
Abstract
Periostin is a protein that plays a key role in development and repair within the biological matrix of the lung. As a matricellular protein that does not contribute to extracellular matrix structure, periostin interacts with other extracellular matrix proteins to regulate the composition of the matrix in the lung and other organs. In this review, we discuss the studies exploring the role of periostin to date in chronic respiratory diseases, namely asthma and idiopathic pulmonary fibrosis. Asthma is a major health problem globally affecting millions of people worldwide with significant associated morbidity and mortality. Periostin is highly expressed in the lungs of asthmatic patients, contributes to mucus secretion, airway fibrosis and remodeling and is recognized as a biomarker of Th2 high inflammation. Idiopathic pulmonary fibrosis is a fatal interstitial lung disease characterized by progressive aberrant fibrosis of the lung matrix and respiratory failure. It predominantly affects adults over 50 years of age and its incidence is increasing worldwide. Periostin is also highly expressed in the lungs of idiopathic pulmonary fibrosis patients. Serum levels of periostin may predict clinical progression in this disease and periostin promotes myofibroblast differentiation and type 1 collagen production to contribute to aberrant lung fibrosis. Studies to date suggest that periostin is a key player in several pathogenic mechanisms within the lung and may provide us with a useful biomarker of clinical progression in both asthma and idiopathic pulmonary fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Periostin is a member of the matricellular family of proteins. Matricellular proteins are defined by the ability to bind both to extracellular matrix (ECM) and to cell surface receptors. Periostin is known to occupy a role in airway development and alveolar epithelial repair and is notably up-regulated in infants with bronchopulmonary dysplasia [1]. Periostin influences cellular behavior via interactions with integrin receptors and can influence production and localization of fibrogenic cytokines and growth factors [2, 3]. It also facilitates tissue remodeling and collagen crosslinking through its interaction with other ECM proteins and enzymes [4]. It is expressed in several different human tissues including the lung. Periostin is considered as a key factor in the evolution of aberrant airway and parenchymal fibrosis and is implicated in the pathogenesis of several chronic lung diseases including asthma and interstitial lung disease (ILD) [2].
Lung and airway fibrosis
Many chronic lung diseases result in the development of airway or parenchymal fibrosis including asthma, idiopathic pulmonary fibrosis (IPF), hypersensitivity pneumonitis (HP), fibroproliferative disease post-acute respiratory distress syndrome (ARDS), fibrotic non-specific interstitial pneumonia (NSIP), bronchiolitis obliterans syndrome (BOS) and others. The common feature of fibrotic lung diseases is the progressive deposition of extracellular matrix (ECM) proteins (e.g. collagen, fibronectin, and vimentin) that eventually impair lung function [5]. Deposition of “scar” tissue in the interstitium of the lung as occurs in IPF can negatively impact gas exchange because the thickened interstitial space limits the effective diffusion of oxygen and carbon dioxide. Sadly, patients suffering from IPF, which is the most common form of interstitial lung fibrosis [6] often die from respiratory insufficiency within 3–5 years of diagnosis and the prevalence and mortality rates for IPF are increasing globally [7,8,9].
Fibrosis can also occur in the setting of airway-centric diseases like asthma or BOS, but with differing histologic patterns. In asthma, airway remodeling results from deposition of ECM and proliferation of smooth muscle cells around large airways essentially narrowing the airway and constricting the airflow [10]. In BOS, airway remodeling occurs leading to an accumulation of granulation tissue which is submucosal or peribronchiolar in its distribution and results in a constrictive process and airflow limitation [11].
Periostin
Periostin is a member of the matricellular family of proteins. Matricellular proteins are defined by the ability to bind both to ECM and to cell surface receptors. Other matricellular protein family members implicated in lung fibrosis include osteopontin [12], tenascin C [13] and secreted protein acidic and rich in cysteine (SPARC) [14]. Periostin was originally identified as osteoblast-specific factor 2 in a mouse osteoblast cell line [15] and is expressed in the periosteum and in the periodontal ligament. The expression of matricellular proteins can be induced by various cytokines including transforming growth factor (TGF-β), interleukin-4 (IL-4), and IL-13 [16, 17] The periostin molecule is made up of a cysteine-rich domain within the N-terminal region, four fasciclin I domains, and an alternative splicing domain within the C-terminal region. Interestingly, up to nine splice variants have been identified, but the full-length transcript encodes an approximately 90 kDa secreted protein that includes all exons [18]. The functional significance of the splice variants is not well understood.
Periostin is known to bind type I collagen and fibronectin and has been shown to be involved in collagen fibrillogenesis [3] Cells can bind periostin through cellular integrin receptors and stimulation of cells by periostin can influence cell adhesion, proliferation, migration and angiogenesis [15]. Not surprisingly, periostin has been implicated in invasion and metastasis of various tumors [19]. Table 1 highlights actions of periostin on lung epithelial cells and fibroblasts. Work from our laboratory and others have shown that there are increased circulating periostin levels in IPF patients compared to controls and that periostin is found at higher levels in lung tissue of IPF patients [20, 21]. Periostin has recently been shown to be a marker of disease progression in IPF [20,21,22] and asthma [23].
Asthma, airway remodeling and periostin
The form of fibrosis that occurs in asthma is airway remodeling. Deposition of ECM and proliferation of airway smooth muscle cells around the airway can lead to variable narrowing of the airways with airflow obstruction and dyspnea. Asthma often develops in children, but it can persist and even develop de novo in adulthood [24]. Interestingly, periostin is one of the most highly expressed genes in asthma [25]. One study examined the production of periostin by airway epithelial cells either in asthmatic children or in cells from children that were atopic non-asthmatics versus healthy children and noted that periostin was differentially expressed in the airway epithelial cells from these three groups, with the highest expression found in the asthmatic children [26]. This suggests that periostin likely plays an important role in asthmatic airway remodeling.
Asthma can be categorized based on the underlying mechanisms that are driving a shared phenotype, often termed “endotypes”. A common endotype for this disease is one characterized by high Th2 inflammatory cytokines such as IL-4, IL-5 and IL-13 and studies support periostin as a biomarker to distinguish this Th2 endotype of asthma from Th2 low subjects [27, 28]. In fact, periostin levels can be used to predict clinical responses to anti IL-13 based therapies [29]. In addition to periostin being a marker for Th2 cytokine-mediated disease, IL-13 and IL-4 can both stimulate the secretion of periostin from lung fibroblasts [16], and IL-13 can induce epithelial cell production of periostin [30]. Periostin also induces TGF-β signaling which can further promote ECM deposition and airway remodeling [30]. Sub-epithelial periostin promotes adherence and possibly migration of eosinophils in the lung [31, 32]. Other biomarker studies have also found that periostin levels predict features of asthma including older age at onset, eosinophilia and worse pulmonary function [33]; furthermore, high periostin was the best and least variable predictor for airway eosinophilia in the airways or blood [33]. Periostin also had the least intra-patient variability when compared with fractional exhaled nitric oxide (FeNO) and blood eosinophils [34]. Up-regulated levels of serum periostin are also associated with several upper airway disorders that share some common features with asthma including chronic rhinosinusitis with nasal polyps [35] and aspirin exacerbated respiratory disease [36]. Periostin levels have also been studied as a biomarker of immunoglobulin E (IgE)-targeted therapeutic responses. Omalizumab, a monoclonal antibody targeting IgE, has established efficacy in asthma treatment and the data support a trend towards significant clinical responses in the periostin “high” versus “low” group [37].
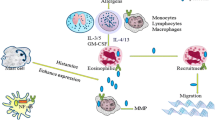
Animal models of asthma and allergen challenge have shown conflicting results regarding periostin. Two different studies have shown that periostin-deficient mice experience worse airway hyperresponsiveness when sensitized and challenged with ovalbumin or Aspergillus fumigatus [38, 39]. In contrast, other studies found a reduction in allergen-induced eosinophil recruitment to the lungs in periostin-deficient mice [32] and demonstrated that periostin-deficient mice are protected from house dust-mite induced allergic disease [40]. These results are more consistent with the human asthmatic data. Taken together, there is accumulating evidence that periostin is an important mediator of allergic airway disease in the lung and that periostin levels in the blood show promise as a biomarker of Th2 response. Consistent with this finding, periostin was the blood biomarker that was best able to predict patients who respond to anti-IL-13 therapies [41]. Figure 1 shows a schematic of periostin actions in remodeling associated with asthma.

Periostin secretion in asthma. Periostin is secreted from activated epithelium in a basolateral mechanism where it interacts with other mediators in the sub-epithelial space to promote airway smooth muscle remodeling and sub-epithelial fibrosis. Periostin also promotes eosinophil recruitment and mucus secretion contributing to the pathophysiology of asthma
Pathobiology of pulmonary fibrosis
The etiologies of progressive fibrotic lung diseases are still very poorly understood, and it is likely that IPF represents a spectrum of “molecular endotypes” where disease may be triggered by different environmental or genetic insults that result in a final common pathway of fibrosis [42]. In the case of IPF, a currently held paradigm is that repetitive injuries to the alveolar epithelial cells potentially caused by viral infections, genetic mutations, inhalation of toxins, inhalation of dusts or radiation cause damage to the epithelium disrupting the homeostatic crosstalk between epithelial cells and mesenchymal cells [7, 42]. Epithelial cells secrete anti-fibrotic, homeostatic mediators like prostaglandin E2 (PGE2) [43]. Loss of epithelial cells can result in lower levels of PGE2, which in turn, can allow resident fibroblasts to proliferate and become activated to differentiate into alpha-smooth muscle actin (αSMA) positive myofibroblasts [44]. Myofibroblasts are believed to be the major pathogenic cell type in IPF because they are highly secretory cells producing abundant ECM and highly contractile, causing distortion of the alveolar architecture [45]. Additionally, release of TGF-β, the most potent pro-fibrotic growth factor studied to date [46] can have devastating effects by triggering the “apoptosis paradox” which means that TGF-β promotes apoptosis of epithelial cells while simultaneously preventing apoptosis in lung fibroblasts [47]. The result of this apoptosis paradox is to allow resident fibroblasts to accumulate and become myofibroblasts. Finally, injurious insults are often associated with release of chemokines which can attract inflammatory cells including fibrocytes [48]. As will be discussed below, fibrocytes are important sources of periostin and regulate lung fibrogenesis [17, 20].
Thus far, a number of genetic mutations and polymorphisms have been associated with development or progression of IPF. These include mutations in surfactant protein C genes and telomerase genes [49,50,51]. Mechanistically, these mutations can lead to misfolded proteins or increased cellular senescence which can cause endoplasmic reticulum stress and epithelial cell apoptosis [52, 53]. There are also a number of mutations that are believed to impact host defense that also occur in patients with IPF. These include mutations in toll like receptor 3 (TLR3), mucin 5b (MUC5b) and toll interacting protein (TOLLIP) [54,55,56]. The speculation in the case of these mutations is that impaired or altered host defense may impair pathogen clearance and allow damage to the lung epithelium. Our recent work has also demonstrated that levels of circulating proteins associated with host defense response are downregulated in IPF patients further supporting impaired immunity as a key feature of disease pathology [57]. Given this supposition, it is especially interesting that recent studies have suggested that the lung microbiome is altered in patients with IPF. Patients with IPF show evidence of increased bacterial load in the lung [58], loss of diversity within the lung bacterial community, outgrowth of potentially pathogenic species [59] and evidence of gene expression profile changes that correlate with altered microbial communities in IPF lungs [60]. Additionally, there is evidence that certain exposures may exacerbate the disease. One exposure that is believed to contribute to disease pathology is gastroesophageal reflux where microaspiration of stomach acid is believed to repetitively injure lung epithelium [61]. Another potential exposure that causes damage to epithelial cells is viral infection [62]. Finally, there is a condition known as an acute exacerbation of IPF that is defined by the absence of a clinical infection, but which results in rapid disease progression with high mortality [63]. Interestingly, in patients experiencing acute exacerbation of IPF, higher numbers of circulating fibrocytes have been noted [64].
Fibrocytes
Fibrocytes play a significant role in the development of lung fibrosis. Recently a circulating fibroblast precursor cell has been identified in the bone marrow and circulation which expresses CD45 and the collagen receptor DDR2 [65]. It is likely that these cells are the precursors to fibrocytes. Fibrocytes are circulating cells derived from hematopoietic precursors and they are functionally defined by co-expression of both hematopoietic (CD45) and mesenchymal markers (Col 1). When expanded ex vivo from lung mince digests, fibrocytes express a number of ECM proteins including collagen 1, collagen 3 and fibronectin [66–69]. In addition, fibrocytes express chemokine receptors, including CXCR4, CCR7 and CCR2, which may contribute to the recruitment and activation of fibrocytes in the lung [70–72]. Fibrocytes can express col 1 mRNA and protein, but have also been shown to ingest col 1 protein [73–75]. The exact role fibrocytes play in production vs. uptake of collagen is still debatable.
In vitro work has clearly shown that fibrocytes can differentiate into myofibroblasts [70, 72, 76, 77]; however, whether this occurs in vivo is much less clear. Clonal transplantation of circulating fibroblast precursors does give rise to fibroblasts in the lung [65] and lineage tracing studies have suggested that hematopoietic precursors can become lung fibroblasts, but these fibroblasts derived from bone marrow sources rarely become myofibroblasts [78, 79]. In fact, it is not clear that circulating precursors even contribute significantly to the ECM pool in lung fibrosis. For example, deleting the ability of hematopoietic cells to produce collagen using a transgenic approach in mice had no impact on the outcome of fibrosis and collagen deposition following challenge with bleomycin [75]. Thus, fibrocytes are likely to contribute to the development of fibrosis via mechanisms that are not related to intrinsic myofibroblast differentiation.
Our past studies have shown that adoptive transfer of fibrocytes 5 days post-administration of fluorescein isothiocyanate (FITC) or bleomycin (two well-studied exposures that lead to development of fibrosis following intratracheal instillation in rodents) results in enhanced fibrosis by day 21 [17, 70, 71]. In addition, several studies show correlations between increased numbers of fibrocytes and worse disease progression in both humans and animal models [17, 71, 80–83]. Two studies using either the bleomycin model or the TGFα overexpression model have demonstrated convincingly that most fibrocytes do not differentiate into fibroblasts in vivo, but rather appear to promote fibrosis via secretion of paracrine mediators [17, 84]. This is consistent with human studies using samples from patients with hypersensitivity pneumonitis which demonstrated that co-culture of fibrocytes with lung fibroblasts led to a significant increase in the expression of ECM, matrix metalloprotease 1 and pro-fibrotic platelet-derived growth factor β and that fibrocytes could influence the release of the chemokine CCL2 from lymphocytes [82]. When considering the mediators that fibrocytes might produce to regulate other cell functions in a paracrine manner, it is interesting to note that previous work showed that fibrocytes in circulation of IPF patients are major producers of the matricellular protein, periostin [20].
Periostin and interstitial fibrosis
Periostin is highly expressed in the lungs and is found at increased levels in the circulation of patients with IPF [20, 21]. While the significance of alternative splicing in formation of periostin mRNA is not known, it has been noted that periostin utilizes different exons in IPF patients than normal volunteers [85]. The expression of periostin localizes to areas of active fibrosis, including areas known as fibrotic foci, which are believed to be the hallmark of the histopathologic diagnosis of IPF [20]. This same study found periostin expressed in subepithelial and subendothelial regions of the IPF lung. In terms of circulating levels, two studies have shown that elevated levels of periostin in the serum or plasma can predict a decrease in lung function over 6 months or 48 weeks respectively [20, 21]. Importantly, periostin may be a relevant biomarker for disease activity in these older patients as new research suggests that in normal subjects, periostin levels are stable from age 32 past 70 [86]. This elevation in periostin during IPF disease is not surprising when you consider that two well-known pro-fibrotic mediators, namely TGF-β [87, 88] and IL-13 [89] are also highly expressed in fibrotic lung tissue and are likely to mediate the increase in periostin seen in IPF. It is believed that periostin may play an important role in helping to stiffen the lung ECM. For instance, cross-linking between collagen fibrils is catalyzed by lysyl oxidases. Periostin can activate bone morphogenetic protein-1 to cleave lysyl oxidase; this in turn activates lysyl oxidase while also localizing this active enzyme to the ECM [90]. Ultimately, this leads to crosslinking of the collagen fibers and stiffening of the ECM. This increase in the stiffness of the ECM is believed to promote ongoing fibroblast activation, which may perpetuate the progressive nature of IPF [91].
Given the growing belief that bacterial infections or changes in the microbiome may promote lung fibrosis as discussed above, antibiotic therapy has been suggested as a therapeutic treatment for patients with IPF [92]. Interestingly, a recent study showed that clarithromycin was able to attenuate the expression of periostin, even in the face of IL-13 stimulation [93]. This raises the intriguing possibility that antibiotic effects on lung fibrosis may in fact work in part via inhibition of periostin.
Mouse models of lung fibrosis
While human studies have suggested a role for periostin in the progression of IPF, mechanistic studies require an animal model. There has been significant debate about the usefulness of animal models for IPF research, but they do represent well-characterized models of the evolution of fibrosis from injury to acute inflammation to chronic inflammation and ECM deposition [94]. Furthermore, an American Thoracic Society working group recommended that the bleomycin model be used as an important tool for the preclinical testing of anti-fibrotic agents [95]. With that context, two previous studies have noted that periostin accumulates in the lungs of both Balb/c and C57Bl/6 mice treated with bleomycin [20, 96]. Excitingly, both strains of mice were protected from the eventual development of fibrosis following bleomycin injury. However, there were different conclusions made as to the reason for the protection. In the Balb/c background, protection resulted from impaired production of chemokines by periostin-deficient fibroblasts, and reduced inflammatory responses [96]. In C57Bl/6 mice, inflammatory responses are similar between periostin knockout mice and wild-type littermates, but the mice developed less fibrosis during the fibroproliferative phase [20]. In fact, an antibody, OC-20, that blocks interactions of periostin with integrins, was able to block fibrosis even when given after the inflammatory phase of the disease [20]. Similarly, OC-20 was able to improve outcomes in a murine model of asthma as well [40].
In the C57Bl/6 background, one of the most interesting findings was that bone marrow chimeric mice created to lose expression of periostin in either the hematopoietic or structural cell compartments were both protected compared to chimeric mice expressing periostin in both donor and host cells [20]. This suggested that a circulating source of periostin was also important for the full fibrogenic effect. As mentioned earlier, fibrocytes are a cell type that are known to promote fibrogenesis and have been shown to express periostin in IPF patients [20, 80]. Our laboratory undertook a series of experiments to explore the importance of periostin production from fibrocytes specifically [17]. In these experiments, we showed that adoptive transfer of fibrocytes from wild-type mice augmented development of fibrosis to a greater degree than adoptive transfer of fibrocytes from periostin−/− mice. As we saw no evidence of fibrocytes actually differentiating into myofibroblasts in vivo, we tested the effects of supernatant obtained from cultured fibrocytes derived from bleomycin-treated wild-type and periostin−/− mice. We noted that supernatants from wild-type fibrocytes were more effective at inducing myofibroblast differentiation than those from periostin-deficient cells. We also found evidence that periostin and TGFβ were potent co-regulators of expression of each molecule, but that periostin had impacts on fibrocyte activation that were mediated by the β1 integrin rather than the TGFβ receptor. In concert with the paracrine nature of the periostin effect, fibrocytes from wild-type, but not periostin−/− mice produced more connective tissue growth factor (CTGF) and lysyl oxidase, which likely contribute to the ability to enhance collagen synthesis and crosslinking. Figure 2 shows a schematic of periostin in the pathogenesis of lung fibrosis.

Contributions of Periostin to IPF pathophysiology. In IPF, recurrent idiopathic epithelial cell injury results in inflammation. Periostin is highly expressed in the lungs and subsequently leads to TGF-B activation, increased type 1 collagen production, promotes fiber cross linking and enhanced stiffening of the interstitial matrix. Periostin mediates fibroblast to myofibroblast differentiation and studies from animal models support a role for periostin in epithelial to mesenchymal cell transition
In other animal models, microaspiration of bile acids (a model that mimics gastroesophageal reflux) led to development of fibrosis in rat lungs and this fibrosis was associated with an increase in several growth factors including TGFβ, CTGF and periostin [97]. Additionally in a model of bronchopulmonary dysplasia (BPD) in which neonatal mice are exposed to hyperoxia, it was found that periostin expression increased in the alveolar walls, particularly in areas in which interstitial thickening was noted, and this was similar to the expression pattern seen in infants with BPD where periostin staining co-localized with fibroblasts [1]. Furthermore, hyperoxia-exposed periostin−/− mice did not show the characteristic enlarged airspaces and interstitial thickening of BPD whereas the wild-type mice did. Similar to the findings in Balb/c mice with bleomycin, periostin seemed to promote expression of several chemokines including CXCL1, CXCL2 and CCL4 [1]. Thus, it appears that periostin contributes to fibrosis in both neonate and adult animals.
Conclusions
Periostin is a remarkable master regulator of the extracellular matrix. It plays a key role in maintaining a normal tissue matrix in the lung and abnormalities of periostin contribute significantly to the pathophysiology of several chronic respiratory diseases. In asthma, the levels of periostin are proving particularly efficacious in discriminating between different endotypes of the disease and predicting clinical progression. Importantly, current and ongoing clinical studies continue to explore the role of periostin as a potential biomarker of therapeutic responses in asthmatic patients. Periostin drives several features of the pathophysiology of IPF including myofibroblast differentiation, type 1 collagen production and cross linking of fibers within the lung matrix. It has also been reported as a biomarker of clinical progression. Several further studies are required to elaborate further on the role of periostin, its efficacy as a biomarker in chronic respiratory disease and even its potential candidacy as a therapeutic target in disease.
References
Bozyk PD, Bentley JK, Popova AP, Anyanwu AC, Linn MD, Goldsmith AM, Pryhuber GS, Moore BB, Hershenson MB (2012) Neonatal periostin knockout mice are protected from hyperoxia-induced alveolar simplication. PLoS One 7(2):e31336. doi:10.1371/journal.pone.0031336
Izuhara K, Conway SJ, Moore BB, Matsumoto H, Holweg CT, Matthews JG, Arron JR (2016) Roles of periostin in respiratory disorders. Am J Respir Crit Care Med 193(9):949–956. doi:10.1164/rccm.201510-2032PP
Kudo A (2011) Periostin in fibrillogenesis for tissue regeneration: periostin actions inside and outside the cell. Cell Mol Life Sci 68(19):3201–3207. doi:10.1007/s00018-011-0784-5
Liu AY, Zheng H, Ouyang G (2014) Periostin, a multifunctional matricellular protein in inflammatory and tumor microenvironments. Matrix Biol 37:150–156. doi:10.1016/j.matbio.2014.04.007
Wilson MS, Wynn TA (2009) Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol 2(2):103–121. doi:10.1038/mi.2008.85
American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus (2002) Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 165(2):277–304. doi:10.1164/ajrccm.165.2.ats01
King TE Jr, Pardo A, Selman M (2011) Idiopathic pulmonary fibrosis. Lancet 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB (2014) Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thorac Soc 11(8):1176–1185. doi:10.1513/AnnalsATS.201404-145OC
Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, Flaherty KR, Schwartz DA, Noble PW, Raghu G, Brown KK (2005) The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 142(12 Pt 1):963–967
Trejo Bittar HE, Yousem SA, Wenzel SE (2015) Pathobiology of severe asthma. Annu Rev Pathol 10:511–545. doi:10.1146/annurev-pathol-012414-040343
Kuehnel M, Maegel L, Vogel-Claussen J, Robertus JL, Jonigk D (2017) Airway remodelling in the transplanted lung. Cell Tissue Res 367(3):663–675. doi:10.1007/s00441-016-2529-0
White ES, Xia M, Murray S, Dyal R, Flaherty CM, Flaherty KR, Moore BB, Cheng L, Doyle TJ, Villalba J, Dellaripa PF, Rosas IO, Kurtis JD, Martinez FJ (2016) Plasma surfactant protein-D, matrix metalloproteinase-7, and osteopontin Index distinguishes idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias. Am J Respir Crit Care Med 194(10):1242–1251. doi:10.1164/rccm.201505-0862OC
Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, Lakota K, Budinger GR, Raparia K, Tamaki Z, Varga J (2016) Tenascin-C drives persistence of organ fibrosis. Nat Commun 7:11703. doi:10.1038/ncomms11703
Chang W, Wei K, Jacobs SS, Upadhyay D, Weill D, Rosen GD (2010) SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J Biol Chem 285(11):8196–8206. doi:10.1074/jbc.M109.025684
Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A (1999) Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res 14(7):1239–1249. doi:10.1359/jbmr.1999.14.7.1239
Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, Izuhara K (2006) Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol 118(1):98–104. doi:10.1016/j.jaci.2006.02.046
Ashley SL, Wilke CA, Kim KK, Moore BB (2017) Periostin regulates fibrocyte function to promote myofibroblast differentiation and lung fibrosis. Mucosal Immunol 10(2):341–351. doi:10.1038/mi.2016.61
Morra L, Rechsteiner M, Casagrande S, von Teichman A, Schraml P, Moch H, Soltermann A (2012) Characterization of periostin isoform pattern in non-small cell lung cancer. Lung Cancer 76(2):183–190. doi:10.1016/j.lungcan.2011.10.013
Ruan K, Bao S, Ouyang G (2009) The multifaceted role of periostin in tumorigenesis. Cell Mol Life Sci 66(14):2219–2230. doi:10.1007/s00018-009-0013-7
Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA, Fry CD, White ES, Sisson TH, Tayob N, Carnemolla B, Orecchia P, Flaherty KR, Hershenson MB, Murray S, Martinez FJ, Moore BB (2012) Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 303(12):L1046–L1056. doi:10.1152/ajplung.00139.2012
Okamoto M, Hoshino T, Kitasato Y, Sakazaki Y, Kawayama T, Fujimoto K, Ohshima K, Shiraishi H, Uchida M, Ono J, Ohta S, Kato S, Izuhara K, Aizawa H (2011) Periostin, a matrix protein, is a novel biomarker for idiopathic interstitial pneumonias. Eur Respir J 37(5):1119–1127. doi:10.1183/09031936.00059810
Ohta S, Okamoto M, Fujimoto K, Sakamoto N, Takahashi K, Yamamoto H, Kushima H, Ishii H, Akasaka K, Ono J, Kamei A, Azuma Y, Matsumoto H, Yamaguchi Y, Aihara M, Johkoh T, Kawaguchi A, Ichiki M, Sagara H, Kadota JI, Hanaoka M, Hayashi SI, Kohno S, Hoshino T, Izuhara K (2017) The usefulness of monomeric periostin as a biomarker for idiopathic pulmonary fibrosis. PLoS One 12(3):e0174547. doi:10.1371/journal.pone.0174547
James A, Janson C, Malinovschi A, Holweg C, Alving K, Ono J, Ohta S, Ek A, Middelveld R, Dahlen B, Forsberg B, Izuhara K, Dahlen SE (2017) Serum periostin relates to type-2 inflammation and lung function in asthma: data from the large population-based cohort Swedish GA(2)LEN. Allergy. doi:10.1111/all.13181 (in press)
Sood A, Qualls C, Schuyler M, Arynchyn A, Alvarado JH, Smith LJ, Jacobs DR Jr (2013) Adult-onset asthma becomes the dominant phenotype among women by age 40 years. The longitudinal CARDIA study. Ann Am Thorac Soc 10(3):188–197. doi:10.1513/AnnalsATS.201212-115OC
Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV (2007) Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA 104(40):15858–15863. doi:10.1073/pnas.0707413104
Lopez-Guisa JM, Powers C, File D, Cochrane E, Jimenez N, Debley JS (2012) Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol 129(4):990.e6–997.e6. doi:10.1016/j.jaci.2011.11.035
Stokes JR, Casale TB (2016) Characterization of asthma endotypes: implications for therapy. Ann Allergy Asthma Immunol 117(2):121–125. doi:10.1016/j.anai.2016.05.016
Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV (2009) T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 180(5):388–395. doi:10.1164/rccm.200903-0392OC
Antoniu SA (2016) Lebrikizumab for the treatment of asthma. Expert Opin Investig Drugs 25(10):1239–1249. doi:10.1080/13543784.2016.1227319
Sidhu SS, Yuan S, Innes AL, Kerr S, Woodruff PG, Hou L, Muller SJ, Fahy JV (2010) Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci USA 107(32):14170–14175. doi:10.1073/pnas.1009426107
Johansson MW, Annis DS, Mosher DF (2013) alpha(M)beta(2) integrin-mediated adhesion and motility of IL-5-stimulated eosinophils on periostin. Am J Respir Cell Mol Biol 48(4):503–510. doi:10.1165/rcmb.2012-0150OC
Blanchard C, Mingler MK, McBride M, Putnam PE, Collins MH, Chang G, Stringer K, Abonia JP, Molkentin JD, Rothenberg ME (2008) Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses. Mucosal Immunol 1(4):289–296. doi:10.1038/mi.2008.15
Matsusaka M, Kabata H, Fukunaga K, Suzuki Y, Masaki K, Mochimaru T, Sakamaki F, Oyamada Y, Inoue T, Oguma T, Sayama K, Koh H, Nakamura M, Umeda A, Ono J, Ohta S, Izuhara K, Asano K, Betsuyaku T (2015) Phenotype of asthma related with high serum periostin levels. Allergol Int 64(2):175–180. doi:10.1016/j.alit.2014.07.003
Jia G, Erickson RW, Choy DF, Mosesova S, Wu LC, Solberg OD, Shikotra A, Carter R, Audusseau S, Hamid Q, Bradding P, Fahy JV, Woodruff PG, Harris JM, Arron JR (2012) Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol 130(3):647.e610–654.e610. doi:10.1016/j.jaci.2012.06.025
Wang M, Wang X, Zhang N, Wang H, Li Y, Fan E, Zhang L, Zhang L, Bachert C (2015) Association of periostin expression with eosinophilic inflammation in nasal polyps. J Allergy Clin Immunol 136(6):1700.e1701–1703.e1709. doi:10.1016/j.jaci.2015.09.005
Kim MA, Izuhara K, Ohta S, Ono J, Yoon MK, Ban GY, Yoo HS, Shin YS, Ye YM, Nahm DH, Park HS (2014) Association of serum periostin with aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol 113(3):314–320. doi:10.1016/j.anai.2014.06.014
Hanania NA, Wenzel S, Rosen K, Hsieh HJ, Mosesova S, Choy DF, Lal P, Arron JR, Harris JM, Busse W (2013) Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am J Respir Crit Care Med 187(8):804–811. doi:10.1164/rccm.201208-1414OC
Gordon ED, Sidhu SS, Wang ZE, Woodruff PG, Yuan S, Solon MC, Conway SJ, Huang X, Locksley RM, Fahy JV (2012) A protective role for periostin and TGF-beta in IgE-mediated allergy and airway hyperresponsiveness. Clin Exp Allergy 42(1):144–155. doi:10.1111/j.1365-2222.2011.03840.x
Sehra S, Yao W, Nguyen ET, Ahyi AN, Tuana FM, Ahlfeld SK, Snider P, Tepper RS, Petrache I, Conway SJ, Kaplan MH (2011) Periostin regulates goblet cell metaplasia in a model of allergic airway inflammation. J Immunol 186(8):4959–4966. doi:10.4049/jimmunol.1002359
Bentley JK, Chen Q, Hong JY, Popova AP, Lei J, Moore BB, Hershenson MB (2014) Periostin is required for maximal airways inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol 134(6):1433–1442. doi:10.1016/j.jaci.2014.05.029
Scheerens H, Arron JR, Zheng Y, Putnam WS, Erickson RW, Choy DF, Harris JM, Lee J, Jarjour NN, Matthews JG (2014) The effects of lebrikizumab in patients with mild asthma following whole lung allergen challenge. Clin Exp Allergy 44(1):38–46. doi:10.1111/cea.12220
Goodwin AT, Jenkins G (2016) Molecular endotyping of pulmonary fibrosis. Chest 149(1):228–237. doi:10.1378/chest.15-1511
Lama V, Moore BB, Christensen P, Toews GB, Peters-Golden M (2002) Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am J Respir Cell Mol Biol 27(6):752–758. doi:10.1165/rcmb.4857
Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB (2003) Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 29(5):537–544. doi:10.1165/rcmb.2002-0243OC
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180(4):1340–1355. doi:10.1016/j.ajpath.2012.02.004
Aschner Y, Downey GP (2016) Transforming growth factor-beta: master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol 54(5):647–655. doi:10.1165/rcmb.2015-0391TR
Thannickal VJ, Horowitz JC (2006) Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc 3(4):350–356. doi:10.1513/pats.200601-001TK
Loomis-King H, Moore BB (2013) Fibrocytes in the pathogenesis of chronic fibrotic lung disease. Curr Respir Med Rev 9(1):34–41
Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB, Markin C, Renzoni E, Lympany P, Thomas AQ, Roldan J, Scott TA, Blackwell TS, Phillips JA 3rd, Loyd JE, du Bois RM (2004) Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 59(11):977–980. doi:10.1136/thx.2004.026336
Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE (2007) Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 356(13):1317–1326. doi:10.1056/NEJMoa066157
Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, Rock JR, Looney MR, Wolters PJ (2016) Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 1(14):e86704. doi:10.1172/jci.insight.86704
Bridges JP, Wert SE, Nogee LM, Weaver TE (2003) Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J Biol Chem 278(52):52739–52746. doi:10.1074/jbc.M309599200
Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS (2011) Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 108(26):10562–10567. doi:10.1073/pnas.1107559108
O’Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, Simpson AJ, Millar AB, McGrath EE, Whyte MK, Hirani N, Hogaboam CM, Donnelly SC (2013) The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 188(12):1442–1450. doi:10.1164/rccm.201304-0760OC
Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, Evans CM, Garantziotis S, Adler KB, Dickey BF, du Bois RM, Yang IV, Herron A, Kervitsky D, Talbert JL, Markin C, Park J, Crews AL, Slifer SH, Auerbach S, Roy MG, Lin J, Hennessy CE, Schwarz MI, Schwartz DA (2011) A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 364(16):1503–1512. doi:10.1056/NEJMoa1013660
Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, Richards TJ, Juan-Guardela BM, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae D, Kaminski N, Garcia JG (2013) Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 1(4):309–317. doi:10.1016/S2213-2600(13)70045-6
O’Dwyer DN, Norman KC, Xia M, Huang Y, Gurczynski SJ, Ashley SL, White ES, Flaherty KR, Martinez FJ, Murray S, Noth I, Arnold KB, Moore BB (2017) The peripheral blood proteome signature of idiopathic pulmonary fibrosis is distinct from normal and is associated with novel immunological processes. Sci Rep 7:46560. doi:10.1038/srep46560
Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, Murphy E, Johnston SL, Schwartz DA, Wells AU, Cookson WO, Maher TM, Moffatt MF (2014) The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 190(8):906–913. doi:10.1164/rccm.201403-0541OC
Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, Moore BB, White ES, Flaherty KR, Huffnagle GB, Martinez FJ, Investigators C (2014) Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med 2(7):548–556. doi:10.1016/S2213-2600(14)70069-4
Huang Y, Ma SF, Espindola MS, Vij R, Oldham JM, Huffnagle GB, Erb-Downward JR, Flaherty KR, Moore BB, White ES, Zhou T, Li J, Lussier YA, Han MK, Kaminski N, Garcia JG, Hogaboam CM, Martinez FJ, Noth I, Investigators C (2017) Microbes Associate with Host Innate Immune Response in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. doi:10.1164/rccm.201607-1525OC (in press)
Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, Hayes J, Sillery JK, Pope CE 2nd, Pellegrini CA (2006) High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 27(1):136–142. doi:10.1183/09031936.06.00037005
Moore BB, Moore TA (2015) Viruses in idiopathic pulmonary fibrosis. Etiology and exacerbation. Ann Am Thorac Soc 12(Suppl 2):S186–192. doi:10.1513/AnnalsATS.201502-088AW
Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr, Lasky JA, Loyd JE, Noth I, Olman MA, Raghu G, Roman J, Ryu JH, Zisman DA, Hunninghake GW, Colby TV, Egan JJ, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kondoh Y, Lynch DA, Muller-Quernheim J, Myers JL, Nicholson AG, Selman M, Toews GB, Wells AU, Martinez FJ (2007) Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 176(7):636–643. doi:10.1164/rccm.200703-463PP
Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, O’Byrne PM, Strieter RM, Kolb M (2009) Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179(7):588–594. doi:10.1164/rccm.200810-1534OC
McDonald LT, Mehrotra M, LaRue AC (2015) Hematopoietic origin of murine lung fibroblasts. Stem Cells Int 2015:159713. doi:10.1155/2015/159713
Kleaveland KR, Moore BB, Kim KK (2014) Paracrine functions of fibrocytes to promote lung fibrosis. Expert Rev Respir Med 8(2):163–172. doi:10.1586/17476348.2014.862154
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A (1994) Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1(1):71–81
Chesney J, Bacher M, Bender A, Bucala R (1997) The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc Natl Acad Sci USA 94(12):6307–6312
Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R (1998) Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol 160(1):419–425
Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB (2005) CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol 166(3):675–684. doi:10.1016/S0002-9440(10)62289-4
Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB (2006) The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol 35(2):175–181. doi:10.1165/rcmb.2005-0239OC
Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM (2004) Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 114(3):438–446. doi:10.1172/JCI20997
Madsen DH, Ingvarsen S, Jurgensen HJ, Melander MC, Kjoller L, Moyer A, Honore C, Madsen CA, Garred P, Burgdorf S, Bugge TH, Behrendt N, Engelholm LH (2011) The non-phagocytic route of collagen uptake: a distinct degradation pathway. J Biol Chem 286(30):26996–27010. doi:10.1074/jbc.M110.208033
Bianchetti L, Barczyk M, Cardoso J, Schmidt M, Bellini A, Mattoli S (2012) Extracellular matrix remodelling properties of human fibrocytes. J Cell Mol Med 16(3):483–495. doi:10.1111/j.1582-4934.2011.01344.x
Kleaveland KR, Velikoff M, Yang J, Agarwal M, Rippe RA, Moore BB, Kim KK (2014) Fibrocytes are not an essential source of type I collagen during lung fibrosis. J Immunol 193(10):5229–5239. doi:10.4049/jimmunol.1400753
Abe R, Donnelly SC, Peng T, Bucala R, Metz CN (2001) Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol 166(12):7556–7562
Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S (2003) Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol 171(1):380–389
Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH (2004) Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest 113(2):243–252. doi:10.1172/jci18847
Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE (2009) Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 180(7):657–665. doi:10.1164/rccm.200903-0322OC
Xu J, Kisseleva T (2015) Bone marrow-derived fibrocytes contribute to liver fibrosis. Exp Biol Med (Maywood) 240(6):691–700. doi:10.1177/1535370215584933
Xu J, Cong M, Park TJ, Scholten D, Brenner DA, Kisseleva T (2015) Contribution of bone marrow-derived fibrocytes to liver fibrosis. Hepatobiliary Surg Nutr 4(1):34–47. doi:10.3978/j.issn.2304-3881.2015.01.01
Garcia de Alba C, Buendia-Roldan I, Salgado A, Becerril C, Ramirez R, Gonzalez Y, Checa M, Navarro C, Ruiz V, Pardo A, Selman M (2015) Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am J Respir Crit Care Med 191(4):427–436. doi:10.1164/rccm.201407-1334OC
Moore BB, Kolb M (2014) Fibrocytes and progression of fibrotic lung disease. Ready for showtime? Am J Respir Crit Care Med 190(12):1338–1339. doi:10.1164/rccm.201411-2013ED
Madala SK, Edukulla R, Schmidt S, Davidson C, Ikegami M, Hardie WD (2014) Bone marrow-derived stromal cells are invasive and hyperproliferative and alter transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 50(4):777–786. doi:10.1165/rcmb.2013-0042OC
Nance T, Smith KS, Anaya V, Richardson R, Ho L, Pala M, Mostafavi S, Battle A, Feghali-Bostwick C, Rosen G, Montgomery SB (2014) Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS One 9(3):e92111. doi:10.1371/journal.pone.0092111
Walsh JS, Gossiel F, Scott JR, Paggiosi MA, Eastell R (2017) Effect of age and gender on serum periostin: relationship to cortical measures, bone turnover and hormones. Bone 99:8–13. doi:10.1016/j.bone.2017.03.041
Khalil N, O’Connor R, Unruh H, Warren P, Kemp A, Greenberg A (1991) Enhanced expression and immunohistochemical distribution of transforming growth factor-beta in idiopathic pulmonary fibrosis. Chest 99(3 Suppl):65S–66S
Khalil N, O’Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, Bereznay OH, Greenberg AH (1991) Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 5(2):155–162
Passalacqua G, Mincarini M, Colombo D, Troisi G, Ferrari M, Bagnasco D, Balbi F, Riccio A, Canonica GW (2017) IL-13 and idiopathic pulmonary fibrosis: possible links and new therapeutic strategies. Pulm Pharmacol Ther. doi:10.1016/j.pupt.2017.05.007
Maruhashi T, Kii I, Saito M, Kudo A (2010) Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem 285(17):13294–13303. doi:10.1074/jbc.M109.088864
Tschumperlin DJ, Jones JC, Senior RM (2012) The fibrotic matrix in control: does the extracellular matrix drive progression of idiopathic pulmonary fibrosis? Am J Respir Crit Care Med 186(9):814–816. doi:10.1164/rccm.201208-1561ED
Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, Wilson EC, Twentyman OP, Davison AG, Curtin JJ, Crawford MB, Wilson AM (2013) Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: a randomised controlled trial. Thorax 68(2):155–162. doi:10.1136/thoraxjnl-2012-202403
Komiya K, Ohta S, Arima K, Ogawa M, Suzuki S, Mitamura Y, Nunomura S, Nanri Y, Yoshihara T, Kawaguchi A, Kadota JI, Rubin BK, Izuhara K (2017) Clarithromycin attenuates IL-13-induced periostin production in human lung fibroblasts. Respir Res 18(1):37. doi:10.1186/s12931-017-0519-8
Moore B, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM (2013) Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol 49(2):167–179. doi:10.1165/rcmb.2013-0094TR
Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Konigshoff M, Kolb M, Laurent GJ, Nanthakumar CB, Olman MA, Pardo A, Selman M, Sheppard D, Sime PJ, Tager AM, Tatler AL, Thannickal VJ, White ES (2017) An official American Thoracic Society Workshop Report: use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol 56(5):667–679. doi:10.1165/rcmb.2017-0096ST
Uchida M, Shiraishi H, Ohta S, Arima K, Taniguchi K, Suzuki S, Okamoto M, Ahlfeld SK, Ohshima K, Kato S, Toda S, Sagara H, Aizawa H, Hoshino T, Conway SJ, Hayashi S, Izuhara K (2012) Periostin, a matricellular protein, plays a role in the induction of chemokines in pulmonary fibrosis. Am J Respir Cell Mol Biol 46(5):677–686. doi:10.1165/rcmb.2011-0115OC
Chen B, You WJ, Liu XQ, Xue S, Qin H, Jiang HD (2017) Chronic microaspiration of bile acids induces lung fibrosis through multiple mechanisms in rats. Clin Sci (Lond) 131(10):951–963. doi:10.1042/cs20160926
Akram KM, Samad S, Spiteri MA, Forsyth NR (2013) Mesenchymal stem cells promote alveolar epithelial cell wound repair in vitro through distinct migratory and paracrine mechanisms. Respir Res 14:9. doi:10.1186/1465-9921-14-9
Hong L, Sun H, Lv X, Yang D, Zhang J, Shi Y (2010) Expression of periostin in the serum of NSCLC and its function on proliferation and migration of human lung adenocarcinoma cell line (A549) in vitro. Mol Biol Rep 37(5):2285–2293. doi:10.1007/s11033-009-9721-1
Chetta A, Zanini A, Foresi A, D’Ippolito R, Tipa A, Castagnaro A, Baraldo S, Neri M, Saetta M, Olivieri D (2005) Vascular endothelial growth factor up-regulation and bronchial wall remodelling in asthma. Clin Exp Allergy 35(11):1437–1442. doi:10.1111/j.1365-2222.2005.02360.x
Suzaki I, Kawano S, Komiya K, Tanabe T, Akaba T, Asano K, Suzaki H, Izuhara K, Rubin BK (2017) Inhibition of IL-13-induced periostin in airway epithelium attenuates cellular protein expression of MUC5AC. Respirology 22(1):93–100. doi:10.1111/resp.12873
Acknowledgements
Funded by NIH HL115618 (Moore, B.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
O’Dwyer, D.N., Moore, B.B. The role of periostin in lung fibrosis and airway remodeling. Cell. Mol. Life Sci. 74, 4305–4314 (2017). https://doi.org/10.1007/s00018-017-2649-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2649-z