
Abstract
Isolation of genetically modified cells generated by designed nucleases are challenging, since they are often phenotypically indistinguishable from their parental cells. To efficiently enrich genetically modified cells, we developed two dual-reporter surrogate systems, namely NHEJ-RPG and SSA-RPG based on NHEJ and SSA repair mechanisms, respectively. Repair and enrichment efficiencies of these two systems were compared using different nucleases. In both CRISPR-Cas9- and ZFNs-induced DSB repair studies, we found that the efficiency and sensitivity of the SSA-RPG reporter with direct repeat length more than 200 bp were much higher than the NHEJ-RPG reporter. By utilizing the SSA-RPG reporter, we achieved the enrichment for indels in several endogenous loci with 6.3- to 34.8-fold of non-selected cells. Thus, the highly sensitive SSA-RPG reporter can be used for activity validation of designed nucleases and efficient enrichment of genetically modified cells. Besides, our systems offer alternative enrichment choices either by puromycin selection or FACS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genome editing tools, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) systems, have made genetic modification more efficient, and genome engineering is now at the dawn of the golden age [1]. Both engineered nuclease ZFNs and TALENs facilitate targeted genome editing by creating double-strand breaks (DSBs), using DNA-binding proteins and FokI nucleases to recognize and cleave targeted DNA substrate, respectively [2, 3]. CRISPR-Cas9 system is a RNA-guided DNA cleavage system derived from bacteria immune system [4, 5], a single chimeric-guiding RNA (gRNA or sgRNA) can direct the Cas9 nuclease to target DNA sequence [6], which has been proved to result in target cleavage activity in various eukaryotic species [7–11].
However, lack of an efficient selection approach for genetically modified cells hampers the wide application of designed nucleases in gene therapy and genome engineering. Given the limited cleavage and repair efficiencies of designed nucleases, most of the cells will not be induced with intent mutations at target sites after transfected with designed nuclease plasmids [12], and even the cells harboring mutations would not have significant discernible phenotypes for selection. Thus, a simple method for enriching cells with artificial nuclease-induced mutations will facilitate the usage of these genome engineering tools. When the target sequence on a chromosome was mutated by designed nuclease, the same target sequence on the other homologous chromosome in the same cell would be mutated more frequently than that in other cells [13, 14]. Several surrogate reporters containing target sequences had been devised by Kim et al. to enrich cells with nuclease-induced mutations [12, 15–17]. Fluorescence-activated cell sorting (FACS), magnetic separation and antibiotic selection have been successfully utilized for the enrichment of endogenous gene-modified cells.
Since artificial nucleases-induced DSBs can result in mutations or chromosome rearrangements and eventually cell death; cells have evolved a number of pathways to deal with DSBs including homologous recombination (HR), single-strand annealing (SSA) and non-homologous end joining (NHEJ). To simplify the surrogate reporter system, the NHEJ or SSA surrogate reporter may be the preferential choice, because an additional donor is necessary for the HR pathway. For NHEJ, break ends are rapidly recognized by Ku proteins and DNA-dependent protein kinase, resulting in end resection; and the broken DNA ends are rejoined directly end-to-end, which could result in small insertions and deletions (Indels) [18–20]. If a DSB lies between two homogenous repeat sequences, the SSA pathway will lead to the formation of a deletion between the repeats [21–24]. In several organisms, it had been reported that NHEJ was predominant among these three repair pathways [19, 25–29]. While, it became more possible to anneal via SSA pathway instead of NHEJ under the special condition with homogenous repeats, and the efficiency of SSA could be more than ten times higher than NHEJ [30]. Similar phenomenon also had been found in Drosophila germ cells [31]. Homogenous repeats are critical for the SSA efficiency, which could reach a plateau if the repeat length is enough [32–35]. Nevertheless, additional experiments are needed to determine the generality relationship between NHEJ and SSA, and using reporters with varying direct repeat lengths may be one approach to assess repair in different contexts.
The sensitivity of surrogate reporters is directly related to the efficiency of corresponding repair pathway used, and developing a surrogate reporter with higher repair efficiency could help to enrich more genetically modified cells instead of discarding many true positive cells. The existing surrogate reporters were established mainly based on the NHEJ repair pathway [12, 15, 16]. However, our previous studies suggested that surrogate reporters with SSA direct repeats were efficient for nuclease activity validation and positive cells selection [36–39], and thus the NHEJ and SSA repair pathways remain to be compared for surrogate reporter usage. Besides, different polarities and propensities for DSB ends induced by different nuclease platforms also influence the preference of SSA and NHEJ [30]. ZFNs and TALENs are known as FokI-based enzymes that leave 5′-overhangs [40] and Cas9-based enzymes leave mainly blunt-ended DSBs or in some cases 1 bp overhangs [6, 41]. Therefore, it is necessary to investigate surrogate reporters based on different repair pathways for different nuclease platforms.
Here, we present two DsRed-PuroR-eGFP (RPG) dual-reporter surrogate systems which are based on the NHEJ and SSA repair pathways, and named as NHEJ-RPG and SSA-RPG, respectively (Fig. 1a, b). The DsRed fluorescent marker gene is used for calculating the transfection efficiency, and puromycin resistant (Puro R) and eGFP fluorescent genes are fused by T2A as dual reporter. The T2A is a “self-cleaving” small peptide, which makes a single open reading frame (ORF) encoding two proteins after the translation [38]. After transfection, functional repair of the dual-reporter genes will facilitate the selection for nuclease positive cells by either puromycin selection or FACS, which will further leverage for the enrichment of genetically modified cells (Fig. 2a). The repair efficiencies and sensitivities for the NHEJ-RPG reporter and different SSA-RPG reporters (Fig. 1c) were compared under the two end conditions generated by CRISPR-Cas9 and ZFNs (Fig. 2b, c). The enrichment efficiencies for genetically modified cells with the two reporter systems were also compared, and the SSA-RPG reporter with higher sensitivity was further applied successfully for the enrichment of CRISPR-Cas9- and ZFN-induced genetically modified cells by both puromycin selection and FACS. Our study provided a highly sensitive SSA-RPG surrogate reporter system, which will facilitate the usage of artificial nucleases in biotechnological and transgenic researches.

Schematic drawing for the function principle of the dual surrogate reporters. a Function principle of the NHEJ-RPG reporter. The DsRed marker gene was driven by CMV promoter. The target-disrupted Puro R gene and eGFP fluorescent gene are fused by T2A as dual reporter, and driven by CAG promoter. When the designed nuclease cleaves the target sequence, the mutagenic NHEJ-mediated DNA DSB repair will generate one-third of products with correct ORF, resulting in functional expression of Puro R and eGFP genes. b Function principle of the SSA-RPG reporter. Differently with the NHEJ-RPG reporter, the Puro R gene is interrupted by target sequence flanked with direct repeats as SSA arms. When designed nuclease functions to cleave the target sequence, the SSA-mediated repair will result in the deletion of one repeat, together with the intervening sequence, correcting the ORF for Puro R and eGFP reporter genes. c Different lengths of direct repeats designed for the SSA-RPG reporters. The lengths are set to range from 50 to 350 bp with 50 bp increased each

Schematic drawing of the enrichment principle with dual-reporter surrogate system and the designed nucleases. a Schematic of the enrichment for genetically modified cells. b Schematic of CRISPR-Cas9 system derived from S. thermophilus. c Schematic of the CCR5.ZFN heterodimer nuclease and the target sequence [31]
Results
Cas9-induced DSB repair efficiencies for different surrogate reporters
To understand the Cas9-induced DSB repair preference for different surrogate reporters, 293T cells were co-transfected with CCR5.a.gRNA/Cas9 expression plasmid and corresponding surrogate reporter plasmids. Flow cytometric results indicated that the eGFP functional repair efficiency of the NHEJ-RPG surrogate reporter was about 0.28 %. Given that the NHEJ-mediated functional eGFP expression only reflects one-third of the real DSB repair efficiency, for the two-thirds of frame shift repair which can not result in the expression of reporter genes, the real DSB repair efficiency should be the data (eGFP functional repair efficiency) multiplied by the factor 3 (3*NHEJ), which is still much lower than SSA-mediated repair efficiency when the direct repeat length is 50 bp. As for the SSA-RPG surrogate reporter, a repair efficiency correlation was observed with the direct repeat length increased until it reached plateau at 200 bp, and the efficiency increased by sixfold (from 1.1 to 6.8 %) (Fig. 3a, Supplementary Fig. 2). These suggested that the SSA-RPG surrogate reporter is a preferred choice to evaluate the Cas9-induced DSB repair efficiency and the CRISPR-Cas9 nuclease activity.

Repair efficiency and sensitivity comparison of the NHEJ-RPG and SSA-RPG reporters. a Statistical results of CCR5.a.gRNA/Cas9-induced DSB repair efficiencies for different surrogate reporters. b Statistical results of CCR5.ZFN-induced DSB repair efficiencies for different surrogate reporters. The repair efficiency is calculated as the mean value of three independent experiments. Error bars represent standard deviation. c Visualization of DsRed and eGFP expression by fluorescence microscopy. d Sensitivity comparison of the NHEJ-RPG and SSA-RPG reporters under puromycin treatment. 293T cells were co-transfected with CCR5.a.gRNA/Cas9 expression plasmid and corresponding surrogate reporter plasmids, NHEJ-RPG and SSA-RPG, respectively. Cells were observed and photographed with fluorescence microscope 2 days after transfection, and maintained continuously with puromycin treatment for another 5 days, and photographed. Scale bar 50 μm
ZFN-induced DSB repair efficiencies for different surrogate reporters
To compare DSB repair efficiencies between NHEJ- and SSA-based reporters with ZFN-induced cohesive ends, 293T cells were co-transfected with CCR5.ZFNL/CCR5.ZFNR expression plasmids and corresponding surrogate reporter plasmids. Results demonstrated that the NHEJ-mediated DSB repair efficiency (3*NHEJ) was equal to that SSA mediated when the direct repeat length was 100 bp (Fig. 3b). However, the SSA-mediated homologous recombination repair efficiency increased with the augment of the direct repeat length until it reached a plateau at approximately 200 bp. The efficiency increased precipitously by near sevenfold when the direct repeat length increased from 50 to 200 bp. The extra length of direct repeats beyond 200 bp did not increase the repair efficiency significantly (Fig. 3b, Supplementary Fig. 3). This experiment also indicated that the SSA-RPG reporter, when the length of direct repeats reaches 200 bp, still possesses higher sensitivity than the NHEJ-RPG reporter to validate the nuclease activity of ZFNs.
Sensitivities of the NHEJ-RPG and SSA-RPG surrogate reporters
As demonstrated above, for both Cas9- and ZFN-induced DSB repair, we found that efficiency of the SSA-RPG surrogate reporter with direct repeat length more than 200 bp was much higher than the NHEJ-RPG reporter. In general, CCR5.ZFN-induced DSB repair efficiencies were about twofold of that generated by CCR5.a.gRNA/Cas9 congruously with different SSA-RPG surrogate reporters. However, for the NHEJ-RPG reporter, CCR5.a.gRNA/Cas9-induced DSB repair efficiency was obviously inhibited, which was more than fivefold lower than that of CCR5.ZFN-induced cohesive ends (Fig. 3a, b). Taken together, these results suggested that, for the activity validation of both Cas9 and ZFNs, the SSA-RPG reporter is higher sensitive than the NHEJ-RPG reporter, when the length of direct repeats reaches its optimal 200 bp. These observations were also confirmed by microscopic examination. For CCR5.a.gRNA/Cas9-transfected cells, the fluorescence microscope observations showed prominently more eGFP+ cells with the SSA-RPG reporter with direct repeats of 200 bp (Fig. 3c), and further puromycin selection also generated much more positive colonies than the NHEJ-RPG reporter (Fig. 3d). For the control group, the majority of cells died after puromycin treatment for 5 days, suggesting that puromycin also can be used as an efficient selection antibiotic. Similar results were observed with AAVS1.gRNA/Cas9-, CCR2.gRNA/Cas9-, CCR5.b.gRNA/Cas9- and CCR5.ZFN-transfected cells. These indicated that the SSA-RPG reporter can be highly sensitive for the selection and enrichment of nuclease-functioned positive cells by both FACS and puromycin selection.
Enrichment efficiencies of the NHEJ-RPG and SSA-RPG surrogate reporters
As the reported NHEJ-HygroR-based surrogate reporter [12, 16, 17], we used Puro R gene for fast and efficient enrichment of genetically modified cells. Based on the results described above, the SSA-RPG surrogate reporter with direct repeats of 200 bp, compared with the NHEJ-RPG reporter, was chosen for further enrichment investigation. 293T cells were co-transfected with AAVS1.gRNA/Cas9 expression plasmid and corresponding surrogate reporter plasmids, NHEJ-RPG and SSA-RPG, respectively. Subsequent T7E1 assay demonstrated that mutation frequencies (Indels, %) within AAVS1 locus for SSA-RPG and NHEJ-RPG groups were 43.9 and 37.9 %, 34.8-fold and 29.1-fold of the unselected groups (1.2 and 1.3 %), respectively (Fig. 4a). Further sequencing results revealed that the mutation frequencies for puromycin-selected cells in SSA-RPG and NHEJ-RPG groups were 86.6 and 64.7 %, 21.1-fold and 18.5-fold, respectively, of the unselected groups (Fig. 4b, c). In conclusion, the SSA-RPG and NHEJ-RPG surrogate reporters share similar enrichment efficiencies for genetically modified cells, indicating that the genomic modification within selected positive cells is independent of the repair of surrogate reporters. However, the SSA-RPG surrogate reporter did help to yield efficient number of genetically modified cells, exhibiting a higher sensitivity upon the NHEJ-RPG reporter.

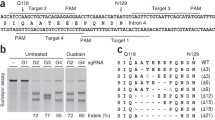
Enrichment of AAVS1.gRNA/Cas9-induced genetically modified mutants by puromycin selection. 293T cells were co-transfected with AAVS1.gRNA/Cas9 expression plasmid and corresponding surrogate reporter plasmids, NHEJ-RPG and SSA-RPG, respectively, and were subjected to puromycin treatment before indel detection. a T7E1 assay for the indels within unselected and puromycin-selected cells (PuroR+). Arrows indicate the expected fragments. Mutation frequencies (Indels, %) at the bottom were calculated by measuring the band intensities. b Sequencing results of indels within SSA-RPG based unselected and puromycin-selected cells. c Sequencing results of indels within NHEJ-RPG based unselected and puromycin-selected cells. Dashes and lower-case letters in green indicate deleted and inserted base pairs, respectively (the inserted or deleted base pairs are numbered in the parentheses). The number (×2 or ×3) represents the multiple occurrences for indicated clones
Enrichment of genetically modified cells with SSA-RPG based puromycin selection
To further verify the efficiency and sensitivity of the SSA-RPG surrogate reporter for enrichment of genetically modified cells, three additional CRISPR-Cas9 targets, CCR2, CCR5.a and CCR5.b, were subsequently investigated. T7E1 assay demonstrated that mutation frequencies for puromycin-selected cells were 57.8, 27.1 and 24.7 %, which were 13.8-fold, 12.3-fold and 6.3-fold of the unselected cells (4.2, 2.2 and 3.9 %), respectively (Table 1; Supplementary Fig. 4a, c, e). The enrichment was further verified by the sequencing results as 72.7, 30.8 and 45 %, 34.6-fold, 18.1-fold and 11.8-fold of the unselected groups (2.1, 1.7 and 3.8 %), respectively (Table 1; Supplementary Fig. 4b, d, f). To ask whether the SSA-RPG reporter is also efficient for the enrichment of cohesive-ending ZFN-induced genetically modified cells, 293T cells co-transfected with CCR5.ZFNL/CCR5.ZFNR expression plasmids and corresponding SSA-RPG surrogate reporter plasmid were also subjected to puromycin selection and mutation detection. T7E1 assay and sequencing results suggested mutation frequencies for puromycin-selected cells as 34.2 and 26.3 %, which were >34.2-fold and 18.8-fold of the unselected cells (Table 1; Supplementary Fig. 4g, h). Taken together, the efficiency and sensitivity of the SSA-RPG surrogate reporter are significantly efficient for the enrichment of both Cas9- and ZFN-induced genetically modified cells by puromycin selection.
Enrichment of genetically modified cells with SSA-RPG-based FACS
To improve the feasibility of the surrogate reporter system, the eGFP fluorescence gene was fused with the target/SSA arms-interrupted Puro R gene by T2A sequence as dual reporter (Fig. 1). FACS was supposed as an alternative to the puromycin selection for the enrichment of genetically modified cells. 293T cells co-transfected with CCR5.a.gRNA/Cas9 or CCR5.ZFNL/CCR5.ZFNR expression plasmids and corresponding SSA-RPG surrogate reporter plasmid, were sorted 3 days (~72 h) after transfection for DsRed+ eGFP+ cells. Subsequent T7E1 assay and sequencing detection revealed that intent mutation frequencies for flow cytometric sorted cells were 31.4 and 23.5 %, 13.2-fold and 11.2-fold of the unsorted cells (2.4 and 2.1 %) for CCR5.a.gRNA/Cas9 (Table 2; Supplementary Fig. 5a, b), and as for CCR5.ZFN, 27.7 and 20.8 %, which were >27.7-fold and 14.9-fold of the unsorted cells (Table 2; Supplementary Fig. 5c, d). These results suggested that the SSA-RPG surrogate reporter could be used for the enrichment of genetically modified cells not only by puromycin selection but also by FACS.
Detection of off-target effect after puromycin selection and FACS
To determine if there are any off-target events within the enriched cells, the genome DNA prepared after puromycin selection or flow cytometry sorting in the above studies was also used for the off-target detection (T7E1 assay). The off-target sites were predicted according to Cradick’s method for ZFNs [43] and the online software (http://crispr.mit.edu/) for CRISPR-Cas9 system, respectively. We selected and tested three top off-target sites with the highest potential for each intent target site (CCR5.a.gRNA/Cas9 and CCR5.ZFN). We did not detect any obvious off-target effects even after puromycin selection and flow cytometric enrichment (Supplementary Figs. 6, 7), suggesting that a conclusion can be made in a certain extent that our surrogate system does not exacerbate the off-target effect obviously and can be used for efficient enrichment of genetically modified cells.
Discussion
In this work, we designed two dual-reporter surrogate systems to facilitate the enrichment of genetically modified cells by either puromycin selection or FACS. By comparing the DSB repair efficiencies of the two surrogate reporter systems under different end conditions generated by Cas9 and ZFNs, we found that the efficiency and sensitivity of the SSA-RPG surrogate reporter with direct repeat length more than 200 bp were much higher than the NHEJ-RPG reporter. The enrichment efficiencies for genetically modified cells were also compared to be independent of the repair efficiency of surrogate reporters, but the SSA-RPG surrogate reporter did increase obviously positive cells and exhibited higher sensitivity, which was further applied successfully for the enrichment of CRISPR-Cas9- and ZFN-induced genetically modified cells by both puromycin selection and FACS. Thus, the SSA-RPG surrogate reporter provides a useful tool for efficient activity validation and enrichment of genetically modified cells, especially when the nuclease activity is very low.
For a long time, NHEJ has been considered as the predominant mechanism among NHEJ, SSA and HR repair pathways [19, 25–29]. There were only few reports claiming that the frequency of SSA was higher than that of NHEJ [30, 31]. We found that the length of the direct repeats introduced would influence the SSA-based reporter repair efficiency distinctly in a certain range. Both Cas9- and ZFN-induced SSA-mediated repair efficiencies were positively correlated with the length of the direct repeats until they reached plateaus, suggesting that the length of direct repeats is a major factor to determine the SSA repair pathway. For NHEJ-mediated DSB repair, Cas9-induced DSBs showed lower efficiency, compared with the ZFN-induced cohesive ends. Although the SSA-mediated repair efficiency was much higher than NHEJ when the length of direct repeats as SSA arms is 200 bp, when the length is less than 50 bp for Cas9-induced DSBs, or less than 100 bp for ZFN-induced cohesive ends, the SSA-based surrogate reporters possess no superiority on the NHEJ-based reporter (Fig. 3a, b). Difference in repair pathway utilization between I-SceI (Producing 3′-overhangs) and TALENs (Producing 5′-overhangs) was also reported, due to intrinsic properties of the nuclease reagents [30]. Here, we compared the DNA repair pathway preference between different DSB conditions generated by Cas9 and ZFNs, respectively, and difference was only observed when the length of direct repeats for the SSA-based reporter is less than 100 bp. Whether the I-SceI nuclease involves different repair pathway preference remains elusive.
In conclusion, both the SSA-RPG and NHEJ-RPG dual reporters are proved to be efficient for the enrichment of genetically modified 293T cells. However, the sensitivity for the SSA-RPG reporter with the direct repeat length of 200 bp is much higher because of higher repair efficiency, which could contribute to yield more positive cells through puromycin selection or FACS. Similar results have been achieved in another project of our team with mouse C2C12 cell line for Rosa26 and Vitamin D loci (Data not shown). Thus, the SSA-RPG reporter should be the prior choice, which can help to save the time and cost to enrich enough genetically modified cells for the purposes of the gene therapy, gene function analysis and generation of genome-edited animals. Nevertheless, for a new cell line, pre-experimental comparison of the two reporters is strongly recommended as we did, if possible.
Materials and methods
Construction of NHEJ-RPG surrogate reporter
The NHEJ-RPG surrogate reporter was constructed with two independent expression cassettes using standard molecular biology techniques. The DsRed expression cassette consists of the DsRed gene driven by CMV promoter and terminated by the rabbit beta-globin gene polyA signal, which is used for measuring transfection efficiency. The Puro R gene and eGFP fluorescent gene are fused by T2A (GSGEGRGSLLTCGDVEENPGP) as dual-reporter [42], and the cassette is driven by CAG promoter and terminated by the SV40 polyA signal. A multiple cloning site (MCS) containing NotI/BamHI sites is introduced just following the initiation codon ATG of Puro R gene for nuclease target sequence cloning (Fig. 1a, Supplementary Fig. 1a). The Puro R and eGFP genes could be expressed successfully for wide type NHEJ-RPG reporter, but the open reading frame (ORF) will be disrupted once a nuclease target sequence was inserted. When the designed nuclease cleaves the target sequence, the mutagenic NHEJ-mediated DNA DSB repair will generate one-third of products with correct ORF, resulting in functional expression of Puro R and eGFP genes. Then, PuroR and eGFP positive cells can be subjected to puromycin selection or flow cytometry sorting. The oligonucleotides-annealing products of CCR2/CCR5.a.CRISPR and CCR5.ZFN target sequences (Supplementary Table 1) were inserted into the NotI/BamHI sites to generate corresponding surrogate reporter vectors. All the plasmids were confirmed by sequencing.
Construction of the SSA-RPG surrogate reporter
Differently with the NHEJ-RPG system, for the SSA-RPG surrogate reporter, the MCS with NotI/BamHI sites is flanked with direct repeats as SSA arms and inserted into the middle of Puro R gene to interrupt the ORF (Fig. 1b, Supplementary Fig. 1b). The direct repeat sequences derived from Puro R gene are designed to flank the MCS site, and the lengths are set to range from 50 to 350 bp with 50 bp increased each (Fig. 1c). When designed nuclease functions to cleave the target sequence inserted within the surrogate reporter, the SSA-mediated repair will result in the deletion of one repeat, together with the intervening sequence, correcting the ORF for Puro R and eGFP reporter genes. The oligonucleotides-annealing products of CCR2/CCR5.a.CRISPR, CCR5.b.CRISPR and AAVS1.CRISPR and CCR5.ZFN target sequences (Supplementary Table 1) were also inserted into the parental SSA-RPG plasmid using NotI/BamHI sites to generate corresponding surrogate reporter vectors. All the plasmids were confirmed by sequencing.
Construction of the designed nuclease expression vectors
The CRISPR-Cas9 system used in this work was the Streptococcus thermophilus CRISPR3-Cas (StCas9) system as we previously reported [38] (Fig. 2b). Target sequences were selected from human CCR2, CCR5 and AAVS1 loci. The gRNA and humanized S. thermophilus Cas9 protein are expressed within one pll3.7 vector driven by mU6 and CMV promoter, respectively (Supplementary Fig. 1c).
The ZFN pair designed to target CCR5 locus as shown in Fig. 2c, were synthesized (Biosune, Inc. China) directly, based on the sequences reported previously [13], and inserted into the eukaryotic expression vector pst1374-MCS to construct pst1374-CCR5.ZFNL and pst1374-CCR5.ZFNR, respectively (Supplementary Fig. 1d). To improve performance of the ZFN nuclease, FokI within ZFNL was introduced with Sharky and ELD mutations, and that within ZFNR introduced with the Sharky and KKR mutations [44]. These mutations were generated by a multiple site-directed mutagenesis method as reported [45].
Cell culture
Human embryonic kidney 293T (HEK293T) cells were routinely maintained in Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin and 10 % (v/v) fetal bovine serum (FBS, Hyclone), at 37 °C and 5 % CO2. About 1 × 104 293T cells were seeded into 0.5 mL of culture medium per well of 24-well plate and incubated for about 24 h before transfection.
Cell transfection
The transfection was conducted with related plasmids (Total of 1.6 μg plasmid DNA per transfection) within 24-well plates using Sofast transfection reagent (Xiamen Sunma Biotechnology Co., Ltd. China) according to the user manual. For the ZFN groups, the mole ration of ZFNL and ZFNR expression vectors and surrogate reporter was 1.2:1.2:1, and the ZFN parental plasmid pst1374-MCS was used as the control. Meanwhile, for the CRISPR-Cas9 groups, the mole ration of gRNA/Cas9 expression vector and surrogate reporter was 1.2:1, and the Cas9 expression vector without gRNA was used as the control. For each transfection, more than three independent replicates were performed.
Detection of surrogate reporter repair efficiency in 293T
The cells were observed and photographed with fluorescence microscope 1, 2 and 3 days after transfection, respectively. Then, cells were harvested and subjected to flow cytometric counting. 30,000 cells were counted to number the DsRed+ cells and DsRed+ eGFP+ positive cells. The percentage of DsRed+ cells represents the efficiency of transfection, and the percentage of DsRed+ eGFP+ cells compared with DsRed+ cells was calculated to estimate the repair efficiency of surrogate reporter and the activity of designed nucleases. Compensation experiments with 293T cells transfected with independent eGFP or DsRed expression vectors were performed to decrease the errors. Data were analyzed using FCS Express 4 flow cytometric data analysis software.
Puromycin selection
The cells within half number of wells were harvested and directly used for genomic DNA extraction with E.Z.N.A.DNA Kit (OMEGA Bio-Tek) by standard procedure 3 days after transfection. Meanwhile, the other wells were maintained with puromycin (Sigma) treatment 2 days after transfection at a final concentration of 3 μg/mL for 5 days, and the medium was changed every day. After the treatment, selected cells within positive colonies were collected and genomic DNA was extracted for subsequent T7E1 assay. Untransfected cells, cells transfected with surrogate reporter alone and cells co-transfected with nuclease parental vector and surrogate reporter were used as controls.
Fluorescence-activated cell sorting
3 days after transfection, cells were harvested and half were directly subjected to genomic DNA extraction for subsequent T7E1 assay, and the remaining cells were used for flow cytometric sorting before genomic DNA extraction. Briefly, single-cell suspensions were analyzed and cells with strong DsRed and eGFP signals were sorted using the FACSAria II cell sorter (BD Biosciences, USA) to enrich cells harboring functional surrogate reporter. Untransfected cells, cells transfected with surrogate reporter alone and cells co-transfected with nuclease parental vector and surrogate reporter were used as controls.
Detection of nuclease-induced mutations in genomic level
T7E1 assay was performed as previously described [12, 14] to detect the nuclease-induced mutations in genomic level. Generally, the genomic DNAs prepared form different experiment groups were PCR amplified for DNA fragments containing intent target sequences with appropriate primers (Supplementary Table 2). 100 ng amplified DNA fragment was denatured by heating and annealed to form heteroduplex DNA, subsequently treated with 5 units of T7 nuclease I (New England Biolabs) for 20 min at 37 °C and finally analyzed using 2 % agarose gel electrophoresis. Mutation frequencies were calculated as previously described based on the band intensities using ImageJ software and the following equation: mutation frequency (%) = 100 × [1 − (1 − fraction cleaved)1/2], where the fraction cleaved is the total relative density of the cleavage bands divided by the sum of the relative density of the cleavage bands and uncut bands [46]. PCR-amplified fragments were also inserted into the pGEM-T vector by T-A cloning and 20 or more clones were sequenced.
References
Segal DJ, Meckler JF (2013) Genome engineering at the dawn of the golden age. Annu Rev Genom Hum G 14:135–158. doi:10.1146/annurev-genom-091212-153435
Cathomen T, Joung JK (2008) Zinc-finger nucleases: the next generation emerges. Mol Ther 16(7):1200–1207. doi:10.1038/Mt.2008.114
Pennisi E (2012) The tale of the TALEs. Science 338(6113):1408–1411
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327(5962):167–170. doi:10.1126/science.1179555
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482(7385):331–338. doi:10.1038/Nature10886
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821. doi:10.1126/science.1225829
Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823. doi:10.1126/science.1231143
Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, Calarco JA (2013) Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods 10(8):741. doi:10.1038/Nmeth.2532
Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O’Connor-Giles KM (2013) Genome engineering of drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194(4):1029. doi:10.1534/genetics.113.152710
Hwang WY, Fu YF, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JRJ, Joung JK (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31(3):227–229. doi:10.1038/Nbt.2501
Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826. doi:10.1126/science.1232033
Kim H, Um E, Cho SR, Jung C, Kim H, Kim JS (2011) Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat Methods 8(11):941–943. doi:10.1038/nmeth.1733
Perez EE, Wang JB, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH (2008) Establishment of HIV-1 resistance in CD4(+) T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26(7):808–816. doi:10.1038/Nbt1410
Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS (2009) Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19(7):1279–1288. doi:10.1101/gr.089417.108
Kim H, Kim MS, Wee G, Lee CI, Kim H, Kim JS (2013) Magnetic separation and antibiotics selection enable enrichment of cells with ZFN/TALEN-induced mutations. PLoS One. doi:10.1371/journal.pone.0056476
Ramakrishna S, Cho SW, Kim S, Song M, Gopalappa R, Kim JS, Kim H (2014) Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. Nat Commun. doi:10.1038/Ncomms4378
Kim YH, Ramakrishna S, Kim H, Kim JS (2014) Enrichment of cells with TALEN-induced mutations using surrogate reporters. Methods 69(1):108–117. doi:10.1016/j.ymeth.2014.04.012
Wang C, Lees-Miller SP (2013) Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Radiat Oncol Biol Phys 86(3):440–449. doi:10.1016/j.ijrobp.2013.01.011
Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211. doi:10.1146/annurev.biochem.052308.093131
Ramsden DA, Gellert M (1998) Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J 17(2):609–614. doi:10.1093/emboj/17.2.609
Fishman-Lobell J, Rudin N, Haber JE (1992) Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12(3):1292–1303
Lin FL, Sperle K, Sternberg N (1984) Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol Cell Biol 4(6):1020–1034
Lin FL, Sperle K, Sternberg N (1990) Repair of double-stranded DNA breaks by homologous DNA fragments during transfer of DNA into mouse L cells. Mol Cell Biol 10(1):113–119
Maryon E, Carroll D (1991) Characterization of recombination intermediates from DNA injected into Xenopus laevis oocytes: evidence for a nonconservative mechanism of homologous recombination. Mol Cell Biol 11(6):3278–3287
Liu JG, Gong L, Chang CQ, Liu C, Peng JR, Chen J (2012) Development of novel visual-plus quantitative analysis systems for studying DNA double-strand break repairs in zebrafish. J Genet Genomics 39(9):489–502. doi:10.1016/j.jgg.2012.07.009
Knoll A, Fauser F, Puchta H (2014) DNA recombination in somatic plant cells: mechanisms and evolutionary consequences. Chromosome Res 22(2):191–201. doi:10.1007/s10577-014-9415-y
Iyama T, Wilson DM (2013) DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 12(8):620–636. doi:10.1016/j.dnarep.2013.04.015
Bennardo N, Cheng A, Huang N, Stark JM (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. doi:10.1371/journal.pgen.1000110
Jeggo PA (1998) Identification of genes involved in repair of DNA double-strand breaks in mammalian cells. Radiat Res 150(5 Suppl):S80–S91
Kuhar R, Gwiazda KS, Humbert O, Mandt T, Pangallo J, Brault M, Khan I, Maizels N, Rawlings DJ, Scharenberg AM, Certo MT (2014) Novel fluorescent genome editing reporters for monitoring DNA repair pathway utilization at endonuclease-induced breaks. Nucleic Acids Res. doi:10.1093/nar/gkt872
Preston CR, Flores CC, Engels WR (2006) Differential usage of alternative pathways of double-strand break repair in Drosophila. Genetics 172(2):1055–1068. doi:10.1534/genetics.105.050138
Perez C, Guyot V, Cabaniols JP, Gouble A, Micheaux B, Smith J, Leduc S, Paques F, Duchateau P (2005) Factors affecting double-strand break-induced homologous recombination in mammalian cells. Biotechniques 39(1):109–115. doi:10.2144/05391gt01
Ira G, Haber JE (2002) Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol 22(18):6384–6392. doi:10.1128/Mcb.22.18.6384-6392.2002
Sugawara N, Ira G, Haber JE (2000) DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol 20(14):5300–5309. doi:10.1128/Mcb.20.14.5300-5309.2000
Ren C, Yan Q, Zhang Z (2014) Minimum length of direct repeat sequences required for efficient homologous recombination induced by zinc finger nuclease in yeast. Mol Biol Rep. doi:10.1007/s11033-014-3579-6
Wang L, Lin J, Zhang T, Xu K, Ren C, Zhang Z (2013) Simultaneous screening and validation of effective zinc finger nucleases in yeast. PLoS One 8(5):e64687. doi:10.1371/journal.pone.0064687
Ren C, Yan Q, Zhang Z (2014) Minimum length of direct repeat sequences required for efficient homologous recombination induced by zinc finger nuclease in yeast. Mol Biol Rep 41(10):6939–6948. doi:10.1007/s11033-014-3579-6
Xu K, Ren C, Liu Z, Zhang T, Zhang T, Li D, Wang L, Yan Q, Guo L, Shen J, Zhang Z (2014) Efficient genome engineering in eukaryotes using Cas9 from Streptococcus thermophilus. Cell Mol Life Sci CMLS. doi:10.1007/s00018-014-1679-z
Zhang C, Xu K, Hu L, Wang L, Zhang T, Ren C, Zhang Z (2014) A suicidal zinc finger nuclease expression coupled with a surrogate reporter for efficient genome engineering. Biotechnol Lett. doi:10.1007/s10529-014-1690-3
Morton J, Davis MW, Jorgensen EM, Carroll D (2006) Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. P Natl Acad Sci USA 103(44):16370–16375. doi:10.1073/pnas.0605633103
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. P Natl Acad Sci USA 109(39):E2579–E2586. doi:10.1073/pnas.1208507109
Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF (2004) Vignali DAA (2004) Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector (vol 22, pg 589). Nat Biotechnol 22(12):1590. doi:10.1038/nbt1204-1590b
Cradick TJ, Ambrosini G, Iseli C, Bucher P, McCaffrey AP (2011) ZFN-Site searches genomes for zinc finger nuclease target sites and off-target sites. BMC Bioinform. doi:10.1186/1471-2105-12-152
Guo J, Gaj T, Barbas CF 3rd (2010) Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J Mol Biol 400(1):96–107. doi:10.1016/j.jmb.2010.04.060
Seyfang A, Jin JHQ (2004) Multiple site-directed mutagenesis of more than 10 sites simultaneously and in a single round. Anal Biochem 324(2):285–291. doi:10.1016/J.Ab.2003.10.012
Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ (2010) A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649:247–256. doi:10.1007/978-1-60761-753-2_15
Acknowledgments
The authors would like to thank all the colleagues in Professor Zhang’s lab for their excellent technical assistance and helpful discussions. This work was supported by grants from National Natural Science Foundation of China (NSFC) [31171186], National Science and Technology Major Project of China [2014ZX0801009B] and Natural Science Basic Research Plan in Shaanxi Province of China [2013JQ3009].
Conflict of interest
The authors declare that no competing interests exist.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Ren and K. Xu contributed equally to this work and should be regarded as joint First Authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ren, C., Xu, K., Liu, Z. et al. Dual-reporter surrogate systems for efficient enrichment of genetically modified cells. Cell. Mol. Life Sci. 72, 2763–2772 (2015). https://doi.org/10.1007/s00018-015-1874-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1874-6