
Abstract
Major depressive disorder (MDD) is a common psychiatric disorder effecting approximately 121 million people worldwide and recent reports from the World Health Organization (WHO) suggest that it will be the leading contributor to the global burden of diseases. At present, the most commonly used treatment strategies are still based on the monoamine hypothesis that has been the predominant theory in the last 60 years. Clinical observations show that only a subset of depressed patients exhibits full remission when treated with classical monoamine-based antidepressants together with the fact that patients exhibit multiple symptoms suggest that the pathophysiology leading to mood disorders may differ between patients. Accumulating evidence indicates that depression is a neural circuit disorder and that onset of depression may be located at different regions of the brain involving different transmitter systems and molecular mechanisms. This review synthesises findings from rodent studies from which emerges a role for different, yet interconnected, molecular systems and associated neural circuits to the aetiology of depression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major depressive disorders (MDD) are projected to become the leading contributor to the global burden of disease according to the world health organisation (WHO) and other studies [1–3]. There have been dramatic advances in the treatments for various diseases that target the immune and cardiovascular systems. In comparison, treatments for depressive disorders, predominantly based on the monoamine hypothesis, still relies on approaches developed over 60 years ago [4, 5]. Depression is a diverse and complex mental illness with different antecedent causes and manifestation of symptoms. The major drawback of classical antidepressant treatment is that less than 50 % of patients respond positively to current antidepressant medications while alleviation of depressive symptoms can take weeks or months for those who do respond to treatment [6, 7]. The limited understanding of the neural circuitry and molecular mechanisms involved in depression is one of the key reasons for the lack of more effective treatments. Recent groundbreaking studies showing rapid alleviation of depression with deep brain stimulation (DBS) strongly suggests that depression is a neural circuit disorder [8–11]. Advances in molecular studies have highlighted the role of specific molecular signalling and epigenetic changes and the associated modifications in neuronal development of depression [12–17]. It is likely that these differential molecular and functional responses in various brain regions may be responsible for the wide spectrum of symptoms observed in depressive patients.
Studies in depression have benefited from the availability of various technical approaches enabling researchers to investigate molecular changes at the level of the single cell while others take a more expansive systems-level approach to investigate functional changes at the level of the neural circuit. In this review article, we try to highlight the major points to give the reader a flavour of the current standings in different aspects of research in depression. Since various biological processes effect different systems such that, for example, molecular changes can then lead to changes in neural circuit activity, it will be evident that description of certain processes will overlap with other sections within the review. We begin this review by first describing evidence for molecular changes linked to depression followed by genetic modifications and associated neurogenesis leading to structural changes associated with depression-related synaptic plasticity. We then focus on the more expansive studies showing evidence for the role of the immune system in depression followed by findings from systems-level approaches describing functional changes of the neural circuit in depression. Finally, we further emphasise functional circuit-level complexity of depression by describing the role of two distinct regions of the brain, the circadian and sleep/wake centres, in depression.
Molecular signalling
Brain-derived neurotrophic factor (BDNF) has numerous functions in the developing and mature brain [18, 19]. Among these is signalling within the mesolimbic brain reward centre, ventral tegmental area-nucleus accumbens (VTA-NAc) dopaminergic circuit, in encoding for depression related behaviour. For example conditional deletion of BDNF expression in the VTA, but not NAc, prevents the development of social defeat stress-induced behavioural abnormality [12, 20]. Furthermore, evidence suggests that the increase in BDNF signalling in the VTA-NAc circuit is stress-context dependent since the phasic activity of VTA-NAc dopamine neurons leads to increased BDNF levels in the NAc of social stress experienced, but not stress naïve mice [21]. In the NAc the stress induced neuropeptide corticotropin-releasing factor (CRF) is functional at both the pre- and post-synaptic terminals [22], and increased CRF signalling in the NAc increases motivation for cued rewards [23]. It is hypothesised that stress-induced CRF gates BDNF signalling in the NAc [21]. In this model, CRF signalling in NAc activates its receptors located presynaptically on VTA DA neurons and works together with arriving phasic spikes from these DA neurons to release BDNF unto medium spiny neurons (MSNs) in the NAc. In addition to the NAc, exposure to stress increases BDNF levels in the amygdala in a time dependent manner [24, 25]. BDNF mRNA levels were shown to be increased in the BLA and CeM, but not medial amygdala, 2-h following stress exposure while 24-h post-stress exposure BDNF levels were shown to be reduced [24, 26, 27]. In contrast, decreased BDNF levels were found in the postmortem amygdala of female subjects with MDD [28]. The differential temporal expression of BDNF mRNA expression indicates that stress-induced changes in BDNF expression are transient in the amygdala. Evidence supports the notion that the role of BDNF in mood disorders varies between different brain regions. For example, elevated NAc BDNF increases the depressive phenotype, while findings from the hippocampus and PFC indicate increased BDNF levels reverses the depressive phenotype. In humans studies, lower levels of BDNF were found in the hippocampus and PFC of subject who had committed suicide while levels were elevated in patients treated with antidepressants [29–32]. Recent studies further confirmed that chronic antidepressant treatment increases BDNF and TrkB receptor mRNA expression in the hippocampus [33]. In contrast, post-mortem analysis of depressed patients found higher levels of BNDF in the NAc [12]. Though the mechanism of how antidepressant treatment increases BDNF-TrkB expression is unknown, a possible mechanism may involve increased monoaminergic transmission [33, 34]. Findings that antidepressants induced increases in BDNF overlaps closely with increases in both CREB mRNA expression and CREB-phosphorylation state within the hippocampus have led to the suggestion that CREB may contribute to the antidepressant-induced increase in BDNF expression [33]. The mechanisms of action of BDNF in the context of depression in these various brain regions are still unclear. The differential interaction of pre- and post-synaptic BNDF-TrkB pathway with various intra and extracellular signalling systems such as second messenger pathways (including phospholipase C, PI3K and MAPK/ERK pathways), inflammatory cytokines, excitatory glutamatergic neurotransmitters and stress hormones could in part result in the contrasting observations of BDNF levels in the different regions of the brain in relation to depression [35].
Previous studies have implicated changes in the level of the intracellular signalling molecule calcium/calmodulin-dependent protein kinase II (CaMKII) following stress and antidepressant responses [36–38]. βCaMKII is known to regulate activity of various intracellular pathways and channels that regulate neuronal activity [39]. The lateral habenula (LHb) is an evolutionary conserved nucleus that integrates inputs from various limbic forebrain and motor systems involved in regulating motivational behaviours [40]. Furthermore, aberration in LHb function has been implicated in depression [41–43]. Recent evidence has shown that stress-induced increased activity of LHb neurons is the result of up-regulation of βCaMKII activity leading to increased membrane insertion of the excitatory glutamate receptor GluR1 [44]. In contrast, treatment with antidepressants down-regulated CaMKII expression in the LHb [44].
Clinical studies consistently demonstrate that a single sub-psychotomimetic dose of Ketamine, an ionotropic glutamatergic NMDAR (N-methyl-d-aspartate receptor) antagonist, produces rapidly acting antidepressant responses in patients suffering from depression [45–47]. Ketamine and other NMDAR antagonists together with metabotropic GluR2/3 receptor antagonists also produce rapid antidepressant-like effects in mouse behaviour [48–50]. By determining the degree of immobility, a measure of helplessness associated with the depressive phenotype, in the forced swim test (FST) paradigm, NMDAR antagonist ketamine, CPP and MK-801 were shown to alleviate depressive behaviour within 30 min of drug administration [48]. The antidepressant effects of ketamine lasted 24-h post administration, much longer than its 2-h half-life, indicating that ketamine induces longer-term changes. In line with its putative role in inducing longer-term plastic changes, administration of ketamine increased BDNF levels in the hippocampus [48]. How is it that blocking excitatory NMDAR activity elicits plasticity in the hippocampus? Prior work had demonstrated that NMDAR block maintains eukaryotic elongation factor (eEF2) in the dephosphorylated (active) state [13]. Mechanistically, ketamine blockade of NMDA receptors deactivates CaMKII (also known as eukaryotic elongation factor 2 kinase—eEF2k) activity and the subsequent decrease in phosphorylation of eEF2 leads to rapid hippocampal BDNF synthesis by removal of baseline phosphorylated-eEF2 suppression of BDNF synthesis [48, 51, 52]. The antidepressant actions of ketamine are blocked by pretreatment with glutamate-AMPA receptor antagonist [53]. Detailed analysis showed that acute suppression of NMDA receptor mediated spontaneous neurotransmitter release leads to rapid BDNF protein synthesis resulting in increased AMPA receptors surface expression and subsequent increases in potentiation of synaptic responses in the hippocampus [52]. Activation of NMDA receptors is believed to regulate mTOR activity via a variety of putative intracellular signalling mechanisms such as the phosphatidylinositol 3 kinase—AKT (PI3K-AKT) and calcineurin pathways [54–56]. The mammalian target of rapamycin (mTOR) signalling pathway is a serine/threonine kinase, that regulates the initiation of protein translation, is expressed in dendritic development that controls new protein synthesis [57, 58]. Changes in activity of various signalling processes such as BDNF, NMDA and their downstream target mTOR are possible mechanisms that underlie changes in synaptic plasticity leading to depression [59]. For example, changes in homeostatic plasticity in regions such as the PFC are putatively linked in part to aberrations in mTOR signalling, which has been hypothesised to lead to the expression of the depressive phenotype [59]. Convergent evidence over the past 20 years indicates that prolonged stress leads to overall neuronal atrophy and synaptic depression in the PFC and hippocampus [59–62] while regions such as the amygdala and NAc exhibit changes consistent with neuronal hypertrophy and synaptic potentiation [59, 63, 64]. Why stress induces opposing effects in different brain regions is still unclear and much more work is required to delineate these molecular differences. Some of the pathways involving stress-induced changes in BDNF and NMDA-mTOR signalling have been better studied in the PFC. For example, synaptic deficits in the PFC following stress has been shown to be precipitated by a initial increase in glutamate release and reduced glutamate uptake leading to increased glutamate excitotoxicity, thus resulting in neuronal atrophy through dendritic retraction, reduced dendritic arborisation, decreased spine density and reduced synaptic strength [59]. Emergence of such synaptic dysconnectivity can potentially lead to the reduction of neurotrophic factors such as BDNF and the overall decrease in NMDA signalling and the inhibition of mTOR signalling that subsequently leads to the expression of depressive-like behaviour [59, 65, 66]. This putative mechanism by which stress induces synaptic changes in the PFC highlights the significance of cortical mTOR signalling underlying ketamine-mediated antidepressant responses [67, 68]. Thus, at the micro-circuit synaptic level, a possible mechanism, by which Ketamine-induced NMDA blockade can reverse depressive behaviours may initially involve inhibition of pre-synaptic NMDA receptors at GABAergic interneurons leading to decreased inhibitory tone and subsequent net increase in glutamatergic surge; while inhibition of, excitotoxic, extrasynaptic NMDA receptors on the post-synaptic neurons increases cell survival. Furthermore, increased net glutamatergic surge leads to increased postsynaptic AMPA receptor activation of neuroplasticity-related signalling pathways involving BDNF and mTOR, resulting in overall synaptogenesis and synaptic potentiation [48, 59, 68, 69]. The complex interactions between various pathways are evidenced by findings that activation of L-type voltage sensitive calcium channels (VSCC) resulted in AMPA receptor stimulation leading to mTOR pathway activation [70]. In line with this, pretreatment with L-type VSCC antagonist blocked the antidepressant effects of ketamine [71]. Hence, another possible mechanism by which ketamine induces antidepressant effects may involve activation of both L-type VSCC and AMPA receptors subsequently leading to downstream activation of the mTOR pathway.
The neuroendocrine system consisting of the hypothalamic-pituitary-adrenal axis (HPA axis) controls reactions to stress and is involved in homeostatic regulation of processes such as digestion, immune system, mood and emotions, energy storage and expenditure. The HPA axis plays a pivotal role in regulating physiological responses induced by stress events where its activation prepares the body for the fight/flight response. Depression and anxiety disorders are often characterised by altered HPA-axis [72–74]. During periods of stress the HPA axis releases corticotrophin-releasing hormone (CRH) from the hypothalamus, which stimulates adrenocorticotropin (ACTH) hormone secretion from the anterior pituitary, which then stimulates secretion of cortisol and other glucocorticoids from the adrenal cortex. Chronic (long-term), but not acute (short-term), unpredictable stress paradigm induced impairment in adrenal medullary function as measured by robust decreases in numerous proteins such as catecholamines, and SNARE proteins [75]. Increased cortisol concentration in plasma, urine and cerebrospinal fluid, and exaggerated cortisol response to ACTH has been reported in MDD patients [76–79]. Furthermore, increased levels of CRH have been found in cerebrospinal fluid, locus coeruleus and PFC of depressed and suicide patients [79–83]. Animal studies strongly corroborate the role of the HPA axis in depression where early life stressors such as maternal separation induce increased CHR mRNA expression in hypothalamic, paraventricular nucleus, central nucleus of the amygdala, bed nucleus of the stria terminalis and locus coeruleus [84]. Direct ACTH administration increased both the serum cortisol levels and the depressive phenotype as measured by the forced swim test and latency to feed in a novelty suppressed feeding test [85]. Furthermore, ACTH administration decreased hippocampal BDNF levels, evidence that correlates well with observations of decreased BDNF levels in the hippocampus of animals expressing the depressed phenotype. As will be discussed later mood disorders are associated with disruptions in homeostatic plasticity and associated with this, recent data suggest that stress changes neural connections. Chronic stress reduces spine density in hippocampus, mPFC, medial amygdala, while in the basolateral amygdala spine density was increased [86, 87]. Glucocorticoids exert both direct and non-direct genomic effects that can regulate intracellular signalling and synaptic plasticity [87]. Acute cortisol challenges given to stress naïve or chronically stressed rats revealed highly differential gene profiles depending on the stress history of the animals, where 200 of the genes altered by cortisol was similar in both groups of rats; however, 500 genes were differentially altered following chronic stress [87, 88]. These findings would indicate that though the brain is able to recover following exposure to acute stress, stress history leads to changes that alter future molecular reactivity to subsequent stress that may increase susceptibility to depression. Recent findings have shown that the levels of presynaptic auto-inhibitory mGluR2 receptors are decreased in stress susceptible mice and that the antidepressant drug acetyl-L-carnitine, via epigenetic mechanisms, induces expression of presynaptic mGluR2 receptors leading then to the normalisation of glutamate transmission and the subsequent reversal of the depressive phenotype [89]. A recent follow up study showed differences in mGluR2 expression following CUS exposure where hippocampal mGluR2 expression was decreased only in stress susceptible mice while PFC mGluR2 expression was decreased in both stress resilient and stress susceptible mice [90]. In addition, evidence for the differential effects of stress duration was observed where mice exposed to acute stress still exhibited decreased hippocampal mGluR2 expression but no effect was observed in the PFC [90]. Furthermore, mineralocorticoid receptor, but not glucocorticoid, levels in the hippocampus were lower in stress resilient mice while there was no difference in the PFC [90]. Using pharmacological methods, the authors postulate that glucocorticoid activation regulates mGLuR2 expression via mineralocorticoid receptor activation [90]. The differential molecular response in mGlu2 expression in stress resilient and susceptible mice further highlights the complex responses to stressors in varying regions of the brain. Furthermore, a better understanding of the interaction within the neuroendocrine system in stress will help elucidate novel therapeutic targets for antidepressant treatment. The complexity of biochemical cross-talk at the molecular level further highlights potential variability in the activation of signalling pathways in various brain regions and circuits that may underlie the large variation in symptoms of depressive patients.
Epigenetic mechanisms
Genetics alone does not account for MDD due to the high discordance rate (50 %) observed in monozygotic twins [91]. Environmental factors, particularly exposure to stressful life events increases the likelihood of the onset of depression [92, 93]. However, most people exposed to environmental stress do not exhibit MDD [94]. This variability in vulnerability to environmental stress has lead to the hypothesis that epigenetic mechanisms may determine risk for depression throughout life [95]. Epigenetics is a molecular phenomenon describing heritable changes in gene expression that does not involve changes in DNA sequence [96]. Briefly, DNA wrapped around histone protein makes up the nucleosome—the unit of chromatin. Genes within tightly spaced nucleosomes are actively transcribed while genes within tightly packed nucleosomes are silenced. Complex post-translational processes such acetylation or methylation on histone tails lead to changes in nucleosome spacing that result in modulation of gene expression [97]. Histone acetylation, catalysed by histone acetyltransferase (HATS) and reversed by histone deacetylase (HDACs), generally drives an open state of the chromatin resulting in increased gene expression. Histone methylation, catalysed by histone methyltransferase (HMTs) and reversed by histone demethylase (HDMs), can activate or reverse gene expression. In addition, DNA methylation, catalysed by DNA methyltransferase (DNMTs) generally, but not always, drives gene repression. Post-translational modification of these acetylation and methylation are carried out by numerous ‘reader’ and ‘eraser’ proteins resulting in change of chromatin structure and gene transcription [96, 97]. Growing evidence supports the hypothesis that epigenetics is a mechanism by which exposure to environmental stimuli such as stress lead to changes in gene expression in vulnerable, genetically predisposed, individuals resulting in MDD. One of the earliest studies to report epigenetic modifications in an animal model of depression demonstrated transient decrease followed by persistent increase of histone acetylation in the NAc following chronic social defeat (CSD) stress [98]. Furthermore, direct infusion of HDAC inhibitors reversed the depressive phenotype in previously depressive mice [98]. More recently, conditional inhibition of HDAC2 function in the NAc induced the stress-resilient phenotype in mice exposed to chronic stress [99]. Conversely, decreasing HDAC5 function leads to a stronger expression of depressive-like behaviour following CSD stress [100]. In contrast, HDAC5 expression was significantly elevated in the hippocampus of rats following chronic mild stress [101]. Epigenetic changes exhibit differing temporal dynamics in different brain regions following exposure to CSD stress. For example, in the hippocampus of stress-susceptible mice histone acetylation is initially increased followed by a persistent decrease [102] while in the NAc increased histone acetylation was observed [100]. Levels of histone methyltransferase G9a, GLP and SUV39h1 was decreased in the NAc of mice susceptible to CSD stress, while in resilient mice the levels were increased [103]. Furthermore, overexpression of G9a in the NAc exerted antidepressant-like behaviours, whereas knockdown of G9a induced pro-depressant-like effects [103]. A more recent study using engineered transcription factors (zinc finger proteins—ZFP) to site selectively remodel chromatin found that ZFP-induced enrichment of H3K9me2 at fosb gene in the NAc reduced both fosb/delta fosb expression and also induced anxiety and depressive-like behaviours following subthreshold social defeat stress [104]. Classical antidepressant medications, such as selective serotonin reuptake inhibitor (SSRI), imipramine was shown to increase NMDA receptor 2B (NR2B) subunit expression via mechanisms involving increased histone acetylation on the NR2B promoters and decreased activity of HDAC [105]. As mentioned previously expression of neurotropic factor BDNF is implicated in animal models of depression [12, 20]. Recent clinical studies have extended these findings in the context of epigenetics where patients with MDD exhibited increased levels of methylation on the BDNF promoter while treatments with antidepressant decreased BDNF promoter methylation [106]. Furthermore, it has been suggested that DNA methylation of BDNF in the peripheral blood can be used as a biomarker of epigenetic changes in the brain of patients susceptible to depression [107]. It is likely that these differential epigenetic responses in various brain regions may be responsible for the wide spectrum of symptoms observed in depressive patients.
Exposure to early life stress and subsequent epigenetic programming in the brain is hypothesised to be a potential cause of MDD later in life [108]. Postnatal adversity following maternal separation in rodents has been shown to lead to broad changes in histone modifying enzymes and to post-translational histone modifications. Rodents exposed to maternal separation display reduced levels of HDAC mRNA in the PFC [109], while some studies revealed elevated levels of histone acetylation [110, 111]. In addition, early life stress following maternal separation induces epigenetic changes in DNA methylation within the hypothalamus, a brain region associated with motivated behaviours [112]. Exposure to stressors prior to birth can also induce epigenetic changes potentially leading to the development of depression later in life. For example, significant differences in DNA methylation were observed in neonate umbilical cord blood sample taken from mothers suffering from depression and anxiety disorders [113]. Earlier studies had shown that early life experiences could have either beneficial or negative consequences to stress susceptibility in later life. For example, postnatal handling of young rodents decreased both stress reactivity and associated basal stress hormone levels in adulthood while maternal separation resulted in increased stress reactivity [114]. In addition to epigenetic changes associated with early life stress, there is growing literature suggesting that early life stress increases vulnerability in offsprings of these animals to stress [114, 115]. Furthermore transmission of susceptibility of stress and mood disorders has been observed in human subjects, where for example maternal, but not paternal, exposure to PTSD resulted in increased risk of PTSD in children of holocaust survivors [116]. Early animals studies of stress transmission had demonstrated that variation in maternal care served as a basis for a nongenomic behavioural transmission of individual differences in stress susceptibility across several generations [114, 115]. For instance when biological offsprings of mothers (dams) who exhibited low maternal caring behaviour were placed under the care of dams that exhibited high maternal caring behaviour, these offsprings exhibited significantly less fearful responses to novelty than the offsprings of dams who exhibited high maternal caring behaviour that had been subsequently placed under the care of dams exhibiting low maternal caring behaviour [114, 115]. Furthermore early life experience of maternal care was shown to induce differential expression of receptors associated with stress sensitivity, such as glucocorticoid, benzodiazepine and corticotropin-releasing factor (CRF) receptor mRNA [115]. A recent study had demonstrated transmission of stress vulnerability via epigenetic mechanisms by changes in the germ cells that subsequently influence the behaviour of the offsprings for three generation in a gender dependent manner [117]. In addition to increased expression of the anxiety and depressive phenotype in mice that had undergone early life stress, epigenetic changes such as DNA methylation of several candidate genes associated with depression were found in the sperm germ line of these mice [117]. Strikingly, the study showed similar changes in DNA methylation in the brains of the female progeny that also exhibited both anxiety and depressive phenotypes [117]. These findings are extremely interesting as it demonstrates a potential mechanism by which stressful environmental factors can alter DNA methylation in the germline and that these alterations can be partly maintained across generations. The story, however, appears to be far more complex and subtle. In a study where mice had undergone CSD, offsprings, following natural breeding, of both stress-susceptible and stress-resilient mice were equally more vulnerable to stress [118]. Furthermore, male offsprings of fathers who had undergone CSD also exhibited significant disturbances in neuroendocrine signalling, specifically corticosterone and vestibular endothelial growth factor (VGEF), pathways previously shown to be associated with depressive-like phenotypes [119–121]. Since offsprings of males that had undergone CSD stress exposure were more vulnerable to stress in vitro fertilisation (IVF) experiments were performed in order to investigate whether the behavioural phenotype was directly transmissible through the sperm of the defeated mice. Sperm from defeated mice were used to impregnate normal female mice and the subsequent offsprings were put through a battery of behavioural measures of depression and anxiety. IVF derived mice from defeated fathers did not exhibit robust anxiety or depressive phenotypes on most measures except for the forced swim test paradigm [118]. With data from this study, together with observations that female rodents are able to adjust their reproductive investments depending on their interaction with the male, the authors concluded that the bulk of the vulnerabilities to stress are passed onto subsequent generations through behavioural mechanisms, possibly on the basis of the female detecting that she had procreated with an impaired male [118]. Given that FST alone was significantly different in IVF-derived mice [118] together with previous findings that epigenetic changes in DNA methylation was observed moderately in offsprings of stress vulnerable mice [117] could suggest that each gene possibly contributes to a small part of the behavioural phenotype and that these subtle differences were not robustly transmitted during IVF in the CSD paradigm.
In general, the evidence thus far suggests that global histone acetylation is pro-adaptive in response to stress and depression while aspects of histone methylation are pro-depressant. Evidence linking early life stress with biological changes bridges the field of social and biological sciences and highlights the importance of developing a better social system for high-risk youths. Furthermore, advances in understanding epigenetic changes following stress exposure and possible mechanisms of transmission through the generations may lead to the potential development of specific tests to determine high-risk individuals and early treatment options.
Neurogenesis
The hypothesis that antidepressant medications might alleviate depression by increasing adult hippocampal neurogenesis [122] comes from observations that classical antidepressant therapy such as SSRIs take 3–4 weeks to have efficacy and that antidepressant treatment increases the numbers of adult-born neurons [123, 124], which take about 4 weeks to form synaptic connections [125]. Numerous chronic stressors such as repeated restraint stress, chronic unpredictable mild stress, social defeat stress, social isolation and corticosterone administration that induce depressive and anxiety-like behaviours all result in impaired adult hippocampal neurogenesis [126–130]. A recent study, however, did not find any evidence of hippocampal neurogenesis in mice following exposure to numerous stress-paradigms [131]. Since it is unclear in this study whether exposure to stressors induced depressive or anxiety-like behaviours in the mice (as they did not check for this), it has been hypothesised that chronic stress impairs hippocampal neurogenesis only in those animals that exhibit depressive or anxiety-like behaviours [132]. Hypersecretion and sustained elevations of the stress hormone cortisol is neurotoxic [133]. Furthermore, elevation of cortisol suppresses BDNF, which in turn leads to neurodegeneration and contributes to symptoms of depression [134]. The neurotropic factor BDNF is involved in neurogenesis by regulating neuronal differentiation and growth [135, 136]. Human post-mortem studies have shown decreased BDNF levels in the hippocampus of depressed patients [30]. It is likely that the decreased hippocampal volume in depressed patients may be associated with decreased BDNF levels [137–139]. Antidepressants in animals have been shown to reverse cortisol- or stress-induced suppression of cell proliferation [140–142]. In addition the SSRI sertraline was recently shown to increase neurogenesis in hippocampal progenitor cell lines by increasing protein kinase A signalling, glucocorticoid receptor phosphorylation and the subsequent activation of a specific subset of genes [143]. Regulation of glucocorticoid release following stress exposure serves numerous beneficial homeostatic functions while dysregulation of glucocorticoids is associated with cognitive and depressive disorders [86, 144]. Within the hippocampus, a region abundant in glucocorticoid receptors that is also functionally important in cognitive and emotional behavioural processes, stress and elevated glucocorticoid levels inhibits neurogenesis [145]. Further evidence for the link between neurogenesis and stress come from recent findings that conditional inhibition of hippocampal neurogenesis, in a transgenic mouse line, lead to significant elevation of glucocorticoid levels following exposure to acute stress [146]. Furthermore, behavioural analysis on transgenic mice lacking the ability for neurogenesis found that these mice exhibited both baseline depressive phenotypes and greater susceptibility to stress when compared to wild-type controls [146]. Conversely, in experiments using an inducible transgenic mouse line in which adult neurogenesis could be increased demonstrated that these mice were resilient to chronic treatment with corticosterone, a mouse model of stress [147]. Neurogenesis may exhibit temporal characteristics in relation to depression. For example, immediately following exposure to CSD-stress, both stress-resilient and stress-susceptible mice exhibited increased levels of stress hormone and concomitant transient decrease in hippocampal neurogenesis that was normalised within 24 h [148]. Surprisingly, however, hippocampal neurogenesis was significantly elevated in stress-susceptible mice 4 weeks post-CSD [148]. Observations that ablation of hippocampal neurogenesis prior to CSD stress induced the stress-resilient phenotype strongly suggests that the compensatory enhancement of hippocampal neurogenesis is related to long-term individual differences in maladaptive stress responses [148]. The studies described above are interesting and point to a new avenue of research for treatments in mental disorder; however, the evidence for the role of neurogenesis and depression is still unclear. For example, neurogenesis does not occur in a significant amount in the elderly, yet antidepressants have been shown to be effective in treating elderly depressed patients [149]. Thus, although numerous studies have shown that stress decreases neurogenesis it should be noted that various studies have also reported no correlation between stress and neurogenesis [131]. For example, at the cellular level administration of antidepressant SSRIs have shown to ‘demature’ older granule cells to more plastics functional states without actually promoting neurogenesis [150, 151]. Furthermore, there are reports of antidepressants at behaviourally active or clinically relevant doses that do not exert any effect on processes involving neurogenesis [131]. Though, it should be noted that these analysis were performed on non-stressed, and presumably, non-depressed, animals. Other behavioural studies have highlighted a mixed bag of results. For example, one report, suggested inescapable shock decreased neurogenesis only after the development of contextual fear association, thus strongly suggesting that the important factor in modulation neurogenesis is not stress or fear, but the emotionally charged learning [152]. However, another study using a similar paradigm did not find any changes in neurogenesis even after the occurrence of substantial learning [153]. Another important complication in the findings is that the majority of the experiments in neurogenesis, and depression as a whole, are performed on males. Experiments utilising both males and females have found vastly different results between the sexes [154, 155]. Overall, the neurogenesis hypothesis of depression is supported by numerous studies; however, the existence of contradictory findings suggests that neurogenesis can be effected by stress and antidepressants under certain conditions but that these effects do not appear in all conditions of psychological stress, depression and antidepressant treatment [131]. It is clear that much more work is needed to clarify the role of neurogenesis and depression in different stress conditions and between the sexes.
Neural circuits
Deep brain stimulation (DBS) is a well-accepted procedure for the treatment of movement disorders such as Parkinson’s disease [156]. Recent groundbreaking studies have shown that DBS, which involves delivery of focal electrical current to specific neural structures within the brain, rapidly alleviates symptoms of depression [9, 10]. The effectiveness of DBS in alleviating depression in patients when targeted to various brain regions suggests that depression is a neural circuit disorder [9, 157–161]. Figure 1 shows a representation of some of the regions of the brains and their connections that have been implicated in depression. DBS as a therapeutic option for treatment-resistant patients is based on the novel hypothesis that depression come about as a result of aberrations in communications between specific neural structures of the brain. Thus, focal neuromodulation with DBS is believed to ‘reset’ neural activity of aberrant circuits associated with depression. At present the mechanism by which the pathophysiology of neural circuits lead to depression is unclear. However, recent approaches combining animal models of depression together with electrophysiological, optogenetic and molecular analysis have begun to reveal a complex interplay between various neural circuits and cell types in encoding for depression [15, 162–168]. The mesolimbic dopaminergic pathway composed of dopaminergic (DA) neurons in the ventral tegmental area (VTA) and their projections to the nucleus accumbens (NAc) is crucial for the recognition of emotionally salient stimuli such as reward [169] and aversion [170]. In addition DA neurons of the VTA-NAc circuit play a key role in modulating depression-related behaviours [6, 12, 18, 20, 21, 163, 168, 171, 172]. Earlier work has shown that the in vitro firing rate and in vivo phasic firing events of VTA DA neurons in the brain’s reward system is significantly increased in mice exhibiting the susceptible (depressed) phenotype following exposure to the chronic social defeat paradigm [12, 171, 173, 174]. Conversely in vivo recordings of rats susceptible to the learned helplessness paradigm exhibited decreased VTA DA neuron activity and that the antidepressant ketamine both rescued those rats previously susceptible to the learned helplessness paradigm and increased VTA DA activity [175]. Two recent optogenetic studies directly demonstrated the role of VTA DA neurons in depression [163, 168]. In one study optogenetic induction of phasic, but not tonic, firing of VTA DA neurons was shown to rapidly induce the susceptible (depressed) phenotype in mice that had previously undergone a subthreshold social defeat stress paradigm [163]. Conversely, the second study showed the opposite effects where phasic activity of VTA DA neurons rescued stress-induced depressive-like behaviour in mice that had undergone chronic mild stress [168]. The discrepancy between the two studies has been discussed [176, 177] and may highlight differential coding processes of VTA DA neurons for strong or weak stressful stimuli [166, 178], which is highly consistent with the fine context-detecting functions of VTA DA neurons [21]. It has recently been shown that rats put through a strong stressful paradigm (restrained stress) exhibited increased firing in the VTA while those put through a weaker stress paradigm (mild inescapable stress) exhibited decreased activity [179]. A possible explanation for the differential coding for weak or strong stressors might lie in the existence of functionally distinct populations of VTA DA neurons. Those VTA DA neurons that exhibited decreased firing following weak stress exposure were located primarily in the medial and central portions of the VTA [179]. Furthermore, VTA DA neurons located in ventral VTA are excited by noxious footshock while dorsal VTA DA neural activity is inhibited [180]. Differential activity coding has also been demonstrated in mice exposed to the same stressor. VTA neurons projecting to the NAc (VTA-NAc) exhibit increased firing while those projecting to mPFC (VTA-mPFC) exhibit decreased firing in mice susceptible to social defeat stress [163]. Likewise, rapid induction of the depressed phenotype was observed by (1) optical induction of phasic activity in the VTA-NAc circuit and (2) optical inhibition of the VTA-mPFC circuit [163]. These projection-specific DA neurons exhibit differing physiological properties. DA neurons projecting to NAc exhibit robust I h currents (hyperpolarisation-activated non-selective cation channel mediated currents) while DA neurons projecting to mPFC lack robust I h currents [172, 181]. The degree of complexity of these circuits is evident by observations that subpopulations of VTA DA neurons receive synaptic inputs from different nuclei in the brain. For example, neurons from the laterodorsal tegmentum (LDTg) synapses primarily on VTA DA neurons projecting to the NAc, while neurons from the lateral habenula (LHb) synapse either onto VTA DA neurons projecting to the mPFC or to GABAergic neurons in a portion of the VTA also known as the rostromedial tegmental nucleus—RMTg [182]. In addition, these circuits encode opposing behaviours where for example selective activation of LTD and LHb inputs to the VTA elicit reward and aversive behaviours respectively [182]. The LHb, a nucleus that integrates signalling from the basal frontal cortical areas and midbrain monoaminergic nuclei, has a functional role in motivated behaviours [183] and LHb neurons projecting specifically to the RMTg mediate behavioural avoidance [184]. Studies have shown increased activity of LHb neurons projecting to the VTA in mice exhibiting the depressed-phenotype following exposure to a learned helplessness model of depression [43] and to a chronic social defeat stress paradigm [185]. Furthermore, excitatory basal ganglia input to the LHb, that has been shown to encode aversive behaviour, is suppressed by serotonin [186]. This is interesting as it highlights a putative circuit that may be a target for classical monoaminergic uptake blocker antidepressants. The majority of neurons of the VTA are dopaminergic though a small percentage are glutamatergic and GABAergic [187]. The LHb is a key neuroanatomical regulator of midbrain reward circuits and increased activity of LHb projections to VTA is known to encode for depression [43, 185], it is likely, therefore, that aberration in the VTA-LHb-VTA loop may lead to depression-like behaviours. VTA DA neurons receive potent inhibitory inputs from the Ventral Pallidum (VP) while the basolateral amygdala, a region associated with stress and fear learning, sends excitatory glutamatergic inputs to the VP [188, 189]. As previously described in relation to stress, DA and depression, acute stressors activate the DA system [51] followed 24-h later by potent attenuation of DA activity [16]. A recent study had shown that the BLA-VP circuit mediates the decreased VTA DA neural activity in stress susceptible rats following exposure to CMS [190]. Evidence for the role of the BLA-VP-VTA circuit in modulating depression-related behaviours comes from the findings showing (a) the decreased VTA DA activity in CMS susceptible rats was restored by blockade of excitatory glutamatergic inputs to the VP, and (b) pharmacological activation of BLA decreases VTA DA activity [190].

Simplified schematic of the major neural circuit connections involved in encoding for depression-related behaviours. The network displays the complex interplay between numerous neurotransmitters in regulating cellular activity within various brain nuclei. 5-HT serotonin, ACh acetylcholine, DA dopamine, GABA gamma-aminobutyric acid, Glu glutamate, NE norepinephrine, VIP vasoactive intestinal peptide
Disregulation in signalling of the monoamine neurotransmitter, serotonin, in depression has been known for the last 60 years with the accidently discovery that inhibitors of monoamine neurotransmitter breakdown alleviate depression. The dorsal raphe nucleus (DRN), the main source of the brains serotonin (5-HT), is implicated in the pathophysiology and therapeutics of mental disorders such as autism, anxiety and depression [191, 192]. The DRN has reciprocal projections with various regions of the brains involved in regulating emotions such as the LHb, hippocampus, hypothalamus, amygdala and cerebral cortex [191]. Furthermore, DRN, neurons are precisely regulated, both phasically and tonically, by glutamatergic and inhibitory GABAergic neural inputs from various regions implicated in mood disorders [193]. In line with the monoamine hypothesis of depression, 5-HT levels were decreased in mice exhibiting the depressed phenotype following chronic mild stress [194]. Furthermore, evidence suggests a close link between the LHb and DRN circuit in depression since lesion of the LHb alleviated symptoms of depression by increasing 5-HT levels [194]. The LHb and mPFC are critical regulators of DRN activity, where glutamatergic projections from these regions attenuate DRN activity via a feedforward inhibitory mechanism involving inhibitory interneurons [195–197]. Investigations into the micro-circuitry of DRN in encoding for depression showed increased excitability of GABAergic DRN neurons in mice susceptible, but not those resilient, to social defeat stress [198]. Furthermore, this study found associated decreases in 5-HT neuron activity in the DRN of susceptible, but not resilient mice, implicating that in the DRN, encoding for depression leads to increased GABA activity followed by the subsequent decrease in 5-HT neuron activity [198]. Follow-up studies showed a direct functional correlation between mPRC-DRN circuit activity and depression-related behaviours where optogenetic modulation of mPFC neurons projecting to DRN functionally increased DRN GABAergic interneuron activity and increased or decreased depressive-like behaviour in mice that undergone social defeat stress [197].
The locus coeruleus (LC), the major noradrenergic nuclei in the brain, is a vital component of the stress response. In rodents, stress increases LC activity [199] and treatments with antidepressants such as SSRIs, reduce LC activity, possibly via pre-synaptic autoreceptor activation [200]. Anatomically the LC has reciprocal connections with various regions of the brains also known to be associated with depression [201]. Significant increases in gene expression levels for NMDA and mGluR subunits were found in the LC of post-mortem depressed patients [16] supporting the notion that disrupted glutamatergic-noradrenergic interactions in the LC leads to depression. Electrophysiological measures of basal firing rates of LC neurons in the Wistar Kyoto (WKY) rat, a strain that exhibits baseline depressive and anxiety-like behaviours, was shown to be significantly higher compared to the standard Wistar strain of rats [202]. In addition LC neurons from the WKY were found to be less responsive to the inhibitory effect of Alpha2-adrenoreceptor activation [203]. Furthermore, a role for LC dysfunction in depression comes from observations of robust glutamatergic projections from the PFC and LHb, two regions known to exhibit increased activity in depression [43, 44, 204, 205]. Neural processing of fear learning has recently been shown to pass from the lateral hypothalamus to the amygdala via the LC [206]. This study demonstrated that orexin (hypocretin) fibres from the lateral hypothalamus directly depolarize LC neurons via rapid co-release of glutamate and orexin leading to activation of NMDA and orexin-1 receptors, respectively. Furthermore, orexin activation of LC neurons leads to increased noradrenergic signalling, via beta adrenergic receptor in the lateral nucleus of the amygdala resulting in enhanced fear memory formation. Disregulation of this LH (orexinergic)-LC (noradrenergic)-amygdala circuit may be another possible mechanism by which depression occurs. Recently two populations of projection-specific neurons were identified in the basolateral amygdala that encode for reward and aversion [207]. The authors demonstrate that basal lateral amygdala (BLA) neurons projecting to the NAc exhibit increased synaptic plasticity in mice following reward training while BLA neurons projecting to the central amygdala (CeM) exhibited increased synaptic plasticity in mice that had undergone fear conditioning [207]. This finding further extends the notions that distinct population of neurons exists within a nuclei encoding for opposing behavioural phenotypes, and that aberration in balance between neural circuits may lead to mental disorders such as depression. Exposure to stress has shown shifts in the balance of network inputs from cortical and thalamic regions to the BLA. For example, activation of NE beta receptors following fear conditioned learning shifts the responses of LA neurons more robustly to the faster acting thalamic input over the slower cortical inputs [208]. The BLA receives inputs from the auditory thalamus and auditory association cortex where thalamic input encodes sensory information of the conditioned stimulus while the cortical pathway provides a more processed representation of the stimulus [209]. A recent neural circuit study extended the putative mechanistic role of DA regulation of networks within the BLA following exposure to stress. Specifically D2 receptor activation exhibited stronger net excitatory modulation of the auditory pathway and a stronger net inhibitory modulation of the cortical pathway [210]. The study of NE Beta and DA receptor modulation [208, 210] showing shifts in input towards subcortical pathways is believed to be an evolutionary advantageous fight-or-flight response in animals during acute stress while the slower cortical inputs is involved in evaluating complex environmental stimulus [208, 209]. Thus in relation to the two dynamically different processing pathways, it is hypothesised that chronic stress induces maladaptive changes in the brain [86] where, for example, repeated chronic stress could induce shifts in stronger inputs from cortical inputs where organisms become more ruminative instead of proactive, as is proposed to occur in depression [211].
Increasing evidence shows that the NAc, a region typically associated with reward-related behaviours, has a critical role in depression symptomatology including reduced motivation and anhedonia [12, 14, 15, 20, 212, 213]. Medium spiny neurons (MSNs) of the NAc and dorsal striatum are enriched in D1 or D2 receptors and they send distinct projections to basal ganglia and reward structures. NAc D1-MSNs send projections to ventral pallidum, globus pallidum, VTA and substantia nigra (SN), while NAc D2-MSNs send projections to ventral pallidum [214, 215]. These two neural populations work in concert to promote normal behaviour while imbalance in one sub-type can promote dysfunctional motivational states [216–219]. The network balance model demonstrates that activation of D1-MSNs leads to positive reward behaviour while activation of D2-MSNs leads to aversive behaviours [217, 220–222]. Exposure to chronic social defeat was shown to differentially induce expression of the transcription factor delta FosB in the NAc MSNs. Mice susceptible to CSD stress expressed elevation of delta FosB in D2-MSNs while those resilient to CSD stress expressed elevation of delta FosB in D1-MSNs [223]. Furthermore, anhedonia following restrained stress is mediated by decreased excitatory synaptic strength of NAc D1-, but not D2-MSNs [224]. These findings were further extended in a recent study where mice susceptible to CSD exhibited decreased excitatory synaptic inputs into D1, but not D2-MSNs and that chronic chemogenetic attenuation of D1-MSNs, but not D2-MSNs, activity induced depressive-like behaviours in mice previously resilient to CSD stress [164]. Furthermore, repeated optogenetic activation of D2-MSNs induced depressive-like behaviour in mice exposed to a subthreshold social defeat paradigm [164]. These findings are exciting as it demonstrates: (a) two distinct circuit mechanisms within the NAc encode for depressive-like behaviours; and (b) changes in synaptic signalling in the NAC MSNs circuit most likely requires long-term molecular changes in order for the expression of depression-like behaviours since chronic, but not acute, optogenetic and chemogenetic manipulations were required to induce susceptibility to stress. Analysis of immediate early gene expression as a measure of neural activity found that expression levels of Egr1 was decreased in the ventral hippocampus (vHIP) of mice resilient to CSD stress while Arc expression was decreased in mice susceptible to CSD [225]. Furthermore, physiological measures showed differential circuit specific synaptic adaptations following CSD where, in resilient mice, vHIP afferents projecting to NAc exhibited decreased glutamate release while mPFC afferents projecting to NAc exhibited increased glutamate release [225]. The transcription factor delta FosB regulates transcription of numerous genes in the NAc [226, 227]. Two target genes of delta FosB, AMPA glutamate receptor subunit GluR2 and Sparc-like 1 (SC1) are upregulated in the NAc of mice resilient to CSD stress [212]. Furthermore, CHIPsec analysis showed significant binding of delta FosB on the GluR2 promoter and qPCR analysis revealed sustained GluR2 mRNA in NAc of resilient mice [212]. GluR2 subunit has profound effects on AMPA receptor function, where GluR2-lacking AMPA receptors are Ca2+-permeable and show greater receptor conductance and strong inward rectification, as compared to GluR2-containing receptors [228]. This switch to GluR2-lacking AMPA receptors increases neuronal excitability [229]. Electrophysiological measure showed decrease in GluR2-mediated currents and increased inward rectification in stress susceptible, but not stress resilient mice, further implicating that increased excitability, in a subset of NAc neurons, encodes for depressive-like behaviour [212]. Several brain regions that mediate aspects of motivated, goal-directed, behaviours such as the ventral subiculum of the hippocampus, amygdala and prefrontal cortex send overlapping projections to the NAc where these inputs are integrated under dopaminergic modulatory control [230]. Furthermore, systems-level analysis has shown that DA receptor subtypes differentially regulate inputs to the NAc from the limbic system and PFC, where for example, tonic D2 receptor activation selectively attenuated inputs from the mPFC while phasic DA activity increases NAc neurons responsiveness to limbic inputs via activation of D1 receptors [231–234]. Aberration in the balance between these various inputs to the NAc is hypothesised to lead to the pathophysiology of motivated behaviours such as addiction and depression. A recent study using in vivo electrophysiology recordings in rats that had previously undergone a learned helplessness, depression, paradigm found decreased synaptic input from the vSub into the NAc of rats susceptible to, but not those resilient, to the depressive phenotype [175]. Furthermore, administration of the novel antidepressant ketamine both rescued the depressed phenotype in rats previously susceptible to the learned helplessness paradigm and also increased the vSub synaptic input to the NAc in these rats [175]. At the microcircuit level, decreased synaptic activity of vSub projections to the NAc shell, but not NAc core, specifically induced the depressed phenotype [175].
Dendritic spine plasticity is a critical element of experience-dependent reorganisation of the brain circuits and that maladaptive changes in dendritic spine development are suggested to underlie neuropsychiatric disorders such as depression, anxiety and addiction [6, 235–238]. Various stress paradigms lead to significant alterations in neuronal morphology in different cell types [239–241]. Furthermore, BDNF and its downstream targets such as IKB kinase (IKK) and the transcription factor nuclear factor kB (NFkB) have been shown to be important regulators of neuronal structure [242, 243]. Mice susceptible to CSD stress exhibit dendritic remodelling (increased stubby spine formation) together with increased excitatory input into MSNs [14]. Furthermore, molecular analysis found increases in both IKK and the associated phosphorylated IkB proteins in the NAC of mice susceptible to social stress [14]. Evidence linking stress regulated changes in spine morphology and activation of IKK activity comes from approaches involving conditional knock down of IKK. Viral mediated conditional knock down of IKK by expression of IKK dominant negative (IKKdn) protein reversed the formation of stubby spines in mice susceptible to CSD stress [14]. Experience-dependent plasticity and the associated increases in synaptic input are known to lead to changes in spine morphology [236]. The observations that VTA DA neurons projecting to NAc exhibit increased phasic firing in stress susceptible mice [163] and that, the corticotropin stress hormone gates BDNF signalling in NAc MSNs [21], provides a mechanism by which increased BDNF induces changes in IKK enzyme activity leading to associated changes in dendritic spine morphology in the NAc of stress susceptible mice [14].
Homeostatic adaptations
Recent advances have highlighted possible mechanisms that determine the brain’s ability to cope with stress. Multiple lines of evidence implicate dysregulation in the brain’s reward circuit in depression [12, 20, 163, 168, 244]. Increased activity of VTA DA neurons has been causally linked to depression-related behaviours [12, 163, 171]. Increased activity of VTA DA neuron, in stress susceptible mice, is intrinsically induced by up-regulation of I h, an excitatory driving force of VTA DA neurons [171, 245, 246] while pharmacological reduction of increased I h in susceptible mice reverses depression [171]. Furthermore chronic antidepressant treatment with fluoxetine normalizes this hyperexcitability and decreases I h in these neurons [171]. Together, these observations suggest that VTA DA neuron hyperactivity and increased excitatory I h are both pathophysiological adaptations in mice susceptible to stress. A recent study confirmed that upregulation of I h current in VTA DA neurons induced increased activity in these neurons in stress susceptible mice [172]. Surprisingly, while in resilient mice activity of these neurons was found to be normal, I h current was even higher, which was observed in parallel with increased potassium (K+) current [172]. The role of I h current on mediating the resilient phenotype was determined by overexpression of the I h current related HCN channels in VTA DA neurons in susceptible mice. Here overexpression of HCN2 channel, thus further increasing I h current in previously susceptible mice, induced the resilient phenotype [172]. In primary neuronal cultures, excessive hyperactivity has been shown to induce homeostatic upregulation of inhibitory driving force K+-mediated current [247]. Repeated optogenetic activation of VTA DA neurons in susceptible mice, that presumably exhibit increased activity in these cells, was shown to induce the resilient phenotype together with associated decreased spontaneous activity and increased K+-currents in these cells [172]. Furthermore, these observations were observed specifically in VTA DA neurons projecting to NAc but not mPFC, which correlates with previous projecting specific roles in encoding for depression [163]. Homeostatic plasticity plays a fundamental role in stabilising neuronal activity in response to excessive perturbations under both physiological [248, 249] and disease [250] conditions. Previous molecular analysis had shown that mice resilient to CSD stress exhibited normalised firing activity in VTA DA neurons associated with a corresponding increase in genes coding for subtype of K+ channels [12]. Observations that VTA DA neurons of resilient mice exhibit upregulation of the excitatory driving force I h and inhibitory driving force K+-current suggests homeostatic plasticity plays a fundamental role in stabilising neural activity in promoting natural resilience to stress. Further investigations into homeostatic adaptive mechanisms leading to natural resilience have potential implication in the development of more naturalistic treatment strategies for mental disorders such as depression.
Immune response
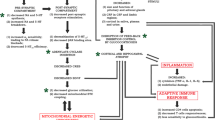
Accumulating evidence suggests that inflammatory processes play a key role in the pathophysiology of depression [251, 252]. Post-mortem samples from patients with a history of depression express elevated levels of pro-inflammatory cytokines in the frontal cortex [253]. Rodents exposed to chronic mild stress exhibit neuroinflammatory markers [254] while administration of cytokine or lipopolysaccharide (a cytokine inducer) induced depressive-like behaviour [255, 256]. Extracellular signal regulated kinase (ERK1/2) is one of several mitogen-activated protein kinases (MAPK) involved in numerous cellular processes including long-term neural plasticity, cell maintenance, survival and immune responses [257–259]. ERK1/2 is also implicated in adaptive responses to stress and antidepressant treatment [260–262]. Acute and chronic exposure to chronic unpredictable stress increases phosphorylation of ERK1/2 and of two downstream targets (ribosomal S6 kinase and mitogen- and stress-activated protein kinase 1) within the VTA [263]. In addition, pro-inflammatory cytokines modulate various MAPK including ERK [264]. Though depression and anxiety have been linked to depression, it is not clear if inflammatory status predates disease onset or whether it is a consequence of mood disorders. Recent studies, however, suggest that inflammatory status predates mental disease. Elevated levels of peripheral cytokines, in particular, interleukin-6 (IL-6), were found to be a good predicator of susceptibility to stress in mice exposed to both physical and purely emotional stressors [265]. Furthermore, bone marrow hematopoietic cell transplants from the susceptible donor mice to a stress naïve mice induced stress-susceptibility in the naïve mice when exposed to both physical and emotional stressors [265]. The observation that replacing the peripheral immune system of a stress-naïve animal with that of a stressed animal increases susceptibility to stressors suggests that the immune status of an animal with markers such as IL-6 can be used a predicator of susceptibility to stress. Conversely, findings that transgenic mice lacking IL-6 (IL-6−/−) are resilient to stress suggest another potential strategy for the treatment of mood disorders [265]. Though it is intuitive to link inflammatory processes following stress/physical injury with depression, the data is still unclear. For example, a recent study in rodents and humans found that administration of anti-inflammatory drugs following electrode implantation attenuated the antidepressive effects of deep brain stimulation [266]. Future animal studies investigating the effect of manipulating molecular pathways involved in the immune response on animal models of depression together with human studies on immune-compromised patients with depression should help clarify this interesting field of study.
Circadian/sleep rhythm and depression
Though it has been known since the 1950s that daily rhythms are disrupted in patients suffering from MDD [267, 268], the molecular mechanisms linking aberration in circadian/sleep rhythms and mood disorders is still not well understood. In healthy humans, mood and reward are modulated by circadian phase [269, 270]. Mood disorders involve deficits in reward processing and motivation where perception in reward is blunted with a corresponding reduction in motivation to pursue hedonic goals. Circadian oscillation of gene expression, neural firing, neurotransmitter levels and receptor expression have been discovered in various brain regions implicated in mood-regulation and reward of both rodents and humans [271]. The Social Zeitgeber theory of mood disorders proposes that stressful life events changes sleep/wake schedule that alters molecular and cellular rhythms in vulnerable individuals, leading to mood disorders [272]. In light of the close interaction between the circadian and sleep/wake rhythms, it is, therefore, not surprising that sleep deprivation therapy (SDT) rapidly alleviates depressive symptoms [273]. Studies over several decades have confirmed that SDT rapidly (within 24-h) reduces depressive symptoms in 40–60 % of patients [268, 274–276]. However, the drawback with SDT is that the majority of patients report relapses of depressive symptoms after the first bout of sleep [277, 278]. Though not yet determined, it is thought that SDT resets the aberrant circadian clock in depressive patients resulting in alleviation of symptoms [277, 278]. Accumulating clinical evidence highlights potential changes in circadian clock gene expression in depressed patients. Molecular analysis of clock genes in suicide vs non-suicides in postmortem found significant down regulation of per1 in suicide victims [279]. Furthermore, microarray analysis of circadian clock gene expression in brain postmortem tissue taken from control and MDD human subjects found robust sinusoidal 24-h rhythms in control but not in MDD patients [280]. Mice susceptible to social defeat stress exhibit, fragmentation in sleep patterns and anxiety [281]. Expression of circadian gene mper1 and mper2 was decreased in the NAc of mice that exhibited increased anxiety-like behaviour [282]. Furthermore, viral knock down of mper1/2 gene alone induced anxiety-like behaviour in mice, suggesting a causal link between aberration of circadian clock gene expression and anxiety/depression. Stress exposed mice also exhibited flattened diurnal locomotor activity returning to normal rhythms 7 days post-termination of stress [283]. Social defeat in the middle of the light phase increased both NREM sleep intensity and duration and decreased REM sleep [284]. Though limited in numbers, the few of studies of sleep deprivation (SD) on depression/anxiety have been promising. SD has been shown to reverse CSD stress-induced anxiety [285] and induce phase shifts in circadian clock gene expression [286, 287]. Molecular analyses of clock gene expression in mice following SD found elevated Per1 and Per2 levels in cortex, basal forebrain and hypothalamus which returned to normal levels following sleep recovery [288, 289]. Furthermore, SD has been shown to alter A) DNA binding of specific clock proteins BMAL, CLOCK and NPAS2 [290] and B) expression of various proteins known to be associated with depression such as GSK-3β, AMPA, Glutamate and mTOR [278]. Furthermore the rapidly acting antidepressant ketamine alters circadian gene expression [291], intracellular signalling molecule and neurotransmitter levels [278] and decreases aspects of REM sleep [292]. As mentioned previously, VTA DA neurons play a key role in modulating depression-related behaviours [12, 163, 171, 172] and it is likely that aberration in clock gene expression in the VTA may play a role in depression-related behaviours. Manipulation of Clock gene expression in the VTA modulates circadian rhythms, VTA neural activity and the depressive phenotype [293]. A more recent study extended the role of Clock gene and the regulation of anxiety related behaviours. Clock mutant mice were found to exhibit rapid mood cycling across the light–dark cycle such that the Clock mutants exhibited robust manic-like behaviour compared to wild-type controls when tested in an open field-, forced swim- and sucrose-preference test during the day [294]. Furthermore Clock mutant mice exhibited robust increases in both daytime tyrosine hydroxylase expression and VTA DA neuron activity [294]. Moreover, further evidence that the molecular components of the circadian timing system have a critical role in mood regulation comes from numerous findings that mutations of clock genes Bmal1 and Per2 induce mania-like behaviours in mice while knock-out of Cryptochrome 1 and 2 genes induces altered anxiety-like behaviours [295–297]. The circadian nuclear receptor REV-ERBα, an important constituent of molecular circadian signalling that serves as transcriptional repressors of the Bmal RNA, has recently been shown to negatively regulate midbrain DA neuronal function via circadian modulation of tyrosine hydroxylase mRNA transcription [298]. REV-ERBα knockout mice exhibit altered circadian rhythmicity in mood related behaviours while conditional inhibition of REV-ERBα produces mania-like behaviour [298]. These recent findings further identify molecular connections between the circadian timing system and mood regulation that may potentially help specific targeting in the treatment of circadian-related mood disorders. Social interaction, a rewarding phenomenon in social animals such as mice, follows circadian rhythmicity [299] and since depression-like behaviour leads to aberration in neural processing in the reward system, it is likely that aberration in the circadian system results in aberration in reward processing in the reward centre leading to depression-related behaviours. Understanding the mechanism by which the circadian and sleep wake centres regulate depressive behaviours has great potential for the development of rapidly acting therapies for depression.
Conclusion
Depression is a complex mental illness where patients can express vastly different symptoms. It is evident from the literature reviewed above that the multiplicity of symptoms related to depression most likely is the result of aberrations in different aspects of normal neural functions that can range from the molecular up to the neural circuit. The observation that classical antidepressant medications only work on a subset of patients indicates that depressive patients express aberration in different neural processes. Thus, to develop more effective and faster acting treatments for depression much work is still needed in understanding how exposure to stress lead to the sequence of changes in molecular, genetic/epigenetic processes and eventually neural circuit signalling. With the development of novel technologies to study molecular and genetic changes together with more sophisticated neural circuit analysis, it will be possible to better understand complex changes in the brain that ultimately lead to depression.
Abbreviations
- 3V:
-
Third ventricle
- 4V:
-
Fourth ventricle
- 5-HT:
-
Serotonin
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Arc:
-
Activity-regulated cytoskeleton-associated protein gene
- BDNF:
-
Brain-derived neurotrophic factor
- BLA:
-
Basolateral amygdala
- Ca2+ :
-
Calcium
- CaMKII:
-
Calcium/calmodulin-dependent protein kinase II
- CeM:
-
Central amygdala
- CPP:
-
3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid
- CRF:
-
Corticortrophin-releasing factor
- CSD:
-
Chronic social defeat
- CUS:
-
Chronic unpredictable stress
- D1:
-
Dopamine 1 receptor
- D2:
-
Dopamine 2 receptor
- D3V:
-
Dorsal third ventricle
- DBS:
-
Deep brain stimulation
- delta FosB:
-
Delta FBJ murine osteosarcoma viral oncogene homolog B
- DMH:
-
Dorsomedial nucleus of the hypothalamus
- DNMT:
-
DNA methyltransferase
- DRN:
-
Dorsal raphe nucleus
- eEF2:
-
Eukaryotic elongation factor
- Egr1:
-
Early growth response protein 1 gene
- ERK1/2:
-
Extracellular signal regulated kinase
- FosB:
-
FBJ murine osteosarcoma viral oncogene homolog B
- G9a:
-
Histone H3 lysine 9 methyltransferase
- GABA:
-
Gamma-aminobutyric acid
- GLP:
-
Histone H3 lysine 9 methyltransferase
- GluR1:
-
Glutamate receptor subunit 1
- GluR2:
-
Glutamate receptor subunit 2
- GluR3:
-
Glutamate receptor subunit 3
- GPo:
-
Globus pallidum
- H3K9me2:
-
Histone H3 dimethyl Lys9
- HATS:
-
Histone acetyltransferase
- HCN:
-
Hyperpolarisation-activated cyclic nucleotide-gated channel
- HDAC:
-
Histone deacetylase
- HDM:
-
Histone demethylase
- HMT:
-
Histone methyltransferase
- I h :
-
Hyperpolarisation-activated non-selective cation current
- IKK:
-
IKB kinase
- IKKdn:
-
IKK dominant negative
- IVF:
-
In vitro fertilisation
- K+ :
-
Potassium
- LC:
-
Locus coeruleus
- LDTg:
-
Laterodorsal tegmental nucleus
- LH:
-
Lateral hypothalamus
- LHb:
-
Lateral habenula
- LTD:
-
Laterodorsal tegmentum
- MAPK:
-
Mitogen-activated protein kinases
- MDD:
-
Major depressive disorder
- mGluR2:
-
Metabotropic glutamate receptor 2
- MK-801:
-
5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine
- mper1/2:
-
Period gene 1 and 2
- mPFC:
-
Medial prefrontal cortex
- MSN:
-
Medium spiny neurons
- mTOR:
-
Mammalian target of rapamycin
- NAc:
-
Nucleus accumbens
- NFkB :
-
Nuclear factor kB
- NMDAR:
-
N-methyl-d-aspartate receptor
- NR2B:
-
NMDAR 2B subunit
- PPTg:
-
Pedunculopontine tegmental nucleus
- REM:
-
Rapid eye movement
- RMTg:
-
Rostromedial tegmental nucleus
- SC1:
-
Sparc-like 1
- SCN:
-
Suprachiasmatic nucleus
- SD:
-
Sleep deprivation
- SDT:
-
Sleep deprivation therapy
- SN:
-
Substrantia nigra
- SPZ:
-
Subparaventricular zone
- SSRI:
-
Serotonin selective reuptake inhibitor
- SUV39H1:
-
Histone-lysine N-methyltransferase
- vHIP:
-
Ventral hippocampus
- VP:
-
Ventral pallidum
- VSCC:
-
Voltage sensitive calcium channels
- VTA:
-
Ventral tegmental area
- WKY:
-
Wistar Kyoto
- ZFP:
-
Zing-finger protein
References
Ferrari AJ, Charlson FJ, Norman RE, Flaxman AD, Patten SB, Vos T, Whiteford HA (2013) The epidemiological modelling of major depressive disorder: application for the Global Burden of Disease Study 2010. PLoS One 8:e69637. doi:10.1371/journal.pone.0069637
Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, Charlson FJ, Norman RE, Flaxman AD, Johns N, Burstein R, Murray CJ, Vos T (2013) Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 382:1575–1586. doi:10.1016/S0140-6736(13)61611-6
Toseeb U, Brage S, Corder K, Dunn VJ, Jones PB, Owens M, St Clair MC, van Sluijs EM, Goodyer IM (2014) Exercise and depressive symptoms in adolescents: a longitudinal cohort study. JAMA Pediatrics 168:1093–1100. doi:10.1001/jamapediatrics.2014.1794
Nestler EJ (1998) Antidepressant treatments in the 21st century. Biol Psychiatry 44:526–533. pii: S000632239800095X
Hyman SE (2014) Revitalizing psychiatric therapeutics. Neuropsychopharmacology 39:220–229. doi:10.1038/npp.2013.181
Berton O, Nestler EJ (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 7:137–151. doi:10.1038/nrn1846
Mathew SJ, Manji HK, Charney DS (2008) Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology 33:2080–2092. doi:10.1038/sj.npp.1301652
Mayberg HS (2009) Targeted electrode-based modulation of neural circuits for depression. J Clin Invest 119:717–725. doi:10.1172/JCI38454
Holtzheimer PE 3rd, Mayberg HS (2010) Deep brain stimulation for treatment-resistant depression. Am J Psychiatry 167:1437–1444. doi:10.1176/appi.ajp.2010.10010141
Zarate C, Duman RS, Liu G, Sartori S, Quiroz J, Murck H (2013) New paradigms for treatment-resistant depression. Ann N Y Acad Sci 1292:21–31. doi:10.1111/nyas.12223
Merkl A, Neumann WJ, Huebl J, Aust S, Horn A, Krauss JK, Dziobek I, Kuhn J, Schneider GH, Bajbouj M, Kuhn AA (2015) Modulation of beta-band activity in the subgenual anterior cingulate cortex during emotional empathy in treatment-resistant depression. Cereb Cortex. doi:10.1093/cercor/bhv100
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ (2007) Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131:391–404. doi:10.1016/j.cell.2007.09.018
Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM (2007) Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 55:648–661. doi:10.1016/j.neuron.2007.07.030
Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, Krishnan V, Reyes CM, Han MH, Ables JL, Eisch AJ, Dietz DM, Ferguson D, Neve RL, Greengard P, Kim Y, Morrison JH, Russo SJ (2011) IkappaB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci 31:314–321. doi:10.1523/JNEUROSCI.4763-10.2011
Russo SJ, Nestler EJ (2013) The brain reward circuitry in mood disorders. Nat Rev Neurosci 14:609–625. doi:10.1038/nrn3381
Chandley MJ, Szebeni A, Szebeni K, Crawford JD, Stockmeier CA, Turecki G, Kostrzewa RM, Ordway GA (2014) Elevated gene expression of glutamate receptors in noradrenergic neurons from the locus coeruleus in major depression. Int J Neuropsychopharmacol 17:1569–1578. doi:10.1017/S1461145714000662
Lopizzo N, Bocchio Chiavetto L, Cattane N, Plazzotta G, Tarazi FI, Pariante CM, Riva MA, Cattaneo A (2015) Gene-environment interaction in major depression: focus on experience-dependent biological systems. Frontiers in psychiatry 6:68. doi:10.3389/fpsyt.2015.00068
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34:13–25. pii: S0896627302006530
Park H, Poo MM (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14:7–23. doi:10.1038/nrn3379
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ (2006) Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311:864–868. doi:10.1126/science.1120972
Walsh JJ, Friedman AK, Sun H, Heller EA, Ku SM, Juarez B, Burnham VL, Mazei-Robison MS, Ferguson D, Golden SA, Koo JW, Chaudhury D, Christoffel DJ, Pomeranz L, Friedman JM, Russo SJ, Nestler EJ, Han MH (2014) Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci 17:27–29. doi:10.1038/nn.3591
Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ, Chavkin C, Phillips PE (2012) Severe stress switches CRF action in the nucleus accumbens from appetitive to aversive. Nature 490:402–406. doi:10.1038/nature11436
Pecina S, Schulkin J, Berridge KC (2006) Nucleus accumbens corticotropin-releasing factor increases cue-triggered motivation for sucrose reward: paradoxical positive incentive effects in stress? BMC Biol 4:8. doi:10.1186/1741-7007-4-8
Yu H, Chen ZY (2011) The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol Sin 32:3–11. doi:10.1038/aps.2010.184
Autry AE, Monteggia LM (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64:238–258. doi:10.1124/pr.111.005108
Pizarro JM, Lumley LA, Medina W, Robison CL, Chang WE, Alagappan A, Bah MJ, Dawood MY, Shah JD, Mark B, Kendall N, Smith MA, Saviolakis GA, Meyerhoff JL (2004) Acute social defeat reduces neurotrophin expression in brain cortical and subcortical areas in mice. Brain Res 1025:10–20. doi:10.1016/j.brainres.2004.06.085
Fanous S, Hammer RP Jr, Nikulina EM (2010) Short- and long-term effects of intermittent social defeat stress on brain-derived neurotrophic factor expression in mesocorticolimbic brain regions. Neuroscience 167:598–607. doi:10.1016/j.neuroscience.2010.02.064
Guilloux JP, Douillard-Guilloux G, Kota R, Wang X, Gardier AM, Martinowich K, Tseng GC, Lewis DA, Sibille E (2012) Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry 17:1130–1142. doi:10.1038/mp.2011.113
Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT (2001) Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 50:260–265
Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN (2003) Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60:804–815. doi:10.1001/archpsyc.60.8.804
Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R (2005) Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res 136:29–37. doi:10.1016/j.molbrainres.2004.12.020
Duclot F, Kabbaj M (2015) Epigenetic mechanisms underlying the role of brain-derived neurotrophic factor in depression and response to antidepressants. J Exp Biol 218:21–31. doi:10.1242/jeb.107086
Chourbaji S, Brandwein C, Gass P (2011) Altering BDNF expression by genetics and/or environment: impact for emotional and depression-like behaviour in laboratory mice. Neurosci Biobehav Rev 35:599–611. doi:10.1016/j.neubiorev.2010.07.003
Tapia-Arancibia L, Rage F, Givalois L, Arancibia S (2004) Physiology of BDNF: focus on hypothalamic function. Front Neuroendocrinol 25:77–107. doi:10.1016/j.yfrne.2004.04.001
Cai S, Huang S, Hao W (2015) New hypothesis and treatment targets of depression: an integrated view of key findings. Neurosci Bull 31:61–74. doi:10.1007/s12264-014-1486-4
Suenaga T, Morinobu S, Kawano K, Sawada T, Yamawaki S (2004) Influence of immobilization stress on the levels of CaMKII and phospho-CaMKII in the rat hippocampus. Int J Neuropsychopharmacol 7:299–309. doi:10.1017/S1461145704004304
Almeida RC, Souza DG, Soletti RC, Lopez MG, Rodrigues AL, Gabilan NH (2006) Involvement of PKA, MAPK/ERK and CaMKII, but not PKC in the acute antidepressant-like effect of memantine in mice. Neurosci Lett 395:93–97. doi:10.1016/j.neulet.2005.10.057
Robison AJ, Vialou V, Sun HS, Labonte B, Golden SA, Dias C, Turecki G, Tamminga C, Russo S, Mazei-Robison M, Nestler EJ (2014) Fluoxetine epigenetically alters the CaMKIIalpha promoter in nucleus accumbens to regulate DeltaFosB binding and antidepressant effects. Neuropsychopharmacology 39:1178–1186. doi:10.1038/npp.2013.319
Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002) alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36:1103–1114
Meye FJ, Lecca S, Valentinova K, Mameli M (2013) Synaptic and cellular profile of neurons in the lateral habenula. Front Hum Neurosci 7:860. doi:10.3389/fnhum.2013.00860
Sartorius A, Kiening KL, Kirsch P, von Gall CC, Haberkorn U, Unterberg AW, Henn FA, Meyer-Lindenberg A (2010) Remission of major depression under deep brain stimulation of the lateral habenula in a therapy-refractory patient. Biol Psychiatry 67:e9–e11. doi:10.1016/j.biopsych.2009.08.027
Aizawa H, Cui W, Tanaka K, Okamoto H (2013) Hyperactivation of the habenula as a link between depression and sleep disturbance. Front Hum Neurosci 7:826. doi:10.3389/fnhum.2013.00826
Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R (2011) Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 470:535–539. doi:10.1038/nature09742
Li K, Zhou T, Liao L, Yang Z, Wong C, Henn F, Malinow R, Yates JR 3rd, Hu H (2013) betaCaMKII in lateral habenula mediates core symptoms of depression. Science 341:1016–1020. doi:10.1126/science.1240729
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354. pii: S0006-3223(99)00230-9
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK (2006) A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864. doi:10.1001/archpsyc.63.8.856
Price RB, Nock MK, Charney DS, Mathew SJ (2009) Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry 66:522–526. doi:10.1016/j.biopsych.2009.04.029
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95. doi:10.1038/nature10130
Dwyer JM, Lepack AE, Duman RS (2013) mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J Mol Psychiatry 1:15. doi:10.1186/2049-9256-1-15
Iadarola ND, Niciu MJ, Richards EM, Vande Voort JL, Ballard ED, Lundin NB, Nugent AC, Machado-Vieira R, Zarate CA Jr (2015) Ketamine and other N-methyl-d-aspartate receptor antagonists in the treatment of depression: a perspective review. Ther Adv Chronic Dis 6:97–114. doi:10.1177/2040622315579059
Gideons ES, Kavalali ET, Monteggia LM (2014) Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci USA 111:8649–8654. doi:10.1073/pnas.1323920111
Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, Kavalali ET (2013) Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci 33:6990–7002. doi:10.1523/JNEUROSCI.4998-12.2013
Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK (2008) Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63:349–352. doi:10.1016/j.biopsych.2007.05.028
Hou L, Klann E (2004) Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci 24:6352–6361. doi:10.1523/JNEUROSCI.0995-04.2004
Gong R, Park CS, Abbassi NR, Tang SJ (2006) Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem 281:18802–18815. doi:10.1074/jbc.M512524200
Yu JJ, Zhang Y, Wang Y, Wen ZY, Liu XH, Qin J, Yang JL (2013) Inhibition of calcineurin in the prefrontal cortex induced depressive-like behavior through mTOR signaling pathway. Psychopharmacology 225:361–372. doi:10.1007/s00213-012-2823-9
Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18:1926–1945. doi:10.1101/gad.1212704
Duman RS, Li N, Liu RJ, Duric V, Aghajanian G (2012) Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 62:35–41. doi:10.1016/j.neuropharm.2011.08.044
Abdallah CG, Sanacora G, Duman RS, Krystal JH (2015) Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med 66:509–523. doi:10.1146/annurev-med-053013-062946
Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, Lepack A, Majik MS, Jeong LS, Banasr M, Son H, Duman RS (2012) Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med 18:1413–1417. doi:10.1038/nm.2886
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, Almeida OF, Sousa N (2009) The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 14(764–773):739. doi:10.1038/mp.2008.119
Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z (2012) Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73:962–977. doi:10.1016/j.neuron.2011.12.033
Bessa JM, Morais M, Marques F, Pinto L, Palha JA, Almeida OF, Sousa N (2013) Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl Psychiatry 3:e266. doi:10.1038/tp.2013.39
Vyas A, Pillai AG, Chattarji S (2004) Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience 128:667–673. doi:10.1016/j.neuroscience.2004.07.013
Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59:1116–1127. doi:10.1016/j.biopsych.2006.02.013
Ota KT, Liu RJ, Voleti B, Maldonado-Aviles JG, Duric V, Iwata M, Dutheil S, Duman C, Boikess S, Lewis DA, Stockmeier CA, DiLeone RJ, Rex C, Aghajanian GK, Duman RS (2014) REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat Med 20:531–535. doi:10.1038/nm.3513
Liu RJ, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK (2013) GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology 38:2268–2277. doi:10.1038/npp.2013.128
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. doi:10.1126/science.1190287
Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK (2012) Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry 71:996–1005. doi:10.1016/j.biopsych.2011.09.030
Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M (2009) Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci 29:8688–8697. doi:10.1523/JNEUROSCI.6078-08.2009
Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS (2015) BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol. doi:10.1093/ijnp/pyu033
Hasler G, Drevets WC, Manji HK, Charney DS (2004) Discovering endophenotypes for major depression. Neuropsychopharmacology 29:1765–1781. doi:10.1038/sj.npp.1300506
Lamers F, Vogelzangs N, Merikangas KR, de Jonge P, Beekman AT, Penninx BW (2013) Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol Psychiatry 18:692–699. doi:10.1038/mp.2012.144
Sotnikov S, Wittmann A, Bunck M, Bauer S, Deussing J, Schmidt M, Touma C, Landgraf R, Czibere L (2014) Blunted HPA axis reactivity reveals glucocorticoid system dysbalance in a mouse model of high anxiety-related behavior. Psychoneuroendocrinology 48:41–51. doi:10.1016/j.psyneuen.2014.06.006
Santana MM, Rosmaninho-Salgado J, Cortez V, Pereira FC, Kaster MP, Aveleira CA, Ferreira M, Alvaro AR, Cavadas C (2015) Impaired adrenal medullary function in a mouse model of depression induced by unpredictable chronic stress. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2015.06.013
Holsboer F, Barden N (1996) Antidepressants and hypothalamic–pituitary–adrenocortical regulation. Endocr Rev 17:187–205. doi:10.1210/edrv-17-2-187
De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M (1998) Brain corticosteroid receptor balance in health and disease. Endocr Rev 19:269–301. doi:10.1210/edrv.19.3.0331
Holsboer F (2000) The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23:477–501. doi:10.1016/S0893-133X(00)00159-7
Chang HS, Won ES, Lee HY, Ham BJ, Kim YG, Lee MS (2015) The association of proopiomelanocortin polymorphisms with the risk of major depressive disorder and the response to antidepressants via interactions with stressful life events. J Neural Transm 122:59–68. doi:10.1007/s00702-014-1333-9
Banki CM, Bissette G, Arato M, O’Connor L, Nemeroff CB (1987) CSF corticotropin-releasing factor-like immunoreactivity in depression and schizophrenia. Am J Psychiatry 144:873–877
Bissette G, Klimek V, Pan J, Stockmeier C, Ordway G (2003) Elevated concentrations of CRF in the locus coeruleus of depressed subjects. Neuropsychopharmacology 28:1328–1335. doi:10.1038/sj.npp.1300191
Merali Z, Kent P, Du L, Hrdina P, Palkovits M, Faludi G, Poulter MO, Bedard T, Anisman H (2006) Corticotropin-releasing hormone, arginine vasopressin, gastrin-releasing peptide, and neuromedin B alterations in stress-relevant brain regions of suicides and control subjects. Biol Psychiatry 59:594–602. doi:10.1016/j.biopsych.2005.08.008
Lloyd RB, Nemeroff CB (2011) The role of corticotropin-releasing hormone in the pathophysiology of depression: therapeutic implications. Curr Top Med Chem 11:609–617
Plotsky PM, Thrivikraman KV, Nemeroff CB, Caldji C, Sharma S, Meaney MJ (2005) Long-term consequences of neonatal rearing on central corticotropin-releasing factor systems in adult male rat offspring. Neuropsychopharmacology 30:2192–2204. doi:10.1038/sj.npp.1300769
Antunes MS, Ruff JR, de Oliveira Espinosa D, Piegas MB, de Brito ML, Rocha KA, de Gomes MG, Goes AT, Souza LC, Donato F, Boeira SP, Jesse CR (2015) Neuropeptide Y administration reverses tricyclic antidepressant treatment-resistant depression induced by ACTH in mice. Horm Behav 73:56–63. doi:10.1016/j.yhbeh.2015.05.018
McEwen BS (2007) Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 87:873–904. doi:10.1152/physrev.00041.2006
McEwen BS, Gray JD, Nasca C (2015) 60 years of neuroendocrinology: redefining neuroendocrinology: stress, sex and cognitive and emotional regulation. J Endocrinol 226:T67–T83. doi:10.1530/JOE-15-0121
Datson NA, van den Oever JM, Korobko OB, Magarinos AM, de Kloet ER, McEwen BS (2013) Previous history of chronic stress changes the transcriptional response to glucocorticoid challenge in the dentate gyrus region of the male rat hippocampus. Endocrinology 154:3261–3272. doi:10.1210/en.2012-2233
Nasca C, Xenos D, Barone Y, Caruso A, Scaccianoce S, Matrisciano F, Battaglia G, Mathe AA, Pittaluga A, Lionetto L, Simmaco M, Nicoletti F (2013) l-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA 110:4804–4809. doi:10.1073/pnas.1216100110
Nasca C, Bigio B, Zelli D, Nicoletti F, McEwen BS (2015) Mind the gap: glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol Psychiatry 20:755–763. doi:10.1038/mp.2014.96
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 102:10604–10609. doi:10.1073/pnas.0500398102
Kessler RC (1997) The effects of stressful life events on depression. Annu Rev Psychol 48:191–214. doi:10.1146/annurev.psych.48.1.191
Hammen C (2005) Stress and depression. Annu Rev Clin Psychol 1:293–319. doi:10.1146/annurev.clinpsy.1.102803.143938
Dudley KJ, Li X, Kobor MS, Kippin TE, Bredy TW (2011) Epigenetic mechanisms mediating vulnerability and resilience to psychiatric disorders. Neurosci Biobehav Rev 35:1544–1551. doi:10.1016/j.neubiorev.2010.12.016
Vialou V, Feng J, Robison AJ, Nestler EJ (2013) Epigenetic mechanisms of depression and antidepressant action. Annu Rev Pharmacol Toxicol 53:59–87. doi:10.1146/annurev-pharmtox-010611-134540
Nestler EJ (2014) Epigenetic mechanisms of depression. JAMA Psychiatry 71:454–456. doi:10.1001/jamapsychiatry.2013.4291
Sun H, Kennedy PJ, Nestler EJ (2013) Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 38:124–137. doi:10.1038/npp.2012.73
Covington HE 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ (2009) Antidepressant actions of histone deacetylase inhibitors. J Neurosci 29:11451–11460. doi:10.1523/JNEUROSCI.1758-09.2009
Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, Suzuki T, Miyata N, Watanabe Y (2011) Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69:359–372. doi:10.1016/j.neuron.2010.12.023
Renthal W, Maze I, Krishnan V, Covington HE 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ (2007) Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56:517–529. doi:10.1016/j.neuron.2007.09.032
Liu D, Qiu HM, Fei HZ, Hu XY, Xia HJ, Wang LJ, Qin LJ, Jiang XH, Zhou QX (2014) Histone acetylation and expression of mono-aminergic transmitters synthetases involved in CUS-induced depressive rats. Exp Biol Med (Maywood) 239:330–336. doi:10.1177/1535370213513987
Covington HE 3rd, Vialou VF, LaPlant Q, Ohnishi YN, Nestler EJ (2011) Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci Lett 493:122–126. doi:10.1016/j.neulet.2011.02.022
Covington HE 3rd, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, Dietz DM, Lobo MK, Ghose S, Mouzon E, Neve RL, Tamminga CA, Nestler EJ (2011) A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 71:656–670. doi:10.1016/j.neuron.2011.06.007
Heller EA, Cates HM, Pena CJ, Sun H, Shao N, Feng J, Golden SA, Herman JP, Walsh JJ, Mazei-Robison M, Ferguson D, Knight S, Gerber MA, Nievera C, Han MH, Russo SJ, Tamminga CS, Neve RL, Shen L, Zhang HS, Zhang F, Nestler EJ (2014) Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci 17:1720–1727. doi:10.1038/nn.3871
Nghia NA, Hirasawa T, Kasai H, Obata C, Moriishi K, Mochizuki K, Koizumi S, Kubota T (2015) Long-term imipramine treatment increases N-methyl-d-aspartate receptor activity and expression via epigenetic mechanisms. Eur J Pharmacol 752:69–77. doi:10.1016/j.ejphar.2015.02.010
Dell’Osso B, D’Addario C, Carlotta Palazzo M, Benatti B, Camuri G, Galimberti D, Fenoglio C, Scarpini E, Di Francesco A, Maccarrone M, Altamura AC (2014) Epigenetic modulation of BDNF gene: differences in DNA methylation between unipolar and bipolar patients. J Affect Disord 166:330–333. doi:10.1016/j.jad.2014.05.020
Kundakovic M, Gudsnuk K, Herbstman JB, Tang D, Perera FP, Champagne FA (2014) DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci USA. doi:10.1073/pnas.1408355111
Pena CJ, Bagot RC, Labonte B, Nestler EJ (2014) Epigenetic signaling in psychiatric disorders. J Mol Biol 426:3389–3412. doi:10.1016/j.jmb.2014.03.016
Blaze J, Roth TL (2013) Exposure to caregiver maltreatment alters expression levels of epigenetic regulators in the medial prefrontal cortex. Int J Dev Neurosci 31:804–810. doi:10.1016/j.ijdevneu.2013.10.001
Levine A, Worrell TR, Zimnisky R, Schmauss C (2012) Early life stress triggers sustained changes in histone deacetylase expression and histone H4 modifications that alter responsiveness to adolescent antidepressant treatment. Neurobiol Dis 45:488–498. doi:10.1016/j.nbd.2011.09.005
Xie L, Korkmaz KS, Braun K, Bock J (2013) Early life stress-induced histone acetylations correlate with activation of the synaptic plasticity genes Arc and Egr1 in the mouse hippocampus. J Neurochem 125:457–464. doi:10.1111/jnc.12210
Mueller BR, Bale TL (2008) Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci 28:9055–9065. doi:10.1523/JNEUROSCI.1424-08.2008
Non AL, Binder AM, Kubzansky LD, Michels KB (2014) Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics 9:964–972. doi:10.4161/epi.28853
Meaney MJ (2001) Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci 24:1161–1192. doi:10.1146/annurev.neuro.24.1.1161
Francis D, Diorio J, Liu D, Meaney MJ (1999) Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science 286:1155–1158
Yehuda R, Bell A, Bierer LM, Schmeidler J (2008) Maternal, not paternal, PTSD is related to increased risk for PTSD in offspring of Holocaust survivors. J Psychiatr Res 42:1104–1111. doi:10.1016/j.jpsychires.2008.01.002
Franklin TB, Russig H, Weiss IC, Graff J, Linder N, Michalon A, Vizi S, Mansuy IM (2010) Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry 68:408–415. doi:10.1016/j.biopsych.2010.05.036
Dietz DM, Laplant Q, Watts EL, Hodes GE, Russo SJ, Feng J, Oosting RS, Vialou V, Nestler EJ (2011) Paternal transmission of stress-induced pathologies. Biol Psychiatry 70:408–414. doi:10.1016/j.biopsych.2011.05.005
Warner-Schmidt JL, Duman RS (2008) VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol 8:14–19. doi:10.1016/j.coph.2007.10.013
Greene J, Banasr M, Lee B, Warner-Schmidt J, Duman RS (2009) Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment: pharmacological and cellular characterization. Neuropsychopharmacology 34:2459–2468. doi:10.1038/npp.2009.68
de Jong IE, de Kloet ER (2004) Glucocorticoids and vulnerability to psychostimulant drugs: toward substrate and mechanism. Ann N Y Acad Sci 1018:192–198. doi:10.1196/annals.1296.022
Duman RS, Malberg J, Nakagawa S (2001) Regulation of adult neurogenesis by psychotropic drugs and stress. J Pharmacol Exp Ther 299:401–407
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20:9104–9110. pii: 20/24/9104
Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, Arango V (2009) Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34:2376–2389. doi:10.1038/npp.2009.75
Toni N, Laplagne DA, Zhao C, Lombardi G, Ribak CE, Gage FH, Schinder AF (2008) Neurons born in the adult dentate gyrus form functional synapses with target cells. Nat Neurosci 11:901–907. doi:10.1038/nn.2156
Cameron HA, Gould E (1994) Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 61:203–209
Pham K, Nacher J, Hof PR, McEwen BS (2003) Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci 17:879–886
Lee KJ, Kim SJ, Kim SW, Choi SH, Shin YC, Park SH, Moon BH, Cho E, Lee MS, Choi SH, Chun BG, Shin KH (2006) Chronic mild stress decreases survival, but not proliferation, of new-born cells in adult rat hippocampus. Exp Mol Med 38:44–54. doi:10.1038/emm.2006.6
Van Bokhoven P, Oomen CA, Hoogendijk WJ, Smit AB, Lucassen PJ, Spijker S (2011) Reduction in hippocampal neurogenesis after social defeat is long-lasting and responsive to late antidepressant treatment. Eur J Neurosci 33:1833–1840. doi:10.1111/j.1460-9568.2011.07668.x
Dranovsky A, Picchini AM, Moadel T, Sisti AC, Yamada A, Kimura S, Leonardo ED, Hen R (2011) Experience dictates stem cell fate in the adult hippocampus. Neuron 70:908–923. doi:10.1016/j.neuron.2011.05.022
Hanson ND, Owens MJ, Boss-Williams KA, Weiss JM, Nemeroff CB (2011) Several stressors fail to reduce adult hippocampal neurogenesis. Psychoneuroendocrinology 36:1520–1529. doi:10.1016/j.psyneuen.2011.04.006
Miller BR, Hen R (2015) The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol 30:51–58. doi:10.1016/j.conb.2014.08.012
Sapolsky RM, Uno H, Rebert CS, Finch CE (1990) Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci 10:2897–2902
Kunugi H, Hori H, Adachi N, Numakawa T (2010) Interface between hypothalamic–pituitary–adrenal axis and brain-derived neurotrophic factor in depression. Psychiatry Clin Neurosci 64:447–459. doi:10.1111/j.1440-1819.2010.02135.x
Benraiss A, Chmielnicki E, Lerner K, Roh D, Goldman SA (2001) Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J Neurosci 21:6718–6731
Pencea V, Bingaman KD, Wiegand SJ, Luskin MB (2001) Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci 21:6706–6717
Duman RS, Heninger GR, Nestler EJ (1997) A molecular and cellular theory of depression. Arch Gen Psychiatry 54:597–606
Murakami S, Imbe H, Morikawa Y, Kubo C, Senba E (2005) Chronic stress, as well as acute stress, reduces BDNF mRNA expression in the rat hippocampus but less robustly. Neurosci Res 53:129–139. doi:10.1016/j.neures.2005.06.008
Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF (2008) TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59:399–412. doi:10.1016/j.neuron.2008.06.023
Malberg JE, Duman RS (2003) Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 28:1562–1571. doi:10.1038/sj.npp.1300234
Chen H, Pandey GN, Dwivedi Y (2006) Hippocampal cell proliferation regulation by repeated stress and antidepressants. NeuroReport 17:863–867. doi:10.1097/01.wnr.0000221827.03222.70
Crupi R, Cambiaghi M, Spatz L, Hen R, Thorn M, Friedman E, Vita G, Battaglia F (2010) Reduced adult neurogenesis and altered emotional behaviors in autoimmune-prone B-cell activating factor transgenic mice. Biol Psychiatry 67:558–566. doi:10.1016/j.biopsych.2009.12.008
Anacker C, Zunszain PA, Cattaneo A, Carvalho LA, Garabedian MJ, Thuret S, Price J, Pariante CM (2011) Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol Psychiatry 16:738–750. doi:10.1038/mp.2011.26
Holsboer F, Ising M (2010) Stress hormone regulation: biological role and translation into therapy. Annu Rev Psychol 61(81–109):C101–C111. doi:10.1146/annurev.psych.093008.100321
Mirescu C, Gould E (2006) Stress and adult neurogenesis. Hippocampus 16:233–238. doi:10.1002/hipo.20155
Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA (2011) Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 476:458–461. doi:10.1038/nature10287
Hill AS, Sahay A, Hen R (2015) Increasing adult hippocampal neurogenesis is sufficient to reduce anxiety and depression-like behaviors. Neuropsychopharmacology. doi:10.1038/npp.2015.85
Lagace DC, Donovan MH, DeCarolis NA, Farnbauch LA, Malhotra S, Berton O, Nestler EJ, Krishnan V, Eisch AJ (2010) Adult hippocampal neurogenesis is functionally important for stress-induced social avoidance. Proc Natl Acad Sci USA 107:4436–4441. doi:10.1073/pnas.0910072107
Lucassen PJ, Stumpel MW, Wang Q, Aronica E (2010) Decreased numbers of progenitor cells but no response to antidepressant drugs in the hippocampus of elderly depressed patients. Neuropharmacology 58:940–949. doi:10.1016/j.neuropharm.2010.01.012
Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, Malenka RC (2004) Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 42:535–552
Wiskott L, Rasch MJ, Kempermann G (2006) A functional hypothesis for adult hippocampal neurogenesis: avoidance of catastrophic interference in the dentate gyrus. Hippocampus 16:329–343. doi:10.1002/hipo.20167
Pham K, McEwen BS, Ledoux JE, Nader K (2005) Fear learning transiently impairs hippocampal cell proliferation. Neuroscience 130:17–24. doi:10.1016/j.neuroscience.2004.09.015
Van der Borght K, Meerlo P, Luiten PG, Eggen BJ, Van der Zee EA (2005) Effects of active shock avoidance learning on hippocampal neurogenesis and plasma levels of corticosterone. Behav Brain Res 157:23–30. doi:10.1016/j.bbr.2004.06.004
Westenbroek C, Snijders TA, den Boer JA, Gerrits M, Fokkema DS, Ter Horst GJ (2005) Pair-housing of male and female rats during chronic stress exposure results in gender-specific behavioral responses. Horm Behav 47:620–628. doi:10.1016/j.yhbeh.2005.01.004
Shors TJ, Mathew J, Sisti HM, Edgecomb C, Beckoff S, Dalla C (2007) Neurogenesis and helplessness are mediated by controllability in males but not in females. Biol Psychiatry 62:487–495. doi:10.1016/j.biopsych.2006.10.033
Larson PS (2014) Deep brain stimulation for movement disorders. Neurotherapeutics 11:465–474. doi:10.1007/s13311-014-0274-1
Sartorius A, Henn FA (2007) Deep brain stimulation of the lateral habenula in treatment resistant major depression. Med Hypotheses 69:1305–1308. doi:10.1016/j.mehy.2007.03.021
Schneider TM, Beynon C, Sartorius A, Unterberg AW, Kiening KL (2013) Deep brain stimulation of the lateral habenular complex in treatment-resistant depression: traps and pitfalls of trajectory choice. Neurosurgery 72:ons184–ons193. doi:10.1227/NEU.0b013e318277a5aa (discussion ons193)
Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P (2014) Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord 159:31–38. doi:10.1016/j.jad.2014.02.016
Bogod NM, Sinden M, Woo C, Defreitas VG, Torres IJ, Howard AK, Ilcewicz-Klimek MI, Honey CR, Yatham LN, Lam RW (2014) Long-term neuropsychological safety of subgenual cingulate gyrus deep brain stimulation for treatment-resistant depression. J Neuropsychiatry Clin Neurosci 26:126–133. doi:10.1176/appi.neuropsych.12110287
Dobrossy MD, Furlanetti LL, Coenen VA (2015) Electrical stimulation of the medial forebrain bundle in pre-clinical studies of psychiatric disorders. Neurosci Biobehav Rev 49:32–42. doi:10.1016/j.neubiorev.2014.11.018
Han MH, Friedman AK (2012) Virogenetic and optogenetic mechanisms to define potential therapeutic targets in psychiatric disorders. Neuropharmacology 62:89–100. doi:10.1016/j.neuropharm.2011.09.009
Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, Ferguson D, Tsai HC, Pomeranz L, Christoffel DJ, Nectow AR, Ekstrand M, Domingos A, Mazei-Robison MS, Mouzon E, Lobo MK, Neve RL, Friedman JM, Russo SJ, Deisseroth K, Nestler EJ, Han MH (2013) Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493:532–536. doi:10.1038/nature11713
Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, Brooks JM, Iniguez SD, O’Donnell P, Kravitz A, Lobo MK (2015) Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry 77:212–222. doi:10.1016/j.biopsych.2014.07.021
Francis TCCD, Lobo MK (2014) Optogenetics: illuminating the neural basis of rodent behavior. Open Access Anim Physiol 6:33–51
Lammel S, Lim BK, Malenka RC (2014) Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology 76 Pt B:351–359. doi:10.1016/j.neuropharm.2013.03.019
Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K (2011) Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 471:358–362. doi:10.1038/nature09820
Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, Kim SY, Adhikari A, Thompson KR, Andalman AS, Gunaydin LA, Witten IB, Deisseroth K (2013) Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493:537–541. doi:10.1038/nature11740
Koob GF (2008) A role for brain stress systems in addiction. Neuron 59:11–34. doi:10.1016/j.neuron.2008.06.012
Wenzel JM, Rauscher NA, Cheer JF, Oleson EB (2015) A role for phasic dopamine release within the nucleus accumbens in encoding aversion: a review of the neurochemical literature. ACS Chem Neurosci 6:16–26. doi:10.1021/cn500255p
Cao JL, Covington HE 3rd, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, Nestler EJ, Han MH (2010) Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci 30:16453–16458. doi:10.1523/JNEUROSCI.3177-10.2010
Friedman AK, Walsh JJ, Juarez B, Ku SM, Chaudhury D, Wang J, Li X, Dietz DM, Pan N, Vialou VF, Neve RL, Yue Z, Han MH (2014) Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science 344:313–319. doi:10.1126/science.1249240
Anstrom KK, Miczek KA, Budygin EA (2009) Increased phasic dopamine signaling in the mesolimbic pathway during social defeat in rats. Neuroscience 161:3–12. doi:10.1016/j.neuroscience.2009.03.023
Razzoli M, Andreoli M, Michielin F, Quarta D, Sokal DM (2011) Increased phasic activity of VTA dopamine neurons in mice 3 weeks after repeated social defeat. Behav Brain Res 218:253–257. doi:10.1016/j.bbr.2010.11.050
Belujon P, Grace AA (2014) Restoring mood balance in depression: ketamine reverses deficit in dopamine-dependent synaptic plasticity. Biol Psychiatry 76:927–936. doi:10.1016/j.biopsych.2014.04.014
Lammel S, Tye KM, Warden MR (2014) Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav 13:38–51. doi:10.1111/gbb.12049
Marton TF, Sohal VS (2015) Of mice, men, and microbial opsins: how optogenetics can help hone mouse models of mental illness. Biol Psychiatry. doi:10.1016/j.biopsych.2015.04.012
Walsh JJ, Han MH (2014) The heterogeneity of ventral tegmental area neurons: projection functions in a mood-related context. Neuroscience 282C:101–108. doi:10.1016/j.neuroscience.2014.06.006
Valenti O, Gill KM, Grace AA (2012) Different stressors produce excitation or inhibition of mesolimbic dopamine neuron activity: response alteration by stress pre-exposure. Eur J Neurosci 35:1312–1321. doi:10.1111/j.1460-9568.2012.08038.x
Brischoux F, Chakraborty S, Brierley DI, Ungless MA (2009) Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci USA 106:4894–4899. doi:10.1073/pnas.0811507106
Lammel S, Ion DI, Roeper J, Malenka RC (2011) Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 70:855–862. doi:10.1016/j.neuron.2011.03.025
Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC (2012) Input-specific control of reward and aversion in the ventral tegmental area. Nature 491:212–217. doi:10.1038/nature11527
Lecca S, Meye FJ, Mameli M (2014) The lateral habenula in addiction and depression: an anatomical, synaptic and behavioral overview. Eur J Neurosci 39:1170–1178. doi:10.1111/ejn.12480
Stamatakis AM, Stuber GD (2012) Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci 15:1105–1107. doi:10.1038/nn.3145
Dipesh Chaudhury HZ, Juarez B, Friedman A, Ku S, Han M-H (2014) Lateral habenula projections to a subset of ventral tegmental area neurons rapidly encodes for susceptibility to social defeat stress. Soc Neurosci Abstr
Shabel SJ, Proulx CD, Trias A, Murphy RT, Malinow R (2012) Input to the lateral habenula from the basal ganglia is excitatory, aversive, and suppressed by serotonin. Neuron 74:475–481. doi:10.1016/j.neuron.2012.02.037
Ungless MA, Grace AA (2012) Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci 35:422–430. doi:10.1016/j.tins.2012.02.003
Napier TC, Maslowski-Cobuzzi RJ (1994) Electrophysiological verification of the presence of D1 and D2 dopamine receptors within the ventral pallidum. Synapse 17:160–166. doi:10.1002/syn.890170304
Sesack SR, Grace AA (2010) Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology 35:27–47. doi:10.1038/npp.2009.93
Chang CH, Grace AA (2014) Amygdala-ventral pallidum pathway decreases dopamine activity after chronic mild stress in rats. Biol Psychiatry 76:223–230. doi:10.1016/j.biopsych.2013.09.020
Michelsen KA, Prickaerts J, Steinbusch HW (2008) The dorsal raphe nucleus and serotonin: implications for neuroplasticity linked to major depression and Alzheimer’s disease. Prog Brain Res 172:233–264. doi:10.1016/S0079-6123(08)00912-6
Dolen G, Darvishzadeh A, Huang KW, Malenka RC (2013) Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 501:179–184. doi:10.1038/nature12518
Soiza-Reilly M, Commons KG (2014) Unraveling the architecture of the dorsal raphe synaptic neuropil using high-resolution neuroanatomy. Front Neural Circuits 8:105. doi:10.3389/fncir.2014.00105
Yang LM, Hu B, Xia YH, Zhang BL, Zhao H (2008) Lateral habenula lesions improve the behavioral response in depressed rats via increasing the serotonin level in dorsal raphe nucleus. Behav Brain Res 188:84–90. doi:10.1016/j.bbr.2007.10.022
Wang RY, Aghajanian GK (1977) Inhibiton of neurons in the amygdala by dorsal raphe stimulation: mediation through a direct serotonergic pathway. Brain Res 120:85–102
Ferraro G, Montalbano ME, Sardo P, La Grutta V (1996) Lateral habenular influence on dorsal raphe neurons. Brain Res Bull 41:47–52
Challis C, Beck SG, Berton O (2014) Optogenetic modulation of descending prefrontocortical inputs to the dorsal raphe bidirectionally bias socioaffective choices after social defeat. Front Behav Neurosci 8:43. doi:10.3389/fnbeh.2014.00043
Challis C, Boulden J, Veerakumar A, Espallergues J, Vassoler FM, Pierce RC, Beck SG, Berton O (2013) Raphe GABAergic neurons mediate the acquisition of avoidance after social defeat. J Neurosci 33(13978–13988):13988a. doi:10.1523/JNEUROSCI.2383-13.2013
Curtis AL, Bello NT, Connolly KR, Valentino RJ (2002) Corticotropin-releasing factor neurones of the central nucleus of the amygdala mediate locus coeruleus activation by cardiovascular stress. J Neuroendocrinol 14:667–682
West CH, Ritchie JC, Boss-Williams KA, Weiss JM (2009) Antidepressant drugs with differing pharmacological actions decrease activity of locus coeruleus neurons. Int J Neuropsychopharmacol 12:627–641. doi:10.1017/S1461145708009474
Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10:211–223. doi:10.1038/nrn2573
Bruzos-Cidon C, Miguelez C, Rodriguez JJ, Gutierrez-Lanza R, Ugedo L, Torrecilla M (2014) Altered neuronal activity and differential sensitivity to acute antidepressants of locus coeruleus and dorsal raphe nucleus in Wistar Kyoto rats: a comparative study with Sprague Dawley and Wistar rats. Eur Neuropsychopharmacol 24:1112–1122. doi:10.1016/j.euroneuro.2014.02.007
Bruzos-Cidon C, Llamosas N, Ugedo L, Torrecilla M (2015) Dysfunctional inhibitory mechanisms in locus coeruleus neurons of the wistar kyoto rat. Int J Neuropsychopharmacol. doi:10.1093/ijnp/pyu122
Covington HE 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E, Ghose S, Tamminga CA, Neve RL, Deisseroth K, Nestler EJ (2010) Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci 30:16082–16090. doi:10.1523/JNEUROSCI.1731-10.2010
Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, Cai X (2015) An excitatory synapse hypothesis of depression. Trends Neurosci 38:279–294. doi:10.1016/j.tins.2015.03.003
Sears RM, Fink AE, Wigestrand MB, Farb CR, de Lecea L, Ledoux JE (2013) Orexin/hypocretin system modulates amygdala-dependent threat learning through the locus coeruleus. Proc Natl Acad Sci USA 110:20260–20265. doi:10.1073/pnas.1320325110
Namburi P, Beyeler A, Yorozu S, Calhoon GG, Halbert SA, Wichmann R, Holden SS, Mertens KL, Anahtar M, Felix-Ortiz AC, Wickersham IR, Gray JM, Tye KM (2015) A circuit mechanism for differentiating positive and negative associations. Nature 520:675–678. doi:10.1038/nature14366
Johnson LR, Hou M, Prager EM, Ledoux JE (2011) Regulation of the fear network by mediators of stress: norepinephrine alters the balance between cortical and subcortical afferent excitation of the lateral amygdala. Front Behav Neurosci 5:23. doi:10.3389/fnbeh.2011.00023
LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23:155–184. doi:10.1146/annurev.neuro.23.1.155
Chang CH, Grace AA (2015) Dopaminergic modulation of lateral amygdala neuronal activity: differential D1 and D2 receptor effects on thalamic and cortical afferent inputs. Int J Neuropsychopharmacol. doi:10.1093/ijnp/pyv015
Belzung C, Willner P, Philippot P (2015) Depression: from psychopathology to pathophysiology. Curr Opin Neurobiol 30:24–30. doi:10.1016/j.conb.2014.08.013
Vialou V, Robison AJ, Laplant QC, Covington HE 3rd, Dietz DM, Ohnishi YN, Mouzon E, Rush AJ 3rd, Watts EL, Wallace DL, Iniguez SD, Ohnishi YH, Steiner MA, Warren BL, Krishnan V, Bolanos CA, Neve RL, Ghose S, Berton O, Tamminga CA, Nestler EJ (2010) DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci 13:745–752. doi:10.1038/nn.2551
Lim LW, Prickaerts J, Huguet G, Kadar E, Hartung H, Sharp T, Temel Y (2015) Electrical stimulation alleviates depressive-like behaviors of rats: investigation of brain targets and potential mechanisms. Transl Psychiatry 5:e535. doi:10.1038/tp.2015.24
Nicola SM (2007) The nucleus accumbens as part of a basal ganglia action selection circuit. Psychopharmacology 191:521–550. doi:10.1007/s00213-006-0510-4
Smith RJ, Lobo MK, Spencer S, Kalivas PW (2013) Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol 23:546–552. doi:10.1016/j.conb.2013.01.026
Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12:366–375
Maia TV, Frank MJ (2011) From reinforcement learning models to psychiatric and neurological disorders. Nat Neurosci 14:154–162. doi:10.1038/nn.2723
Kravitz AV, Kreitzer AC (2012) Striatal mechanisms underlying movement, reinforcement, and punishment. Physiology 27:167–177. doi:10.1152/physiol.00004.2012
Lenz JD, Lobo MK (2013) Optogenetic insights into striatal function and behavior. Behav Brain Res 255:44–54. doi:10.1016/j.bbr.2013.04.018
Carlezon WA Jr, Thomas MJ (2009) Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology 56(Suppl 1):122–132. doi:10.1016/j.neuropharm.2008.06.075
Lobo MK, Nestler EJ (2011) The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat 5:41. doi:10.3389/fnana.2011.00041
Freeze BS, Kravitz AV, Hammack N, Berke JD, Kreitzer AC (2013) Control of basal ganglia output by direct and indirect pathway projection neurons. J Neurosci 33:18531–18539. doi:10.1523/JNEUROSCI.1278-13.2013
Lobo MK, Zaman S, Damez-Werno DM, Koo JW, Bagot RC, DiNieri JA, Nugent A, Finkel E, Chaudhury D, Chandra R, Riberio E, Rabkin J, Mouzon E, Cachope R, Cheer JF, Han MH, Dietz DM, Self DW, Hurd YL, Vialou V, Nestler EJ (2013) DeltaFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J Neurosci 33:18381–18395. doi:10.1523/JNEUROSCI.1875-13.2013
Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC (2012) Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487:183–189. doi:10.1038/nature11160
Bagot RC, Parise EM, Pena CJ, Zhang HX, Maze I, Chaudhury D, Persaud B, Cachope R, Bolanos-Guzman CA, Cheer J, Deisseroth K, Han MH, Nestler EJ (2015) Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nature communications 6:7062. doi:10.1038/ncomms8062
McClung CA, Nestler EJ (2003) Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci 6:1208–1215. doi:10.1038/nn1143
Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE 3rd, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ (2009) Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 62:335–348. doi:10.1016/j.neuron.2009.03.026
Bredt DS, Nicoll RA (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40:361–379
Li DP, Byan HS, Pan HL (2012) Switch to glutamate receptor 2-lacking AMPA receptors increases neuronal excitability in hypothalamus and sympathetic drive in hypertension. J Neurosci 32:372–380. doi:10.1523/JNEUROSCI.3222-11.2012
Grace AA, Floresco SB, Goto Y, Lodge DJ (2007) Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci 30:220–227. doi:10.1016/j.tins.2007.03.003
Goto Y, Grace AA (2005) Dopamine-dependent interactions between limbic and prefrontal cortical plasticity in the nucleus accumbens: disruption by cocaine sensitization. Neuron 47:255–266. doi:10.1016/j.neuron.2005.06.017
Goto Y, Grace AA (2005) Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-directed behavior. Nat Neurosci 8:805–812. doi:10.1038/nn1471
O’Donnell P, Grace AA (1994) Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res 634:105–112. pii: 0006-8993(94)90263-1
West AR, Grace AA (2002) Opposite influences of endogenous dopamine D1 and D2 receptor activation on activity states and electrophysiological properties of striatal neurons: studies combining in vivo intracellular recordings and reverse microdialysis. J Neurosci 22:294–304. pii:22/1/294
Kauer JA, Malenka RC (2007) Synaptic plasticity and addiction. Nat Rev Neurosci 8:844–858. doi:10.1038/nrn2234
Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10:647–658. doi:10.1038/nrn2699
Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ (2010) The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci 33:267–276. doi:10.1016/j.tins.2010.02.002
Luscher C, Malenka RC (2012) NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a005710
Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, McEwen BS, Morrison JH (2006) Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex 16:313–320. doi:10.1093/cercor/bhi104
Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, Morrison JH (2009) Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 164:798–808. doi:10.1016/j.neuroscience.2009.08.053
Shansky RM, Hamo C, Hof PR, McEwen BS, Morrison JH (2009) Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb Cortex 19:2479–2484. doi:10.1093/cercor/bhp003
Chakravarthy S, Saiepour MH, Bence M, Perry S, Hartman R, Couey JJ, Mansvelder HD, Levelt CN (2006) Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proc Natl Acad Sci USA 103:1071–1076. doi:10.1073/pnas.0506305103
Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolanos CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, Nestler EJ (2009) Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J Neurosci 29:3529–3537. doi:10.1523/JNEUROSCI.6173-08.2009
Nestler EJ, Carlezon WA Jr (2006) The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59:1151–1159. doi:10.1016/j.biopsych.2005.09.018
Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A (2008) Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol 586:2157–2170. doi:10.1113/jphysiol.2007.150078
Neuhoff H, Neu A, Liss B, Roeper J (2002) I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci 22:1290–1302
Zhang J, Shapiro MS (2012) Activity-dependent transcriptional regulation of M-Type (Kv7) K(+) channels by AKAP79/150-mediated NFAT actions. Neuron 76:1133–1146. doi:10.1016/j.neuron.2012.10.019
Destexhe A, Marder E (2004) Plasticity in single neuron and circuit computations. Nature 431:789–795. doi:10.1038/nature03011
Whalley K (2013) Synaptic plasticity: balancing firing rates in vivo. Nat Rev Neurosci 14:820–821. doi:10.1038/nrn3637
Dickman DK, Davis GW (2009) The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 326:1127–1130. doi:10.1126/science.1179685
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–741. doi:10.1016/j.biopsych.2008.11.029
Hashimoto K (2015) Inflammatory biomarkers as differential predictors of antidepressant response. Int J Mol Sci 16:7796–7801. doi:10.3390/ijms16047796
Shelton RC, Claiborne J, Sidoryk-Wegrzynowicz M, Reddy R, Aschner M, Lewis DA, Mirnics K (2011) Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol Psychiatry 16:751–762. doi:10.1038/mp.2010.52
Himmerich H, Fischer J, Bauer K, Kirkby KC, Sack U, Krugel U (2013) Stress-induced cytokine changes in rats. Eur Cytokine Netw 24:97–103. doi:10.1684/ecn.2013.0338
O’Connor MF, Irwin MR, Wellisch DK (2009) When grief heats up: pro-inflammatory cytokines predict regional brain activation. Neuroimage 47:891–896. doi:10.1016/j.neuroimage.2009.05.049
Kaster MP, Gadotti VM, Calixto JB, Santos AR, Rodrigues AL (2012) Depressive-like behavior induced by tumor necrosis factor-alpha in mice. Neuropharmacology 62:419–426. doi:10.1016/j.neuropharm.2011.08.018
Hetman M, Gozdz A (2004) Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem 271:2050–2055. doi:10.1111/j.1432-1033.2004.04133.x
Subramaniam S, Unsicker K (2010) ERK and cell death: ERK1/2 in neuronal death. FEBS J 277:22–29. doi:10.1111/j.1742-4658.2009.07367.x
Gantke T, Sriskantharajah S, Sadowski M, Ley SC (2012) IkappaB kinase regulation of the TPL-2/ERK MAPK pathway. Immunol Rev 246:168–182. doi:10.1111/j.1600-065X.2012.01104.x
Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G (2003) The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci 23:7311–7316
Dwivedi Y, Rizavi HS, Conley RR, Pandey GN (2006) ERK MAP kinase signaling in post-mortem brain of suicide subjects: differential regulation of upstream Raf kinases Raf-1 and B-Raf. Mol Psychiatry 11:86–98. doi:10.1038/sj.mp.4001744
Gourley SL, Wu FJ, Kiraly DD, Ploski JE, Kedves AT, Duman RS, Taylor JR (2008) Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol Psychiatry 63:353–359. doi:10.1016/j.biopsych.2007.07.016
Iniguez SD, Vialou V, Warren BL, Cao JL, Alcantara LF, Davis LC, Manojlovic Z, Neve RL, Russo SJ, Han MH, Nestler EJ, Bolanos-Guzman CA (2010) Extracellular signal-regulated kinase-2 within the ventral tegmental area regulates responses to stress. J Neurosci 30:7652–7663. doi:10.1523/JNEUROSCI.0951-10.2010
Kaminska B (2005) MAPK signalling pathways as molecular targets for anti-inflammatory therapy–from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta 1754:253–262. doi:10.1016/j.bbapap.2005.08.017
Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, Rebusi N, Heshmati M, Aleyasin H, Warren BL, Lebonte B, Horn S, Lapidus KA, Stelzhammer V, Wong EH, Bahn S, Krishnan V, Bolanos-Guzman CA, Murrough JW, Merad M, Russo SJ (2014) Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci USA 111:16136–16141. doi:10.1073/pnas.1415191111
Perez-Caballero L, Perez-Egea R, Romero-Grimaldi C, Puigdemont D, Molet J, Caso JR, Mico JA, Perez V, Leza JC, Berrocoso E (2014) Early responses to deep brain stimulation in depression are modulated by anti-inflammatory drugs. Mol Psychiatry 19:607–614. doi:10.1038/mp.2013.63
McClung CA (2007) Circadian genes, rhythms and the biology of mood disorders. Pharmacol Ther 114:222–232. doi:10.1016/j.pharmthera.2007.02.003
Wirz-Justice A (2006) Biological rhythm disturbances in mood disorders. Int Clin Psychopharmacol 21(Suppl 1):S11–S15. doi:10.1097/01.yic.0000195660.37267.cf
Boivin DB, Czeisler CA, Dijk DJ, Duffy JF, Folkard S, Minors DS, Totterdell P, Waterhouse JM (1997) Complex interaction of the sleep-wake cycle and circadian phase modulates mood in healthy subjects. Arch Gen Psychiatry 54:145–152
Birchler-Pedross A, Schroder CM, Munch M, Knoblauch V, Blatter K, Schnitzler-Sack C, Wirz-Justice A, Cajochen C (2009) Subjective well-being is modulated by circadian phase, sleep pressure, age, and gender. J Biol Rhythms 24:232–242. doi:10.1177/0748730409335546
Sidor MM, McClung CA (2014) Timing matters: using optogenetics to chronically manipulate neural circuitry and rhythms. Front Behav Neurosci 8:41. doi:10.3389/fnbeh.2014.00041
Ehlers CL, Kupfer DJ (1987) Hypothalamic peptide modulation of EEG sleep in depression: a further application of the S-process hypothesis. Biol Psychiatry 22:513–517
Bunney BG, Bunney WE (2012) Rapid-acting antidepressant strategies: mechanisms of action. Int J Neuropsychopharmacol 15:695–713. doi:10.1017/S1461145711000927
Benedetti F, Colombo C, Serretti A, Lorenzi C, Pontiggia A, Barbini B, Smeraldi E (2003) Antidepressant effects of light therapy combined with sleep deprivation are influenced by a functional polymorphism within the promoter of the serotonin transporter gene. Biol Psychiatry 54:687–692
Wirz-Justice A, Van den Hoofdakker RH (1999) Sleep deprivation in depression: what do we know, where do we go? Biol Psychiatry 46:445–453. pii: S0006-3223(99)00125-0
Wu JC, Bunney WE (1990) The biological basis of an antidepressant response to sleep deprivation and relapse: review and hypothesis. Am J Psychiatry 147:14–21
Bunney BG, Bunney WE (2013) Mechanisms of rapid antidepressant effects of sleep deprivation therapy: clock genes and circadian rhythms. Biol Psychiatry 73:1164–1171. doi:10.1016/j.biopsych.2012.07.020
Bunney BG, Li JZ, Walsh DM, Stein R, Vawter MP, Cartagena P, Barchas JD, Schatzberg AF, Myers RM, Watson SJ, Akil H, Bunney WE (2015) Circadian dysregulation of clock genes: clues to rapid treatments in major depressive disorder. Mol Psychiatry 20:48–55. doi:10.1038/mp.2014.138
Sequeira A, Morgan L, Walsh DM, Cartagena PM, Choudary P, Li J, Schatzberg AF, Watson SJ, Akil H, Myers RM, Jones EG, Bunney WE, Vawter MP (2012) Gene expression changes in the prefrontal cortex, anterior cingulate cortex and nucleus accumbens of mood disorders subjects that committed suicide. PLoS ONE 7:e35367. doi:10.1371/journal.pone.0035367
Li JZ, Bunney BG, Meng F, Hagenauer MH, Walsh DM, Vawter MP, Evans SJ, Choudary PV, Cartagena P, Barchas JD, Schatzberg AF, Jones EG, Myers RM, Watson SJ Jr, Akil H, Bunney WE (2013) Circadian patterns of gene expression in the human brain and disruption in major depressive disorder. Proc Natl Acad Sci USA 110:9950–9955. doi:10.1073/pnas.1305814110
Kinn AM, Gronli J, Fiske E, Kuipers S, Ursin R, Murison R, Portas CM (2008) A double exposure to social defeat induces sub-chronic effects on sleep and open field behaviour in rats. Physiol Behav 95:553–561. doi:10.1016/j.physbeh.2008.07.031
Spencer S, Falcon E, Kumar J, Krishnan V, Mukherjee S, Birnbaum SG, McClung CA (2013) Circadian genes Period 1 and Period 2 in the nucleus accumbens regulate anxiety-related behavior. Eur J Neurosci 37:242–250. doi:10.1111/ejn.12010
Slattery DA, Uschold N, Magoni M, Bar J, Popoli M, Neumann ID, Reber SO (2012) Behavioural consequences of two chronic psychosocial stress paradigms: anxiety without depression. Psychoneuroendocrinology 37:702–714. doi:10.1016/j.psyneuen.2011.09.002
Meerlo P, Turek FW (2001) Effects of social stimuli on sleep in mice: non-rapid-eye-movement (NREM) sleep is promoted by aggressive interaction but not by sexual interaction. Brain Res 907:84–92
Meerlo P, Overkamp GJ, Benning MA, Koolhaas JM, Van den Hoofdakker RH (1996) Long-term changes in open field behaviour following a single social defeat in rats can be reversed by sleep deprivation. Physiol Behav 60:115–119. pii: 0031938495022716
Lavebratt C, Sjoholm LK, Soronen P, Paunio T, Vawter MP, Bunney WE, Adolfsson R, Forsell Y, Wu JC, Kelsoe JR, Partonen T, Schalling M (2010) CRY2 is associated with depression. PLoS ONE 5:e9407. doi:10.1371/journal.pone.0009407
Kavcic P, Rojc B, Dolenc-Groselj L, Claustrat B, Fujs K, Poljak M (2011) The impact of sleep deprivation and nighttime light exposure on clock gene expression in humans. Croat Med J 52:594–603
Wisor JP, O’Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, Edgar DM, Franken P (2002) A role for cryptochromes in sleep regulation. BMC Neurosci 3:20
Wisor JP, Pasumarthi RK, Gerashchenko D, Thompson CL, Pathak S, Sancar A, Franken P, Lein ES, Kilduff TS (2008) Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J Neurosci 28:7193–7201. doi:10.1523/JNEUROSCI.1150-08.2008
Mongrain V, La Spada F, Curie T, Franken P (2011) Sleep loss reduces the DNA-binding of BMAL1, CLOCK, and NPAS2 to specific clock genes in the mouse cerebral cortex. PLoS One 6:e26622. doi:10.1371/journal.pone.0026622
Bellet MM, Vawter MP, Bunney BG, Bunney WE, Sassone-Corsi P (2011) Ketamine influences CLOCK:BMAL1 function leading to altered circadian gene expression. PLoS One 6:e23982. doi:10.1371/journal.pone.0023982
Gottschlich MM, Mayes T, Khoury J, McCall J, Simakajornboon N, Kagan RJ (2011) The effect of ketamine administration on nocturnal sleep architecture. J Burn Care Res 32:535–540. doi:10.1097/BCR.0b013e31822ac7d1
Mukherjee S, Coque L, Cao JL, Kumar J, Chakravarty S, Asaithamby A, Graham A, Gordon E, Enwright JF 3rd, DiLeone RJ, Birnbaum SG, Cooper DC, McClung CA (2010) Knockdown of Clock in the ventral tegmental area through RNA interference results in a mixed state of mania and depression-like behavior. Biol Psychiatry 68:503–511. doi:10.1016/j.biopsych.2010.04.031
Sidor MM, Spencer SM, Dzirasa K, Parekh PK, Tye KM, Warden MR, Arey RN, Enwright JF 3rd, Jacobsen JP, Kumar S, Remillard EM, Caron MG, Deisseroth K, McClung CA (2015) Daytime spikes in dopaminergic activity drive rapid mood-cycling in mice. Mol Psychiatry. doi:10.1038/mp.2014.167
Hampp G, Ripperger JA, Houben T, Schmutz I, Blex C, Perreau-Lenz S, Brunk I, Spanagel R, Ahnert-Hilger G, Meijer JH, Albrecht U (2008) Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr Biol 18:678–683. doi:10.1016/j.cub.2008.04.012
Kondratova AA, Dubrovsky YV, Antoch MP, Kondratov RV (2010) Circadian clock proteins control adaptation to novel environment and memory formation. Aging 2:285–297
De Bundel D, Gangarossa G, Biever A, Bonnefont X, Valjent E (2013) Cognitive dysfunction, elevated anxiety, and reduced cocaine response in circadian clock-deficient cryptochrome knockout mice. Front Behav Neurosci 7:152. doi:10.3389/fnbeh.2013.00152
Chung S, Lee EJ, Yun S, Choe HK, Park SB, Son HJ, Kim KS, Dluzen DE, Lee I, Hwang O, Son GH, Kim K (2014) Impact of circadian nuclear receptor REV-ERBalpha on midbrain dopamine production and mood regulation. Cell 157:858–868. doi:10.1016/j.cell.2014.03.039
Panksepp JB, Wong JC, Kennedy BC, Lahvis GP (2008) Differential entrainment of a social rhythm in adolescent mice. Behav Brain Res 195:239–245. doi:10.1016/j.bbr.2008.09.010
Acknowledgments
This work was supported by the National Institute of Mental Health (MH092306: M.H.H.), Johnson & Johnson/International Mental Health Research Organization (M.H.H.), NARSAD Independent Investigator Award (M.H.H.), and NARSAD Young Investigator Award (D.C.).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chaudhury, D., Liu, H. & Han, MH. Neuronal correlates of depression. Cell. Mol. Life Sci. 72, 4825–4848 (2015). https://doi.org/10.1007/s00018-015-2044-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2044-6