
Abstract
Glutamate is the predominant excitatory neurotransmitter in the central nervous system. Excitatory amino acid transporter 2 (EAAT2) is primarily responsible for clearance of extracellular glutamate to prevent neuronal excitotoxicity and hyperexcitability. EAAT2 plays a critical role in regulation of synaptic activity and plasticity. In addition, EAAT2 has been implicated in the pathogenesis of many central nervous system disorders. In this review, we summarize current understanding of EAAT2, including structure, pharmacology, physiology, and functions, as well as disease relevancy, such as in stroke, Parkinson’s disease, epilepsy, amyotrophic lateral sclerosis, Alzheimer’s disease, major depressive disorder, and addiction. A large number of studies have demonstrated that up-regulation of EAAT2 protein provides significant beneficial effects in many disease models suggesting EAAT2 activation is a promising therapeutic approach. Several EAAT2 activators have been identified. Further understanding of EAAT2 regulatory mechanisms could improve development of drug-like compounds that spatiotemporally regulate EAAT2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glutamate is the predominant excitatory neurotransmitter in the central nervous system (CNS). Glutamate is released from pre-synaptic terminals and diffuses across the synaptic cleft where it binds glutamate receptors. Glutamatergic transmission is terminated once excitatory amino acid transporters (EAATs) take up synaptic glutamate. Five mammalian EAAT isoforms (EAAT1-5) have been characterized (for review see: [1–5]) with each having different nomenclatures, expression patterns, and uptake kinetics (Table 1). EAAT1, 2, and 3 are widely expressed in the CNS, whereas the expression of EAAT4 and 5 is predominately limited to the cerebellum and retina, respectively. The expression of EAAT2 is higher than that of EAAT1 in the forebrain while the inverse is true in the cerebellum. Under normal conditions, EAAT1 and 2 are mainly expressed in astrocytes and localized to the cellular membrane while EAAT3, 4, and 5 are mainly expressed in neurons [6–12]. EAAT2 is the most abundant EAAT and is primarily responsible for glutamate homeostasis in the forebrain [6, 13–16]. Therefore, dysfunction or dysregulation of EAAT2 can lead to excessive glutamate-mediated toxicity [16]. This review describes what is currently known about EAAT2 basic biology and role in disease state.
Structure and pharmacology
EAAT2 splice variants
The EAAT2 gene is composed of 11 exons which form multiple splice variants. EAAT2 transcripts contain a long 3′ UTR (11–12 kb transcript; 3′UTR of ~9.5 kb) and multiple terminal variants (at least two types of N-terminal and three types of C-terminal) which yield a protein product composed of eight transmembrane (TM) domains [3, 17, 18]. EAAT2 also has two N-glycosylation sites located in the extracellular loop between TM domains 3 and 4. For the splice variants, the two N-terminal start sequences are MASTEG- and MVSANN-, while the three C-terminal sequences are -PWKREK (-a type), -DIETCI (-b type), and EYQSWV (-c type). EAAT2b (GLT1b) and EAAT2c (GLT1c) contain a PDZ-binding domain (the last three amino acids as indicated by the underline) whereas EAAT2a (GLT1a) does not. The PDZ-binding domain of EAAT2b is involved in an interaction between EAAT2b and the PDZ domain protein, PICK1 [19, 20]. The interaction modulates EAAT2 functions but it has a minor effect on [3H]glutamate uptake [19, 20]. These results are consistent with a previous report that the pharmacological properties of EAAT2a and EAAT2b are indistinguishable [21].
EAAT2 proteins are almost exclusively expressed in astrocytes under normal conditions and rarely in neurons [7, 8, 22, 23]. EAAT2a in CA1 of the hippocampus is expressed in neuronal axon terminals (as well as in astrocytes) but accounts for approximately 10 % of total EAAT2a hippocampal expression [23]. The percentage of total EAAT2 protein in the adult rat hippocampus is 90 % (EAAT2a), 6 % (EAAT2b), and 1 % (EAAT2c) [24]. Results for EAAT2b protein distribution are inconsistent among researchers. In the early study, some results indicate that the EAAT2b protein in the brain is preferentially expressed in neurons [21, 25, 26], while others show that the expression is restricted to astrocytes [27, 28]. The inconsistencies may be attributed to the following reasons: (1) EAAT2b protein levels are much lower than EAAT2a protein levels and (2) the unique splice region is only 11 amino acids between the two variants. Therefore, the antibodies for EAAT2b may have insufficient affinity to distinguish between isoforms [29]. Recently, the verified EAAT2b antibody, with EAAT2 knockout mice as a negative control, shows EAAT2b protein is preferentially expressed in astrocytes [24]. Interestingly, in situ hybridization demonstrates that both EAAT2a mRNA and EAAT2b mRNA are expressed in neurons and astrocytes in the brain implicating cell-specific translational regulation [30]. For the subcellular mRNA distributions in astrocytes, EAAT2a mRNA is predominant in perisynaptic processes, whereas EAAT2b mRNA is distributed in the cell body [30]. EAAT2c protein is expressed in rat and human retinal neurons [31].
EAAT2 regulation
EAAT2 expression is regulated at the level of transcription (including epigenetic modification), translation, trafficking, transport, and degradation. For regulation at the transcriptional level, endogenous and pharmacological modulators can induce activation or repression, such as EGF, cAMP, PACAP TGFbeta, TNFalpha, ceftriaxone, and estrogen related compounds as well as co-culturing astrocytes with neurons [32–37]. The EAAT2 promoter contains several transcription factor-binding sequences, including NF-κB, Sp1, N-myc, CREB, EGR, and NFAT [37, 38]. Several lines of study have demonstrated that NF-κB plays an important role in the transcriptional regulation of EAAT2 by functioning as an intrinsic activator. EGF-induced EAAT2 transcriptional activation is mediated by NF-κB activation, but not by the canonical enhancement of IκB degradation and nuclear accumulation of the p65 isoform of NF-κB [36, 39]. On the other hand, TNFalpha-mediated EAAT2 repression also utilizes NF-κB activation but also requires N-myc activation which leads to the conversion of NF-κB to a transcriptional repressor [36]. Tamoxifen (antagonist of the estrogen receptor), raloxifene (selective estrogen receptor modulator), and 17beta-estradiol increase EAAT2 expression via activation of both NF-κB and CREB [33–35]. Ceftriaxone, a beta-lactam antibiotic, increases EAAT2 expression via the conventional NF-κB pathway with IκB degradation and p65 nuclear accumulation [40, 41]. It is also notable that basal EAAT2 protein expression in primary astrocyte culture is maintained at a low level, but co-culturing with neurons strongly enhances EAAT2 expression [42, 43]. This induction involves binding of NF-κB to the EAAT2 promoter and activation of kappaB-motif binding phosphor-protein (KBPP) [38, 44]. These results indicate that the NF-κB pathway serves as an important modulator of EAAT2 expression at the transcription level. On the other hand, riluzole, a neuroprotective drug for the treatment of amyotrophic lateral sclerosis (ALS), has many effects which include enhanced EAAT2 uptake activity and protein expression [45, 46]. EAAT2 transcriptional activation by riluzole is mediated by heat shock factor 1 (HSF1) which regulates heat shock proteins (HSPs) that are essential for proper protein folding, trafficking, and degradation in cellular stress responses [46]. EAAT2 expression is also regulated epigenetically. Histone deacetylase (HDAC) inhibitors (valproic acid and trichostatin A) enhance EAAT2 expression in primary astrocytes [47]. Yin Yang 1 (YY1), a ubiquitous transcription factor, decreases EAAT2 promoter activity by recruiting HDACs as co-repressors in primary astrocytes [48]. In addition, the EAAT2 promoter exhibits hypomethylation in cortex relative to the cerebellum, suggesting a potential explanation for why EAAT2 expression is higher in cortex than in cerebellum [49]. The methylation on CpG sites of the EAAT2 promoter is reduced in astrocytes isolated from astrocyte–neuron co-cultures when compared to astrocytes cultured alone [50]. These results indicate that the acetylation and methylation state of the EAAT2 promoter is strongly involved in the regulation of EAAT2 expression.
EAAT2 expression and function are regulated at the post-transcriptional level. EAAT2 translation is controlled by corticosterone in primary astrocyte cell lines, primary cortical neuron–astrocyte mixed cultures, and mice [51]. This regulation involves the 5′-UTR of EAAT2. Neuronal exosomes have been shown to use miR-124a, via a translational regulation mechanism, to increase EAAT2 protein [52]. In addition, EAAT1 and EAAT2 protein, but not mRNA, are increased in ephrin-A3-knockout mice and accompany synaptic changes [53, 54]. Recently, we have developed drug-like, small-molecule pyridazine derivatives which can activate EAAT2 translation [55–57]. One of these compounds increases EAAT2 protein within only 2 h in vivo which is very rapid when compared with transcription activators which require 24–48 h. This translational activation mechanism involves PKC activation and subsequent YB-1 phosphorylation [56]. At the level of post-translational modification, EAAT2 protein undergoes palmitoylation at cysteine 38 (C38) which is required for normal glutamate uptake function [58]. EAAT2 is also constitutively sumoylated in the CNS in vivo. The sumoylated form of EAAT2 is localized to intracellular compartments while non-sumoylated EAAT2 is primarily found at the plasma membrane [59]. Desumoylation in primary astrocytes causes increased EAAT2-mediated glutamate uptake. In addition, EAAT2 is ubiquitinated at the C-terminal which mediates PKC-induced internalization and degradation [60–62]. In addition, the glutamate uptake function of EAAT2 is decreased by reducing membrane cholesterol levels by dissociation from lipid rafts which are microdomains of organized glycosphingolipids, cholesterol, and protein receptors. This indicates that plasma membrane organization of lipid rafts regulate EAAT2 activity [63]. Moreover, the activity of EAAT2 is associated with clustering at the astrocyte perisynapse which is mediated by neuronal activity [64, 65]. There are many potential EAAT2 regulators at the post-transcriptional level.
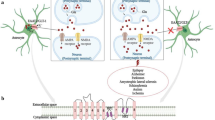
There are pharmacological agents that directly modulate the transport function of EAAT2. Parawixin1, purified from the spider Parawixia bistriata venom, directly enhances the glutamate uptake function of EAAT2 [66, 67]. On the other hand, there are many glutamate transporter inhibitors. The pharmacological properties of these compounds are described in recent review articles [68, 69]. In this review, inhibitors are categorized into two classes: (1) competitive inhibitors (binding to the substrate binding site) and (2) noncompetitive inhibitors (interacting with a site that is different from the substrate binding site) (Fig. 1). The competitive inhibitors (i.e., substrate analogues) include the cyclic molecules l-trans-2,4-pyrrolidinedicarboxylic acid (PDC; nonselective EAATs affinity) [11, 70–73], and Dihydrokainic acid (DHK; a selective inhibitor for EAAT2) [70]. To reduce affinity for the glutamate receptor, a bulky substituent was added to the hydroxyl group of hydroxyl-aspartate, e.g. l-threo-beta-benzyloxyaspartate (TBOA) and (3S)-3-[[3-[[4-(Trifluoromethyl)benzoyl]amino]phenyl]methoxy]-l-aspartic acid (TFB-TBOA). While TBOA is a nonspecific subtype inhibitor, TFB-TBOA has higher affinity for EAATs than TBOA [74, 75]. More recently, N-[4-(2-Bromo-4,5-difluorophenoxy)phenyl]-l-asparagine (WAY-213613) has been developed. WAY-213613 has higher potency and selectivity for EAAT2 over EAAT1 and EAAT3 [76, 77]. HIP-B is a noncompetitive EAATs inhibitor which binds at an allosteric binding site [78, 79]. UCPH-101 is an EAAT1-selective noncompetitive inhibitor that targets a predominantly hydrophobic crevice in the trimerization domain of EAAT1 [80–82]. In sum, these studies show that EAAT2 expression is dynamically regulated at the transcription and post-transcriptional level and that EAAT2 function can be pharmacologically modulated with moderate specificity.

Structure of substrate, activators, and inhibitors
Mechanisms of glutamate transport
Extracellular glutamate transport is achieved by the co-transportation of 3 Na+ and 1 H+ for the antiport of 1 K+. The Na+ gradient drives glutamate transport. The mechanisms of glutamate transport have been assessed by mutagenesis and crystal structure studies (for review see: [5, 83]) based on the archaeal homolog of the EAATs from pyrococcus horikoshii, GltPh [84–86]. The GltPh protein has 37 % conserved identity with human EAAT2. GltPh contains the 8 TM domains and two re-entrant loops (Fig. 2a). Although, there are some differences between the archaeal and the mammalian transporter, including substrate preference (high affinity of l-aspartate when compared with EAATs transport as well as similar efficiency for aspartate and glutamate), the cotransport and countertransport molecules (without 1 H+, 1 K+), and slow transport turnover (Fig. 2b). However, the GltPh structure provides important information for understanding the transport mechanism. The first six TM domains of N-terminal form a scaffold domain. The C-terminal domain contains two helical hairpins (HP1 and HP2), and two TM domains, including the core transport domain. Initiation of transport requires substrate binding on the extracellular side. The tips HP1 and HP2 are proposed to contribute to the substrate binding and transport (Fig. 2c, [85]). The structure of GltPh shows a homotrimeric assembly of subunits containing the substrate-binding site to form a bowl-shaped structure (Fig. 2d). The cavity of the bowl faces the extracellular side with a solvent-filled extracellular basin extending halfway across the membrane bilayer. This trimeric formation is also observed in EAATs protein from biological studies. EAAT1 and EAAT2 are homotrimeric, while EAAT4 forms heterotrimers [15, 87, 88]. Understanding underlying details of the mechanisms of EAAT2 transport will facilitate drug development for EAAT2 modulators.

Transport mechanism and structure of glutamate transporter. There are eight transmembrane (TM) domains and two helical hairpins (HP1 and HP2). a The topology model of an archaeal homolog of the EAATs from pyrococcus horikoshii, GltPh, is shown. TM 1, 2, 4, and 5 are included in the scaffold domain (trimerization domain) while TM 3, 6, 7, and 8, and HP1-2 are included in the core transport domain. b The stoichiometry of transport. EAATs exhibit influx of glutamate/aspartate (Glu), 3 Na+ and 1 H+, and outflux of 1 K+. GltPh utilizes influx of aspartate (Asp) and 3 Na+. c The hypothetical transport mechanism of GltPh. Reyes et al. proposed that the tips HP1 and HP2 contribute to substrate binding and transport [85] and are accompanied with rotation of the core transport domain (red). The trimerization domain is indicated in blue. d The bowl-shaped structure of the GltPh trimer. Each subunit is represented as blue, green, and white. The crystal structure is based on GltPh binding with TBOA (PBD 2NWW, http://www.rcsb.org/pdb/home/home.do)
Physiology and functions
Glutamate uptake
Pre-synaptic nerve terminals release glutamate by synaptic-vesicle exocytosis. The glutamate release elevates glutamate concentration in the synaptic cleft, and the glutamate binds to glutamate receptors (NMDA and AMPA receptors) on the post-synaptic neurons. This binding stimulates the neurons via Ca2+ or Na+ influx. The glutamate is then quickly removed from the synaptic cleft by EAAT2 in astrocytes. EAAT2 is responsible for up to 80–90 % of total extracellular glutamate uptake activities [6, 14–16, 89]. The roles of glutamate uptake by EAAT2 are to modulate glutamate transmission, prevent excitotoxicity, supply glutamate to adjacent neuron via conversion to glutamine, and energy production.
Glutamate uptake by EAATs plays an important role in reducing the glutamate concentration in the extracellular space as there is no strong evidence of a specific enzyme for glutamate degradation here. The baseline concentration of extracellular glutamate is as low as 25 nM in hippocampal slice [90]. The concentration of released glutamate from the pre-synaptic terminal reaches approximately 1 mM, and is rapidly decreased by binding to glutamate transporters (rate constant: 107 M−1 s−1) [91–93]. EAATs then transport glutamate into the intracellular space slowly (~30 glutamate/s) [4, 94–97]. A growing body of evidence shows that EAAT2 effects synaptic transmission. Blocking of EAAT2 with a specific inhibitor, DHK, shows extended NMDA-receptor mediated excitatory post-synaptic current [98]. EAAT2 is responsible for increased glutamate uptake during late-LTP (long-term potentiation) and may also play an ongoing role in hippocampal circuitry to encode and store information [99]. These results indicate that EAAT2 contributes to glutamatergic signal transmission as well as maintenance of synaptic glutamate concentration at a low level.
Excess glutamate diffuses from the synaptic cleft to the extrasynaptic (outside of the synapse) space. The activation of synaptic NMDA receptors promotes cell survival while extrasynaptic NMDA receptor activation promotes cell death which is termed excitotoxicity (for review see: [100, 101]). The primary synaptic NMDA receptor subunit GluN2A (NR2A) and the primary extrasynaptic NMDA receptor GluN2B (NR2B) contribute to neuronal survival and death, respectively [102]. Because of this differential distribution of NMDA receptor subunits, the synaptic and extrasynaptic NMDA receptors have different intracellular signaling pathways. The activation of synaptic NMDA receptors induces anti-apoptotic genes and many transcription factors, including cyclic-AMP response element binding protein (CREB) via nuclear Ca2+ signaling. The activation of CREB subsequently increases BDNF production which has neuroprotective properties [100]. On the other hand, the activation of extrasynaptic NMDA receptors inhibits the neuroprotective signaling promoted by synaptic NMDA receptors which lead to excitotoxicity. Excitotoxicity is postulated to contribute to a myriad of acute and chronic diseases. Acute increased glutamate is involved in many diseases associated with severe neuronal damage such as stroke and traumatic brain injury. Chronic excitotoxicity and/or hyperexcitability are/is related to chronic neurological disease as well as psychiatric disease, including Alzheimer’s disease, Parkinson’s disease, ALS, major depressive disorder, and addiction. Therefore, reduction of glutamate could prevent excitotoxicity in numerous neurological and psychiatric diseases.
Once glutamate is taken up by the astrocyte, it is converted into glutamine via glutamine synthetase. Glutamine is then transported back to the pre-synaptic neuron and then converted back into glutamate via glutaminase. This completes the glutamate–glutamine cycle which was first proposed in the 1970’s [103, 104]. It is notable that these canonical biochemical pathways are not essential for supplying glutamate for neurotransmitter production or release [2]. Blocking the glutamate–glutamine cycle failed to suppress glutamatergic synaptic transmission in organotypic brain slices [105] indicating that the glutamate–glutamine cycle contributes little to glutamate transmission. However, a local astrocyte-dependent glutamate–glutamine cycle is required to maintain glutamate transmission in active neurotransmission at excitatory terminals [106].
Glutamate taken up by EAAT2 can also be oxidized for energy by astrocytes [107]. Glutamate oxidative metabolism occurs at high rates in astrocytes [108–110] and produces ATP. The oxidation of one glutamate is estimated to produce 24–27 ATPs via the TCA cycle and pyruvate-recycling pathway. The sodium–potassium ATPase spends one ATP to maintain membrane sodium–potassium gradients to drive glutamate uptake. Therefore, one glutamate can then net produce 23–26 ATPs [107]. Although EAAT2 is not localized to mitochondria, mitochondria are densely localized at perisynaptic astrocytic processes where EAAT2 is expressed. EAAT2, therefore, has the potential to interact with hexokinase which is a mitochondrial protein and is the first step of glycolysis [111]. These results indicate that some glutamate transported via EAAT2 is taken up by local mitochondria for oxidative energy metabolism.
Reverse transport
EAATs can release glutamate into the extracellular space via reverse transport (for review see: [2, 112]). A driving force (Na+ influx) for the inward transport of glutamate into astrocytes by EAATs is generated by the sodium–potassium ATPase under normal physiological conditions. When the driving force is reduced, such as in a membrane-depolarized condition, EAATs can reverse glutamate transport outward [113]. It is known that sodium–potassium ATPase activity is decreased in an energy deprivation condition due to the lack of ATP synthesis which results in disruption of the sodium and potassium gradients. Indeed, ischemia-induced depolarization causes glutamate release via reverse transport of neuronal EAATs [114]. Interestingly, extrasynaptic NMDA receptors on dendrite membranes are closely apposed to astrocytic processes [115]. Therefore, reversed astrocytic glutamate transport may stimulate extrasynaptic NMDA receptors in disease condition to induce excitotoxicity [116].
Heteroexchange
EAATs can also facilitate substrate exchange of external and internal substrate in a 1:1 ratio (for review see: [2]). It is notable that transportable inhibitor, like PDC, induce release of internal endogenous substrates (i.e., glutamate), thus, exacerbating excitotoxicity [117, 118]. Recently, the results of EAAT2 reconstituted in liposomes shows that heteroexchange is electroneutral but is voltage dependent [119].
Anion conductance
EAATs also exhibit anion conductance that is not coupled to glutamate transport [91, 120–123]. The magnitude of this conductance is inversely related to the pattern of glutamate transport rates (the glutamate transport rate are EAAT1/EAAT2/EAAT3 ≫ EAAT4/EAAT5). EAAT4 and EAAT5 have a high chloride conductance which inhibits glutamate transmission. EAAT4 and EAAT5 are expressed in cerebellar and retinal neurons, respectively. EAAT4 and EAAT5 predominantly serve to inhibit glutamate transmission in these neurons rather than to transport glutamate [124]. EAAT2 exhibits weak anion conductance and mainly plays a role in glutamate uptake.
Disease relevancy
Neurodegenerative disease is primarily characterized by the pathology of neuronal death which occurs in different regions and cell types in a disease specific pattern. Neuronal death is predominant in excitotoxicity. The current therapeutic strategy is to prevent neuronal death (e.g., the prevent excitotoxicity) and delay disease-related symptom progression. Excess glutamate plays an important role in excitotoxicity, and, therefore, glutamate uptake enhancement is one of the most promising drug targets for the prevention of excitotoxicity. On the other hand, psychiatric diseases are characterized by no obvious classical pathological phenomenon. Recently, advances in brain imaging techniques have revealed dysregulation in the glutamate pathway that make it a candidate therapeutic target for psychiatric disease. Here, we review how the glutamate pathway is involved in select neurological (stroke, Parkinson’s disease, epilepsy, ALS, and Alzheimer’s disease) and psychiatric diseases (major depressive disorder and addiction) as well as accumulating evidence that EAAT2 activators are a potential therapeutic strategy for drug development in these diseases.
Stroke
Stroke is the second leading causes of death in both men and women (12 % of total death, WHO data in 2012) and the third leading cause of disability-adjusted life-years (DALYs) worldwide [125, 126]. DALYs indicate how many years of “healthy” life are lost. Stroke patients typically experience a long period of disability associated with the disease. Stroke is caused by either ischemia (80 % of cases) or hemorrhage (20 %) [127]. Ischemic stroke causes a reduction in oxygen and glucose supplies which in turn prevents ATP synthesis. The deprivation of ATP leads to reduced sodium–potassium ATPase activity, disruption of sodium and, potassium gradients, and, in turn, increased synaptic glutamate concentrations via reverse transport of glutamate by neuronal EAATs [102, 114]. Neuronal EAATs involvement here is confirmed by the finding that ischemia-induced glutamate release is blocked by PDC (non-specific glutamate transporter inhibitor) but not DHK (EAAT2 specific inhibitor). EAAT2 uptake function is preserved in the ischemic condition. This is possibly due to the fact that astrocytes are able to metabolize glycogen and glucose [128], as well as produce ATP by oxidation of glutamate to obtain energy for maintenance of glutamate uptake in an acute ischemic condition. Several studies of ischemia animal models show glutamate levels in the brain are increased which subsequently cause excitotoxicity [129–134]. The detailed mechanisms of excitotoxicity in the context of stroke are described in a current review article [102]. The strategy of reducing glutamate by blocking neuronal reverse transport and/or enhancing EAAT2 glutamate uptake is attractive here. Several studies demonstrate that increased EAAT2 provides neuroprotection in ischemia. GFAP-driven EAAT2 expression in astrocytes enhances neuroprotection after moderate oxygen–glucose deprivation in rat hippocampal slice cultures [135]. Ceftriaxone-induced EAAT2 expression shows protective effects in several ischemia animal models [136–141]. These results indicate that EAAT2 is a potential therapeutic target for stroke.
Parkinson’s disease
Parkinson’s disease (PD) is characterized by motor symptoms, including akinesia, bradykinesia, rigidity and tremor. The neuropathological hallmark of PD is the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc). The development of PD motor symptoms is believed to be due to a loss of dopaminergic neurons here. This leads to loss of modulatory input and results in glutamatergic hyperexcitability of the subthalamic nucleus (STN) which projects to the medial globus pallidus and the substantia nigra pars reticulata—the output regions of the basal ganglia [142, 143]. In addition, over-activation of the glutamatergic neurons in the STN can result in excitotoxicity and degeneration of surviving neurons in the SNc. Blocking-enhanced glutamate transmission in this circuit could both alleviate symptoms and delay progression. Both preclinical and clinical studies demonstrate that glutamate receptors are a therapeutic target for PD (see review [142, 144]). Recently, several studies demonstrated that EAAT2 expression is involved in PD. Decreased EAAT2 expression has been reported in animal models of PD, including the 6-hydroxydopamine-lesioned PD model and the acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treated mouse model [145, 146]. Recently, growing evidence shows ceftriaxone has beneficial effects in PD animal model, including the prevention of motor dysfunction, neuronal death, and PD related memory deficits [147–152]. EAAT2 has the potential as a target for PD therapy.
Epilepsy
Epilepsy is a chronic neurological disease characterized by seizures and has a prevalence of 1–2 % worldwide. Epileptic seizures generally fall into two major categories: generalized seizures and partial seizures. Generalized seizures exhibit a widespread electrical discharge in both brain hemispheres and are typically associated with genetic factors. The partial seizures are a local electrical discharge in the brain and are typically caused by brain injury, stroke, or tumor. The molecular mechanisms underlying the development of epilepsy during initial insults (epileptogenesis) are not well understood. The progression of epileptogenesis during the latent period involves differential release of several neurotrophic factors, neuronal sprouting, altered synaptic plasticity, neurogenesis, and excitotoxicity [153, 154] which subsequently result in the development of spontaneous recurrent seizures. Enhanced synaptic glutamate concentration during the initial insult is related to excitotoxicity and is maintained during epileptogenesis [155, 156]. EEG and microdialysis studies in epilepsy patients show that the basal glutamate concentration in epileptogenic areas is 4.7 times higher than in nonepileptogenic areas of the hippocampus [157]. The concentration of glutamate in epileptogenic areas reaches neurotoxic concentration under basal conditions and increases further during seizures [157, 158]. Dysfunction of glutamate transport may contribute to high extracellular glutamate in the epileptogenic hippocampus. Impaired glutamate transport function has been reported in human epilepsy but remains controversial [159–163]. Excessive glutamate released by astrocytes plays a role in the synchronous firing of large populations of neurons during seizures [164–167]. Reducing glutamate-mediated excitotoxicity may prevent epileptogenesis and, subsequently, spontaneous-recurrent seizures. Ceftriaxone shows protective effects by reducing seizure activity and the acute mortality in a pentylenetetrazole model of epilepsy [168], a model of tuberous sclerosis [169], and a traumatic brain injury-induced epilepsy model [170]. We have investigated pilocarpine-induced status epilepticus in EAAT2 transgenic mice and the pyridazine-derivative EAAT2 translational-activator-treated mice [56, 171]. Enhanced EAAT2 expression significantly prevents seizure-induced epileptogenesis and subsequent spontaneous recurrent seizures. Overall, these studies indicate that enhanced EAAT2 protein expression is a potential therapeutic approach for epilepsy.
ALS
Amyotrophic lateral sclerosis (ALS) is a selective motor neuron degenerative disease which exhibits rapid progression in the brain and the spinal cord. The incidence is 2–3/100,000. Sporadic ALS accounts for approximately 90 % of cases and hereditary (familial ALS) accounts for the other 5–10 %. SOD1 mutation accounts for 20 % of familial ALS [172]. While differing or currently unknown causes result in ALS, a similar pathogenesis occurs for all ALS cases, including: oxidative damage, aberrant RNA metabolism, accumulation of intracellular aggregates, growth factor deficiency, defects in axonal transport, mitochondrial dysfunction, glial cell pathology, and glutamate excitotoxicity [173]. A 30–95 % loss of the EAAT2 protein in the motor cortex and spinal cord is observed in approximately 60–70 % of ALS patients [174]. The loss of EAAT2 protein is also observed in a transgenic animal model of mutant SOD1-mediated familial ALS [175–177]. These results suggest that the EAAT2 protein decline is correlated with neuronal loss. Enhanced EAAT2 expression by genetic manipulation and pharmacological treatment in SOD1 mice has shown some beneficial effects [41, 56, 178]. However, ceftriaxone treatment in patients with ALS did not show clinical efficacy [179]. There are many complex conditions in the disease stage that may have led to the lack of efficacy in this trial. It is notable that this study did not include pharmacodynamic results, such as measuring EAAT2 expression pre- and post-treatment. EAAT2 PET imaging is required in future clinical trials to confirm EAAT2 is upregulated as expected.
Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by declarative memory impairments and progressive dementia. The cognitive deficits are more significantly correlated with reduced glutamatergic pre-synaptic-bouton density than with neurofibrillary tangles or amyloid-β burden [180]. In addition, deficiencies in the glutamatergic system (e.g., reduced glutamate uptake) have been observed in AD and are correlated with cognitive decline [181–186]. Reduced glutamate uptake function may result in increased extracellular glutamate levels which, in turn, potentially increase amyloid-β production over time [187–189]. Amyloid-β can directly further enhance glutamate release thus creating a positive feedback cycle that synergistically increases glutamate at the synaptic cleft [190–192]. In addition, amyloid-β has been reported to prevent induction of long-term potentiation (LTP) and promote long-term depression (LTD) [193–196]. This amyloid-β-facilitated LTD is inhibited by addition of an extracellular glutamate scavenger [193]. Overall, homeostatic regulation of extracellular glutamate levels may play a crucial role in the pathogenesis of AD.
EAAT2 plays an essential role in cognitive functions [99, 197, 198]. The loss of EAAT2 protein and function are observed in AD patients [181, 184, 185] and constitutes an early event in disease pathology. This EAAT2 protein decline is likely due to disturbances at the post-transcriptional level because EAAT2 mRNA is not decreased in AD patients [199]. Mice that lack one allele for EAAT2 and crossed with AβPPswe/PS1ΔE9 mice, an animal model of AD, exhibit accelerated cognitive deficits when compared to AβPPswe/PS1ΔE9 mice. These results suggest that decreased EAAT2 levels may contribute to AD pathogenesis. We have investigated whether restored EAAT2 protein could benefit cognitive functions and pathology in APPSw,Ind mice, an animal model of AD. We conducted this investigation using both a transgenic mouse approach by crossing EAAT2 transgenic mice with APPSw,Ind mice and a pharmacological approach using a novel EAAT2 translational activator (LDN/OSU-0212320) [200]. Both approaches resulted in restored EAAT2 protein function which attenuated premature death, memory loss, and AD-like pathology (amyloid β deposition and loss of synaptic integrity) in APPSw,Ind mice. It is notable that LDN/OSU-0212320 could (1) reverse cognitive deficits after a short treatment period, (2) sustain the cognitive benefits even after 1 month of treatment cessation, (3) restore synaptic integrity, and (4) increase EAAT2 expression via the translational, rather than the transcriptional, activation mechanism which resolves the central problem of reduced EAAT2 protein expression. LDN/OSU-0212320 or its derivatives may have therapeutic potential as a drug for AD.
Major depressive disorder
Major depressive disorder (MDD) is estimated to have a lifetime prevalence of 12.8–16.6 % and is predicted to be the 2nd leading cause of disease burden by 2030 [201–203]. Symptoms of depression are characterized by a depressed mood or a loss of interest or pleasure in daily activities consistently for at least 2 weeks. Brain imaging studies have demonstrated abnormalities in the prefrontal–limbic circuit, specifically decreased top-down connectivity and increased bottom–up connectivity. MDD patients show decreased activity in the prefrontal cortex (associated with executive function) while activity in the amygdala (involved in emotional response) and the anterior cingulate cortex (related to decision-making and emotional regulation) is increased [204, 205]. These network changes are postulated to contribute to the clinical phenotypes of depression, but it is not clear what causes these changes. One possibility may be related to a loss of astrocytes in the depressed brain. Astrocyte loss is a prominent feature of mood disorders [206, 207] although there is no unique pathology for depression. Postmortem studies of patients with depression have demonstrated that the density of glial cells is decreased in the amygdala as well as prefrontal, orbitofrontal, and cingulate cortices [208–213]. Astrocyte-related genes, glial fibrillary acidic protein (GFAP), EAAT1, EAAT2, and glutamine synthetase, are reduced in cortical and amygdalar regions of MDD patients [211, 214–217]. In preclinical studies, a reduction in astrocyte number in the hippocampus and frontal cortex region is observed after chronic unpredictable stress [218, 219]. Moreover, the local treatment of the astrocyte toxin, l-alpha-aminoadipic acid (L-AAA), but not the neurotoxin ibotenate, in the prefrontal cortex produces an anhedonia-like behavior (a core clinical feature of depression) and anxiety behavior [218]. These results suggest that dysregulated glutamate transmission, due to pathological astrocytic functional changes, may be involved in depression symptom development. Indeed, the anti-glutamatergic agent, riluzole, has antidepressant effects in depressed patients and animal models of depression [220–223].
EAAT2 expression may play a role in depression-like symptoms. Glutamate release and uptake in the frontal cortex and hippocampus are increased by acute stress [224–226]. Chronic mild, predictable stress also causes increased glutamate release but also results in EAAT2 upregulation [227–230]. This likely accounts for the fact that predictable stress has beneficial effects on depressive- and anxiety-like behaviors [231, 232]. On the other hand, chronic, unpredictable stress reduces EAAT2 expression which is a similar phenomenon observed in MDD patients [215, 216, 233, 234]. A growing body of evidence suggests that loss of EAAT2 function causes depression-like behaviors. DHK treatment in rat prefrontal cortex or cerebral ventricle induces anhedonia behavior as measured by intracranial self-stimulation [197, 235]. DHK treatment in the amygdala reduces social interaction [236]. The inhibition of EAAT2 in the lateral habenula increases susceptibility to chronic stress, including increased anxiety and disinhibition during rapid eye movement sleep [237]. These results indicate that loss of EAAT2 expression contributes to depression-like behavior. In addition, chronic antidepressant treatments increase EAAT2 expression in the hippocampus [233]. Ceftriaxone reduces helplessness behavior in the forced swim and tail suspension tests [238]. It is notable that animal models can reflect only a single or limited range of symptoms of depression. In humans, the symptoms of depression are complex and arise from multi-phenomena. Thus, to determine whether enhanced EAAT2 expression is a target for depression treatment, several models of depression must be examined.
Addiction
Addiction is a behavioral state characterized by altered reward processing, disrupted emotional responses and poor decision-making. The motivationally relevant circuitry involved here is the cortico-striato-thalamic circuit, including prelimbic cortex, nucleus accumbens, and ventral tegmental area. Brain imaging studies have demonstrated that relapse in drug seeking is related to activation of the prefrontal and anterior cingulate cortices which project to the nucleus accumbens [239]. During the late withdraw period, low activities in the prefrontal and anterior cingulate cortices are observed. Animal studies demonstrate that the reinstatement of drug seeking occurs due to an imbalance in glutamate transmission from the prelimbic cortex to the nucleus accumbens core [240]. These impairments lead to disruption of cortico-striato-thalamic processing. Reinstatement of drug seeking in animal models is commonly associated with increased extracellular glutamate levels in the nucleus accumbens [241–244]; however, extracellular glutamate is not increased by the reinstatement of food seeking [244]. Thus, the enhanced glutamate levels in the nucleus accumbens play an important role in drug addiction [240, 245].
Importantly, the expression of EAAT2 is altered in animal models of addiction. Animal studies of cocaine self-administration exhibit reduced glutamate uptake [246, 247] and heroin-reduced EAAT2 expression causes the spillover of synaptic glutamate that leads to the activation of extrasynaptic NMDA receptors in the nucleus accumbens core [248]. Ethanol withdrawal reduces EAAT2 expression in striatum [249]. Ceftriaxone attenuates relapse-like ethanol intake [250–252]. Ceftriaxone was also shown to prevent cue-induced heroin seeking through increased glutamate uptake and a subsequent reduction in synaptic glutamate spillover [248]. Ceftriaxone also prevents morphine physical dependence [253]. In addition, ceftriaxone reduces the reinstatement of drug seeking in animals trained to self-administer cocaine [247, 254–256]. Propentofylline, an adenosine uptake and phosphodiesterase inhibitor, prevents cue-primed cocaine seeking via restored expression of EAAT2 in the nucleus accumbens [257]. Ceftriaxone attenuates the reinstatement of methamphetamine seeking in a condition place preference paradigm [258]. Ceftriaxone also reduces nicotine withdrawal and nicotine-seeking behaviors [259]. These results strongly suggest that enhanced EAAT2 protein expression is a potential therapeutic approach for addiction treatment.
Other diseases
Schizophrenia is associated with dysregulation of the glutamatergic system, including EAAT2 expression (for review see [260, 261]). Briefly, a glutamate receptor blocker, phencyclidine, induces schizophrenic-like behavior. Although the results of EAAT2 expression changes in schizophrenia remain controversial, antipsychotic drug treatments decrease EAAT2 expression. Ceftriaxone exacerbates phencyclidine-induced prepulse inhibition impairment [262]. Traumatic brain injury (TBI), which typically results in glutamate excitotoxicity at and near the lesion site, is expected to be a therapeutic target for enhanced EAAT2 (for review see [263, 264]). Ceftriaxone treatment shows beneficial effects for TBI including neuroprotection [170, 265]. The Huntington’s disease animal model exhibits both decreased EAAT2 protein and mRNA expression; however, dysregulation of EAAT2 expression in patients with Huntington’s disease is controversial (for review see [1, 266, 267]). Ceftriaxone attenuates the Huntington’s disease phenotype in the R6/2 mouse [268]. EAAT2 expression in the spinal cord is decreased in animal models of pain (for review see [261, 269]). Ceftriaxone has anti-nociceptive effects in pain models [270–272]. EAAT2 expression is altered in malignant gliomas (for review see [273]). Multiple sclerosis (MS) exhibits both excitotoxicity and oxidative stress (for review see [263, 265]). Ceftriaxone dampens excitotoxic inflammatory CNS damage in a mouse model of MS, but this may not be due to increased EAAT2 expression [274]. Finally, EAAT2 has also been linked to autism [275, 276]. Certainly, the evidence is overwhelming that EAAT2 dysregulation plays an important role in several neurological and psychiatric diseases.
Future perspectives
Glutamate transmission is essential for normal brain functions, inducing learning and memory. Reduced glutamate transmission affects normal brain functions and produces negative side effects through decreased glutamate receptor occupancy. Several neurological and psychiatric diseases exhibit excess extracellular glutamate and loss of EAAT2 expression. Restored EAAT2 expression levels and function may provide therapeutic benefit. Here, we have described three types of EAAT2 activators at different levels; (1) transcriptional (2) translational, and (3) functional. Each type has advantages and disadvantages. Certain diseases demonstrate decreased EAAT2 protein but not EAAT2 mRNA. In such a case, the transcriptional activator type, such as ceftriaxone, is unlikely to increase EAAT2 at a therapeutic level as EAAT2 dysregulation occurs post-transcriptionally. The translational activator type, such as pyridazine-derivative, is unlikely to increase EAAT2 when there is no EAAT2 mRNA; but this type is a putative fit for an immediate EAAT2 requirement disease such as stroke. The EAAT2 functional activator, such as spider extracts, is unlikely to modulate disease symptoms when there is little EAAT2 protein remaining. A better understanding of disease mechanisms will be essential to design and select therapy types for disease with EAAT2 dysregulation. Accumulation of knowledge in the EAAT2 variant, structure, localization, expression regulation, trafficking, and degradation could produce a therapeutic drug that provides higher spatiotemporal EAAT2 regulation. In addition EAAT2 function can now be monitored by PET probe in human subjects. This may pave the way for tailor-made medicine for disease associated with EAAT2 dysregulation.
Abbreviations
- NF-kB:
-
Nuclear factor kappa B
- Sp1:
-
Specificity protein 1
- NFAT:
-
Nuclear factor of activated T-cells
- YY1:
-
Yin Yang 1
- EGF:
-
Epidermal growth factor
- TGF-alpha:
-
Transforming growth factor alpha
- EGR:
-
Early growth response protein
References
Beart PM, O’Shea RD (2007) Transporters for l-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150:5–17
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Grewer C et al (2014) SLC1 glutamate transporters. Pflugers Arch 466:3–24
Grewer C, Rauen T (2005) Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol 203:1–20
Vandenberg RJ, Ryan RM (2013) Mechanisms of glutamate transport. Physiol Rev 93:1621–1657
Holmseth S et al (2012) The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci 32:6000–6013
Rothstein JD et al (1994) Localization of neuronal and glial glutamate transporters. Neuron 13:713–725
Lehre KP et al (1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15:1835–1853
Dehnes Y et al (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18:3606–3619
Yamada K et al (1996) EAAT4 is a post-synaptic glutamate transporter at Purkinje cell synapses. Neuroreport 7:2013–2017
Arriza JL et al (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA 94:4155–4160
Massie A et al (2008) High-affinity Na+/K+-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J Comp Neurol 511:155–172
Bjornsen LP et al (2014) The GLT-1 (EAAT2; slc1a2) glutamate transporter is essential for glutamate homeostasis in the neocortex of the mouse. J Neurochem 128:641–649
Rothstein JD et al (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16:675–686
Haugeto O et al (1996) Brain glutamate transporter proteins form homomultimers. J Biol Chem 271:27715–27722
Tanaka K et al (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702
Kim SY et al (2003) Cloning and characterization of the 3′-untranslated region of the human excitatory amino acid transporter 2 transcript. J Neurochem 86:1458–1467
Maragakis NJ et al (2004) Altered expression of the glutamate transporter EAAT2b in neurological disease. Ann Neurol 55:469–477
Bassan M et al (2008) Interaction between the glutamate transporter GLT1b and the synaptic PDZ domain protein PICK1. Eur J Neurosci 27:66–82
Sogaard R et al (2013) Functional modulation of the glutamate transporter variant GLT1b by the PDZ domain protein PICK1. J Biol Chem 288:20195–20207
Chen W et al (2002) Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 22:2142–2152
Chaudhry FA et al (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720
Furness DN et al (2008) A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 157:80–94
Holmseth S et al (2009) The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience 162:1055–1071
Kugler P, Schmitt A (2003) Complementary neuronal and glial expression of two high-affinity glutamate transporter GLT1/EAAT2 forms in rat cerebral cortex. Histochem Cell Biol 119:425–435
Schmitt A et al (2002) A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience 109:45–61
Reye P et al (2002) Distribution of two splice variants of the glutamate transporter GLT-1 in rat brain and pituitary. Glia 38:246–255
Sullivan R et al (2004) Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: implications for CNS glutamate homeostasis. Glia 45:155–169
Chen W et al (2004) The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24:1136–1148
Berger UV et al (2005) Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol 492:78–89
Rauen T et al (2004) A new GLT1 splice variant: cloning and immunolocalization of GLT1c in the mammalian retina and brain. Neurochem Int 45:1095–1106
Figiel M, Engele J (2000) Pituitary adenylate cyclase-activating polypeptide (PACAP), a neuron-derived peptide regulating glial glutamate transport and metabolism. J Neurosci 20:3596–3605
Karki P et al (2013) cAMP response element-binding protein (CREB) and nuclear factor kappaB mediate the tamoxifen-induced up-regulation of glutamate transporter 1 (GLT-1) in rat astrocytes. J Biol Chem 288:28975–28986
Karki P et al (2014) Mechanism of raloxifene-induced upregulation of glutamate transporters in rat primary astrocytes. Glia 62:1270–1283
Lee E et al (2012) GPR30 regulates glutamate transporter GLT-1 expression in rat primary astrocytes. J Biol Chem 287:26817–26828
Sitcheran R et al (2005) Positive and negative regulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlled repression. EMBO J 24:510–520
Su ZZ et al (2003) Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc Natl Acad Sci USA 100:1955–1960
Ghosh M et al (2011) Nuclear factor-kappaB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J Neurosci 31:9159–9169
Zelenaia O et al (2000) Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol 57:667–678
Lee SG et al (2008) Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem 283:13116–13123
Rothstein JD et al (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433:73–77
Gegelashvili G et al (1997) Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem 69:2612–2615
Schlag BD et al (1998) Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol 53:355–369
Yang Y et al (2009) Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron 61:880–894
Fumagalli E et al (2008) Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol 578:171–176
Liu AY et al (2011) Neuroprotective drug riluzole amplifies the heat shock factor 1 (HSF1)- and glutamate transporter 1 (GLT1)-dependent cytoprotective mechanisms for neuronal survival. J Biol Chem 286:2785–2794
Perisic T et al (2010) Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology 35:792–805
Karki P et al (2014) Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Mol Cell Biol 34:1280–1289
Perisic T et al (2012) The CpG island shore of the GLT-1 gene acts as a methylation-sensitive enhancer. Glia 60:1345–1355
Yang Y et al (2010) Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia 58:277–286
Tian G et al (2007) Translational control of glial glutamate transporter EAAT2 expression. J Biol Chem 282:1727–1737
Morel L et al (2013) Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J Biol Chem 288:7105–7116
Carmona MA et al (2009) Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc Natl Acad Sci USA 106:12524–12529
Filosa A et al (2009) Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci 12:1285–1292
Colton CK et al (2010) Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen 15:653–662
Kong Q et al (2014) Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest 124:1255–1267
Xing X et al (2011) Structure-activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg Med Chem Lett 21:5774–5777
Huang K et al (2010) Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol Dis 40:207–215
Foran E et al (2014) Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia 62:1241–1253
Garcia-Tardon N et al (2012) Protein kinase C (PKC)-promoted endocytosis of glutamate transporter GLT-1 requires ubiquitin ligase Nedd4-2-dependent ubiquitination but not phosphorylation. J Biol Chem 287:19177–19187
Gonzalez-Gonzalez IM et al (2008) PKC-dependent endocytosis of the GLT1 glutamate transporter depends on ubiquitylation of lysines located in a C-terminal cluster. Glia 56:963–974
Sheldon AL et al (2008) Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem Int 53:296–308
Tian G et al (2010) Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer’s disease. J Neurochem 113:978–989
Benediktsson AM et al (2012) Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia 60:175–188
Poitry-Yamate CL et al (2002) Neuronal-induced and glutamate-dependent activation of glial glutamate transporter function. J Neurochem 82:987–997
Fontana AC et al (2007) Enhancing glutamate transport: mechanism of action of Parawixin1, a neuroprotective compound from Parawixia bistriata spider venom. Mol Pharmacol 72:1228–1237
Fontana AC et al (2003) Purification of a neuroprotective component of Parawixia bistriata spider venom that enhances glutamate uptake. Br J Pharmacol 139:1297–1309
Bridges RJ, Esslinger CS (2005) The excitatory amino acid transporters: pharmacological insights on substrate and inhibitor specificity of the EAAT subtypes. Pharmacol Ther 107:271–285
Bunch L et al (2009) Excitatory amino acid transporters as potential drug targets. Expert Opin Ther Targets 13:719–731
Arriza JL et al (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14:5559–5569
Fairman WA et al (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375:599–603
Griffiths R et al (1994) l-Trans-pyrrolidine-2,4-dicarboxylate and cis-1-aminocyclobutane-1,3-dicarboxylate behave as transportable, competitive inhibitors of the high-affinity glutamate transporters. Biochem Pharmacol 47:267–274
Rauen T et al (1992) Comparative analysis of sodium-dependent l-glutamate transport of synaptosomal and astroglial membrane vesicles from mouse cortex. FEBS Lett 312:15–20
Shimamoto K et al (1998) dl-Threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53:195–201
Shimamoto K et al (2004) Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol 65:1008–1015
Dunlop J et al (2005) Characterization of novel aryl-ether, biaryl, and fluorene aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT2. Mol Pharmacol 68:974–982
Greenfield A et al (2005) Synthesis and biological activities of aryl-ether-, biaryl-, and fluorene-aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT-2. Bioorg Med Chem Lett 15:4985–4988
Callender R et al (2012) Mechanism of inhibition of the glutamate transporter EAAC1 by the conformationally constrained glutamate analogue (+)-HIP-B. Biochemistry 51:5486–5495
Colleoni S et al (2008) Neuroprotective effects of the novel glutamate transporter inhibitor (-)-3-hydroxy-4,5,6,6a-tetrahydro-3aH-pyrrolo[3,4-d]-isoxazole-4-carboxylic acid, which preferentially inhibits reverse transport (glutamate release) compared with glutamate reuptake. J Pharmacol Exp Ther 326:646–656
Abrahamsen B et al (2013) Allosteric modulation of an excitatory amino acid transporter: the subtype-selective inhibitor UCPH-101 exerts sustained inhibition of EAAT1 through an intramonomeric site in the trimerization domain. J Neurosci 33:1068–1087
Erichsen MN et al (2010) Structure-activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-Amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chrom ene-3-carbonitrile (UCPH-101). J Med Chem 53:7180–7191
Jensen AA et al (2009) Discovery of the first selective inhibitor of excitatory amino acid transporter subtype 1. J Med Chem 52:912–915
Jiang J, Amara SG (2011) New views of glutamate transporter structure and function: advances and challenges. Neuropharmacology 60:172–181
Boudker O et al (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445:387–393
Reyes N et al (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462:880–885
Yernool D et al (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431:811–818
Gendreau S et al (2004) A trimeric quaternary structure is conserved in bacterial and human glutamate transporters. J Biol Chem 279:39505–39512
Nothmann D et al (2011) Hetero-oligomerization of neuronal glutamate transporters. J Biol Chem 286:3935–3943
Danbolt NC et al (1992) An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 51:295–310
Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741
Otis TS, Jahr CE (1998) Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci 18:7099–7110
Tong G, Jahr CE (1994) Block of glutamate transporters potentiates postsynaptic excitation. Neuron 13:1195–1203
Wadiche JI, Kavanaugh MP (1998) Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci 18:7650–7661
Diamond JS, Jahr CE (1997) Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17:4672–4687
Otis TS, Kavanaugh MP (2000) Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci 20:2749–2757
Tzingounis AV, Wadiche JI (2007) Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 8:935–947
Wadiche JI et al (1995) Kinetics of a human glutamate transporter. Neuron 14:1019–1027
Lozovaya NA et al (1999) Enhancement of glutamate release uncovers spillover-mediated transmission by N-methyl-d-aspartate receptors in the rat hippocampus. Neuroscience 91:1321–1330
Pita-Almenar JD et al (2012) Relationship between increase in astrocytic GLT-1 glutamate transport and late-LTP. Learn Mem 19:615–626
Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696
Parsons MP, Raymond LA (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82:279–293
Lai TW et al (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188
Hertz L (1979) Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Prog Neurobiol 13:277–323
van den Berg CJ, Garfinkel D (1971) A stimulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem J 123:211–218
Kam K, Nicoll R (2007) Excitatory synaptic transmission persists independently of the glutamate–glutamine cycle. J Neurosci 27:9192–9200
Tani H et al (2014) A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 81:888–900
McKenna MC (2013) Glutamate pays its own way in astrocytes. Front Endocrinol (Lausanne) 4:191
Hertz L, Hertz E (2003) Cataplerotic TCA cycle flux determined as glutamate-sustained oxygen consumption in primary cultures of astrocytes. Neurochem Int 43:355–361
McKenna MC (2012) Substrate competition studies demonstrate oxidative metabolism of glucose, glutamate, glutamine, lactate and 3-hydroxybutyrate in cortical astrocytes from rat brain. Neurochem Res 37:2613–2626
Sonnewald U et al (1993) Metabolism of [U-13C]glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J Neurochem 61:1179–1182
Genda EN et al (2011) Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 31:18275–18288
Grewer C et al (2008) Glutamate forward and reverse transport: from molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 60:609–619
Szatkowski M et al (1990) Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348:443–446
Rossi DJ et al (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403:316–321
Petralia RS et al (2010) Organization of NMDA receptors at extrasynaptic locations. Neuroscience 167:68–87
Gouix E et al (2009) Reverse glial glutamate uptake triggers neuronal cell death through extrasynaptic NMDA receptor activation. Mol Cell Neurosci 40:463–473
Blitzblau R et al (1996) The glutamate transport inhibitor l-trans-pyrrolidine-2,4-dicarboxylate indirectly evokes NMDA receptor mediated neurotoxicity in rat cortical cultures. Eur J Neurosci 8:1840–1852
Volterra A et al (1996) The competitive transport inhibitor l-trans-pyrrolidine-2, 4-dicarboxylate triggers excitotoxicity in rat cortical neuron-astrocyte co-cultures via glutamate release rather than uptake inhibition. Eur J Neurosci 8:2019–2028
Zhou Y et al (2014) EAAT2 (GLT-1; slc1a2) glutamate transporters reconstituted in liposomes argues against heteroexchange being substantially faster than net uptake. J Neurosci 34:13472–13485
Billups B et al (1996) Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. J Neurosci 16:6722–6731
Grewer C et al (2000) Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci USA 97:9706–9711
Mim C et al (2005) The glutamate transporter subtypes EAAT4 and EAATs 1-3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J Gen Physiol 126:571–589
Gameiro A et al (2011) The discovery of slowness: low-capacity transport and slow anion channel gating by the glutamate transporter EAAT5. Biophys J 100:2623–2632
Veruki ML et al (2006) Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat Neurosci 9:1388–1396
Lozano R et al (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128
Murray CJ et al (2012) Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2197–2223
Donnan GA et al (2008) Stroke. Lancet 371:1612–1623
Brown AM et al (2004) Energy transfer from astrocytes to axons: the role of CNS glycogen. Neurochem Int 45:529–536
Dawson LA et al (2000) Characterization of transient focal ischemia-induced increases in extracellular glutamate and aspartate in spontaneously hypertensive rats. Brain Res Bull 53:767–776
Mitani A et al (1990) Gerbil hippocampal extracellular glutamate and neuronal activity after transient ischemia. Brain Res Bull 25:319–324
Globus MY et al (1988) Effect of ischemia on the in vivo release of striatal dopamine, glutamate, and gamma-aminobutyric acid studied by intracerebral microdialysis. J Neurochem 51:1455–1464
Hagberg H et al (1985) Ischemia-induced shift of inhibitory and excitatory amino acids from intra- to extracellular compartments. J Cereb Blood Flow Metab 5:413–419
Drejer J et al (1985) Cellular origin of ischemia-induced glutamate release from brain tissue in vivo and in vitro. J Neurochem 45:145–151
Benveniste H et al (1984) Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43:1369–1374
Weller ML et al (2008) Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience 155:1204–1211
Chu K et al (2007) Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke 38:177–182
Hu YY et al (2015) Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J Neurochem 132:194–205
Inui T et al (2013) Neuroprotective effect of ceftriaxone on the penumbra in a rat venous ischemia model. Neuroscience 242:1–10
Ouyang YB et al (2007) Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci 27:4253–4260
Thone-Reineke C et al (2008) The beta-lactam antibiotic, ceftriaxone, dramatically improves survival, increases glutamate uptake and induces neurotrophins in stroke. J Hypertens 26:2426–2435
Verma R et al (2010) Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur J Pharmacol 638:65–71
Amalric M (2015) Targeting metabotropic glutamate receptors (mGluRs) in Parkinson’s disease. Curr Opin Pharmacol 20:29–34
Blandini F et al (2000) Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol 62:63–88
Gardoni F, Di Luca M (2015) Targeting glutamatergic synapses in Parkinson’s disease. Curr Opin Pharmacol 20:24–28
Chung EK et al (2008) Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J Comp Neurol 511:421–437
Holmer HK et al (2005) l-dopa-induced reversal in striatal glutamate following partial depletion of nigrostriatal dopamine with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience 136:333–341
Bisht R et al (2014) Ceftriaxone mediated rescue of nigral oxidative damage and motor deficits in MPTP model of Parkinson’s disease in rats. Neurotoxicology 44:71–79
Chotibut T et al (2014) Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson’s model. Mol Neurobiol 49:1282–1292
Ho SC et al (2014) Effects of ceftriaxone on the behavioral and neuronal changes in an MPTP-induced Parkinson’s disease rat model. Behav Brain Res 268:177–184
Hsu CY et al (2015) Ceftriaxone prevents and reverses behavioral and neuronal deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Neuropharmacology 91:43–56
Kelsey JE, Neville C (2014) The effects of the beta-lactam antibiotic, ceftriaxone, on forepaw stepping and L-DOPA-induced dyskinesia in a rodent model of Parkinson’s disease. Psychopharmacology 231:2405–2415
Leung TC et al (2012) Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. ACS Chem Neurosci 3:22–30
McNamara JO et al (2006) Molecular signaling mechanisms underlying epileptogenesis. Sci STKE 2006:re12
Pitkanen A, Lukasiuk K (2011) Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol 10:173–186
Coulter DA, Eid T (2012) Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60:1215–1226
Jabs R et al (2008) Astrocytic function and its alteration in the epileptic brain. Epilepsia 49(Suppl 2):3–12
Cavus I et al (2005) Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol 57:226–235
During MJ, Spencer DD (1993) Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341:1607–1610
Bjornsen LP et al (2007) Changes in glial glutamate transporters in human epileptogenic hippocampus: inadequate explanation for high extracellular glutamate during seizures. Neurobiol Dis 25:319–330
Mathern GW et al (1999) Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 52:453–472
Proper EA et al (2002) Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125:32–43
Sarac S et al (2009) Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 117:291–301
Tessler S et al (1999) Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience 88:1083–1091
Benarroch EE (2009) Astrocyte-neuron interactions: implications for epilepsy. Neurology 73:1323–1327
Binder DK, Steinhauser C (2006) Functional changes in astroglial cells in epilepsy. Glia 54:358–368
Tian GF et al (2005) An astrocytic basis of epilepsy. Nat Med 11:973–981
Wetherington J et al (2008) Astrocytes in the epileptic brain. Neuron 58:168–178
Jelenkovic AV et al (2008) Beneficial effects of ceftriaxone against pentylenetetrazole-evoked convulsions. Exp Biol Med (Maywood) 233:1389–1394
Zeng LH et al (2010) Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis complex. Neurobiol Dis 37:764–771
Goodrich GS et al (2013) Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma 30:1434–1441
Kong Q et al (2012) Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis 47:145–154
Kiernan MC et al (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955
Rothstein JD (2009) Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 65(Suppl 1):S3–S9
Rothstein JD et al (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84
Bendotti C et al (2001) Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J Neurochem 79:737–746
Bruijn LI et al (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338
Howland DS et al (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA 99:1604–1609
Guo H et al (2003) Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet 12:2519–2532
Cudkowicz ME et al (2014) Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol 13:1083–1091
Bell KF et al (2007) Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci 27:10810–10817
Jacob CP et al (2007) Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis 11:97–116
Kashani A et al (2008) Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging 29:1619–1630
Kirvell SL et al (2006) Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J Neurochem 98:939–950
Masliah E et al (1996) Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol 40:759–766
Scott HA et al (2011) Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging 32:553 e1–553 e11
Sokolow S et al (2012) Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer’s disease cortex. Neurobiol Dis 45:381–387
Bordji K et al (2010) Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-β production. J Neurosci 30:15927–15942
Kim SH et al (2010) Group II metabotropic glutamate receptor stimulation triggers production and release of Alzheimer’s amyloid(beta)42 from isolated intact nerve terminals. J Neurosci 30:3870–3875
Lesne S et al (2005) NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci 25:9367–9377
Chin JH et al (2007) Amyloid beta protein modulates glutamate-mediated neurotransmission in the rat basal forebrain: involvement of presynaptic neuronal nicotinic acetylcholine and metabotropic glutamate receptors. J Neurosci 27:9262–9269
Kabogo D et al (2010) β-amyloid-related peptides potentiate K+-evoked glutamate release from adult rat hippocampal slices. Neurobiol Aging 31:1164–1172
Talantova M et al (2013) Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA 110:E2518–E2527
Li S et al (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801
Li S et al (2011) Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–6638
Shankar GM et al (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14:837–842
Wang Q et al (2004) Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 24:3370–3378
Bechtholt-Gompf AJ et al (2010) Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology 35:2049–2059
Heo S et al (2012) Hippocampal glutamate transporter 1 (GLT-1) complex levels are paralleling memory training in the Multiple T-maze in C57BL/6J mice. Brain Struct Funct 217:363–378
Li S et al (1997) Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol 56:901–911
Takahashi K et al (2015) Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J Exp Med 212:319–332
Alonso J et al (2004) Prevalence of mental disorders in Europe: results from the European Study of the Epidemiology of Mental Disorders (ESEMeD) project. Acta Psychiatr Scand 109(Suppl 420):21–27
Kessler RC et al (2005) Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 62:593–602
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3:e442
Hamilton JP et al (2012) Functional neuroimaging of major depressive disorder: a meta-analysis and new integration of base line activation and neural response data. Am J Psychiatry 169:693–703
Siegle GJ et al (2007) Increased amygdala and decreased dorsolateral prefrontal BOLD responses in unipolar depression: related and independent features. Biol Psychiatry 61:198–209
Rajkowska G, Stockmeier CA (2013) Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets 14:1225–1236
Sanacora G, Banasr M (2013) From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry 73:1172–1179
Bowley MP et al (2002) Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry 52:404–412
Cotter D et al (2002) Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex 12:386–394
Cotter D et al (2001) Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry 58:545–553
Gittins RA, Harrison PJ (2011) A morphometric study of glia and neurons in the anterior cingulate cortex in mood disorder. J Affect Disord 133:328–332
Ongur D et al (1998) Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA 95:13290–13295
Rajkowska G et al (1999) Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry 45:1085–1098
Altshuler LL et al (2010) Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar Disord 12:541–549
Choudary PV et al (2005) Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci USA 102:15653–15658
Miguel-Hidalgo JJ et al (2010) Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J Affect Disord 127:230–240
Si X et al (2004) Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology 29:2088–2096
Banasr M, Duman RS (2008) Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry 64:863–870
Czeh B et al (2006) Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 31:1616–1626
Banasr M et al (2010) Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry 15:501–511
Sanacora G et al (2007) Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry 61:822–825
Takahashi K et al (2011) Riluzole rapidly attenuates hyperemotional responses in olfactory bulbectomized rats, an animal model of depression. Behav Brain Res 216:46–52
Zarate CA Jr et al (2004) An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry 161:171–174
Gilad GM et al (1990) Region-selective stress-induced increase of glutamate uptake and release in rat forebrain. Brain Res 525:335–338
Musazzi L et al (2010) Acute stress increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: the dampening action of antidepressants. PLoS One 5:e8566
Satoh E, Shimeki S (2010) Acute restraint stress enhances calcium mobilization and glutamate exocytosis in cerebrocortical synaptosomes from mice. Neurochem Res 35:693–701
Autry AE et al (2006) Glucocorticoid regulation of GLT-1 glutamate transporter isoform expression in the rat hippocampus. Neuroendocrinology 83:371–379
Fontella FU et al (2004) Repeated restraint stress alters hippocampal glutamate uptake and release in the rat. Neurochem Res 29:1703–1709
Reagan LP et al (2004) Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci USA 101:2179–2184
Zschocke J et al (2005) Differential promotion of glutamate transporter expression and function by glucocorticoids in astrocytes from various brain regions. J Biol Chem 280:34924–34932
Parihar VK et al (2011) Predictable chronic mild stress improves mood, hippocampal neurogenesis and memory. Mol Psychiatry 16:171–183
Suo L et al (2013) Predictable chronic mild stress in adolescence increases resilience in adulthood. Neuropsychopharmacology 38:1387–1400
Chen JX et al (2014) Glutamate transporter 1-mediated antidepressant-like effect in a rat model of chronic unpredictable stress. J Huazhong Univ Sci Technolog Med Sci 34:838–844
Zink M et al (2010) Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology 58:465–473
John CS et al (2012) Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology 37:2467–2475
Lee Y et al (2007) Glia mechanisms in mood regulation: a novel model of mood disorders. Psychopharmacology 191:55–65
Cui W et al (2014) Glial dysfunction in the mouse habenula causes depressive-like behaviors and sleep disturbance. J Neurosci 34:16273–16285
Mineur YS et al (2007) Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry 61:250–252
Goldstein RZ, Volkow ND (2002) Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 159:1642–1652
Kalivas PW (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10:561–572
Gass JT et al (2011) Alcohol-seeking behavior is associated with increased glutamate transmission in basolateral amygdala and nucleus accumbens as measured by glutamate-oxidase-coated biosensors. Addict Biol 16:215–228
Gipson CD et al (2013) Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci USA 110:9124–9129
LaLumiere RT, Kalivas PW (2008) Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci 28:3170–3177
McFarland K et al (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23:3531–3537
Kalivas PW, Volkow ND (2011) New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry 16:974–986
Fischer-Smith KD et al (2012) Differential effects of cocaine access and withdrawal on glutamate type 1 transporter expression in rat nucleus accumbens core and shell. Neuroscience 210:333–339
Knackstedt LA et al (2010) Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry 67:81–84
Shen HW et al (2014) Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J Neurosci 34:5649–5657
Abulseoud OA et al (2014) Attenuation of ethanol withdrawal by ceftriaxone-induced upregulation of glutamate transporter EAAT2. Neuropsychopharmacology 39:1674–1684
Alhaddad H et al (2014) Effects of ceftriaxone on ethanol intake: a possible role for xCT and GLT-1 isoforms modulation of glutamate levels in P rats. Psychopharmacology 231:4049–4057
Qrunfleh AM et al (2013) Ceftriaxone, a beta-lactam antibiotic, attenuates relapse-like ethanol-drinking behavior in alcohol-preferring rats. J Psychopharmacol 27:541–549
Sari Y et al (2011) Ceftriaxone, a beta-lactam antibiotic, reduces ethanol consumption in alcohol-preferring rats. Alcohol Alcohol 46:239–246
Rawls SM et al (2010) beta-Lactam antibiotic inhibits development of morphine physical dependence in rats. Behav Pharmacol 21:161–164
Sari Y et al (2009) Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci 29:9239–9243
Sondheimer I, Knackstedt LA (2011) Ceftriaxone prevents the induction of cocaine sensitization and produces enduring attenuation of cue- and cocaine-primed reinstatement of cocaine-seeking. Behav Brain Res 225:252–258
Trantham-Davidson H et al (2012) Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. J Neurosci 32:12406–12410
Reissner KJ et al (2014) Chronic administration of the methylxanthine propentofylline impairs reinstatement to cocaine by a GLT-1-dependent mechanism. Neuropsychopharmacology 39:499–506
Abulseoud OA et al (2012) Ceftriaxone upregulates the glutamate transporter in medial prefrontal cortex and blocks reinstatement of methamphetamine seeking in a condition place preference paradigm. Brain Res 1456:14–21
Alajaji M et al (2013) Effects of the beta-lactam antibiotic ceftriaxone on nicotine withdrawal and nicotine-induced reinstatement of preference in mice. Psychopharmacology 228:419–426
Lauriat TL, McInnes LA (2007) EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders. Mol Psychiatry 12:1065–1078
Nakagawa T, Kaneko S (2013) SLC1 glutamate transporters and diseases: psychiatric diseases and pathological pain. Curr Mol Pharmacol 6:66–73
Melone M et al (2009) GLT-1 up-regulation enhances the effect of PCP on prepulse inhibition of the startle reflex in adult rats. Schizophr Res 109:196–197
Lin CL et al (2012) Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med Chem 4:1689–1700
Yi JH, Hazell AS (2006) Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int 48:394–403
Cui C et al (2014) Neuroprotective effect of ceftriaxone in a rat model of traumatic brain injury. Neurol Sci 35:695–700
Maragakis NJ, Rothstein JD (2004) Glutamate transporters: animal models to neurologic disease. Neurobiol Dis 15:461–473
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51:333–355
Miller BR et al (2008) Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience 153:329–337
Gegelashvili G, Bjerrum OJ (2014) High-affinity glutamate transporters in chronic pain: an emerging therapeutic target. J Neurochem 131:712–730
Hu Y et al (2010) An anti-nociceptive role for ceftriaxone in chronic neuropathic pain in rats. Pain 148:284–301
Ramos KM et al (2010) Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience 169:1888–1900
Stepanovic-Petrovic RM et al (2014) Antihyperalgesic/antinociceptive effects of ceftriaxone and its synergistic interactions with different analgesics in inflammatory pain in rodents. Anesthesiology 120:737–750
Robert SM, Sontheimer H (2014) Glutamate transporters in the biology of malignant gliomas. Cell Mol Life Sci 71:1839–1854
Melzer N et al (2008) A beta-lactam antibiotic dampens excitotoxic inflammatory CNS damage in a mouse model of multiple sclerosis. PLoS One 3:e3149
Autism Genome Project C et al (2007) Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 39:319–328
Purcell AE et al (2001) Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57:1618–1628
Acknowledgments
This review article was supported by US National Institutes of Health grants R01NS064275 and U01NS074601, the BrightFocus Foundation, the Alzheimer’s Drug Discovery Foundation, and the Thome Memorial Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Takahashi, K., Foster, J.B. & Lin, CL.G. Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 72, 3489–3506 (2015). https://doi.org/10.1007/s00018-015-1937-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1937-8