
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease. Although a major cause of AD is the accumulation of amyloid-β (Aβ) peptide that induces neuronal loss and cognitive impairments, our understanding of its neurotoxic mechanisms is limited. Recent studies have identified putative Aβ-binding receptors that mediate Aβ neurotoxicity in cells and models of AD. Once Aβ interacts with a receptor, a toxic signal is transduced into neurons, resulting in cellular defects including endoplasmic reticulum stress and mitochondrial dysfunction. In addition, Aβ can also be internalized into neurons through unidentified Aβ receptors and induces malfunction of subcellular organelles, which explains some part of Aβ neurotoxicity. Understanding the neurotoxic signaling initiated by Aβ-receptor binding and cellular defects provide insight into new therapeutic windows for AD. In the present review, we summarize the findings on Aβ-binding receptors and the neurotoxicity of oligomeric Aβ.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: Aβ oligomers in neurotoxicity
Extracellular plaques and neurofibrillary tangles (NFTs) are histological hallmarks found in the brains of patients with AD and are mainly composed of Aβ and tau proteins, respectively. AD is characterized by learning and memory deficits largely attributed to the neuronal degeneration and cell death of affected neurons in the hippocampus and cerebral cortex. Aβ is a 4-kDa peptide that is a proteolytic product of amyloid precursor protein (APP). The extracellular region of APP is cleaved by a group of metalloproteases called α-secretases and the remaining fragment undergoes intramembrane proteolysis by the γ-secretase protein complex [1]. This process generates the peptide fragment p3 that is not toxic. In contrast, APP is sequentially processed by β-site APP cleaving enzyme (BACE) and γ-secretase in AD brains [2]. This “amyloidogenic pathway” liberates Aβ, which is regarded as a main culprit in AD etiology because it forms insoluble deposits by self-aggregating. Mutations in APP and presenilin (PS), a catalytic unit of γ-secretase, elicit familial AD by driving the amyloidogenic pathway [3]. Cleavage of Aβ by γ-secretase determines its amino acid length from 37 to 43 amino acids long [4]. Among them, Aβ40 and Aβ42 are the major Aβ species. Longer Aβ42 is more prone to form aggregates than Aβ40 and is regarded as a major mediator of neurotoxicity. An elevated ratio of Aβ42 to Aβ40 is also found in AD brains [5]. Mutations located immediately after the C-terminus of Aβ induce greater Aβ42 production and result in familial AD [6, 7]. Together, strong evidence points to Aβ42 as the critical Aβ isoform in AD pathology.
Interestingly, recent reports support the idea that Aβ oligomers, which assemble with a few Aβ monomers less than 20, exert more toxicity to neurons than fibrillar Aβ deposits [8]. Aβ oligomers are produced in vitro by incubating synthetic Aβ under certain conditions [9, 10]. Although molecular features of synthetic Aβ oligomers are consistent and adjustable, oligomeric conformations differ from each other depending on the preparation conditions. Furthermore, it is unclear whether synthetic Aβ oligomers accurately reflect the features of Aβ found endogenously in AD brains, such as mutations in Aβ sequences, posttranslational modification including phosphorylation and pyroglutamylation, and interaction with divalent metal ion [11–13]. Nevertheless, physiological Aβ oligomers mimicking natural Aβ found in vivo can be prepared from cells and AD tissues. Mutant APP-expressing cells secrete Aβ oligomers that impair neurons and brain tissues [14]. Soluble Aβ oligomers extracted from the brains of AD mouse models and postmortem AD brain tissue also damage neurons [15, 16]. On the contrary, insoluble amyloid prepared from AD brains fails to impair neuronal function in brain slices, underscoring the role of soluble Aβ oligomers in AD pathology [16]. To delineate the mechanism(s) of Aβ neurotoxicity, we first provide an overview of Aβ-binding partners.
Aβ-binding receptors in AD pathology
Because Aβ peptide is generated and released into extracellular region, it first challenges to generate toxic signal into neurons passing through plasma membrane. Aβ itself can directly bind to cell membranes and form ion channels or pores that induce membrane disruption and thus neuronal damage. Many observations show pore-like structure of Aβ in vitro and in the cell membrane of the AD brains and mice [17–20]. In addition, soluble Aβ oligomers, but not monomers or fibrils, increase membrane permeability and thus dysregulate Ca2+ signals for neurotoxicity [21]. More recently, emerging insight into the mechanistic link between Aβ and its binding proteins highlights the potential role of “Aβ receptors” in AD. A number of Aβ-binding proteins have been identified on the plasma membrane of neurons that may have an important role in Aβ-induced neurotoxicity. These proteins include the receptor for advanced glycation end products (RAGE), N-methyl-d-aspartate receptor (NMDAR), α7-nicotinic acetylcholine receptor (α7 nAChR), cellular prion protein (PrPc), ephrin type B receptor 2, immunoglobulin G Fc gamma receptor IIb (FcγRIIb), and paired immunoglobulin-like receptor B (PirB) (Fig. 1) [22–28].

Aβ-binding receptors in neurotoxicity. Aβ low-n or high-n oligomers bind to cognate Aβ receptors, such as RAGE, NMDAR (its direct binding is not clear), α7 nAChR, FcγRIIb, PirB, PrPc, or EphB2. This Aβ-receptor interaction generates and transduces neurotoxic signal into neurons, which causes cellular defects, such as mitochondrial dysfunction and ER stress response. In addition, some Aβ receptors are most likely to internalize Aβ into neurons to display distinct cellular defect. Please see main text for details
RAGE
RAGE is a multi-ligand receptor that binds to advanced glycation end product (AGE), amphoterin and S100/calgranulins [29], and AGE is observed in senile plaque and NFTs in AD brains [33]. RAGE is also known as a cell surface receptor for Aβ in neurons and microglia that mediates AD-related Aβ neurotoxicity, including oxidative stress, synaptic dysfunction, and eventually neuronal cell death [24, 30]. Indeed, the expression of RAGE is significantly increased in the brains of patients with AD, especially in blood vessels [24, 31, 32]. In genetic studies, AD mice (PDAPP J20) crossed with RAGE transgenic mice show early abnormalities in spatial learning and memory, while the mice harboring dominant-negative forms of RAGE are resistant to such neuropathological alterations [34]. RAGE also functions in Aβ transport across the blood–brain barrier (BBB) and Aβ accumulation in the brain by binding to soluble Aβ [31].
Treatment of AD mice with soluble RAGE or a RAGE-specific antibody not only improves impaired long-term potentiation (LTP) and cognitive dysfunction, but also prevents the entry of Aβ into the brain [31, 35]. Moreover, a multimodal RAGE-Aβ interaction blocker reduces the level of Aβ in the brain and neuroinflammatory response and thus prevents cognitive impairment in AD mice [36], indicating that the interaction between RAGE and Aβ is critical for AD pathogenesis. Currently, RAGE is considered as an advanced therapeutic target among Aβ receptors. The orally bioavailable and BBB-permeable PF-04494700, which inhibits the interaction between RAGE and Aβ, is tested for phase II clinical trial. Although low-dose (5 mg/day) test shows a good safety profile and decreased decline on the Alzheimer’s disease assessment scale-cognitive (ADAS-cog) in mild AD patients, it still needs further investigation because of high dropout and discontinuation rates [37].
NMDAR and α7 nAChR
Several reports suggest that Aβ interacts with NMDARs at postsynaptic terminals. Antibodies against the GluN1 or GluN2B subunit of NMDARs markedly block the binding of Aβ oligomer to neurons [38, 39] and Aβ oligomers partially colocalize with GluN2B subunits of NMDARs at the cell surface [13]. Indeed, through NMDAR activation, Aβ oligomers induce Ca2+ dysregulation, neuronal death [40], and synaptic dysfunction [41, 42]. However, it is still unclear whether Aβ directly binds to NMDAR subunits [43, 44]. In addition, Aβ oligomers promote the endocytosis of NMDARs, which requires the activation of α7 nAChR signaling [45]. The α7 nAChR is another candidate Aβ-binding receptor and binds to soluble Aβ with high affinity [23, 45]. The α7 nAChR mediates Aβ-induced tau phosphorylation via ERK and JNK [46]. Although α7 nAChR-expressing neuroblastoma cells are susceptible to Aβ-induced toxicity in vitro [47], the in vivo neurotoxic role of this receptor is inconsistent. For instance, α7 nAChR deficiency improves cognitive deficits and synaptic pathology in PDAPP J9 mouse model of AD, while it exacerbates AD pathology in Tg2576 mouse model [48, 49].
PrPc
PrPc was identified to have a high-affinity binding site for Aβ oligomers [25]. Subsequently, it was shown that PrPc deficiency prevents Aβ oligomer-induced neuronal cell death [50] and inhibits Aβ oligomer-induced LTP blockade [25]. The role of PrPc in the inhibition of LTP was also illustrated using synthetic Aβ oligomers called Aβ-derived diffusible ligands (ADDL) and Aβ oligomers derived from human AD brains [51, 52]. In addition, the deletion of PrPc expression in APPswe/PS1ΔE9 mice rescues the loss of synaptic markers and the impairment of spatial learning and memory [53]. Further, treatment of APPswe/PS1 M146L mice with anti-PrPc antibodies, which block the binding of Aβ oligomer to PrPc, rescues the decreased synapse density and cognitive deficits [54].
Because PrPc is anchored to the cell surface with a glycosylphosphatidylinositol anchor, Aβ-induced neurotoxic signaling is unlikely to be transduced only by PrPc itself. Recently, the metabotropic glutamate receptor mGluR5 was identified as a neurotoxic mediator at the postsynaptic density that couples the Aβ–PrPc complex with Fyn and disrupts neuronal function [55, 56]. Fyn interacts with and localizes tau to the dendritic compartment and facilitates NMDAR–PSD95 interaction, thereby mediating Aβ neurotoxicity at the postsynaptic membrane in AD [57]. In contrast, there is a report showing that PrPc may not be essential for Aβ neurotoxicity. Kessels et al. [58] observed that PrPc is not required for Aβ-induced synaptic depression, reduction in spine density, and blockade of LTP. In addition, the ablation or overexpression of PrPc has no effect on the impairment of hippocampal synaptic plasticity in APPswe/PS1 L166P or PDAPP J20 AD mice [59, 60]. Further, cognitive impairment is not ameliorated in Aβ-injected mice lacking PrPc [61]. Thus, the role of PrPc in Aβ neurotoxicity remains controversial.
FcγRIIb and PirB
Recently, two immune receptors, FcγRIIb and PirB which were originally believed to function exclusively in the immune system, were shown to have neuropathic roles as Aβ receptors in AD brains [27, 28, 62]. Kam and Song et al. showed that FcγRIIb binds to oligomeric Aβ with high affinity (K d = 56.7 nM) in vitro and in the brains of patients with AD. They also found that the expression of FcγRIIb is increased in the brains of AD mice and patients with AD and that FcγRIIb deficiency rescues Aβ-induced neurotoxicity, including cell death, decreased LTP, spine density, as well as memory impairment in AD mice (PDAPP J20). Inhibiting FcγRIIb–Aβ interaction using synthetic peptides also prevents Aβ-induced neurotoxicity in cultured neurons and memory impairment in the mice as assayed with intracerebroventricular-injection [27]. Similar to FcγRIIb, PirB deletion in mice suppresses the deleterious activity of Aβ oligomers on LTP and rescues impaired ocular dominance plasticity and behavioral deficits in AD mice (APP/PS1) [28].
Interestingly, these two proteins show similarity in their structure and in the binding affinity with Aβ oligomers. Both have immunoglobulin (Ig) domains on their extracellular regions and immunoreceptor tyrosine-based inhibitory motifs (ITIM) on their intracellular regions. FcγRIIb has two Ig domains and an ITIM, whereas PirB has six Ig domains and four ITIMs. FcγRIIb interacts with low-n oligomers via its second Ig domain and PirB binds to high-n oligomers via its first two Ig domains. Like FcγRIIb, PirB binds to Aβ with high affinity (K d = 110 nM). One major difference between FcγRIIb and PirB is the requirement of ITIM in the neurotoxic signaling. While tyrosine phosphorylation in the ITIM of FcγRIIb mediates Aβ neurotoxicity, it is apparently not involved in Aβ signaling in the case of PirB. It will be interesting to examine this difference in ITIM to mediate the neurotoxicity.
Overall, we now require more detailed studies to clarify distinct roles and signaling of these Aβ receptors in Aβ neurotoxicity as well as their neuronal expression patterns. Unlike FcγRIIb and PrPc that bind to Aβ low-n oligomers and high n-oligomers, respectively, other receptors have not been characterized for their binding preferences to those Aβ. The binding regions in the receptors as well as in Aβ have not been identified in most cases. In addition, a possibility for protein–protein interaction among those receptors that may function together in Aβ neurotoxicity or for the roles of those receptors in various cell types remains to be addressed.
Cellular defects in Aβ neurotoxicity
Endoplasmic reticulum stress response for Aβ toxicity
ER senses and responds to various changes in cellular circumstances to maintain the protein folding capacity through the unfolded protein response (UPR) [63]. The UPR is a cellular recovery system in response to ER stress and relieves ER overload. The UPR is composed of three main pathways induced by inositol requiring kinase 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6). Among the three arms of UPR, PERK phosphorylates eukaryotic translation initiation factor 2 subunit α (eIF2α) and this phosphorylation prevents recycling of the eIF2 complex to its active GTP-bound form [64], lowering overall protein translation and ER overload. On the other hand, prolonged activation of PERK elicits cell death by expressing C/EBP-homologous protein that inhibits the transcription of anti-apoptotic B cell lymphoma 2 (Bcl-2) [65]. Therefore, tight regulation of the PERK pathway is required for appropriate modulation of ER stress. The effect of the PERK pathway on AD pathogenesis is controversial. Administration of salubrinal, a selective inhibitor of protein phosphatase 1 that counteracts PERK by dephosphorylating eIF2α, is protective against Aβ neurotoxicity [66, 67]. On the contrary, forebrain-specific knockout of PERK in APP/PS1 AD mice recovers cognitive defects [68]. The latter study identified systematic aspects of the PERK pathway on protein translation, especially synaptic proteins, reflecting different patterns of UPR modulated by the duration of Aβ toxicity.
In AD, the ER in neurons is also burdened by other pathologic conditions, such as Ca2+ dysregulation. Because the function of ER chaperones is affected by ER Ca2+ level, disrupted ER Ca2+ triggers ER stress [69]. These features are connected to genetic factors of AD. For example, mutant PS1 upregulates ER ryanodine receptor 3 (RyR3), which mediates ER Ca2+-induced Ca2+ release; mutant PS1-expressing PC12 cells and cortical neurons exhibit increased levels of RyR3 and concomitant enhanced responses to intracellular Ca2+ [70]. The increased expression of RyR3 is also seen in AD model mice harboring mutant PS1 [71]. Interestingly, the level of RyR3 is elevated in TgCRND8 mice containing no PS mutation but KM670/671NL and V717F mutant APP transgenes [72]. In addition, Aβ neurotoxicity is prevented by decreased expression of RyR3 through X-box binding protein 1 (XBP1), which undergoes alternative splicing by IRE1 during ER stress [73]. It is likely that PS and Aβ regulate the expression of RyR3 to affect ER stress responses. In addition to ER RyR3, inositol 1, 4, 5-triphosphate receptor (IP3R) is also linked to ER Ca2+ release by Aβ [74].
Another factor involved in Aβ neurotoxicity and mediating ER stress is ER-resident caspase-12. Sustained ER stress over the capacity of UPR induces cell death independent of typical intrinsic cell death pathways. While ER stress as well as Aβ stimulates murine caspase-12, cell death-inducing stimuli usually do not. Primary neurons from caspase-12-knockout mice show resistance to Aβ neurotoxicity [75]. Mechanistically, proteolytic activation of caspase-12 is achieved by the Ca2+-activated protease calpain and tumor necrosis factor-associated factor 2 under IRE1 [76, 77]. In response to Aβ-induced ER stress, E2-25K, an E2 conjugating enzyme in ubiquitin–proteasome system (UPS), activates calpain to process caspase-12 [78]. Unlike in rodents, however, caspase-12 in the human genome cannot be translated due to a frame-shift mutation and premature stop codon in the transcripts of all variants [79]. Interestingly, sequence comparison analysis among caspases illustrates that human caspase-4 is a homolog of murine caspase-12 with 57 % sequence identity. Consistently, human caspase-4 was shown to be involved in intracellular Aβ-induced neuronal cell death with ER stress [80]. Like caspase-12, human caspase-4 is activated by calpain through increased intracellular Ca2+ triggered by Aβ [78, 81]. It is now clear that the prolonged and aberrant ER stress response mediates Aβ neurotoxicity by triggering Ca2+ dysregulation and ER caspase activation.
The studies on Aβ receptors that induce neurotoxic ER stress, deregulation of Ca2+ flux, and ER-caspase activation have not been active yet, while these signals are strengthened by the interaction of Aβ with its receptors. Currently, limited information on the receptors is available. For Ca2+ dysregulation and ER stress, it was reported that Aβ oligomers induce plasma membrane localization of the GluN2B subunit of NMDAR and leads to Ca2+ dysregulation and neuronal death through activation of the ionotropic glutamate receptors [40]. Aβ oligomer also leads to clustered assembly of mGluR5 cluster, which is possibly mediated by interaction with PrPc [56, 82]. In addition, FcγRIIb was recently shown to play an essential role in the activation of ER-resident caspase-12 during Aβ neurotoxicity [21].
Mitochondrial dysfunction
Mitochondria generate cellular energy in most cells, and in neurons, mitochondria use glucose sources almost exclusively. Interestingly, mitochondrial defects are found in the neurons of patients with AD and in many cases of Aβ-treated neural cells and AD mice, and the key enzymes involved in glucose metabolism and the respiratory chain in mitochondria are impaired. For example, the enzyme activities of pyruvate dehydrogenase and α-ketoglutaraldehyde dehydrogenase in the citric acid cycle and cytochrome C oxidase, and the expression of respiratory chain complexes I, IV, and V are all reduced [83–87]. However, it is uncertain what causes their reduction in the mitochondria of AD neurons. In addition, the expression of enzymes mediating antioxidant functions like catalase is also altered [88]. All these features are associated with metabolic abnormalities of mitochondria, impairing energy production frequently observed during AD pathogenesis.
The presynaptic terminal demands high levels of energy required for sustained neurotransmitter release [89] and requires well-organized Ca2+ regulation machinery for activity-dependent synaptic transmission [90]. To meet these challenges, neuronal mitochondria are moved to the synapse by anterograde axonal transport and build a synaptic mitochondrial pool [91]. Therefore, tight regulation of anterograde mitochondrial axonal transport is critical for adequate synaptic output as well. Consequently, dysfunction of axonal transport is coupled with many neurological disorders and Aβ often induces impairment of anterograde mitochondrial movement [92, 93]. While it is not much known, Aβ likely inhibits axonal transport through NMDA receptor and glycogen synthase kinase 3β (GSK3β) [94] and impairs cargo recognition of microtubules by phosphorylating kinesin light chain through casein kinase 2 [95]. Collectively, these studies delineate the role of Aβ in the failure of axonal delivery of mitochondria in AD pathogenesis.
Besides the impairment in metabolism and axonal transport, alteration in structural dynamics of mitochondria is also observed in AD. In most studies, Aβ shortens mitochondrial length and increases the amount of fragmented mitochondria by modulating the expression of mitochondrial fusion/fission-related proteins [96, 97]. In the brains of patients with AD, phosphorylation and S-nitrosylation of dynamin-related protein 1 (DRP1), which is a critical factor for mitochondrial fission, is increased, likely impacting mitochondrial structure [96, 98]. In addition, mortalin seems to function in Aβ-mediated mitochondrial fragmentation and dysfunction through DRP1 [99]. On the other hand, a recent report showed an opposite result that elongated mitochondria may contribute to neurodegeneration [100]; mislocalization of DRP1 triggered by tau-mediated F-actin stabilization leads to elongated mitochondria to promote neurodegeneration. This inconsistent effect of mitochondria dynamics on the neurotoxicity needs to be clarified. In addition, coupling of Aβ membrane receptors to mitochondrial damage remains to be addressed.
Intracellular Aβ and neurotoxicity
For a long time, extracellular Aβ generating neurotoxic signals through the aforementioned receptors has been blamed as the major cause of AD. However, a growing body of evidence suggests that intracellular accumulation of Aβ also has a potential role in AD pathogenesis. Aβ immunoreactivity was first observed inside neurons with the neurofibrillary tangles of both patients with AD and normal individuals [101]. Intracellular Aβ is widely detected in patients with mild cognitive impairment, AD [102] and down’s syndrome [103]. The accumulation of intracellular Aβ precedes the formation of Aβ deposits and the development of pathologies in these diseases [104]. Consistently, accumulation of intracellular Aβ appears prior to neuronal degeneration and neurofibrillary tangle formation in AD mice, including APP/PS1 [105], 3xTg-AD [106], and 5xFAD [107]. Especially, age-related loss of synaptophysin-immunoreactive presynaptic boutons within the hippocampus occurs before extracellular Aβ deposits are observed in APP/PS1 mice [108]. In addition, intraneuronal accumulation of Aβ is also observed in 4-month-old 3xTg-AD mice which have no detectable Aβ plaques and hyperphosphorylated tau yet but are in the beginnings of cognitive deficits [109], implicating that accumulation of the intraneuronal Aβ is an early event in the progression of AD.
Receptors for Aβ internalization
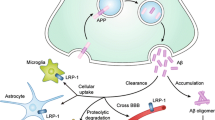
Because APP localizes to several subcellular compartments, including ER, endosomes, and plasma membrane, Aβ could accumulate intracellularly after its production inside cells. However, it is known that most Aβ produced at the plasma membrane or secretory vesicles is secreted extracellulary [110]. Thus, it is reasonable to believe that the main source of the intracellular Aβ pool would result from internalization of the extracellular Aβ, though clear evidence for this is insufficient yet. As a possible way for the internalization of Aβ, it was shown that Aβ might directly interact with lipids, cholesterols, or proteoglycans in extracellular regions and that membrane-bound Aβ oligomers are recruited into lipid rafts by a fyn-dependent manner [11, 112, 113]. In addition, reduction of cellular cholesterol and sphingolipid levels decrease Aβ uptake [114]. More directly, treatment of lipid raft-dependent endocytosis inhibitor or inhibition of clathrin-dependent endocytosis decreases Aβ uptake [47, 115, 116]. Collectively, such direct interaction with lipid rafts and clathrin-mediated endocytosis may provide way(s) for Aβ uptake.
Alternatively, Aβ can actively be uptaken by Aβ-binding proteins, including α7 nAChRs, LRP1, and RAGE. Intracellular Aβ colocalizes with α7 nAChRs in AD brains and overexpression of α7 nAChR in neuroblastoma cells leads to intracellular accumulation of Aβ [47]. LRP1, a classic endocytosis receptor that uptakes extracellular ligands, also internalizes Aβ into cultured neurons [116] and AD mice [117]. Interestingly, LRP1 cooperates with PrPc to internalize Aβ oligomers for cytotoxicity [118]. In addition, RAGE colocalizes with intraneuronal Aβ in the hippocampus of AD mice and RAGE-knockout neurons display reduced uptake of Aβ [119]. However, the route of Aβ uptake into neurons is still unresolved. While Aβ internalized by RAGE accumulates in mitochondria and thus induces mitochondrial dysfunction, Aβ internalized by other receptors, such as α7 nAChRs, localizes to endosomal or lysosomal compartments [47, 119, 120].
Moreover, whether the receptors responsible for Aβ uptake in neurons or non-neurons function for either Aβ neurotoxicity or clearance remains to be further clarified. For example, microglial Toll-like receptor (TLR) 2 and 4 are also known as potential Aβ receptors which directly interact with Aβ and mediate microglial activation [121, 122]. These interactions can lead to either neuronal death through TLR-mediated neuroinflammatory response or neuroprotection by clearing the intracellular Aβ after its uptake [123, 124]. Unlike neuronal Aβ receptors whose inhibition prevents neuronal uptake of Aβ and neurotoxicity, the destructive mutation of TLR4 in AD mice exhibits a decrease of Aβ uptake in microglia and an increase of Aβ deposits in brains, thus leading to cognitive dysfunction [125, 126]. In addition, similar function of TLR2 in Aβ phagocytosis is shown in TLR2-deficient AD mice which accelerate memory impairments with the increases of Aβ load [122, 127]. Thus, Aβ receptors found in different cell types display distinct functions in the progression of AD pathogenesis.
Cellular defects by intracellular Aβ
How the intraneuronal accumulation of Aβ causes neurotoxicity and AD neuropathology is largely unknown. Most studies indicate that intracellular Aβ leads to the malfunction of many intracellular organelles. The stable expression of human intracellular Aβ increases the number of Golgi apparatus elements, lysosomes, and lipofuscin bodies in the hippocampus of APP/PS1 double mutant transgenic rats [128]. Endosomal and lysosomal accumulation of Aβ leads to increase of lysosomal membrane permeability, resulting in the release of lysosomal proteases, especially cathepsins, to trigger neuronal cell death [129]. Mitochondria are another subcellular compartment for Aβ accumulation and neuronal dysfunction in AD [130, 131], as damaged and dysfunctional mitochondria are frequently observed in the AD brain. In particular, interactions between Aβ and mitochondrial resident proteins, such as Aβ-binding alcohol dehydrogenase (ABAD) and cyclophilin D, were reported to mediate mitochondrial and neuronal stress exerted by Aβ [130, 131]. In addition, intraneuronal Aβ42 accumulates in multivesicular bodies (MVB) in transgenic mice and AD brains and thus impairs the MVB sorting pathway in AD [102, 132]. Intracellular Aβ is also observed in the nucleus and increases neuronal apoptosis [133]. Because of these compelling findings, it is now crucial to uncover the receptors driving Aβ internalization and the pathological significance of the internalized Aβ, in parallel to the intense study on Aβ receptors for neurotoxic signaling cascade.
Concluding remarks
Extracellular Aβ interacts with several recently identified receptors to transduce neurotoxicity in cultured neurons and AD mice. With recent advances in identifying those receptors, we now better understand the neurotoxicity of Aβ which elicits diverse cellular defects, including ER stress and damage to mitochondria. However, the connection of those receptor functions to the cellular defects, the signal selectivity and cell-type specificity of the receptors, and cooperative interactions among the receptors need more characterization. In addition, a role of intracellular Aβ in neurotoxicity and AD pathogenesis, which further complicates AD pathogenesis, remains ripe for investigation.
References
Kojro E, Fahrenholz F (2005) The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem 38:105–127
Haass C (2004) Take five-BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J 23(3):483–488
Bertram L, Tanzi RE (2008) Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 9(10):768–778
Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci 25(2):436–445
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356
Cruts M, Dermaut B, Rademakers R, Van den Broeck M, Stögbauer F, Van Broeckhoven C (2003) Novel APP mutation V715A associated with presenile Alzheimer’s disease in a German family. J Neurol 250(11):1374–1375
Suárez-Calvet M, Belbin O, Pera M, Badiola N, Magrané J, Guardia-Laguarta C, Muñoz L, Colom-Cadena M, Clarimón J, Lleó A (2014) Autosomal-dominant Alzheimer’s disease mutations at the same codon of amyloid precursor protein differentially alter Aβ production. J Neurochem 128(2):330–339
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8(2):101–112
Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO (2010) Structural conversion of neurotoxic amyloid-β1-42 oligomers to fibrils. Nat Struct Mol Biol 17(5):561–567
Ryan DA, Narrow WC, Federoff HJ, Bowers WR (2010) An improved method for generating consistent soluble amyloid-beta oligomer preparations for in vitro neurotoxicity studies. J Neurosci Methods 190(2):171–179
Kumar S, Rezaei-Ghaleh N, Terwel D, Thal DR, Richard M, Hoch M, Mc Donald JM, Wüllner U, Glebov K, Heneka MT, Walsh DM, Zweckstetter M, Walter J (2011) Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J 30(11):2255–2265
Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Rönicke R, Reymann K, Hutter-Paier B, Alexandru A, Jagla W, Graubner S, Glabe CG, Demuth HU, Bloom GS (2013) Prion-like behavior and tau-dependent cytotoxicity of pyroglutamylated β-amyloid. Nautre 485(7400):651–655
Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J (2009) A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci 29(13):4004–4015
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31(18):6627–6638
Lesné S, Koh MT, Ktilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440(7082):352–357
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8):837–842
Bhatia R, Lin H, Lal R (2000) Fresh and globular amyloid beta protein (1-42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. FASEB J 14(9):1233–1243
Kawahara M, Negishi-Kato M, Sadakane Y (2009) Calcium dyshomeostasis and neurotoxicity of Alzheimer’s beta-amyloid protein. Expert Rev Neurother 9(5):681–693
Arispe N, Pollard HB, Rojas E (1993) Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [AβP-(1-40)] in bilayer membranes. Proc Natl Acad Sci USA 90(22):10573–10577
Inoue S (2008) In situ Aβ pores in AD brain are cylindrical assembly of Aβ protofilaments. Amyloid 15(4):223–233
Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280(17):17294–17300
Cowburn RF, Wiehager B, Trief E, Li-Li M, Sundstrom E (1997) Effects of beta-amyloid-(25-35) peptides on radioligand binding to excitatory amino acid receptors and voltage-dependent calcium channels: evidence for a selective affinity for the glutamate and glycine recognition sites of the NMDA receptor. Neurochem Res 22(12):1437–1442
Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB (2000) beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem 275(8):5626–5632
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382(6593):685–691
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457(7233):1128–1132
Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, Ho K, Yu GQ, Mucke L (2011) Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469(7328):47–52
Kam TI, Song S, Gwon Y, Park H, Yan JJ, Im I, Choi JW, Choi TY, Kim J, Song DK, Takai T, Kim YC, Kim KS, Choi SY, Choi S, Klein WL, Yuan J, Jung YK (2013) FcgammaRIIb mediates amyloid-beta neurotoxicity and memory impairment in Alzheimer’s disease. J Clin Invest 123(7):2791–2802
Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ (2013) Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341(6152):1399–1404
Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108(7):949–955
Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Munch G (2011) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32(5):763–777
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9(7):907–913
Jeynes B, Provias J (2008) Evidence for altered LRP/RAGE expression in Alzheimer lesion pathogenesis. Curr Alzheimer Res 5(5):432–437
Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N, Koike T, Wakayama I, Yanagihara R, Garruto R, Amano N, Makita Z (1998) Advanced glycation end products in Alzheimer’s disease and other neurodegenerative disease. Am J Pathol 153(4):1149–1155
Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS (2004) RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J 23(20):4096–4105
Chen X, Walker DG, Schmidt AM, Arancio O, Lue LF, Yan SD (2007) RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer’s disease. Curr Mol Med 7(8):735–742
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M, Zarcone T, Fritz G, Friedman AE, Miller BL, Zlokovic BV (2012) A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer’s disease. J Clin Invest 122(4):1377–1392
Galasko D, Bell J, Mancuso JY, Kupiec JW, Sabbagh MN, van Dyck C, Thomas RG, Aisen PS (2014) Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurology 82(17):1536–1542
Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, Oliveira CR, Rego AC, Pereira CM (2012) Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-D-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell 11(5):823–833
De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL (2007) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 282(15):11590–11601
Alberdi E, Sanchez-Gomez MV, Cavaliere F, Perez-Samartin A, Zugaza JL, Trullas R, Domercq M, Matute C (2010) Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 47(3):264–272
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27(11):2866–2875
Ronicke R, Mikhaylova M, Ronicke S, Meinhardt J, Schroder UH, Fandrich M, Reiser G, Kreutz MR, Reymann KG (2011) Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging 32(12):2219–2228
Decker H, Jurgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST (2010) N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-beta peptide oligomers. J Neurochem 115(6):1520–1529
Patel AN, Jhamandas JH (2012) Neuronal receptors as targets for the action of amyloid-beta protein (Abeta) in the brain. Expert Rev Mol Med 14:e2
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8(8):1051–1058
Wang HY, Li W, Benedetti NJ, Lee DH (2003) Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J Biol Chem 278(34):31547–31553
Nagele RG, D’Andrea MR, Anderson WJ, Wang HY (2002) Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110(2):199–211
Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF (2009) Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J Neurosci 29(27):8805–8815
Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT (2010) Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J Neurosci 30(7):2442–2453
Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB, Lee HG (2012) Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum Mol Genet 21(5):1138–1144
Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J (2011) Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2:336
Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ (2011) Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci 31(20):7259–7726
Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 30(18):6367–6374
Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T (2010) Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci 11:130
Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM (2012) Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15(9):1227–1235
Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79(5):887–902
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142(3):387–397
Kessels HW, Nguyen LN, Nabavi S, Malinow R (2010) The prion protein as a receptor for amyloid-beta. Nature 466(7308):E3–E4 discussion E4–5
Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A (2010) Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2(8):306–314
Cisse M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L (2011) Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 31(29):10427–10431
Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G (2010) Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci USA 107(5):2295–2300
Fernandez-Vizarra P, Lopez-Franco O, Mallavia B, Higuera-Matas A, Lopez-Parra V, Ortiz-Munoz G, Ambrosio E, Egido J, Almeida OF, Gomez-Guerrero C (2012) Immunoglobulin G Fc receptor deficiency prevents Alzheimer-like pathology and cognitive impairment in mice. Brain 135(Pt 9):2826–2837
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7):519–529
Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15(5):481–490
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001) GADD153 sensitizes cells to endoplasmic reticulum stress by down- regulating BCL2 and perturbing the cellular redox state. Mol Cell Biol 21(4):1249–1259
Shim S, Lee W, Chung H, Jung YK (2011) Amyloid β-induced FOXRED2 mediates neuronal cell death via inhibition of proteasome activity. Cell Mol Life Sci 68(12):2115–2127
Huang X, Chen Y, Zhang H, Ma Q, Zhang YW, Xu H (2012) Salubrinal attenuates β-amyloid-induced neuronal death and microglial activation by inhibition of the NF-κB pathway. Neurobiol Aging 33(5):e9–e17
Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E (2013) Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci 16(9):1299–1305
Michalak M, Robert Parker JM, Opas M (2002) Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 32(5–6):269–278
Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP (2000) Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem 275(24):18195–18200
Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 26(19):5180–5189
Supnet C, Grant J, Kong H, Westaway D, Mayne M (2006) Amyloid-β-(1-42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J Biol Chem 281(50):38440–38447
Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P (2011) The ER stress factor XBP1 prevents amyloid-β neurotoxicity. Hum Mol Genet 20(11):2144–2160
Demuro A, Parker I (2013) Cytotoxicity of intracellular Aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J Neurosci 33(9):3824–3833
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner B, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403(6765):98–103
Sanges D, Comitato A, Tammaro R, Marigo V (2006) Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci USA 103(46):17366–17371
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276(17):13935–13940
Song S, Lee H, Kam TI, Tai ML, Lee JY, Noh JY, Shim SM, Seo SJ, Kong YY, Nakagawa T, Chung CW, Choi DY, Oubrahim H, Jung YK (2008) E2-25K/Hip-2 regulates caspase-12 in ER stress-mediated Aβ neurotoxicity. J Cell Biol 182(4):675–684
Fischer H, Koenig U, Eckhart L, Tschachler E (2002) Human caspase 12 has acquired deleterious mutations. Biochem Biophy Res Comm 293(2):722–726
Nishitsuji K, Tomiyama T, Ishibashi K, Ito K, Teraoka R, Lambert MP, Klein WL, Mori H (2009) The E693Δ mutation in amyloid precursor protein increases intracellular accumulation of amyloid β oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am J Pathol 174(3):957–969
Matsuzaki S, Hiratsuka T, Kuwahara R, Katayama T, Tohyama M (2010) Caspase-4 is partially cleaved by calpain via the impairment of Ca2+ homeostasis under the ER stress. Neurochem Int 56(2):352–356
Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A (2010) Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66(5):739–754
Pavlov PF, Hansson Petersen C, Glaser E, Ankarcrona M (2009) Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med 13(10):4137–4145
Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA (2002) Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80(1):91–100
Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, Kraut N, Hengerer B (2007) Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 7(4):605–616
Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA (2005) Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci 25(3):672–679
Richards JG, Higgins GA, Ouagazzal AM, Ozmen L, Kew JN, Bohrmann B, Malherbe P, Brockhaus M, Loetscher H, Czech C, Huber G, Bluethmann H, Jacobsen H, Kemp JA (2003) PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci 23(26):8989–9003
Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT (2009) Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J 23(8):2459–2466
Perkins GA, Tjong J, Brown JM, Poquiz PH, Scott RT, Kolson DR, Ellisman MH, Spirou GA (2010) The micro-architecture of mitochondria at active zones: electron tomography reveals novel anchoring scaffolds and cristae structured for high-rate metabolism. J Neurosci 30(3):1015–1026
Cai Q, Sheng ZH (2009) Mitochondrial transport and docking in axons. Exp Neurol 218(2):257–267
Saxton WM, Hollenbeck PJ (2012) The axonal transport of mitochondria. J Cell Sci 125(Pt9):2095–2104
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci USA 107(43):18670–18675
Wang X, Perry G, Smith MA, Zhu X (2010) Amyloid-beta derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener Dis 7(1–3):56–59
Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA (2010) Amyloid-β peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3β in primary cultured hippocampal neurons. J Neurosci 30(27):9166–9171
Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S (2009) Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci USA 106(14):5907–5912
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29(28):9090–9103
Wang X, Su B, Fujioka H, Zhu X (2008) Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol 173(2):470–482
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324(5923):102–105
Park SJ, Shin JH, Jeong JI, Song JH, Jo YK, Kim ES, Lee EH, Hwang JJ, Lee EK, Chung SJ, Koh JY, Jo DG, Cho DH (2014) Down-regulation of mortalin exacerbates Aβ-mediated mitochondrial fragmentation and dysfunction. J Biol Chem 289(4):2195–2204
DuBoff B, Götz J, Feany MB (2012) Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75(4):618–632
Grundke-Iqbal I, Iqbal K, George L, Tung YC, Kim KS, Wisniewski HM (1989) Amyloid protein and neurofibrillary tangles coexist in the same neuron in Alzheimer disease. Proc Natl Acad Sci USA 86(8):2853–2857
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161(5):1869–1879
Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA (2002) Intraneuronal Abeta42 accumulation in down syndrome brain. Amyloid 9(2):88–102
Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA (2004) Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and down syndrome. Neurobiol Aging 25(10):1263–1272
Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306(1–2):116–120
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39(3):409–421
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26(40):10129–10140
Rutten BP, Van der Kolk NM, Schafer S, van Zandvoort MA, Bayer TA, Steinbusch HW, Schmitz C (2005) Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am J Pathol 167(1):161–173
Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM (2005) Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45(5):675–688
Koo EH, Squazzo SL (1994) Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 269(26):17386–17389
Matsuzaki K, Kato K, Yanagisawa K (2010) Abeta polymerization through interaction with membrane gangliosides. Biochim Biophys Acta 1801(8):868–877
Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G (2011) Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci 31(5):1644–1651
Williamson R, Usardi A, Hanger DP, Anderton BH (2008) Membrane-bound β-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J 22(5):1552–1559
Saavedra L, Mohamed A, Ma V, Kar S, de Chaves EP (2007) Internalization of beta-amyloid peptide by primary neurons in the absence of apolipoprotein E. J Biol Chem 282(49):35722–35732
Singh TD, Park SY, Bae JS, Yun Y, Bae YC, Park RW, Kim IS (2010) MEGF10 functions as a receptor for the uptake of amyloid-β. FEBS Lett 584(18):3936–3942
Fuentealba RA, Liu Q, Zhang J, Kanekiyo T, Hu X, Lee JM, LaDu MJ, Bu G (2010) Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS One 5(7):e11884
Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G (2006) Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem 281(47):36180–36186
Rushworth JV, Griffiths HH, Watt NT, Hooper NM (2013) Prion protein-mediated toxicity of amyloid-beta oligomers requires lipid rafts and the transmembrane LRP1. J Biol Chem 288(13):8935–8951
Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, McKhann G, Funatsu Y, Nakamichi N, Nagai T, Mizoguchi H, Ibi D, Hori O, Ogawa S, Stern DM, Yamada K, Yan SS (2009) RAGE-meidated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proc Natl Acad Sci USA 106(47):20021–20026
Lai AY, McLaurin J (2010) Mechanisms of amyloid-Beta Peptide uptake by neurons: the role of lipid rafts and lipid raft-associated proteins. Int J Alzheimers Dis 2011:548380
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE (2009) CD14 and Toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci 29(38):11982–11992
Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, Menger MD, Fassbender K (2012) TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol 188(3):1098–1107
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K (2007) Role of the Toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem 20(6):947–956
Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM (2006) Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem 281(6):3651–3659
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K (2006) Role of Toll-like receptor signalling in Aβ uptake and clearance. Brain 129(11):3006–3019
Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, Fukuchi K (2011) TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J Neuroinflammation 8:92
Richard KL, Filali M, Prefontaine P, Rivest S (2008) Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J Neurosci 28(22):5784–5793
Lopez EM, Bell KF, Ribeiro-da-Silva A, Cuello AC (2004) Early changes in neurons of the hippocampus and neocortex in transgenic rats expressing intracellular human a-beta. J Alzheimers Dis 6(4):421–431 discussion 443–429
Ditaranto K, Tekirian TL, Yang AJ (2001) Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiol Dis 8(1):19–31
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H (2004) ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304(5699):448–452
Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14(10):1097–1105
Almeida CG, Takahashi RH, Gouras GK (2006) β-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci 26(16):4277–4288
Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T (2005) Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. FASEB J 19(2):255–257
Acknowledgments
This work was support by the CRI Grant (NRF-2013R1A2A1A01016896) and the Open Research Program (KIST to YKJ, 2E24582-14-071) funded by the Ministry of Education, Science and Technology.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kam, TI., Gwon, Y. & Jung, YK. Amyloid beta receptors responsible for neurotoxicity and cellular defects in Alzheimer’s disease. Cell. Mol. Life Sci. 71, 4803–4813 (2014). https://doi.org/10.1007/s00018-014-1706-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1706-0