylation and reluctance to switch to glycolytic metabolism under mitochondrial stress, preclude the occurrence of mitophagy. Consistent with this possibility, mitochondrial depolarization triggered by the potassium ionophore valinomycin did not lead to appreciable loss of mitochondrial markers in human iPSc-derived neurons, even following overexpression of Parkin (Rakovic et al. 2013). However, reductions in mtDNA copy number were reported under these conditions, a phenomenon that was not observed in neurons from patients with PINK1 mutations (Seibler et al. 2011). Others have confirmed that mitochondrial proteins are also cleared to a certain extent in human iPSc-derived neurons treated with CCCP (Soutar et al. 2018). Careful inspection of the culture conditions across the different studies (Grenier et al. 2013) revealed that authors reporting massive mitochondrial Parkin translocation or signs of mitophagy in neurons had supplemented the medium with apoptosis inhibitors (Cai et al. 2012) or antioxidants (Joselin et al. 2012). Thus, massive neuronal death may mask the occurrence of mitophagy, which is in line with the recent discovery that Parkin not only promotes mitophagy but also sensitized towards apoptosis under conditions of acute mitochondrial stress (Carroll et al. 2014; Zhang et al. 2014).
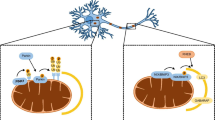
Global depolarization of the mitochondrial network is unlikely to occur under physiological conditions. In such conditions, mitophagy would rather occur as a local process removing depolarized mitochondrial fragments. To mimic such a situation, some researchers used the mitochondrion-targeted red fluorescent protein mt-KillerRed to photosensitize small subsets of mitochondria in the axonal compartment of rat hippocampal neurons cultured in microfluidic devices (Ashrafi et al. 2014). Using time-lapse video microscopy, they observed local recruitment of GFP-LC3-positive autophagic vesicles and LAMP1-YFP-positive lysosomes to damaged mitochondria as early as 20 min following irradiation. In some cases, they also reported disappearance of single mitochondria from these structures. These events were observed in the absence of antioxidants in the culture medium and occurred to a much lesser extent in neurons from Parkin- or PINK1-deficient mice, demonstrating reliance on the PINK1/Parkin pathway. This study demonstrated that mitophagy occurs locally in the distal axonal compartment. Similar observations were made more recently by Hsieh and colleagues in iPSC-derived neurons exposed to the mitochondrial complex III inhibitor antimycin A (Hsieh et al. 2016). These authors also showed a delay in axonal mitophagy in neurons from PD patients with mutations in LRRK2 or from sporadic PD patients. This contrasts with the conclusion drawn by Cai and colleagues, who observed accumulation of Parkin-positive mitochondria and associated autophagosomes essentially in the somatodendritic compartment of neurons treated with CCCP for 24 h (Cai et al. 2012). Based on these observations, it was suggested that autophagic vesicles containing damaged mitochondria are transported back to the soma to be degraded by cytoplasmic lysosomes. This later response may however reflect a compensatory mechanism due to the overwhelming of the axonal lysosomes.
All studies mentioned above investigated mitophagy under conditions of acute and severe mitochondrial stress, but what about neuronal mitophagy under conditions of mild mitochondrial stress, which is more relevant to the chronic progressive mitochondrial dysfunction that characterizes neurodegenerative diseases? Lin and colleagues (Lin et al. 2017) recently proposed that mitophagy may not be the main mechanism by which defective mitochondria are removed from axons under conditions of reversible mitochondrial depolarization triggered by low doses of antimycin A. In this case, damaged mitochondria were removed from axons by retrograde transport to the soma, a process that was enhanced by the release from mitochondria of the axonal mitochondrial-anchoring protein syntaphilin. Finally, basal mitophagy was explored in cultured hippocampal neurons, using the ratiometric mitochondrion-targeted pH-sensitive biosensor mt-Keima, enabling differentiation between mitochondria in the cytoplasm and mitochondria in the acidic microenvironment of the lysosome (Bingol et al. 2014). This study showed progressive accumulation of the biosensor in lysosomes, mostly in the neuronal soma, consistent with ongoing mitophagy in the absence of Parkin overexpression or toxins. The delivery of mitochondria to lysosomes was reduced upon silencing of PINK1 or Parkin using small hairpin RNAs, suggesting that these proteins are required for basal mitophagy, in addition to stress-induced mitophagy. However, this view has been challenged by recent in vivo studies, as will be discussed in the next section.
In Vivo Mitophagy in the Nervous System: A Controversial Issue
Early attempts to determine the turnover rates of mitochondria in different tissues in the 1960s were based on the in vivo radiolabeling of protein and lipid components in rats. Although some of these reports investigated the turnover of different categories of mitochondrial proteins, e.g., water-soluble, water-insoluble, structural, and contractile, and/or included the analysis of cytochrome c (Fletcher and Sanadi 1961; Beattie et al. 1967), in general, these studies did not take into account the half-lives of individual mitochondrial proteins. Based on the observation that insoluble and soluble protein, lipid, and cytochrome c from rat liver mitochondria turned over at near identical rates, Fletcher and Sanadi proposed for the first time that mitochondria are synthesized and broken down as discrete entities (Fletcher and Sanadi 1961). Later reports of some heterogeneity in the turnover of mitochondrial components in different tissues, including the brain, lend support to the possibility that individual mitochondrial components may be degraded independently, but did not fundamentally challenge the idea that a mitochondrial structural unit is turned over as an entity (Beattie et al. 1967; Cuzner et al. 1966; Gross and Rabinowitz 1968). Together with first observations of mitochondria inside lysosomes by electron microscopy (Ashford and Porter 1962; De Duve and Wattiaux 1966; Novikoff 1959; Novikoff and Essner 1962; Swift and Hruban 1964), these pioneering studies anticipated the discovery of mitophagy and strongly supported the existence of a mechanism for the degradation of mitochondrial entities in vivo. This issue was reinvestigated more recently in Drosophila melanogaster, in a proteomic assay based on the feeding of adult flies with deuterated leucine and the use of mass spectrometry analyses to monitor simultaneously the half-lives of numerous mitochondrial and non-mitochondrial proteins (Vincow et al. 2013). Parallel studies in wild-type, parkin null, and autophagy-deficient Atg7 null flies showed prolonged half-lives for nearly 150 mitochondrial proteins in both mutant strains and significant correlation between the effects of Atg7 and parkin mutations, specifically for this set of proteins, and not for proteins targeted to other organelles known to be degraded by autophagy. This study provided the first evidence for a role of parkin in the regulation of mitophagy in vivo. Intriguingly, the turnover of a subset of 40 mitochondrial proteins, including 19 subunits of respiratory chain components representative of the five respiratory complexes, appeared to depend more on Parkin than on Atg7, and there was strong correlation between the effect of parkin mutation and the effect of pink1 mutation on the turnover of these components (Vincow et al. 2013). This suggested the existence of a degradation process dependent on PINK1 and Parkin but independent of autophagy, possibly involving the MDV pathway (Vincow et al. 2013). On the other hand, no change in mitochondrial density was observed in Drosophila larval motor neuron axons or cell bodies, as would have been predicted by the reported role of the PINK1/Parkin pathway in mitophagy (Devireddy et al. 2015). Abnormally large, discrete mitochondria did, however, accumulate in the neuronal cell bodies, suggesting that mitochondrial degradation may be limited to this neuronal compartment in the nervous system in vivo.
Several groups made efforts towards clarifying the relevance of PINK1/Parkin-dependent mitophagy to dopaminergic neurons in mouse models, specifically in the context of accelerated accumulation of mtDNA defects (Pickrell et al. 2015; Pinto et al. 2018; Song et al. 2017; Sterky et al. 2011). The rationale for these studies came from the observation that nigral DA neurons in humans are more prone to accumulate mtDNA deletions than neurons in other brain regions (Bender et al. 2006; Kraytsberg et al. 2006). Such defects are compensated for by an increase in the number of normal mtDNA copies in neurologically healthy individuals but not in PD patients (Dolle et al. 2016), possibly due to alterations in mitochondrial biogenesis (Grunewald et al. 2016). It has thus been suggested that PINK1/Parkin-dependent mitophagy may play a key role in counterselecting mitochondria with high mtDNA mutational loads.
To address the relevance of this mechanism in vivo, Parkin-knockout mice were crossed with different transgenic mice modeling the accumulation of mtDNA deletions or mutations: the MitoPark mouse, with SN DA neuron-specific knockout of mitochondrial transcription factor A, an integral component of the basal mitochondrial transcription machinery with an additional role in the regulation of mtDNA copy number (Sterky et al. 2011); the mutator mouse, homozygous for a mutation affecting the proofreading activity of DNA polymerase γ, responsible for mtDNA replication (Pickrell et al. 2015); a mouse model expressing a mutant version of the Twinkle helicase, involved in mtDNA replication, specifically in SN DA neurons (Song et al. 2017); and the PD-mito-Pst I mouse, in which a mitochondrion-targeted version of the restriction enzyme PstI is selectively expressed in SN DA neurons (Pinto et al. 2018). While the MitoPark, mutant Twinkle, and PD-mito-Pst I models developed age-dependent PD-like phenotypes associated with progressive degeneration of SN DA neurons and defects in locomotor behavior (Ekstrand et al. 2007; Pickrell et al. 2011; Song et al. 2012), the accumulation of somatic mtDNA mutations in mutator mice led to premature aging in the absence of overt neurodegeneration (Kujoth et al. 2005; Trifunovic et al. 2004). Surprisingly, Parkin deficiency affected differently the phenotypes in these mice. There was no modification of the mitochondrial morphological alterations or neurodegenerative process characteristic of the MitoPark model (Sterky et al. 2011). In contrast, mtDNA mutation load or predicted pathogenicity, mitochondrial dysfunction, DA neurodegeneration, and behavioral defects were anticipated or exacerbated in the three other models. Despite different interpretations about whether the observed effects reflected or not a physiological role of Parkin in the clearance of mitochondria with high mtDNA mutation loads, none of these studies were based on the direct exploration of the mitophagy process.
Direct investigation of mitophagy in vivo has only recently been rendered possible by the development of specific fluorescent reporters with acid-labile components, including the already mentioned mt-Keima and mito-QC, a mitochondrion-targeted tandem mCherry-GFP protein (Rodger et al. 2018). These reporters have been used to generate transgenic Drosophila and mouse models and explore the presence of mitochondria within lysosomes under basal conditions across organs and tissues (Cornelissen et al. 2018; Lee et al. 2018; McWilliams et al. 2016; Sun et al. 2015). It should be noted, however, that these reporters cannot formally discriminate between autophagy-dependent and autophagy-independent events, such as those mediated by the MDV pathway, unless systematic parallel analyses are performed in corresponding autophagy-deficient models (Table 1). Nevertheless, in all these studies, quenching of the acid-labile fluorescent component in the lysosome has been exclusively interpreted in terms of mitophagy. In Drosophila, such structures were abundant in various tissues during development, including in the nervous tissue, and were not observed following deletion of Atg5 or overexpression of a kinase-dead version of Atg1 (Cornelissen et al. 2018; Lee et al. 2018). Importantly, in the adult brain, mitophagy events were readily detected in DA neuron clusters reported to degenerate in pink1 and parkin mutant flies, albeit with some differences depending on the reporter used. For example, using mito-QC, the steady-state levels of the supposed mitophagy were found to be stable during aging in the DA neurons from the PPL1 cluster of the posterior inferior lateral protocerebrum (Lee et al. 2018), whereas in mt-Keima flies, the mitophagy index increased by 30% between 1 and 4 weeks of age (Cornelissen et al. 2018). In addition, in mito-QC flies, mitophagy events were not detectable in muscle tissue, which is also affected in pink1 and parkin mutants (Lee et al. 2018). In contrast, mitolysosomes were observed in the indirect flight muscles of mt-Keima flies, where they were also found to increase fourfold in abundance between week 1 and week 4 (Cornelissen et al. 2018). These discrepancies highlight potential differences in the sensitivities of the biosensors used to track mitochondria in lysosomes. Consistent with this possibility, the use of mt-Keima revealed a 20-fold higher mitophagy index in the DA neurons of the PPL1 cluster than that in the indirect flight muscles (Cornelissen et al. 2018), indicating that mitophagy occurs only rarely in muscles, where it may remain below the detection limit when monitored with mito-QC (Lee et al. 2018). Notably, mt-Keima is a mitochondrial matrix protein, whereas mito-QC is targeted to the outer mitochondrial membrane. As reported for proteins of this submitochondrial compartment (Karbowski and Youle 2011), mito-QC may thus be subject to degradation by the ubiquitin-proteasome system in addition to mitophagy, which could act as another source of bias. For example, mitophagy may be artificially underestimated, as a subset of mitochondrial units, potentially undergoing mitophagy will be missed because of prior degradation of the fusion protein. On the other hand, mt-Keima cannot be analyzed in fixed samples, because fixation compromises the lysosomal pH gradient, and this may engender signal variability. Moreover, there is partial overlap in the Keima excitation spectrums for red and green fluorescence, which can complicate the interpretation of the data obtained with this probe.
In mice, events interpreted as mitophagy were detected using the same reporters in a range of tissues with high metabolic activity, such as the heart and skeletal muscle, kidney, liver, pancreas, and brain (McWilliams et al. 2016, 2018b; Sun et al. 2015). Here, mitolysosomes were particularly abundant in the Purkinje cell layer of the cerebellum and in regions enriched with neural stem cells, such as the lateral ventricle and the dentate gyrus in the hippocampus (McWilliams et al. 2016; Sun et al. 2015). Moreover, mitolysosomes were abundant in different DA neuron populations, including the highly vulnerable A9 SNc neurons that degenerate in PD, but also the less susceptible A10 DA neurons of the VTA, and the A16 periglomerular DA neurons of the olfactory bulb, known to increase in number in PD (McWilliams et al. 2018b). In these neurons, mitolysosomes were rare in the axonal arbors, whereas they were enriched in the somata and axon initial segments. In slices of the mesencephalon prepared ex vivo from mice following viral vector–mediated delivery of mt-Keima, the basal rate of mitophagy turned out to be significantly higher in DA neurons of the SNc than in DA neurons of the VTA, or in other basal ganglia neurons (Guzman et al. 2018). Remarkably, chronic treatment of the mice with the Cav1 channel inhibitor isradipine significantly reduced basal mitophagy in SNc DA neurons. Cav1 channel-mediated calcium entry into the somatodendritic compartment stimulates mitochondrial intermediary metabolism and oxidative phosphorylation in these neurons to sustain the bioenergetic demand associated with their characteristic autonomous pacemaking activity; this occurs at the cost of an increase in the generation of mitochondrial reactive oxygen species (Dragicevic et al. 2015). In addition to normalizing basal mitophagy, isradipine mitigated oxidative stress in SNc neuron, suggesting that this mechanism acts as a key determinant of vulnerability in SNc DA neurons (Guzman et al. 2018).
In mito-QC mice, the levels of basal mitophagy did not appear to be affected by loss-of-function mutations of PINK1 or PARK2, supporting the idea that the PINK1/Parkin-dependent mitochondrial clearance program is not active under basal conditions (McWilliams et al. 2018a, b). Alternatively, complementary mitophagy pathways may be activated in the absence of PINK1, as previously suggested (Dagda et al. 2009; Vincow et al. 2013). However, future studies will have to more carefully investigate the possible impact of PINK1/Parkin deficiency on basal mitophagy, particularly during aging and under stress. Sun et al. reported a 70% decrease in mitophagy in dentate gyrus neurons in 21-month-old mice compared with 3-month-old animals (Sun et al. 2015), but mitophagy in the absence of PINK1 was only investigated up to 9.5 months of age (McWilliams et al. 2018b). Moreover, although Lee and colleagues did not observe changes in basal mitophagy in the absence of PINK1 in Drosophila (Lee et al. 2018), Cornelissen and his team reported impairment of the age-dependent increase in mitolysosome abundance in flight muscles and dopaminergic neurons of pink1 and parkin mutant flies (Cornelissen et al. 2018).
Conclusions
Since early studies in the 1960s postulating the existence of a mechanism ensuring the degradation of mitochondria as discrete entities based on classical in vivo radiolabeling approaches in rodents to investigate the turnover of protein and lipid components, this hypothesis has been verified in the past 15 years by the identification of specific programs for the autophagy-dependent destruction of mitochondria. One of these programs is activated in cell culture by mitochondrial stress and regulated by the protein products of two genes responsible for familial forms of one of the most common neurodegenerative diseases, providing the impetus for the direct investigation of mitophagy in neuronal cells. Despite many debates, fueled by the idea that the metabolic and bioenergetic properties of neurons are not compatible with such a mechanism, several laboratories have provided evidence for its occurrence, not only in cultured neurons exposed to toxins but also in neurons in the brain of model organisms under basal conditions. Many open questions still persist: what is the relative contribution of mitophagy to mitochondrial quality control in neurons, compared with other mechanisms involving degradation of mitochondrial components by intramitochondrial proteases, or direct delivery to the lysosome through the MDV pathway (Sugiura et al. 2014)? What are the physiological conditions that activate this process in neurons, and what are the mechanisms that regulate basal versus evoked mitophagy? How is mitophagy interconnected with mitochondrial biogenesis and how is it linked to mitochondrial transport fusion and fission in neurons? In which neuronal compartments does it occur primarily? Does it contribute in any way to the modulation of neuronal activity, for example, by limiting energy supply to fundamental processes, such as synaptic vesicle recycling?
Finally, with respect to PINK1/Parkin-dependent mitophagy, we still need to understand in how far this pathway is central to neuronal degeneration in Parkinson’s disease. This will imply solving present controversies as to the involvement of this specific mechanism in whole organisms, particularly during aging, which is acknowledged to be the greatest risk factor for Parkinson’s disease (Collier et al. 2017), or under stress conditions. These studies will have to be paralleled by the in-depth investigation of the mitophagy-independent functions of the PINK1/Parkin signaling pathway (Jang et al. 2018; Palikaras et al. 2018). PINK1 and Parkin have been reported to exert other functions in relation to mitochondrial maintenance, be it cooperatively or independently, including in mitochondrial biogenesis and the derepression of transcripts encoding specific mitochondrial respiratory chain subunits on the outer mitochondrial membrane, the regulation of the activity of mitochondrial respiratory chain complex I and the MDV pathway (reviewed by Alves da Costa and Checler 2012, Charan and LaVoie 2015, Scarffe et al. 2014, Sugiura et al. 2014, Voigt et al. 2016, Winklhofer 2014, Mouton-Liger et al. 2017, Chu 2018). Importantly, PINK1 and Parkin play also central roles in maintaining neuronal viability in response to stress by various, only partially elucidated mechanisms, independent of their roles in mitochondrial quality control (Alves Alves da Costa and Checler 2012, Charan and LaVoie 2015, Winklhofer 2014), and they can even exert proapoptotic functions under specific circumstances (Carroll et al. 2014, Zhang et al. 2014). Considering this complexity, there is still a long way to go before we can fully appreciate the relative contribution of each of these specific mechanisms to the pathophysiology of Parkinson’s disease.
References
Al Rawi S et al (2011) Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 334:1144–1147. https://doi.org/10.1126/science.1211878
Alves da Costa C, Checler F (2012) Parkin: much more than a simple ubiquitin ligase. Neurodegener Dis 10:49–51. https://doi.org/10.1159/000332803
Amiri M, Hollenbeck PJ (2008) Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol 68:1348–1361. https://doi.org/10.1002/dneu.20668
Aschrafi A, Kar AN, Gale JR, Elkahloun AG, Vargas JNS, Sales N, Wilson G, Tompkins M, Gioio AE, Kaplan BB (2016) A heterogeneous population of nuclear-encoded mitochondrial mRNAs is present in the axons of primary sympathetic neurons. Mitochondrion 30:18–23. https://doi.org/10.1016/j.mito.2016.06.002
Ashford TP, Porter KR (1962) Cytoplasmic components in hepatic cell lysosomes. J Cell Biol 12:198–202. https://doi.org/10.1083/jcb.12.1.198
Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206:655–670. https://doi.org/10.1083/jcb.201401070
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145. https://doi.org/10.1097/00004647-200110000-00001
Barini E, Miccoli A, Tinarelli F, Mulholland K, Kadri H, Khanim F, Stojanovski L, Read KD, Burness K, Blow JJ, Mehellou Y, Muqit MMK (2018) The anthelmintic drug niclosamide and its analogues activate the Parkinson’s disease associated protein kinase PINK1. Chembiochem 19:425–429. https://doi.org/10.1002/cbic.201700500
Beattie DS, Basford RE, Koritz SB (1967) The turnover of the protein components of mitochondria from rat liver, kidney, and brain. J Biol Chem 242:4584–4586
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. https://doi.org/10.1038/ng1769
Berridge MJ (1998) Neuronal calcium signaling. Neuron 21:13–26
Berridge MV, Schneider RT, McConnell MJ (2016) Mitochondrial transfer from astrocytes to neurons following ischemic insult: guilt by association? Cell Metab 24:376–378. https://doi.org/10.1016/j.cmet.2016.08.023
Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K (2014) Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J Neurosci 34:14304–14317. https://doi.org/10.1523/JNEUROSCI.0930-14.2014
Bertolin G, Ferrando-Miguel R, Jacoupy M, Traver S, Grenier K, Greene AW, Dauphin A, Waharte F, Bayot A, Salamero J, Lombès A, Bulteau AL, Fon EA, Brice A, Corti O (2013) The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy 9:1801–1817. https://doi.org/10.4161/auto.25884
Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510:370–375. https://doi.org/10.1038/nature13418
Bolam JP, Pissadaki EK (2012) Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord 27:1478–1483. https://doi.org/10.1002/mds.25135
Byrd RA, Weissman AM (2013) Compact Parkin only: insights into the structure of an autoinhibited ubiquitin ligase. EMBO J 32:2087–2089. https://doi.org/10.1038/emboj.2013.158
Cai Q, Zakaria HM, Simone A, Sheng ZH (2012) Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 22:545–552. https://doi.org/10.1016/j.cub.2012.02.005
Carroll RG, Hollville E, Martin SJ (2014) Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep 9:1538–1553. https://doi.org/10.1016/j.celrep.2014.10.046
Caulfield TR, Fiesel FC, Springer W (2015) Activation of the E3 ubiquitin ligase Parkin. Biochem Soc Trans 43:269–274. https://doi.org/10.1042/BST20140321
Charan RA, LaVoie MJ (2015) Pathologic and therapeutic implications for the cell biology of parkin. Mol Cell Neurosci 66(Pt A):62–71. https://doi.org/10.1016/j.mcn.2015.02.008
Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H (2011) Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J 30:2853–2867. https://doi.org/10.1038/emboj.2011.204
Chen H, Chan DC (2010) Physiological functions of mitochondrial fusion. Ann N Y Acad Sci 1201:21–25. https://doi.org/10.1111/j.1749-6632.2010.05615.x
Chu CT (2018) Multiple pathways for mitophagy: a neurodegenerative conundrum for Parkinson’s disease. Neurosci Lett 697:66–71. https://doi.org/10.1016/j.neulet.2018.04.004
Collier TJ, Kanaan NM, Kordower JH (2017) Aging and Parkinson’s disease: different sides of the same coin? Mov Disord 32:983–990. https://doi.org/10.1002/mds.27037
Cornelissen T, Haddad D, Wauters F, van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, de Strooper B, Verstreken P, Vandenberghe W (2014) The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23:5227–5242. https://doi.org/10.1093/hmg/ddu244
Cornelissen T, Vilain S, Vints K, Gounko N, Verstreken P, Vandenberghe W (2018) Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. Elife 7 doi:https://doi.org/10.7554/eLife.35878
Corti O, Lesage S, Brice A (2011) What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev 91:1161–1218. https://doi.org/10.1152/physrev.00022.2010
Cuzner ML, Davison AN, Gregson NA (1966) Turnover of brain mitochondrial membrane lipids. Biochem J 101:618–662
Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284:13843–13855. https://doi.org/10.1074/jbc.M808515200
De Duve C, Wattiaux R (1966) Functions of lysosomes. Annu Rev Physiol 28:435–492. https://doi.org/10.1146/annurev.ph.28.030166.002251
Devireddy S, Liu A, Lampe T, Hollenbeck PJ (2015) The organization of mitochondrial quality control and life cycle in the nervous system in vivo in the absence of PINK1. J Neurosci 35:9391–9401. https://doi.org/10.1523/JNEUROSCI.1198-15.2015
Dolle C et al (2016) Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 7:13548. https://doi.org/10.1038/ncomms13548
Dragicevic E, Schiemann J, Liss B (2015) Dopamine midbrain neurons in health and Parkinson’s disease: emerging roles of voltage-gated calcium channels and ATP-sensitive potassium channels. Neuroscience 284:798–814. https://doi.org/10.1016/j.neuroscience
Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, Gros P, Shaler TA, Faubert D, Coulombe B, Fon EA (2014) USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J 33:2473–2491. https://doi.org/10.15252/embj.201489729
Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG (2007) Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A 104:1325–1330. https://doi.org/10.1073/pnas.0605208103
Exner N, Lutz AK, Haass C, Winklhofer KF (2012) Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J 31:3038–3062. https://doi.org/10.1038/emboj.2012.170
Fiesel FC, James ED, Hudec R, Springer W (2017) Mitochondrial targeted HSP90 inhibitor Gamitrinib-TPP (G-TPP) induces PINK1/Parkin-dependent mitophagy. Oncotarget 8:106233–106248. https://doi.org/10.18632/oncotarget.22287
Fletcher MJ, Sanadi DR (1961) Turnover of rat-liver mitochondria. Biochim Biophys Acta 51:356–360
Gautier CA, Erpapazoglou Z, Mouton-Liger F, Muriel MP, Cormier F, Bigou S, Duffaure S, Girard M, Foret B, Iannielli A, Broccoli V, Dalle C, Bohl D, Michel PP, Corvol JC, Brice A, Corti O (2016) The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet 25:2972–2984. https://doi.org/10.1093/hmg/ddw148
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131. https://doi.org/10.1038/ncb2012
Gelmetti V, de Rosa P, Torosantucci L, Marini ES, Romagnoli A, di Rienzo M, Arena G, Vignone D, Fimia GM, Valente EM (2017) PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13:654–669. https://doi.org/10.1080/15548627.2016.1277309
Gladkova C, Maslen SL, Skehel JM, Komander D (2018) Mechanism of parkin activation by PINK1. Nature 559:410–414. https://doi.org/10.1038/s41586-018-0224-x
Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW 2nd (2015) Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350:aad2459. https://doi.org/10.1126/science.aad2459
Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13:378–385. https://doi.org/10.1038/embor.2012.14
Grenier K, McLelland GL, Fon EA (2013) Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front Neurol 4:100. https://doi.org/10.3389/fneur.2013.00100
Gross N, Rabinowitz M (1968) Thymidine content and turnover in the rat. Biochim Biophys Acta 157:648–651
Grunewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM (2016) Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann Neurol 79:366–378. https://doi.org/10.1002/ana.24571
Guzman JN, Ilijic E, Yang B, Sanchez-Padilla J, Wokosin D, Galtieri D, Kondapalli J, Schumacker PT, Surmeier DJ (2018) Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J Clin Invest 128:2266–2280. https://doi.org/10.1172/JCI95898
Hall CN, Klein-Flugge MC, Howarth C, Attwell D (2012) Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci 32:8940–8951. https://doi.org/10.1523/JNEUROSCI.0026-12.2012
Harbauer AB (2017) Mitochondrial health maintenance in axons. Biochem Soc Trans 45:1045–1052. https://doi.org/10.1042/BST20170023
Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75:762–777. https://doi.org/10.1016/j.neuron.2012.08.019
Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, Martin SE, Youle RJ (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504:291–295. https://doi.org/10.1038/nature12748
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555. https://doi.org/10.1038/nature18928
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60:7–20. https://doi.org/10.1016/j.molcel.2015.08.016
Hollenbeck PJ, Saxton WM (2005) The axonal transport of mitochondria. J Cell Sci 118:5411–5419. https://doi.org/10.1242/jcs.02745
Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St. Lawrence E, Schüle B, Krainc D, Palmer TD, Wang X (2016) Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19:709–724. https://doi.org/10.1016/j.stem.2016.08.002
Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N (2012) Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 125:1488–1499. https://doi.org/10.1242/jcs.094110
Jang JY, Blum A, Liu J, Finkel T (2018) The role of mitochondria in aging. J Clin Invest 128:3662–3670. https://doi.org/10.1172/JCI120842
Jin SM, Youle RJ (2013) The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9:1750–1757. https://doi.org/10.4161/auto.26122
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942. https://doi.org/10.1083/jcb.201008084
Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung YH, Mak TW, Shen J, Slack RS, Park DS (2012) ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet 21:4888–4903. https://doi.org/10.1093/hmg/dds325
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205:143–153. https://doi.org/10.1083/jcb.201402104
Karbowski M, Youle RJ (2011) Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23:476–482. https://doi.org/10.1016/j.ceb.2011.05.007
Kazlauskaite A, Muqit MM (2015) PINK1 and Parkin - mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson's disease. FEBS J 282:215–223. https://doi.org/10.1111/febs.13127
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MMK (2014) Parkin is activated by PINK1-dependent phophorylation of ubiquitin at Ser65. Biochem J 460:127–139. https://doi.org/10.1042/BJ20140334
Kim I, Rodriguez-Enriquez S, Lemasters JJ (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462:245–253. https://doi.org/10.1016/j.abb.2007.03.034
Kissova I, Deffieu M, Manon S, Camougrand N (2004) Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 279:39068–39074. https://doi.org/10.1074/jbc.M406960200
Koenig E, Giuditta A (1999) Protein-synthesizing machinery in the axon compartment. Neuroscience 89:5–15
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MMK (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2:120080. https://doi.org/10.1098/rsob.120080
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510:162–166. https://doi.org/10.1038/nature13392
Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38:518–520. https://doi.org/10.1038/ng1778
Kujoth GC et al (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481–484. https://doi.org/10.1126/science.1112125
Kumar A, Chaugule VK, Condos TEC, Barber KR, Johnson C, Toth R, Sundaramoorthy R, Knebel A, Shaw GS, Walden H (2017a) Parkin-phosphoubiquitin complex reveals cryptic ubiquitin-binding site required for RBR ligase activity. Nat Struct Mol Biol 24:475–483. https://doi.org/10.1038/nsmb.3400
Kumar A, Tamjar J, Waddell AD, Woodroof HI, Raimi OG, Shaw AM, Peggie M, Muqit MMK, van Aalten DMF (2017b) Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. Elife 6. https://doi.org/10.7554/eLife.29985
Langston JW, Ballard PA Jr (1983) Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med 309:310
Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22:320–333. https://doi.org/10.1016/j.devcel.2011.12.014
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. https://doi.org/10.1038/nature14893
Lee JJ, Sanchez-Martinez A, Zarate AM, Beninca C, Mayor U, Clague MJ, Whitworth AJ (2018) Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol 217:1613–1622. https://doi.org/10.1083/jcb.201801044
Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8:3–5. https://doi.org/10.1089/rej.2005.8.3
Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, Sheng ZH (2017) Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94:595–610 e596. https://doi.org/10.1016/j.neuron.2017.04.004
Matsuda N (2016) Phospho-ubiquitin: upending the PINK-Parkin-ubiquitin cascade. J Biochem 159:379–385. https://doi.org/10.1093/jb/mvv125
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T (2009) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci 29:444–453. https://doi.org/10.1523/JNEUROSCI.4029-08.2009
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221. https://doi.org/10.1083/jcb.200910140
McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33:282–295. https://doi.org/10.1002/embj.201385902
McLelland GL, Lee SA, McBride HM, Fon EA (2016) Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol 214:275–291. https://doi.org/10.1083/jcb.201603105
McLelland GL et al. (2018) Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 7 doi:https://doi.org/10.7554/eLife.32866
McWilliams TG, Muqit MM (2017) PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol 45:83–91. https://doi.org/10.1016/j.ceb.2017.03.013
McWilliams TG et al (2016) Mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214:333–345. https://doi.org/10.1083/jcb.201603039
McWilliams TG et al (2018a) Phosphorylation of Parkin at serine 65 is essential for its activation in vivo. Open Biol 8:180108. https://doi.org/10.1098/rsob.180108
McWilliams TG et al (2018b) Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 27:439–449 e435. https://doi.org/10.1016/j.cmet.2017.12.008
Mink JW, Blumenschine RJ, Adams DB (1981) Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Phys 241:R203–R212. https://doi.org/10.1152/ajpregu.1981.241.3.R203
Misgeld T, Schwarz TL (2017) Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96:651–666. https://doi.org/10.1016/j.neuron.2017.09.055
Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW (2007) Imaging axonal transport of mitochondria in vivo. Nat Methods 4:559–561. https://doi.org/10.1038/nmeth1055
Mouton-Liger F, Jacoupy M, Corvol JC, Corti O (2017) PINK1/Parkin-dependent mitochondrial surveillance: from pleiotropy to Parkinson’s disease. Front Mol Neurosci 10:120. https://doi.org/10.3389/fnmol.2017.00120
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. https://doi.org/10.1083/jcb.200809125
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. https://doi.org/10.1371/journal.pbio.1000298
Novikoff AB (1959) The proximal tubule cell in experimental hydronephrosis. Biophysic Biochem Cytol 6:136–138. https://doi.org/10.1083/jcb.6.1.136
Novikoff AB, Essner E (1962) Cytolysosomes and mitochondrial degeneration. J Cell Biol 15:140–146. https://doi.org/10.1083/jcb.15.1.140
Oh CK, Sultan A, Platzer J, Dolatabadi N, Soldner F, McClatchy DB, Diedrich JK, Yates JR III, Ambasudhan R, Nakamura T, Jaenisch R, Lipton SA (2017) S-Nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models. Cell Rep 21:2171–2182. https://doi.org/10.1016/j.celrep.2017.10.068
Okatsu K, Iemura SI, Koyano F, Go E, Kimura M, Natsume T, Tanaka K, Matsuda N (2012) Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem Biophys Res Commun 428:197–202. https://doi.org/10.1016/j.bbrc.2012.10.041
Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N (2013) A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288:36372–36384. https://doi.org/10.1074/jbc.M113.509653
Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol 209:111–128. https://doi.org/10.1083/jcb.201410050
Okatsu K, Sato Y, Yamano K, Matsuda N, Negishi L, Takahashi A, Yamagata A, Goto-Ito S, Mishima M, Ito Y, Oka T, Tanaka K, Fukai S (2018) Structural insights into ubiquitin phosphorylation by PINK1. Sci Rep 8:10382. https://doi.org/10.1038/s41598-018-28656-8
Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56:360–375. https://doi.org/10.1016/j.molcel.2014.09.007
Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, Harper JW (2015) Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A 112:6637–6642. https://doi.org/10.1073/pnas.1506593112
Ordureau A, Paulo JA, Zhang W, Ahfeldt T, Zhang J, Cohn EF, Hou Z, Heo JM, Rubin LL, Sidhu SS, Gygi SP, Harper JW (2018) Dynamics of PARKIN-dependent mitochondrial ubiquitylation in induced neurons and model systems revealed by digital snapshot. Proteomics Mol Cell 70:211–227 e218. https://doi.org/10.1016/j.molcel.2018.03.012
Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, Trudeau LE (2015) Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol 25:2349–2360. https://doi.org/10.1016/j.cub.2015.07.050
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20:1013–1022. https://doi.org/10.1038/s41556-018-0176-2
Pickrell AM, Pinto M, Hida A, Moraes CT (2011) Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 31:17649–17658. https://doi.org/10.1523/JNEUROSCI.4871-11.2011
Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ (2015) Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87:371–381. https://doi.org/10.1016/j.neuron.2015.06.034
Pilling AD, Horiuchi D, Lively CM, Saxton WM (2006) Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell 17:2057–2068. https://doi.org/10.1091/mbc.e05-06-0526
Pinto M, Nissanka N, Moraes CT (2018) Lack of Parkin anticipates the phenotype and affects mitochondrial morphology and mtDNA levels in a mouse model of Parkinson’s disease. J Neurosci 38:1042–1053. https://doi.org/10.1523/JNEUROSCI.1384-17.2017
Pissadaki EK, Bolam JP (2013) The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front Comput Neurosci 7:13. https://doi.org/10.3389/fncom.2013.00013
Plucinska G, Paquet D, Hruscha A, Godinho L, Haass C, Schmid B, Misgeld T (2012) In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J Neurosci 32:16203–16212. https://doi.org/10.1523/JNEUROSCI.1327-12.2012
Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S (2010) Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci U S A 107:14508–14513. https://doi.org/10.1073/pnas.1006551107
Rakovic et al (2013) Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem 288:2223–2237 https://doi.org/10.1074/jbc.M112.391680
Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156:825–835. https://doi.org/10.1016/j.cell.2013.12.042
Riley et al (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun 4:1982. https://doi.org/10.1038/ncomms2982
Rodger CE, McWilliams TG, Ganley IG (2018) Mammalian mitophagy - from in vitro molecules to in vivo models. FEBS J 285:1185–1202. https://doi.org/10.1111/febs.14336
Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ (2006) Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2:39–46
Rugarli EI, Langer T (2012) Mitochondrial quality control: a matter of life and death for neurons. EMBO J 31:1336–1349. https://doi.org/10.1038/emboj.2012.38
Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454:232–235. https://doi.org/10.1038/nature07006
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376. https://doi.org/10.1038/nature12043
Sato M, Sato K (2011) Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 334:1141–1144. https://doi.org/10.1126/science.1210333
Sauve V et al (2018) Mechanism of parkin activation by phosphorylation. Nat Struct Mol Biol 25:623–630. https://doi.org/10.1038/s41594-018-0088-7
Saxton WM, Hollenbeck PJ (2012) The axonal transport of mitochondria. J Cell Sci 125:2095–2104. https://doi.org/10.1242/jcs.053850
Scarffe LA, Stevens DA, Dawson VL, Dawson TM (2014) Parkin and PINK1: much more than mitophagy. Trends Neurosci 37:315–324. https://doi.org/10.1016/j.tins.2014.03.004
Schapira AH, Gegg M (2011) Mitochondrial contribution to Parkinson’s disease pathogenesis. Parkinsons Dis 2011:159160. https://doi.org/10.4061/2011/159160
Schubert AF, Gladkova C, Pardon E, Wagstaff JL, Freund SMV, Steyaert J, Maslen SL, Komander D (2017) Structure of PINK1 in complex with its substrate ubiquitin. Nature 552:51–56. https://doi.org/10.1038/nature24645
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 104:19500–19505. https://doi.org/10.1073/pnas.0708818104
Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D (2011) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31:5970–5976. https://doi.org/10.1523/JNEUROSCI.4441-10.2011
Seirafi M, Kozlov G, Gehring K (2015) Parkin structure and function. FEBS J 282:2076–2088. https://doi.org/10.1111/febs.13249
Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2:1002. https://doi.org/10.1038/srep01002
Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, Holt CE (2016) Dynamic axonal translation in developing and mature visual circuits. Cell 166:181–192. https://doi.org/10.1016/j.cell.2016.05.029
Shutt TE, McBride HM (2013) Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta 1833:417–424. https://doi.org/10.1016/j.bbamcr.2012.05.024
Sin J, Andres AM, Taylor DJR, Weston T, Hiraumi Y, Stotland A, Kim BJ, Huang C, Doran KS, Gottlieb RA (2016) Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 12:369–380. https://doi.org/10.1080/15548627.2015.1115172
Song L, Shan Y, Lloyd KC, Cortopassi GA (2012) Mutant Twinkle increases dopaminergic neurodegeneration, mtDNA deletions and modulates Parkin expression. Hum Mol Genet 21:5147–5158. https://doi.org/10.1093/hmg/dds365
Song L, McMackin M, Nguyen A, Cortopassi G (2017) Parkin deficiency accelerates consequences of mitochondrial DNA deletions and Parkinsonism. Neurobiol Dis 100:30–38. https://doi.org/10.1016/j.nbd.2016.12.024
Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM (2012) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22:135–141. https://doi.org/10.1016/j.cub.2011.11.057
Soutar MPM, Kempthorne L, Miyakawa S, Annuario E, Melandri D, Harley J, O’Sullivan GA, Wray S, Hancock DC, Cookson MR, Downward J, Carlton M, Plun-Favreau H (2018) AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci Rep 8:8855. https://doi.org/10.1038/s41598-018-26949-6
Spratt DE, Walden H, Shaw GS (2014) RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem J 458:421–437. https://doi.org/10.1042/BJ20140006
Sterky FH, Lee S, Wibom R, Olson L, Larsson NG (2011) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A 108:12937–12942. https://doi.org/10.1073/pnas.1103295108
Sugiura A, McLelland GL, Fon EA, McBride HM (2014) A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO 33:2142–2156. https://doi.org/10.15252/embj.201488104
Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmström KM, Fergusson MM, Yoo YH, Combs CA, Finkel T (2015) Measuring in vivo mitophagy. Mol Cell 60:685–696. https://doi.org/10.1016/j.molcel.2015.10.009
Swift H, Hruban Z (1964) Focal degradation as a biological process. Fed Proc 23:1026–1037
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1138. https://doi.org/10.1083/jcb.201007013
Tolkovsky AM, Xue L, Fletcher GC, Borutaite V (2002) Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie 84:233–240
Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340:1451–1455. https://doi.org/10.1126/science.1237908
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417–423. https://doi.org/10.1038/nature02517
Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W (2017) PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? J Park Dis 7:13–29. https://doi.org/10.3233/JPD-160989
Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB (2011) Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet 20:927–940. https://doi.org/10.1093/hmg/ddq531
Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ (2005) Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47:365–378. https://doi.org/10.1016/j.neuron.2005.06.018
Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ (2013) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A 110:6400–6405. https://doi.org/10.1073/pnas.1221132110
Voigt A, Berlemann LA, Winklhofer KF (2016) The mitochondrial kinase PINK1: functions beyond mitophagy. J Neurochem 139 Suppl 1:232–239. https://doi.org/10.1111/jnc.13655
Voos W, Jaworek W, Wilkening A, Bruderek M (2016) Protein quality control at the mitochondrion. Essays Biochem 60:213–225
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906. https://doi.org/10.1016/j.cell.2011.10.018
Wauer and Komander (2013) Structure of the human Pakin ligase domain in an autoinhibited state. EMBO J 32:2099–20112. https://doi.org/10.1038/emboj.2013.125
Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B (2017) Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168:224–238 e210. https://doi.org/10.1016/j.cell.2016.11.042
Winklhofer KF (2014) Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol 24:332–341. https://doi.org/10.1016/j.tcb.2014.01.001
Wu H, Williams J, Nathans J (2014) Complete morphologies of basal forebrain cholinergic neurons in the mouse. Elife 3:e02444. https://doi.org/10.7554/eLife.02444
Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF, De Camilli P (2017) Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A 114:E4859–E4867. https://doi.org/10.1073/pnas.1701078114
Yamano K, Queliconi BB, Koyano F, Saeki Y, Hirokawa T, Tanaka K, Matsuda N (2015) Site-specific interaction mapping of phosphorylated ubiquitin to uncover Parkin activation. J Biol Chem 290:25199–25211. https://doi.org/10.1074/jbc.M115.671446
Yamano K, Wang C, Sarraf SA, Münch C, Kikuchi R, Noda NN, Hizukuri Y, Kanemaki MT, Harper W, Tanaka K, Matsuda N, Youle RJ (2018) Endosomal Rab cycles regulate Parkin-mediated mitophagy. Elife 7. https://doi.org/10.7554/eLife.31326
Yang JY, Yang WY (2013) Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat Commun 4:2428. https://doi.org/10.1038/ncomms3428
Zhang C, Lee S, Peng Y, Bunker E, Giaime E, Shen J, Zhou Z, Liu X (2014) PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr Biol 24:1854–1865. https://doi.org/10.1016/j.cub.2014.07.014
Funding
Our work is supported by the Institut national de la santé et de la recherche médicale (INSERM), Fondation Institut du Cerveau et de la Moelle épinière and Agence Nationale pour la Recherche (“Investissements d’avenir,” grant ANR-10-IAIHU-06), Fondation de France, Association France Parkinson, and Michael J. Fox Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that she has no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Corti, O. Neuronal Mitophagy: Lessons from a Pathway Linked to Parkinson’s Disease. Neurotox Res 36, 292–305 (2019). https://doi.org/10.1007/s12640-019-00060-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-00060-8