
Abstract
Epigenetic mechanisms play an important role in gene regulation during development. DNA methylation, which is probably the most important and best-studied epigenetic mechanism, can be abnormally regulated in common pathologies, but the origin of altered DNA methylation remains unknown. Recent research suggests that these epigenetic alterations could depend, at least in part, on genetic mutations or polymorphisms in DNA methyltransferases and certain genes encoding enzymes of the one-carbon metabolism pathway. Indeed, the de novo methyltransferase 3B (DNMT3B) has been recently found to be mutated in several types of cancer and in the immunodeficiency, centromeric region instability and facial anomalies syndrome (ICF), in which these mutations could be related to the loss of global DNA methylation. In addition, mutations in glycine-N-methyltransferase (GNMT) could be associated with a higher risk of hepatocellular carcinoma and liver disease due to an unbalanced S-adenosylmethionine (SAM)/S-adenosylhomocysteine (SAH) ratio, which leads to aberrant methylation reactions. Also, genetic variants of chromatin remodeling proteins and histone tail modifiers are involved in genetic disorders like α thalassemia X-linked mental retardation syndrome, CHARGE syndrome, Cockayne syndrome, Rett syndrome, systemic lupus erythematous, Rubinstein–Taybi syndrome, Coffin–Lowry syndrome, Sotos syndrome, and facioescapulohumeral syndrome, among others. Here, we review the potential genetic alterations with a possible role on epigenetic factors and discuss their contribution to human disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
After the elucidation of the genetic code in the late 1950s, it was considered that the information encoded by genetic material (DNA and mRNA nucleotide sequences) would be unchangeable and pass faithfully from one cell to another and from one generation to the next, except for the occurrence of genetic mutations. However, a few decades ago, a new code arose, namely the epigenetic code, which revealed that the message the nucleotide sequence of DNA harbors could be different depending on the epigenetic marks it contained. Thus, epigenetic marks can be defined as modifications affecting the DNA sequence or associated proteins, like histones, others than the DNA sequence, that are heritable through cell division. DNA methylation, histone modifications, non-coding RNAs and other forms of higher-order chromatin remodeling affect how tightly DNA is packaged in chromatin and, consequently, its accessibility to the transcriptional machinery [155].
The epigenome is stable, but also flexible, and can change rapidly during cell development and in response to environmental factors. Therefore, the epigenetic status of a gene is much more dynamic than its DNA nucleotide sequence [360]. Epigenetic marks are considered to be relatively stable during cell growth and development due to somatic maintenance (mitotic transmission), but during reprogramming events in the gametes and early embryos, there is a global erasure of epigenetic marks, which will be further re-established. Some loci can escape from this clearing and the epigenetic pattern will then be transmitted from one cell generation to the next, a process referred to as transgenerational inheritance [60].
Epigenetic changes are crucial for normal development, and disruptions of the epigenetic program can unchain pathogenic processes. In 1983, cancer was the first disease described to have altered epigenetic marks. Colorectal cancers exhibited a global loss of DNA methylation in comparison with their normal counterparts [91]. This hypomethylation resulted in genomic instability, chromosome rearrangements, and induced aberrant activation of certain genes [90]. Subsequent works found out that DNA hypermethylation of the promoter region of tumor suppressor genes was a frequent event in tumors [140]. Later, other epigenetic mechanisms displayed abnormal regulation, like histone modifications [98].
The exact mechanisms that lead to the aberrant epigenetic pattern in cancer remain still unknown. Interestingly, numerous single gene disorders of the epigenetic machinery mediate impaired gene expression, suggesting that mutations in genes coding for epigenetic enzymes might have a major role in the pathogenesis of cancer and other complex diseases. In support of this, epigenetics can elucidate the origin of some non-Mendelian inheritance diseases in which etiology goes further than genetic abnormalities [361]. Hence, both genome and epigenome can contribute to phenotypic variation and disease susceptibility, and the complex outcome of common and rare diseases may be compounded by both genetic and epigenetic anomalies as well as the interaction between them. Currently, analyses of genetic variants within genes with epigenetic function are being carried out in order to find evidence about the particular role of epigenetics in the etiology of a wide range of diseases. Also, genetic association studies represent a very useful tool to identify polymorphisms that show systematic variation among individuals who differ in phenotype and could represent the effects of risk-enhancing or protective alleles towards suffering a certain disease [14].
In this chapter, we will focus on epigenetic diseases associated with (i) the disruption of DNA methylation patterns by genetic defects in DNA methyltransferases, as in the case of the immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome and cancer; (ii) the inability to recognize methylated DNA because of altered functioning of MBD proteins, as is the case in Rett syndrome or systemic lupus erythematosus; (iii) genetic variants of one-carbon metabolism enzymes which coordinate the availability of the methyl group; (iv) other mutations that may have an impact on DNA methylation; (v) erroneous histone modifications originated by mutations in histone-modifying enzymes, which can also cause rare diseases, such as the Rubinstein-Taybi, Coffin-Lowry, and Sotos syndromes; and finally, (vi) the altered distribution and/or function of chromatin remodeling proteins, which might affect chromatin structure with the consequent deregulation of gene expression, as happens in α thalassemia X-linked mental retardation, CHARGE, Cockayne, facioescapulohumeral syndrome, and cancer (Table 1).
Genetic control of DNA methylation
Methylation of DNA at the carbon 5 of cytosine residues that precede guanines (usually referred to as CpG dinucleotides) is the most studied and best-known epigenetic modification in mammals and other vertebrates. In healthy cells, methylated cytosines represent 70–80 % of total cytosines, but they are not randomly distributed. Instead, there are CpG-rich and CpG-poor regions. Those areas of the genome with higher concentrations of CpGs are called CpG islands and are located at the promoter region of approximately 50 % of genes in human and also follow tissue- and cell-specific patterns [79]. In healthy cells, most of the CpG islands are unmethylated, whereas repetitive sequences, intergenic regions and the body of the genes are usually heavily methylated [85]. The methylation outside CpG islands is related to the chromosomal stability maintenance, translocation prevention, and silencing of endoparasitic inserted sequences [26]. In some cases, gene-promoter regions can also show dense methylation in normal physiological status. This would result in transcriptional silencing and could be the case of genes regulated by genomic imprinting, genes located in the X-chromosome in women, and tissue-specific repressed genes. DNA methylation patterns are established in the early stages of embryonic development and further maintained after each round of cell division by the DNA methyltransferases.
Alterations of the normal DNA methylation pattern are associated with human disease [286], like infertility [272], genetic syndromes, autoimmune disorders [226], cancer [90, 165], and aging [27] (Table 1). However, the molecular mechanisms involved in the establishment of aberrant DNA methylation are still poorly understood. Animal models like the agouti and axin-fused alleles in mice and the white gene in Drosophila melanogaster are good examples of epigenetic disturbance with a genetic basis. In addition, genetic defects in the enzymes that transfer methyl groups to DNA, or those that participate in one-carbon metabolism might be possible sources of abnormal DNA methylation, as discussed later.
Animal models of mutation-induced epigenetic changes
The term “metastable epialleles” has been coined to define those alleles that display phenotypic diversity in absence of genetic heterogeneity to distinguish them from “normal” alleles. Thus, the activity of these alleles could be attributed to their epigenetic status [273]. One example of a metastable epiallele is the viable yellow agouti (A vy) allele in murine models. The wild-type murine agouti gene encodes a protein that determines the relative amount of black (eumelanin) and yellow (phaeomelanin) pigment in hair. The transcription start site is located within exon 2, and the expression of the gene varies with development, resulting in a sub-apical yellow band on each black or brown hair, which is the typical brown agouti coat color of wild-type mice. The A vy allele results from the insertion of a contraoriented intracisternal A particle (IAP) retrotransposon upstream of the transcription start site of the agouti gene [74]. As a consequence, constitutive and ectopic expression of agouti is induced not only in hair follicles but in every cell. The murine phenotype of the A vy allele is characterized by yellow fur, obesity, diabetes, and higher susceptibility to tumors [372]. Interestingly, CpG methylation of the newly established A vy IAP promoter is inversely correlated with agouti expression and the degree of methylation causes variation in coat color that ranges from yellow (unmethylated) to pseudoagouti (methylated) among isogenic A vy/a mice. The density of methylated CpG depends on maternal nutrition and environmental exposures during early development [72]. Another metastable epiallele is the axin-fused (Axin Fu), identified in 1937 [278]. The role of the axin protein is to regulate embryonic axis formation in vertebrates [378]. Similarly to the A vy allele, the Axin Fu contains an IAP retrotransposon within intron 6 [343]. The typical phenotype of Axin Fu is the kinked tail, due to axial duplications during embryogenesis. However, the Axin Fu phenotype has variable expression, which ranges from normal to kinked tail and correlates with differential DNA methylation at the IAP [274] and exhibits epigenetic plasticity to maternal diet supplementation [359]. The epigenetic state of both the agouti and Axin Fu alleles can be inherited transgenerationally after parental transmission, maternal in the case of agouti, and both maternal and paternal in the case of axin-fused, resulting in the inheritance of the phenotype, probably due to an inefficient reprogramming of the epigenetic marks during gametogenesis [236, 274].
Also, changes in the activity of a gene or the distribution of its product within a tissue and/or a developmental stage could be due to position effects. H. Muller was the first to describe in 1930 the position-effect variegation in the white gene of Drosophila melanogaster [240], attributing the inactivation of the gene in some cells to its abnormal juxtaposition with heterochromatin. The white gene of Drosophila melanogaster, located at the distal end of the X chromosome and subjected to gene dosage compensation, is related to the pigmentation of Malpighian tubules, testes and eyes of the fruit fly, and expressed in a tissue- and time-specific manner. Chromosome rearrangements place the wild-type gene next to a block of centromeric heterochromatin in some cells, dramatically altering the expression of the gene which, in the case of the eye, gives rise to a variegated phenotype composed of patches from rearranged (colorless) and wild-type (red) alleles. Replacement of the white gene in different euchromatic sites within the genome reversed the effect, demonstrating that the colorless phenotype is due to a position effect and not to mutations within the gene [168].
Epigenetic changes induced by genetic alterations at DNA methyltransferases
DNA methylation is established by a family of proteins called DNA methyltransferases (DNMTs). These enzymes catalyze the addition of methyl groups to cytosine residues at CpG [285], using S-adenosylmethionine (SAM) as the methyl group donor. Three active DNMTs have been described in mammals: DNMT1, DNMT3A, and DNMT3B. DNMT1 exhibits great preference for hemimethylated DNA and, therefore, is responsible for maintaining methylation patterns after DNA replication [18, 24, 126, 271, 314, 346]. DNMT3A and DNMT3B are the de novo methyltransferases, because they can establish the DNA methylation pattern, although they show equal preference for unmethylated or hemimethylated DNA [197, 254]. A third homologue of this family, DNMT3L, lacks the methyltransferase capacity but it is known to assist the other members of the family in methylation reactions by interacting with their catalytic domains [125]. DNMT3L also interacts with histone deacetylases [1]. Each DNMT is essential for life, as homozygous knockout alleles of them have been identified. Both Dnmt1 and Dnmt3b cause embryonic lethality in mice and in the case of Dnmt3a mice die soon after birth [198, 253]. Experiments in deficient Dnmt3l mice have shown that heterozygous progeny of Dnmt3l knockout females exhibits loss of imprinting, stochastic imprinting, and biallelic expression of imprinted genes [31].
Genetic defects in DNMT3B: Immunodeficiency, centromeric instability, and facial anomalies syndrome
The immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome (OMIM #242860) is a rare autosomal recessive disorder that was first described in the 1970s [150, 331], and very few cases have been detected in the world. An ICF child is characterized by variable immunodeficiency, which results in recurrent infections, mild facial anomalies, and chromosomal abnormalities in the juxtapericentromeric heterocromatin of chromosomes 1, 9, and 16 [150, 331], and in the heterochromatic region of the Y and the inactive X chromosomes [136].
The ICF syndrome is, so far, the only disorder known to be caused by germline mutations in one of the genes that encodes for a DNMT gene, the de novo methyltransferase DNMT3B, as first described by Hansen et al. [137]. Nevertheless, two types of ICF syndrome have been described based on the presence or absence of mutations in the DNMT3B gene, namely ICF type 1, which corresponds to ICF patients who exhibit biallelic DNMT3B mutations [160], and ICF type 2, in which mutations in the exons of DNMT3B have not been detected, although most patients showed hypomethylation at the centromeric Satα tandem repeat [160]. Unless otherwise indicated, ICF will be used to denote the type 1 disease.
The typical symptoms of ICF children are probably due to DNA methylation defects and diverge in three aspects. Firstly, important decreases in serum immunoglobulins are seen in spite of the normal presence of B cells, which predispose to recurrent infections responsible for high mortality rates in early childhood [147] although the immunodeficiency can range from mild reduction of the immune response to severe agammaglobulinemia [132]. Secondly, facial anomalies, which are also variable, are not severe but frequent [132]. Typically, an ICF child has a broad flat nasal bridge, very widely spaced eyes, epicanthic folds, and low-set ears [310]. Less frequent but often related to the syndrome are the presence of a small jaw and macroglossia (protrusion and enlargement of the tongue). In addition to facial anomalies, 50 % of the affected fail to thrive properly and have lower weights at birth [132]. About one-third of the patients can also suffer from mental retardation or other neurologic defects affecting motor and cognitive function [311]. Finally, and used for diagnosis, lymphocytes of ICF patients exhibit hypomethylation of satellite DNA sequences (Sat2 and Sat3) in the juxtacentromeric heterochromatin of chromosomes 1, 9, and 16 [158, 334] because of defects in DNMT3B. Acrocentric NBL2 and subtelomeric D4Z4 tandem repeats are also less methylated but still appreciable in comparison with normal cells [80]. This hypomethylation is likely to originate characteristic rearrangements of the chromosomes mentioned above, including breaks, deletions of whole arms and multibranched chromosomes, and chromatin decondensation [78]. These cytogenetic features observed in ICF cells were very similar to the ones induced in normal cells treated with 5-azacytidine, which led researchers to combine efforts in the study of DNA methylation defects in ICF patients [302, 345]. More recently, it has been shown that in ICF-derived cells, the levels of telomeric repeat-containing RNA (TERRA), involved in the formation of telomeric heterochromatin [67], are abnormally elevated and telomeres are particularly shortened. This might be due to mutations in the DNMT3B gene and the consequent defects in DNA methylation [66].
Most of the reported patients with ICF syndrome exhibited heterozygous and varied mutations of the missense type within the part of the gene that contains the ten conserved motifs responsible for catalytic activity the DNMT3B [137, 366]. As a result, catalytic activity may be impaired but there is genetic heterogeneity about how this happens [362]. Occasionally, ICF patients have mutations located in the amino-terminal region of the catalytic domain [337, 365] that do not affect the capacity to transfer methyl groups but do instead affect the interaction with DNMT3L, which enhances DNMT3B for DNA methylation [320]. Other point mutations also affect the localization of the methylase enzyme [337], substrate binding, and oligomerization [232].
Several mice models were designed to better understand the ICF syndrome. A deletion of the catalytic domain of Dnmt3b (null mutation) resulted in embryonic lethality [337], similar to that observed in Dnmt3b-deficient mice [253]. However, other murine models carrying two human ICF-like missense mutations in the catalytic domain allow the embryos to develop to term. Post-natal mice showed an ICF-like phenotype, characterized by low body weight at birth, facial anomalies (shorter nose and wider nasal bridge), and hypomethylation of repeats [337]. These observations lead to the assumption that patients exhibit residual activity of this methyltransferase; otherwise, homozygous null DNMT3B mutations would have lead to early abortions.
To conclude, it appears that deregulation of DNMT3B induces hypomethylation of satellite DNA-rich regions located mainly in the juxtapericentromeric heterochromatin of certain chromosomes, which in turn alters gene expression due to abnormal scavenging of transcription factors, changes in nuclear architecture, and expression of non-coding RNAs, as proposed by several works [80, 106, 159]. Thus, further research needs to focus on the exact function of this region of heterochromatin and the target genes affected, which might be related to immune function, development, and neurogenesis [161] by altered transcription factor binding and other trans-acting effects. Deeper knowledge into the molecular physiopathology of murine ICF syndrome model will definitely shed light on this.
Genetic defects in DNMTs are associated with cancer
One of the first reports that establish a clear connection between DNA methylation and cancer was published by A. Feinberg and B. Vogelstein in 1983, who found extensive hypomethylation in certain genes of tumor samples in comparison with the corresponding normal tissue [91]. DNA hypomethylation affects mainly repetitive sequences dispersed within transposable elements, introns, and coding regions [85]. The functional meaning of this loss of DNA methylation results in chromosome instability and reactivation of transposable elements. When transposable elements are hypomethylated, they would become transcriptionally active and could mediate genomic rearrangements and act as alternative promoters altering the expression of genes implicated in the regulation of cell cycle, apoptosis, and DNA repair. Also, imprinted genes can be reactivated [86]. However, what has really shed light on epigenetic cancer hallmarks is the aberrant site-specific DNA hypermethylation. The de novo hypermethylation of CpG islands located within the promoter region of approximately half of the genes, which are usually unmethylated in normal tissues, results in transcriptional silencing of tumor suppressor genes (TSG), cell cycle control genes, apoptosis-related genes, and DNA repair genes [234], and this is probably a crucial step to consolidate tumor appearance and development. These epigenetic alterations could depend on genetic mutations or polymorphisms in DNMTs and certain genes encoding enzymes of the one-carbon metabolism pathway, altering their normal function.
DNMT genes are usually up-regulated in several types of cancer [162, 204, 261, 287, 303]. In particular, significant over-expression of DNMT3B is observed while over-expression of DNMT1 and DNMT3A is more modest [261, 287]. DNMT1 and DNMT3B seem to be directly implicated in the altered distribution of DNA methylation in cancer cells [20] and chromosomal instability [172]. In particular, lack of Dnmt3b blocks the transition from microadenoma to tumor in the murine Apc Min/+ colon cancer model, demonstrating the vital role of this DNMT in cancer progression [206].
DNMT3B plays an important role in the establishment of aberrant DNA methylation in malignant tumors because it contributes to gene promoter silencing through DNA hypermethylation of tumor suppressor genes and genes involved in cell cycle, apoptosis, and DNA repair. Also, methylation of CpG dinucleotides might facilitate the occurrence of C-to-T transition mutation in genes involved in carcinogenesis [123] and increase susceptibility to environmental carcinogens [68, 374].
The DNMT3B gene is located on chromosome 20q11.2. Its approximately 47 kb of the genomic DNA correspond to 23 exons and 22 introns. DNMT3B has approximately 345 single-nucleotide polymorphisms (SNPs) but only a few, of very low frequency, cause amino acid change [208]. SNPs are the most common form of human genetic variation, and they might increase the risk of an individual to develop a certain disease. Although the functional meaning of these SNPs is still a matter of debate, it has been hypothesized that some variants could influence DNMT3B activity and, consequently, DNA methylation, thereby modulating genetic susceptibility to cancer. Another possibility is that SNPs at the promoter region could alter transcription factors binding sites, which in turn will affect gene expression [368]. Indeed, only three SNPs in the promoter region of DNMT3B, −149 C > T (rs2424913), −283 T > C (rs6058870), and −579 G > T (rs1569686), have been studied as possible genetic susceptibility factors for several types of cancer. The first two SNPs appear to significantly increase the promoter activity of DNMT3B [192, 306], but in regard to the −579 G > T no such evidence has been reported [192]. Anyhow, these SNPs have been evaluated in different populations as risk factors that predispose to cancer.
For instance, carriers of the T allele at the 149 bp from the transcription start site were at significant risk of suffering from lung cancer in a non-Hispanic white population [306]. This −149 C > T polymorphism also increased significantly the genetic susceptibility to prostate cancer [309], head and neck squamous cell carcinoma [208, 355], and hereditary non-polyposis colorectal cancer [164]. Curiously, in the case of breast cancer, the very same T allele exhibited a significant protective effect [235]. However, other studies could not find a significant association between the presence of the −149 T genotype and colorectal cancer (Bao et al. [88], head and neck squamous cell carcinoma [44], hepatocarcinoma [87, 364], gastric cancer [10, 148], gastric cardiac adenocarcinoma [358], and acute leukemia [200].
A similar situation occurs with the −579 G > T polymorphism. Some studies have established a clear link between this SNP and some cancers [16, 88, 129, 143, 148, 192, 306] while others point out that genetic polymorphisms at the DNMT3B gene are not risk-enhancing factors [44, 88, 208, 316].
Little is known about the −283 T > C (from exon 1A transcription start site) SNP. So far, carriers of −283 T genotype were at decreased risk of lung cancer compared with individuals carrying the C allele in a Korean population, due to the fact that the SNP resulted in increased transcriptional activity of the DNMT3B gene [192].
Studies of multiple SNPs have been, in general, inconclusive [45, 371]. That said, SNPs may alter susceptibility to suffering from a certain type of cancer, although cumulative evidence suggests that many other factors have to be taken into consideration together with genetic variants like race, ethnicity, geographic areas, diet, environment factors, etc. The frequency distribution of the different polymorphisms of DNMT3B depends on the population considered. Moreover, SNPs in DNMT genes suffer the consequences of modifier genes that are central for their outcome (DNA methylation) associated to genetic DNMTs gene variants. In addition, different histopathological types of cancers may have different etiologies regarding genetic susceptibility [192]. Furthermore, in the case of DNMT3B, the alternative splice variants that may alter catalytic activity are expressed in a tissue-specific manner [253, 294, 354], so it is important to consider the complex interplay among DNMT3B splice variants and different types of tumors.
Another possible source of altered patterns of DNA methylation in cancer together with SNPs are truncated proteins. DNMT3B transcripts that originate from a promoter within intron 4 correlate with the promoter DNA methylation status of p16 and RASFF1A in non-small-cell lung cancer [354] and some of them encode truncated DNMT3B proteins that could interfere with the normal function of this DNMT [356]. Ostler et al. identified numerous aberrant DNMT3B transcripts in a diverse population of cancer cell lines and primary acute leukemia cells that encode truncated DNMT3B proteins lacking the C-terminal catalytic domain, and were catalytically inactive [255]. They further originated a stable transgenic cell line overexpressing DNMT3B7, the most common transcript overexpressed in the analyzed sample. Microarray expression revealed that the introduction of this aberrant splice variant induced changes in gene expression profile in genes usually located on chromosomes 1, 9, and X. These data led the researchers to conclude that truncated DNMT3B proteins play an important role in the abnormal distribution of DNA methylation in cancer cells. Further investigations need to be done in order to determine the molecular mechanisms, but three hypotheses were launched: (1) that the DNMT3B7-truncated protein may interfere with the normal DNA methylation machinery when binding any of the DNMT3B interacting molecules; (2) that DNMT3B7 could bind itself to DNA impeding adequate DNMTs to act; and (3) that due to altered DNA methylation, histone modifications could be consequently altered [256].
Variations in the DNA methylation profile are a common phenomenon among healthy individuals. These can be gender-related [82], age-related [100], or tissue-specific variations [77], and originated by environmental [96] or genetic factors. In relation to this, DNMTs are highly polymorphic genes. Indeed, in a combined study of polymorphisms in the coding region of DNMT genes and the levels of DNA methylation in a healthy population revealed the existence of 111 SNPs within DNMT genes. Of all genes considered, only one, the rare variant R277Q of DNMT3L, was associated with DNA hypomethylation. This SNP, always present in heterozygosis, leads to the production of a protein with reduced ability to stimulate DNA methylation due to defects that impede adequate DNMT3A-complex formation. The DNA hypomethylation did not correlate with a global loss of DNA methylation but to specific CpG islands repetitive in nature with telomeric location and away from genes (and promoters) or, to a lesser extent, within introns [83]. These data support previous hypotheses that suggest that DNMT3L is involved in the maintaining of the epigenetic identity of telomeres, which in turn constitutes an important factor that regulates telomere length [28]. Although at the time of the study the individual carrying the rare variant R277Q of DNMT3L was in a healthy state, he was young, and further analysis would be necessary to investigate the predisposition to disease at older age. No crucial SNPs affecting the rest of DNMT functions were found, probably because they lead to deleterious effects involved in abnormal development and serious disease, like the ICF syndrome in the case of DNMT3B.
Epigenetic changes induced by genetic alterations in MBD proteins
The information that DNA methylation contains is read and translated into a functional state in chromosomes by three distinct families of proteins: the MBD (methyl-CpG binding domain) family, the SRA family, and the Kaiso and Kaiso-like family [296]. The MBD family is highly conserved and characterized by the presence of an MBD that allows for the interaction with methylated DNA [138]. From all the members, methyl-CpG binding protein 2 (MeCP2) was the first one identified [244], but four more members were included in the family: MBD1, MBD2, MBD3, and MBD4 [138]. MBD proteins play mainly an active role in heterochromatin formation and transcriptional regulation, but recent research suggests that MeCP2 is an especially multifunctional protein involved in the pathogenesis of genetic disorders like Rett syndrome and autoimmume diseases like systemic lupus erythematosus.
Genetic defects in MeCP2 (I): Rett syndrome
The MeCP2 gene was first associated with gene silencing as its gene product binds to methylated DNA and recruits SinA3 (transcriptional repressor) and histone deacetylases (HDACs) [166, 245, 249], thus creating a repressive chromatin environment. The presence of (A/T)n sequences next to methylated cytosines, as well as methylation density and the length of the sequence, promotes high-affinity binding and confers some sequence specificity [180]. However, MeCP2 can also bind to unmethylated DNA (see [135] for a review) and recent findings suggest that it can also: function as a transcriptional enhancer due to its interaction with the transcription factor CREB at the promoters of active genes [41]; mediate RNA splicing by interactions with the RNA binding protein YB1 [375]; induce compaction of chromatin at unmethylated DNA regions [108]; and stabilize large chromatin loops [146]. For instance, there is a dynamic interplay between MeCP2 and linker histone H1 that modulates chromatin structure [113]. Through DNA binding, they both induce chromatin condensation by reduction of the linker DNA entry-exit angle. When expression levels of MeCP2 are lower than those of H1, MeCP2 would be expected to bind to specific sites, but when both genes are equally or similarly expressed, MeCP2 could be exerting both gene-specific and global functions related to chromatin binding [113]. Interestingly, Line 1 retrotranscription is enhanced by MeCP2 absence in murine models, suggesting a new mechanism by which loss of MeCP2 function alters gene expression and could lead to disease [241].
Genome-wide analyses show that MeCP2 has many target genes involved in brain development, like BDNF (brain-derived neurotrophic factor) [49], DLX5 (distal-less homeobox 5) [146], clusterin and cytochrome c oxidase subunit 1 [119], and protocadherins PCDHB1 and PCDH7 [231], among others. These genes must be carefully regulated to ensure the correct development of the neural system, reinforcing the cross-talk between DNA methylation, chromatin remodeling, and gene expression.
In 1999, mutations within the MeCP2 gene were found to originate Rett syndrome (OMIM #312750) [4]. Since then, countless genetic defects in MeCP2 are unequivocally associated with this syndrome that afflicts predominately females. Rett syndrome is an X-linked neurodevelopmental disorder whose symptoms become evident after an uneventful pregnancy and delivery, and normal development until 6–18 months of age, when patients start showing a progressive neurological dysfunction probably due to an impairment of neuronal development and a reduction in brain size [119]. The regression process involves clinical manifestations like mild learning disabilities, autistic behavior, stereotype hand movements, and encephalopathy. Also, severe mental retardation and motor impairments affecting the respiratory tract are commonly seen in Rett syndrome patients [42].
MeCP2 mutations are detected in both sporadic and familial cases of Rett syndrome, the most pathogenic being those affecting the MBD and TRD (transcriptional repression domain) domains [95]. Even though, MeCP2 mutations causing Rett syndrome occur within the whole sequence and are of varied nature: point mutations, missense mutations, frameshift mutations in the C-terminal, etc. Among males, MeCP2 duplication is the most common MeCP2 dysfunction, which is also related with neurological defects and has defined a new syndrome, the MeCP2 duplication syndrome [275]. Often, male individuals carrying mutations in MeCP2 have neurodevelopmental delay, which does not necessarily frame within Rett syndrome. Nonetheless, MeCP2 mutations have different impacts in protein function, depending on where the mutation lies. Precisely, the large phenotypic variability observed in Rett syndrome patients might be due to the specific mutations together with the skewing of the normally expected X chromosome inactivation, which is usually random and balanced [112].
MeCP2, located on chromosome Xq28, is ubiquitously expressed [195], but particularly high levels of the protein are found in the brain, in a time-specific manner depending on neuronal maturation and synaptogenesis [55]. Specific loss of MeCP2 in central nervous system causes Rett syndrome and other autistic-like behaviors, but its overexpression results in profound motor dysfunction [213], progressive neurological disorders and premature death [56, 307, 338], suggesting that MeCP2 must be minutely regulated in order to maintain correct brain functioning. Mice deficient in MeCP2 exhibit a Rett-like phenotype that mimics the human syndrome, a phenotype which can be reverted after restoring MeCP2 expression [130].
Genetic defects in MeCP2 (II): lupus erythematosus
Systemic lupus erythematosus (SLE, OMIM #152700) is an autoimmune and inflammatory disease characterized by the production of autoantibodies against multiple nuclear antigens that affects numerous organs and is associated with high rates of morbidity and mortality. It affects predominantly females [266].
The etiology of SLE remains still elusive. Cumulative evidence suggests that the etiology of SLE is quite complex, and involves not only genetic and environmental factors but also the interaction among them [15, 124, 352, 377]. Association studies in monozygotic twins suffering from SLE showed disease discordance rate and point to epigenetic factors as possible contributors [157]. Indeed, strong evidence supports an important role for aberrant T cell DNA methylation [298], which results in the elevated expression of methylation-sensitive genes like ITGAL (CD11a), TNFSF7 (CD70), PRF1 (perforin), and TNFSF5 (CD40LG) [300]. Similarly, the use of demethylating drugs like 5-azacytidine induces genome-wide hypomethylation and aberrant expression of many genes [282], and also increases autoreactivity in CD4 + cells [280]. Interestingly, when these cells were transferred to mice, they induced an SLE-like disease in mice [376].
In SLE T cells, DNMT1 activity is decreased [65], contributing to the hypomethylation of the aforementioned genes. However, so far, no significant association has been found between SNPs in the DNMT1 gene and susceptibility to SLE [260]. In relation to this, to maintain adequate DNA methylation levels, DNMT1 needs the cooperation of MeCP2 [177]. MeCP2 exerts a vital role in epigenetic transcriptional regulation of methylation-sensitive genes by binding to methylated DNA and recruiting HDACs. As a result, chromatin acquires a configurationally closed state and becomes inaccessible to the transcription machinery [166]. SNPs within the MeCP2 gene showed a strong association with SLE [299], reinforcing the importance this gene plays in this disease. The role of MeCP2 in SLE supports previous data suggesting that an X-linked gene is involved in the pathogenesis. For instance, males with Klinefelter’s syndrome (47 XXY) are at similar risk of suffering from SLE in comparison with females [305].
In addition to this, epigenetics is fundamental in the autoreactivity of SLE CD4+ T cells. The Regulatory Factor X-box 1 (RFX1) binds specifically to regulatory regions in healthy CD4+ T cells, and acts as a transcriptional co-repressor by recruiting SUV39H1, HDAC1, and DNMT1 [383]. Thus, H3K9 tri-methylation, histone hypoacetylation and DNA hypermethylation cooperate to silence RFX1 target genes among which we can find the adhesion molecule lymphocyte function-associated antigen 1 (LFA-1), integrated by CD11a, CD18, and CD70 [251, 281]. The typical downregulation of RFX1 in SLE CD4+ T cells downregulates in turn the aforementioned epigenetic marks, leading to the overexpression of CD11a and CD70, triggering autoimmune responses [383].
Mainly, however, changes in the DNA methylation profile may be associated with different SLE onsets, as reported by Javierre et al. in discordant monozygous twins for SLE [157]. Notably, they provide a list of epigenetically deregulated genes which are relevant in autoimmune inflammatory diseases (cell activation, immune response, cell proliferation, cytokine production), some of which were previously described as altered genes in SLE. Further investigations into these methylation-sensitive genes will probably improve understanding of SLE and other autoimmune diseases.
Epigenetic changes induced by genetic alterations at the one-carbon metabolism enzymes
Not only anomalies in DNMTs and associated proteins, like MBD proteins, represent a possible source of aberrant DNA methylation patterns in human disease, but also anomalies present in the enzymes of the one-carbon metabolism, which regulate SAM supply within cells.
SAM is the main methyl group donor at cellular level and SAM-binding proteins have varied functions, notably those that methylate DNA, RNA, histones, and other proteins and small molecules [209], which may possibly have an important impact on epigenetic regulation. Apart from this, SAM plays a significant role in the regulation of hepatocyte growth, death and differentiation. SAM reactions, included in one-carbon metabolism, are coupled to polyamine synthesis and to the folate cycle. SAM is also a precursor of important metabolites like glutathione, through transsulfuration reactions (Fig. 1). Chronic liver injury results in decreased levels of hepatic SAM, whereas excess of SAM induces liver damage and increase predisposition to steatosis and hepatocellular carcinoma (HCC) [222]. Therefore, it is crucial to maintain SAM levels in liver within a tight range. Methionine adenosyl-transferases (MATs) and glycine-N-methyltransferase (GNMT) are the main enzymes responsible for SAM synthesis and catabolism, respectively [182, 322], and alterations in any of them are related to liver pathologies.

Schematic illustration of the one-carbon metabolism. Highlighted by the grey rectangle is the S-adenosylmethionine cycle, which is coupled to polyamine synthesis, transmethylation and transsulfuration pathways, and folate cycle. MAT deficiency leads to hypermethioninemia and SAM deficiency, causing neurological symptoms and liver injury. GNMT deficiency in humans can be asymptomatic, although some cases have been reported to result in hepatomegalia. MAT methionine adenosyltransferase, SAM S-adenosylmethionine, GNMT glycine N-methyltransferase, SAH S-adenosylhomocysteine, MT methyl transferases, THF tetrahydrofolate, 5′,10′-MTHF 5′,10′-methylene tetrahydrofolate, 5′-MTHF 5′-methyltetrahydrofolate, MTHFR 5′,10′-methylene tetrahydrofolate reductase, MTHFS 5′,10′-methylene tetrahydrofolate synthase, MS methionine synthase, R methyl group acceptors
Genetic variants in MATs lead to SAM deficiency
SAM synthesis occurs in all mammalian cells, but mainly in liver [222]. SAM is originated from methionine and ATP in a reaction catalyzed by MATs. In mammals, MATs are codified by two different genes, MAT1A and MAT2A, whose gene products are MATI and MATIII, and MATII, respectively [221]. MAT1A is primarily expressed in liver and encodes for the α1 subunit found in both native MATI and MATIII. In contrast, MAT2A has a more ubiquitous expression and encodes the α2 subunit found in native MATII [182]. MAT2A is very abundant in fetal liver but, during development, is progressively replaced by MAT1A [120, 145]. MAT1A expression becomes reduced in liver diseases and is almost absent in HCC, while MAT2A is transcriptionally active in HCC [36]; thus, it might facilitate liver cancer cell growth [35]. SAM can influence MAT2A expression: at low levels of SAM, MAT2A expression is induced, inversely, an increase of SAM presence correlates with MAT2A downregulation. However, such effect has not been seen into the case of MAT1A [218, 369].
The most common cause of inherited hypermethioninemia is MAT deficiency (Fig. 1) [58, 107, 176]. Depending on the mutation, the inheritance can be autosomal recessive or dominant. Around 30 different mutations have been described in the MAT1A gene causing MAT deficiency, being R264H the most frequent and of dominant inheritance [17]. MAT deficiency is benign in most cases, but on some occasions symptoms of neurological nature can be seen [17].
Disruption of MAT1A in mice produced high levels of plasma methionine and SAM deficiency in liver, which increase susceptibility of liver to oxidant-cell death, predispose liver to further injury and spontaneous HCC and to impaired liver regeneration, because of an inability to upregulate cyclin D1 [47, 210, 217]. Recently, it was found that forced expression of MAT1A in cells derived from human liver cancer reduced tumorigenesis in vivo, supporting the role of MAT1A deficiency in cancer progression [199]. In addition, MAT1A is necessary to correct VLDL (very low density lipoprotein) assembly and plasma lipid homeostasis in mice, and MAT1A knockout mice model develops non-alcoholic fatty liver disease, which could be mediated, at least in part, by SAM deficiency [37].
Genetic variants in GNMT increase susceptibility of liver disease
SAM transfers the methyl group to a varied range of acceptors, thanks to the action of methyltransferases, and the common product of these reactions is S-adenosylhomocysteine (SAH). Most of SAM-dependent methylation reactions are inhibited by increases in SAH and decreases in SAM [220]. GNMT is considered of particular importance because GNMT deficiency appears to be the only methyltransferase deficiency that causes SAM accumulation [17]. GNMT is the most abundant methyltransferase in liver, but it is also present in prostate and pancreas [252] and it optimizes methylation reactions by maintaining a constant SAM/SAH ratio, which is an indicator of the methylator potential in cells [174]. It is also a major folate-binding protein [57]. Interestingly, a specific form of folate, 5-methyltetrahydrofolate pentaglutamate, which binds to GNMT in vivo, was found to inhibit GNMT activity [351, 373], suggesting that GNMT is an important regulator of the one-carbon metabolism [13]. Indeed, by modifying tissue folate status, GNMT could promote chromosome breakage or abnormal DNA methylation [75]. In addition, it participates in detoxification pathways and might have protective effects against carcinogens exposure by reducing DNA adducts formation [48].
Mutations in the GNMT gene, mapped to chromosome 6p12 [50], have been reported in humans, resulting in GNMT deficiency. Two Italian siblings, who displayed mild hepatomegalia and increased presence of serum transaminases, were diagnosed with GNMT deficiency [238]. Later, Luka et al. demonstrated that both individuals were compound heterozygotes and carried the same two missense mutations within the GNMT gene that affect enzymatic activity [215]. Another patient, reported by Augoustides-Savvopoulou [9] was asymptomatic but, as with the other two patients, he had increased levels of methionine and SAM in plasma and normal SAH and sarcosine [9, 238].
Although GNMT deficiency in humans appears to be a benign disorder, lack of GNMT in mice models leads to different conclusions. Two GNMT knockout mice models have been reported in the literature and phenotypic differences may arise as a consequence of the knock-out strategy [51]. One of them developed chronic liver hepatitis and HCC spontaneously, and glycogen storage disease [203, 207]. 2-D PAGE and real-time experiments revealed that absence of GNMT led to the downregulation of proteins involved in antioxidant/detoxification response, the glycolytic energy metabolisms and the one-carbon metabolisms pathways [202]. As for the other model, it was initially found to be normal [214] but later it was discovered that the lack of GNMT resulted in liver steatosis, fibrosis, and HCC [219], and liver regeneration impairment [341]. In this case, GNMT seemed to modulate DNA and histones methylation of critical carcinogenic pathways, like the JAK/STAT pathway, resulting in epigenetic alterations in HCC [219].
In relation to this, loss of heterozygosity has been reported in approximately 40 % of human HCC. Hence, GNMT has been proposed to be a tumor-susceptibility gene because its expression is diminished in tumorous tissues and in HCC cell lines [333]; it is also downregulated in liver of patients at risk of developing HCC like those with hepatitis C viruses and alcohol-induced cirrhosis [11]. In addition, it is able to bind to benzo[a]pyrene (BaP), reducing the BaP-DNA adducts formation [48].
Thus, depending on the defect that affects SAM levels, treatment with SAM could be considered. For instance, SAM supplementation could be helpful when the liver disease is associated with MATs impairments, accompanied by methionine restriction. However, when the defects affect for instance GNMT, SAM administration is probably useless and might even become toxic as it accumulates [17]. In the latter case, nicotinamide, substrate of nicotinamide N-methyltransferase (present in liver), treatment can prevent SAM accumulation depending on GNMT deficiency and normalize the expression of genes under GNMT regulation involved in diverse pathways that could lead to a pathogenic phenotype [342]. In conclusion, one-carbon metabolism genes need to be carefully regulated and early detection of genetic defects in these enzymes that regulate SAM levels will contribute to the development of adequate treatments and preserve human health.
Epigenetic changes induced by nucleotide expansion and position effect
CGG trinucleotide expansion affects DNA methylation: Fragile X syndrome
The fragile X syndrome (FXS, OMIM #300624) is the most common heritable form of impaired intellectual ability worldwide (Jacquemont 2007). FXS is an X-linked neurodevelopmental disorder of dominant inheritance characterized by cognitive and behavioral difficulties and facial dysmorphisms (elongated face, large and protruding ears), which can manifest in mild to severe forms. Affected females exhibit symptoms but usually to a lesser extent, due to the presence of a normal allele [103].
Cytogenetic analysis revealed that FXS patients have an expansion of a single trinucleotide sequence (CGG) at the promoter of the fragile X mental retardation gene (FMR1), mapped at Xq27.3. Healthy individuals carrying between 5 and 44 CGG repeats, while affected patients with full mutations carry more than 200 repeats. A premutational state has been attributed to carriers of between 55 and 200 repeats and grey zone denotes expansions between 45 and 54 repeats. The larger the number of repeats, the more unstable the alleles are. The length of the expansion also determines the risk of inheriting the full mutation allele [131]. Full mutation and premutation alleles can coexist within an individual, giving rise to a FXS mosaicism, which may make the diagnosis difficult [229].
The expansion of CGG repeats results in the methylation of the affected DNA, which leads in the full mutation alleles to the epigenetic silencing of the FMR1 and the lack of its product, the fragile X mental retardation protein (FMRP) [103]. FMRP is ubiquitously expressed and especially abundant in neurons, where it regulates synaptic plasticity and neuron maturation. FMRP has RNA-binding properties and regulates multiple processes related to RNA metabolism, especially the transport and localization of mRNA to dendrites and synapses [186]. The loss of this protein causes defects in neuronal morphology and physiology, giving rise to the neurodevelopmental delay typical of FXS patients.
Position effects affect DNA methylation: Facioscapulohumeral muscular dystrophy
Facioscapulohumeral muscular dystrophy (FSHD, OMIN #158900) is an autosomal dominant myopathy characterized by progressive, commonly asymmetric, weakness and wasting of the facial, shoulder and upper arm muscles [325]. FHSD patients can also exhibit extramuscular features like hearing loss, retinopathy, mental retardation, and epileptic seizures [326]. Mounting evidence suggests that FSHD is not caused by defects in a single gene; instead, it appears that deregulation of epigenetic mechanisms results in aberrant transcription of multiple disease-related genes.
Most FSHD patients are linked to molecular rearrangements in the subtelomeric region of chromosome 4 long arm (4q35) which maps to a 3.3-kb tandem-repeated macrosatellite, D4Z4 [339]. Healthy individuals carry between 11 and 100 repeats, whereas FSHD patients have a reduced number of copies of D4Z4 that range from 1 to 10. Although the phenotypic variability of FSHD is remarkable, there seems to be a correlation among the number of residual D4Z4 repeats, DNA hypomethylation, and the age of onset and the severity of the disease, being larger deletions related to an earlier onset of the disease and a more rapid progression [340]. Interestingly, the complete deletion of D4Z4 repeat and 200 kb of nearby DNA is not associated to FSHD [335], suggesting that a reduced number of D4Z4 copies implies a gain-of-function mutation probably of the surrounding of the repeat array [33].
D4Z4 is highly methylated in healthy subjects but, interestingly, all FSHD patients show DNA hypomethylation in that region. Sometimes, the DNA hypomethylation correlates with the contraction of the affected area. This fact suggests that the genetic defects observed in FSHD can be dependent (FSHD1 type 1) or independent (FSHD type 2) of D4Z4 deletions [62]. From now on, unless otherwise noted, we will be referring to the FSHD type 1 as FSHD.
On chromosome 10q26, there is a D4Z4 repeat array that shares a 98 % identity with the one on chromosome 4q35 [34]. However, it appears that the short D4Z4 array is not itself responsible of FSHD because almost identical arrays on 10q26 have non-pathogenic effects when they are equally short [194]. In addition, asymptomatic carriers are also associated with reduced D4Z4 DNA methylation in the contracted locus [62]. These observations indicate that D4Z4 hypomethylation is not sufficient to cause FSHD, but that could be altering the expression of nearby genes. Indeed, FSHD patients are characterized by overexpression of FSHD region gene 1 (FRG1), FSHD region gene 2 (FRG2), ANT1, and DUX4, which are located proximal to D4Z4 [102]. Thus, it seems that the role of D4Z4 is to suppress the expression of nearby genes, and the D4Z4 hypomethylation dependent or independent of D4Z4 deletions, results in the up-regulation of candidate genes. Also, several epigenetic mechanisms appear to be involved in the progression of FSHD.
In FSHD, apart from the loss of DNA methylation, histone modifications of D4Z4 on 4q35 in FSHD patients remind of facultative heterochromatin or silent euchromatin more than constitutive heterochromatin itself [381]. D4Z4 has normally both H3K9me3 and H3K27me3 as transcription repressive marks, which are lost in FSHD patients. The loss of H3K9me3 prevents the binding of D4Z4 to the heterochromatin-binding protein HP1γ and the sister chromatoid complex, cohesin, which contributes to the loss of gene repression [379]. Furthermore, D4Z4 and the promoter of the FRG1 are bound by transcription factor YY1, which in turn recruits EZH2 [29]. The latter is a H3K27 methyltransferase that belongs to the Polycomb group proteins involved in transcriptional silencing of higher eukaryotes [308]. As a consequence to the reduced binding of YY1 and EZH2 due to the D4Z4 contraction, the repressive H3K27me3 mark is also depleted [29], which in turns contributes to the loss of repression of genes usually silenced by D4Z4 in healthy individuals.
In addition, Gabellini and colleagues found a 27-bp sequence in each D4Z4 unit that specifically binds to a repressor complex integrated by YY1, HMGB2, and nucleolin [102]. This complex usually binds to D4Z4 mediating transcriptional repression of 4q35 genes, apart from interacting with DNMTs, HDACs, and HP1, which participate in heterochromatin formation and gene silencing. When there is a reduction in the number of D4Z4 repeats, the complex cannot bind and an open state of chromatin is induced, facilitating overexpression of target genes. Moreover, there is a nuclear matrix attachment site in the vicinity of D4Z4, which participates in the organization of DNA loop domains. This matrix attachment is much weaker in cells from FSHD patients than in healthy individuals, altering chromatin structure. The repeat and upstream genes reside in two loops in healthy myoblasts but only in one loop in FSHD myoblasts, enhancing cis-regulation of affected genes [270].
More recently, the group of Gabellini provided a new epigenetic mechanism that contributes to gene silencing. In the healthy population, the Polycomb group targets D4Z4 to repress gene expression. In FSHD patients, the deletion of D4Z4 is associated with reduced Polycomb silencing. In particular, the non-coding RNA DBE-T, which is selectively produced in individuals affected with FSHD, recruits Ash1L to the FSHD locus, remodeling chromatin structure and leading, ultimately, to active transcription of the surrounding genes [32].
Taking the above into consideration, FSHD could thereby arise as a toxic gain-of-function of some genes due to the altered chromatin structure. From candidate genes, researchers first focused on FRG1 because it is located with the D4Z4 repeat array on 4q35, it is overexpressed in FSHD patients [102], and only its induced expression in mice gives rise to a phenotype that resemble human FSHD, which did not appear when other possible candidate genes like FRG2 and ANT1 were induced [101]. The exact function of FRG1 has not been yet elucidated. Hanel et al. recently demonstrated that this protein is a developmentally regulated sarcomeric protein responsible for musculature and vasculature damage in FSHD [133] and this function could be attributable to its involvement in multiple aspects of RNA biogenesis [321]. RNA interference of FRG1 corrects myopathic features in mice expressing toxic levels of FRG1, a promising remedy against dominantly inherited myopathies like FSHD [353]. Thus, the severity of the disease could be mediated, at least in part, by FRG1 gene expression.
Other lines of research have joined efforts on the study of the retrotransposed gene encoding DUX4 (a putative double homeodomain gene) present in each subunit of D4Z4. The DUX4 gene was considered as functionally inactive because it lacks introns and polyadenylation signals and there was no evidence for active in vivo transcription [71]. Interestingly, the open reading frame and its organization within the array are highly conserved. Expression studies detected both DUX4 mRNA and protein in FSHD-derived primary myoblasts but not in healthy myoblasts, suggesting that D4Z4 could influence disease progression by aberrant production of DUX4 [71]. The D4Z4 repeat does not contain a polyadenylation signal and the DUX4 mRNA is generated exclusively by transcription of the most distal region of the array, a pLAM sequence containing itself a polyadenylation signal [71]. The pLAM polyadenylation signal can stabilize DUX4 transcripts and, precisely, a group of FSHD patients were found to carry the same SNP in the pLAM sequence, allowing expression of the DUX4 gene [193]. The DUX4 gene codes for two alternative splice variants. Healthy individuals typically express low levels of a short DUX4 variant in muscles, which encodes a truncated protein, while a full-length DUX4 isoform was found in FSHD cells [313]. The full-length DUX4 mRNA is only expressed in certain developmental stages and suppressed in most somatic tissues [313]. Although little is known about DUX4 function, transfection experiments suggest that it may have pro-apoptotic effects and might affect myogenesis [317].
Epigenetic defects in a specific genetic background trigger FSHD in humans. Although some of the genes mentioned above could create a permissive background for FSHD, a new case of FSHD carrying a D4Z4 deletion in chromosome 10 was recently reported by Lemmers and colleagues [193], pointing out that although FRG1 and DUX4 can contribute to FSHD progression, FSHD-related genes remain currently unknown.
Genetic control of histone modifications
Another important epigenetic factor is histone modifications. Two copies of each of the core histones (H2A, H2B, H3, and H4), around which approximately 146 bp of DNA wrap, constitute the nucleosome, which is the structural subunit of chromatin. Histones not only exert a structural function but also affect chromatin function. Histone tails (the N-terminal region of histone proteins) protrude from the nucleosome and are susceptible of posttranscriptional modifications at different residues, such as acetylation, methylation, phosphorylation, poly-ADP ribosylation, ubiquitination, and glycosylation [184]. The presence of these chemical groups in histone tails severely influences the histones–DNA interactions and also the interaction of non-histonic proteins with chromatin, giving rise to what it has been define as the “histone code” [336] in an attempt to reflect the combination of histone modifications at a certain region of chromatin. As we know, chromatin structure affects gene expression and as it is affected by histones modifications, it is clear that gene expression can be altered by histones modifications.
Modification of histone tails is part of a dynamic and reversible process. Usually, each modification is carried out by a group of enzymes that add the chemical group to the specific residue of the histone tail and removed by an antagonistic group of enzymes. Histone acetylation is the most studied among all histone modifications. Histone acetyltransferases (HATs) are the enzymes that catalyze the addition of acetyl groups using acetyl-coenzyme A as donor. The acetylation occurs mainly in lysine residues of H3 and H4. As a consequence of the acetyl group addition, lysine loses a positive charge and, therefore, DNA will be less attracted due to its negative nature. As is the case of other epigenetic features, histone acetylation can be reverted by HDACs. Residues of lysine (K4, −9, −27, −36 and −79 for H3 and K20 for H4; can also be monomethylated, dimethylated, or even trimethylated) and arginine can become methylated. The methyl group in histone tails is incorporated by histone methyltransferases and can be removed by histone demethylases. In this case, the effect depends on both the modified residue and the extent of methylation. For instance, methylation of histone H3 on lysines 4 and 36 (H3K4 and H3K36) is related to an open chromatin structure and, thus, an active transcriptional state, whereas the contrary occurs in the case of H3K9 and H3K27 methylation [184].
The high number of possible sites for modifications and the varied source of enzymes reveal a complex regulation system which, when altered, can contribute significantly to the onset of numerous diseases (Table 1).
Genetic defects in CBP and EP300: Rubinstein-Taybi syndrome
Rubinstein-Taybi syndrome (RSTS, OMIM #180849) is an autosomal dominant disease with a heterogeneous genetic origin. Affected individuals are characterized by growth and psychomotor development delay, unusual facies including down-slanting palpebral fissures, broad nasal bridge, beaked nose and micrognathia, and skeletal abnormalities in extremities, like radially diverted phalanges and broad and duplicated distal phalanges of thumbs and halluces [290]. Other symptoms, like talon cusps, congenital heart defects, and skin problems can be displayed in RSTS [139]. An increased susceptibility to tumors is also a common feature of RSTS patients [230].
Cytogenetic analyses revealed that de novo mutations in two genes, the cAMP response element-binding protein (CREBBP or CBP), located at 16p13.3 [268], and the E1A-associated protein p300 (EP300), located at 22q13 [289], account for about half of RSTS cases. The mutational spectrum is wide, frameshift, nonsense, splice site and missense mutations often occur in the CBP gene [288], but large deletions, inversions, and translocations have been also reported, although less frequently [267]. Mutations in EP300 are considered rare because of their low incidence [384]. All mutations are heterozygous and even mosaic mutations are found in the literature [110].
Both gene products enhance transcription in numerous processes [121] by recruitment of RNA polymerase II complex to promoter regions and the intrinsic histone acetylation activity they have [43], and although they may overlap in some functions, there is clear evidence that the proteins are different [169]. However, out of all the possible mutations, those affecting the HAT domain of CBP are sufficient to cause RSTS [170, 242], suggesting that chromatin remodeling is a key factor in the development of this disorder and that CBP and EP300, together with other possible candidate genes, may converge in molecular pathways. Homozygous mice deficient in Cbp or Ep300 result in embryonic lethality, as well as the heterozygous double knockout, revealing the fact both genes are needed to achieve normal development [370].
Because of its diverse genetic nature, approximately only 50 % of the patients who suffer from RSTS have a correctly identified genetic lesion. Hence, current studies aim to identify new genes related with RSTS or RSTS-like features [111].
Genetic defects in RSK2: Coffin-Lowry syndrome
Coffin-Lowry syndrome (CLS, OMIM #303600) is a rare form of mental retardation with X-linked inheritance. Affected males show growth and psychomotor developmental delay, peculiar facial characteristics like widely spaced and down-slanting eyes, prominent jaw and thick lips, microcephaly, short stature, and skeletal deformities, like hands and feet with tapering digits [375], as cardinal features of the syndrome. Female carriers are at risk of suffering from impaired learning abilities and physical anomalies.
CLS originates in loss-of-function mutations in the ribosomal protein S6 kinase 2 (RSK2), a serine/threonine kinase which maps to Xp22.2 [332] and acts in the mitogen-activated protein (MAP) kinase pathway, together with the other three members of the RSK family: RSK1, RSK3, and RSK4. RSK proteins are activated by extracellular signal-regulated kinase 1 and 2 (ERK1 and ERK2) in response to a wide range of signals, like stimulation with insulin, growth factors, neurotransmitters, UV-irradiation and malignant transformation, and participate in important cellular routes like proliferation, differentiation, apoptosis, and response to stress by phosphorylation in multiple substrates [154]. Mutations in RSK2 are extremely heterogeneous (missense, nonsense, frameshift, spliced site, short deletions and inversion, and large deletions) [154]. Neurologic symptoms of CLS might be due to reduced or even absent activity of RSK2 that impedes correct hormone release in neurocrine cells [380], provokes dysregulation of neurite growth [93], perturbs the differentiation of neural precursors into neurons [73] and induces AMPA dysfunction of the AMPA glutamate receptor, which mediates fast excitatory synaptic transmission in the central nervous system [227]. By contrast, RSK2 is often upregulated in tumors [52, 54, 171], suggesting that this protein needs to be properly regulated to prevent disease.
The epigenetic nature of this syndrome arises from the fact that RSK2 affects chromatin structure by two independent actions: the direct phosphorylation of histone H3 and the interaction with the CBP, affecting gene expression [61, 228]. In addition, RSK2 can phosphorylate other proteins, so the number of genes under RSK2 regulation could be higher than expected and further research is required to identify them and its role in CLS.
Genetic defects in NSD1: Sotos syndrome
Sotos syndrome (OMIM #117550) is an overgrowth condition in children, firstly described as “cerebral gigantism” by Sotos et al. in 1964 [315]. It was not until 2002 when Kurotaki and colleagues found the main cause of Sotos syndrome, which is mutations and deletions in the NSD1 (nuclear receptor SET domain containing gene 1) gene [188], which belongs to a family of mammalian histone lysine methyltransferases relevant in multiple aspects of development.
Children affected with Sotos syndrome have a large body size and early accelerated growth, advanced bone age, facial gestalt (i.e., macrocephaly, prominent jaw, down-slanting palpebral fissures, high hairline with sparse hair growth) and developmental delay, usually accompanied by learning difficulties. There are also many other features associated with the syndrome, such as neonatal jaundice, hypotonia, seizures, scoliosis, cardiac defects, and genitourinary anomalies. Growth in height and weight tends to normalize at puberty, probably due to the epiphysal fusion and adults are close to normal average height and weight (see [19] for revision). Sotos syndrome, like other overgrowth conditions, is typically associated with higher frequency of neoplasms than that of healthy individuals, recently estimated about 3 % and some patients are prone to develop neural crest tumors, sacrococcygeal teratomas and hematological malignances [324].
The NSD1 gene, located at 5q35, encodes for a histone lysine methyltransferase with multiple functional domains, to highlight the SET (Su[VAR]3-9, enhancer-of-zeste, thritorax) domain, the SAC (SET-associated Cys-rich) domain, five PHD domains and two PWWP domains, all of them related to chromatin regulation, and two nuclear receptor interaction domains, NID-L and NID+L, related to bimodal transcription co-regulators [149, 277]. NSD1 gene is expressed in several tissues among which we find fetal and adult brain, kidney, skeletal muscle, spleen, and thymus [187]. It has recently been reported that NSD1 regulates gene expression by modulating the levels of the various forms of methylation at H3K36 [201, 212]. Target genes, like the bone morphogenetic protein 4 (BMP4), participate in various processes, consistent with the role of NSD1 in certain diseases such as cancer and Sotos syndrome. Indeed, the NSD1-dependent methylation activity promotes recruitment of RNA polymerase II onto promoters, like BMP4, to initiate transcription and elongation [212]. NSD1 also targets methylation in nonnucleosomal targets like K218 and K221 of the p65 subunit of NFKβ [211, 212].
Most of the Sotos syndrome cases are sporadic and originate from haploinsufficiency because of de novo mutations (point mutations, 5q35 microdeletions, and partial deletions) within the whole gene although some cases of autosomal dominant inheritance have been reported in literature [46, 134, 300, 311, 363, 385]. At any rate, the varied phenotype of Sotos syndrome suggests a heterogeneous origin, being NSD1 intragenic mutations the most plausible cause. In any case, mutations, which may vary among populations, lead to truncated proteins that impede the correct functioning of NSD1. Homozygous null mice for Nsd1 cannot complete gastrulation and exhibit apoptosis at early embryonic development [277].
Genetic defects in histone-modifier enzymes lead to altered histone modifications in cancer
Although histone covalent modifications are of varied nature, acetylation and methylation of specific amino acid residues at the tails of histones H3 and H4 remain the most related to cancer. Certain histone marks have been largely described in cancer, like the loss of H4K16Ac and/or H4K20me3 in repeated sequences [97]. Also, aberrant modifications of histones such as deacetylation of histones H3 and H4, loss of H3K4me3 and gain of H3K9me and H3K27me3, in promoter regions lead to a deregulation in gene expression, in conjunction with DNA methylation [167]. Not only are histone marks deregulated in cancer but also the expression pattern of histone-modifying enzymes is different in tumor samples from their normal counterparts, and these differences also depend on the tumor type [256]. This evidence suggests that the questioned histone modifiers could carry out a specific role in cancer.
For instance, altered HAT activity has been reported in hematological and solid cancers, due to mutations within the gene or deregulation in gene expression by viral oncoproteins. In leukemia, chromosomal translocations affecting histone-modifying enzymes, such as HATs, alter their activity. For instance, translocations of HAT or related genes, like TIF2, MOZ and MORF, HDACs, create fusion proteins, which possibly deregulate the expression of target genes by chromatin remodeling [84]. The family of mixed-lineage leukemia (MLL) histone methyltransferases is frequently mutated in human cancer. Rearrangements of MLL1 have been related to deregulation of Hox genes in leukemia [141], and inactivating mutations have been found in MLL2 and MLL3 in primary tumors of medulloblastoma and in non-Hodgkin lymphoma [237, 263]. Specifically, in this type of malignant brain tumor mutation, deletions and amplifications in genes that modulate H3K9 methylation have been previously described [250]. These mutations affect not only other histone methyltransferases, like EHMT1 and SMYD4, but also histone demethylases containing the jmjC domain, such as JMJD2B and JMJD2C [250]. In recent decades, there has been a significant increase in the number of publications providing evidence for a specific role for histone posttranslational modifications in human tumorigenesis.
Epigenetic changes induced by nucleotide expansion
CAG trinucleotide expansion affects histone posttranslational modifications: Huntington disease
Huntington disease (HD, OMIM #143100) is a fatal neurological disease caused by an autosomal dominant mutation on chromosome 4p. The life course of HD lasts approximately 20 years and individuals carrying the mutation are usually asymptomatic until mid-life. Progressively, the brain degenerates and patients show motor impairments (chorea and dystonia), and psychiatric and behavioral disturbances together with a cognitive decline which eventually triggers dementia.
The affected gene that causes HD is IT15, which encodes the protein huntingtin [127], of ubiquitous expression although it is the brain which is the affected organ in this disease, and in particular, the striatum [350]. The mutation originates an expansion of the CAG trinucleotide within exon 1, which is translated into a polyglutamine (polyQ) tract. After this discovery, HD was categorized within a group of neurological disorders called polyQ diseases, all of which are characterized by the presence of CAG repeat expansions [265]. In HD, those unaffected have 5–36 CAG repeats, while expansions of 40 or more CAG repeats imply complete penetrance. The HD repeat length is inversely correlated with age of onset of the disorder and gradually increases from youth to the age of 70 [76]. The presence of 36–39 CAG repeats indicates incomplete penetrance and carriers are at high risk of developing HD [291]. The abnormal polyQ chains hinder normal huntingtin folding with a consequent loss of function [297]. In addition, mutant huntingtin is reluctant to proteolytic cleavage, and, together with the proteins it interacts with, accumulates in protein aggregates, which are toxic to brain cells and trigger brain dysfunction [59, 70].
Even though the genetic origin of HD is well established, not all the phenotypic variation can be explained by genotypic variation. For instance, parent-of-origin effects and genetic anticipation are some of the non-Mendelian features that cannot be explained by conventional genetics [279, 283, 328]. Sabl and Lair postulated in 1992 a hypothesis based on position effect variegation and epigene conversion, which provided a new framework in which the molecular mechanisms underlying HD could be better explained [293]. The role of epigenetics in HD was supported by the fact that monozygotic twins, who, share identical CAG repeats length, exhibit marked differences in HD symptoms [5, 99, 109, 122].
Histone acetylation in this disease represents a robust modulator of genomic stability. By using budding yeast and cultured human astrocytes, Debacker et al. have recently demonstrated that specific HDACs promote CAG repeats expansion while the HAT activity of CBP and p300/CBP-associated factor, which directly bind to mutant huntingtin, inhibit the elongation [63], supporting earlier works in Drosophila [258, 318]. Different HDAC inhibitors administered to transgenic mouse models of HD ameliorate varied symptoms, e.g., improvements in motor performance or reduction of the neural atrophy typically observed [92, 104, 142, 330]. All these reports provide solid evidence for the role of histone modifications in the etiology of HD and the use of HDAC inhibitors represents a promising field for this, so far, incurable disease. However, other mechanisms have also been described in HD, like increased methylation levels in H3K9 in humans and transgenic mice due to abnormally elevated expression of the histone methyltransferase ERG-associated protein with SET domain (ESET) [292]. Also, mutant huntingtin has been found to induce widespread transcriptional dysregulation [6], a phenomena that could be explained by the increased number, and varied nature, of posttranslational modifications to which huntingtin can be subjected, unless mutated [81].
Genetic control of chromatin remodeling
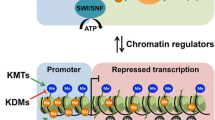
The way DNA and histones are packaged into nucleosomes and higher-order structures is fundamental to maintain a whole genome into the nucleus of a cell but, at the same time, it represents an obstacle for transcription, replication, DNA repair, and recombination [196]. Distinct nuclear machineries are in charge of controlling the interactions within chromatin to allow specific transcription factors to bind to the target sequence in DNA and carry out adequate gene transcription. Chromatin remodeling proteins, together with DNA methylation and histone tail modifiers, thoroughly regulate these processes. Some of the chromatin remodeling proteins are characterized by the presence of a DNA-dependent ATPase domain, which belongs to the SNF2 (sucrose non-fermenting 2) family and allows them to use the energy of ATP hydrolysis to modify interactions between histones and DNA [312]. Depending on the sequence homology of their conserved ATPase domain, these chromatin remodeling proteins have been divided into different families like SWI2/SNF2 (switching/sucrose non-fermenting chromatin remodeling complex), CHD, and so on [94].
Mutations in genes encoding for enzymes that participate in chromatin remodeling severely affect chromatin structure, leading to a deregulation of gene expression and possibly to inadequate protein expression. As a consequence, numerous epigenetic disorders arise, like α thalassemia X-linked mental retardation (ATR-X) syndrome, CHARGE syndrome, and Cockayne B syndrome, and cancer susceptibility increases (Table 1).
Genetic defects in ATRX: α thalassemia X-linked mental retardation syndrome
The α thalassemia X-linked mental retardation syndrome (ATR-X, OMIM #301040) is a rare X-linked recessive developmental disorder that affects mainly males, who are typically characterized by a global developmental delay, facial dysmorphism, mild α thalassemia, and skeletal and urogenital anomalies [114].
Commonly, an ATR-X patient displays a global developmental delay which in 95 % of the cases, accounts for mental retardation. Sometimes, there is also an autistic-like behavior. At any rate, expressive language is limited and patients exhibit hypotonia. Characteristic facial anomalies are often seen, including frontal hair upswept, telecanthus and epicantic folds, flat nasal bridge, small triangular upturned nose, tented upper lip, full and everted lower lip, and protruding tongue. Typically, ATR-X individuals also suffer from skeletal anomalies, probably secondary to the hypotonia, gastroesophageal reflux, and genital anomalies, which range from undescended testes or deficient prepuce to ambiguous genitalia, depending on the ATRX mutations. Mild α thalassemia, characterized by the formation of hemoglobin H inclusion bodies in red blood cells and caused by defects in α globin genes, is not always present as previously thought (reviewed in [114]). Intriguingly, female carriers have usually normal physical and intellectual capabilities. This is due to the skewed X-chromosome inactivation, which is quite common in female carriers of X-linked mutations, and affects preferentially the abnormal bearing allele [117]. This does not occur during early embryogenesis but it is established during development by tissue-specific selection [239].
Most of the reported cases of ATR-X syndrome contain mutations in the ATRX gene, which is located on chromosome Xq13 and encodes a chromatin remodeling protein [118]. The ATRX gene was first described in 1995 [116]. The ATRX gene belongs to the SNF2 family, whose members are involved in the regulation of transcription, cell cycle, DNA repair, and chromosome segregation during mitosis and meiosis; and seem to facilitate these functions by chromatin remodeling [118]. Two domains were clearly identified in the ATRX gene. The ADD domain (ATRX-DNMT3-DNMT3L, because of the sequence homology with the family of de novo DNMTs) at the N-terminus of the protein, which is integrated by N-terminal GATA-like Zn finger, a PHD-like finger and a long C-terminal a helix, giving rise to a globular domain, which may have a DNA binding function [7], as previously suggested by Cardoso et al. [39]. The other main domain is a SWI/SNF-like ATPase/helicase domain, which shows weak chromatin remodeling activity but strong DNA translocase activity in vitro [367].
Mutations in the ATRX gene cluster occur mainly in the ADD domain (50 %) and the helicase domain (30 %) [118]. The majority are missense mutations and those occurring in the ADD domain can take place in buried residues, affecting the structure of the protein; and in surface residues, damaging potential transcription factors binding sites and protein interaction sites [7], but even large intragenic duplications with the consequent loss of function have been detected in ATR-X patients [329].
The ATRX protein is present mainly in pericentromeric heterochromatin [224]. Usually, ATRX is recruited by HP1α [23] through a histone H3-binding module, whose binding is promoted by H3K9me3 and inhibited by H3K4me3 [153] and H3K4me2 [69]. Mutations affecting either ATRX or H3K9me3 result in defective binding and pericentromeric heterochromatin localization [153, 183]. A yeast two-hybrid assay demonstrated the interaction between the Polycomb group protein EZH2, a histone-lysine N-methyltransferase, and ATRX [40]. In promyelocytic leukemia nuclear bodies, ATRX complexes with Daxx, a death-domain-associated protein, and participates in chromatin remodeling for genes that are controlled by Daxx-interacting sequence-specific transcription factors [367]. MeCP2-deficient neurons exhibit ATRX diffuse nuclear distribution suggesting that MeCP2, another chromatin remodeling protein, is responsible for ATRX recruitment in mature neurons [243]. Atrx null mutations in mice result in embryonic lethality and induce DNA methylation changes [105]. Nevertheless, the variability of phenotypic ATR-X syndrome features is influenced by the location of the mutations affecting the ATRX gene but in any case, none of the reported human mutations are true nulls.
Mutations in the human ATRX gene were at first related to decreases in α globin gene expression, which was responsible for the α thalassemia phenotype. However, some ATR-X syndrome patients were completely normal at hematologic level. Interestingly, DNA methylation changes have been observed, affecting mainly rDNA, Y-specific satellites (DYZ2) and subtelomeric repeats [115]. Current knowledge of the ATR-X syndrome establishes a robust link between chromatin remodeling and DNA methylation with gene expression.
Allelic mutations in the ATRX are commonly associated with mental retardation. Indeed, ATR-X syndrome is not the only one caused by mutations in the ATRX gene. Other syndromes related to ATRX dysfunction, which combine mental retardation with dysmorphic features, include the Juberg-Marsidi [348], Carpenter-Waziri [2], Holmes-Gang [319], Smith-Fineman-Myers [347] and Chudley-Lowry [3] syndromes.
So far, several ATRX gene cofactors have been discovered: HP1 [23], EZHZ [40], Daxx [367], MeCP2 [243], and Cohesin [173]. Tang and colleagues have recently identified 12 transcription factors binding sites in the 5′ regulatory region and 5′UTR of the mammalian ATRX gene that could be bona fide candidates for regulation of ATRX gene expression [323]. ATRX is fundamental for proper development not only in the brain and multiple parts of the central nervous system: it also regulates somatic and germ cell function and gametogenesis during both testicular and ovarian development [151]. Altogether, these data suggest that the ATRX gene appears to be more involved in regulating specific target genes rather than behaving as a global regulator. Further experiments are needed to corroborate this hypothesis, which will probably cast light on ATRX-mediated molecular pathogenesis and its relation to mental retardation. At any rate, a clear link is established by ATRX between chromatin remodeling protein that plays an important role in the maintaining and establishment of DNA methylation patterns, although the mechanisms are still elusive.
Genetic defects in CHD7: CHARGE syndrome
CHARGE syndrome (OMIN #214800) is an autosomal dominant complex genetic disorder characterized by the conjunction of multiple birth defects. It was in 1981 when Pagon et al. coined the acronym CHARGE to summarize the main symptoms of the syndrome, which are coloboma of the eyes, heart defects, atresia of the choanae, severe retardation of growth and development, genital abnormalities and/or hypogonadism, and ear abnormalities and/or deafness [257].
The chromodomain helicase DNA-binding protein seven (CHD7), located at 8q12.2, is found to be mutated in two of every three patients [259] and was first identified by Vissers et al. [349]. CHD7 belongs to the CHD superfamily, which groups ATP-dependent chromatin remodeling proteins. The CHD7 protein contains two N-terminal chromodomains, which mediate binding to methylated histones; a central SWI2/SNF2-like ATPase/helicase domain with chromatin remodeling activity; a SANT domain, which is involved in DNA and/or modified histones binding, and two BRK domains with unknown function [382]. Chromatin remodeling enzymes usually exert their function by interacting with other proteins. This also applies to CHD7. It associates with PBAF (Polybromo and BRG-associated factor-containing complex), a chromatin remodeling subcomplex of the SWI/SNF family, which is essential for the activation of the transcriptional program associated with the formation of multipotent migratory neural crest, a transient cell population with a multilineage differential potential [12]. CHD7 is ubiquitously expressed at early stages of development. Then, expression increases in CHARGE syndrome-related organs in both humans and mice, suggesting that the expression pattern of CHD7 seems to be both spatially and temporally regulated [382].
CHD7 is recruited to specific sites of the chromatin depending on the cellular context. The pattern of occupancy is highly correlated with the location of the H3K4me. This, together with the facts that CHD7 binding sites are predominantly in a distal location to the transcription start site, DNAse hypersensitive, frequently conserved and near highly expressed genes, suggests that CHD7 is involved in chromatin remodeling and gene expression by recognizing histone modifications and altering chromatin structure [304]. Recently, it was found that CHD7 binds to hypomethylated rDNA, and could be acting as a positive regulator of rRNA synthesis [381].
Some mutations are missense but the majority are nonsense and frameshift mutations that arise de novo and might result in haploinsufficiency of CHD7, thereby producing a truncated protein. Mice with Chd7 homozygous null mutations die at an early embryonic state but the heterozygous are born with malformations that resemble human CHARGE syndrome [30]. Chromosomal abnormalities have also been reported in patients with CHARGE syndrome, like an interstitial deletion of 8q12.2-q13 [8] and a balanced translocation affecting 8q12 [163]. Mounting evidence points that CHD7 mutations causing sporadic CHARGE syndrome are predominantly of paternal origin [264]. Mutations in CHD7 are also related to other rare genetic syndromes, such as the Kallmann [175] and DiGeorge [295] syndromes.
The clinical manifestations of CHARGE syndrome are extremely variable, and mutations in CHD7 could be of help for diagnosis but may not predict the phenotype [22]. In addition, approximately one-third of the patients do not present CHD7 mutations, suggesting that other chromosomal regions or candidate genes may be involved in the etiology of this disorder. The study of CHD7 target genes and potentially candidate genes will provide vital information with regard to this syndrome.
Genetic defects in CSB: Cockayne syndrome B
The Cockayne syndrome (CS) is a rare autosomal recessive genetic disorder characterized by growth failure within the first years of life, progressive neurological dysfunction, and varied symptoms related with premature aging, like hearing loss, retinal degeneration, and skin problems [246].
CS is classified into two genetic complementation groups, A (CSA, OMIM #216400) and B (CSB, OMIM #133540). The majority of CS patients carry mutations in the CSB gene and in less proportion, in the CSA gene. The CS phenotype can also arise with mutations in xeroderma pigmentosum genes (XPB, XPD, and XPG) [276].
CSA and CSB proteins participate in the nucleotide excision repair mechanism. In particular, the CSB protein is involved in the transcription-coupled repair process. CSB protein, also known as “group 6 excision repair cross-complementing protein” (ERCC6), belongs to the SWI2/SNF2-like-dependent ATPases, which is known to wrap around DNA [21] and acts as a chromatin remodeler [53]. Mutations causing CSB are predominantly located beyond exon 5, affecting ATPase and/or C-terminal regions and producing truncated proteins, which could impede the correct functioning of wild-type CSB protein by hindering CSB interactions [144]. At the cellular level, hypersensibility to UV light [301] and inability to synthesize RNA to normal rates after UV exposure are the main features of cells from CS individuals [223]. After UV exposure, the ATPase and the C-terminal domains are essential for a stable CSB-chromatin association at DNA lesion-stalled transcription sites but in a normal physiological state, the N-terminal domain of CSB inhibits this association by conformational changes [190]. Curiously, Newman and coworkers found that the patterns of gene deregulation in CSB-null cells were very similar to those observed after treatment with epigenetic drugs like HDACs inhibitors and demethylating agents. Furthermore, CSB does not form complexes with histone modifiers like other SWI2/SNF2 ATPase-like chromatin remodeling proteins; instead, it acts directly on chromatin and its effect is seen on particular genes [248].
The piggyBac transposable element 3 (PGBD3) is integrated within the CSB intron 5 and alternative splicing generates a fusion protein, CSB-PGBD3, which is normally expressed in CSB patients and may contribute to the pathogenesis of the disease by unknown mechanisms [247].
Genetic defects in SNF5 are associated with cancer
The SWI/SNF is probably the most studied family of the chromatin remodeling complexes, which is highly conserved from yeast to humans. SNF5 (INI1), present in all variants of the SWI/SNF complex [357], is essential for normal cell viability and its loss due to mutation significantly contribute to tumorigenesis, hence it has been largely considered as tumor suppressor gene. Unfortunately, the molecular basis through which loss of SNF5 leads to cancer is still to be elucidated.
Inactivating mutations in SNF5 were first described in malignant rhabdoid tumors, one of the most aggressive and lethal of childhood tumors [25, 344]. Also, familial cancers arise from inherited mutant alleles of SNF5, a condition termed “rhabdoid predisposition syndrome” [327]. Apart from specific biallelic mutations in the SNF5 gene, tumor cells have normal karyotypes, and this genomic stability in SNF5 deficient cancers suggests that transcription changes of epigenetic origin are key drivers of malignant rhabdoid carcinogenesis [225].
In Drosophila, loss of this gene within the context of Brahma (SWI/SNF) complex lead to alterations in RNA polymerase elongation, pre-mRNA splicing regulation and chromatine accessibility of ecdysone hormone regulated genes, demonstrating that multiple pathways could be affected by loss of SNF5 function [386]. In mice, homozygous deletion of Snf5 causes embryonic lethality, and although the heterozygous genotype appears normal, early in postnatal life tumors develop [128, 178, 284]. In mouse embryonic fibroblasts, inactivation of the Snf5 gene leads to cell cycle arrest and apoptosis [152]. Conditional silencing of Snf5 and p53 accelerates tumor formation, suggesting that Snf5 tumor suppressor activity could be mediated by p53 [64, 152, 179]. In light of these findings, it has been assumed that antitumorigenic activity of SNF5 could be mediated by its role in cell cycle regulation. In this support, further works have shown that other cell cycle-related genes, like p21 [189] and Aurora Kinase A [191] could also mediate SNF5 function. Taking into account that malignant rhadoid tumor cell lines are highly invasive, Caramel and colleagues reported that inhibition of migration is crucial for the protective role of SNF5 against cancer [38]. Another molecular pathway that has been linked to oncogenic processes under loss of SNF5 is the Hedgehog pathway, which is activated during tumorigenesis [156].
Genetic defects within SNF5 gene sequence have been found in other types of malignancies [181, 185, 205, 233]. The different target genes whose expression is regulated by SNF5, together with the cellular context of each tumor type, are major obstacles to determining the exact mechanisms by which SNF5 mutations lead to cancer, but cumulative evidence points to the cell cycle deregulation, which follows SNF5 loss of function perhaps being a vital event for tumor development.
Concluding remarks
It is well known that variation in the DNA sequence can contain genetic differences that predispose to disease, and this fact has allowed scientists to decipher the molecular origin of many illnesses of Mendelian inheritance. However, the mechanisms underlying complex and non-Mendelian disorders are more difficult to elucidate. Adopting an epigenetic perspective could be of help at the time of interpreting the molecular processes that might account for this type of malignances (Fig. 2).

Representative illustration of several genes which have epigenetic function and whose deregulation leads to disease
Nevertheless, several reasons make it difficult to establish a robust link between epigenetics and complex diseases. Firstly, there is a physiological epigenetic variation as a response to internal and external stimuli. Thus, the epigenetic differences observed may not necessarily account for the disease. In addition, the epigenetic status of cells is tissue-specific. This means that the epigenetic profile varies significantly among different cell types. Thus, the epigenome of easily accessible tissues like blood or bucal cells may not reflect the epigenome of the functional organ of interest. Thus, we should focus on the tissues and organs that contribute to the phenotype being studied. Additional problems are the difficulties at the time of acquiring a sample of those tissues and that the tissue of interest may contain a variety of cell populations, and relevant epigenetic changes can be missed because of cellular heterogeneity. However, it should also be noted that the disease itself and associated factors like the treatment can further induce epigenetic variation, because epigenetic changes, in contrast to genetic ones, can be reverted. Hence, it would be interesting to study not only the organ of interest but also the unaffected ones in healthy individuals as well as in those carrying the disease in order to exclude normal phenotypic variation [269]. Moreover, since most epigenetic modifications are reversible, the field of epigenetic drugs, such as demethylating agents or HDACs inhibitors, is rapidly growing and becoming a therapeutic approach to treat or prevent epigenetic-related diseases. Although DNA methylation and histone acetylation have been thoroughly studied, the field of epigenetics is expanding and very little is known about aberrations in the rest of the epigenetic layers, like other histone modifications, chromatin remodeling proteins, and microRNAs.
The emerging field of “epigenetic epidemiology”, as recently proposed by Feinberg [89] to describe the measurement and cataloguing of the epigenetic variations within and across populations and to characterize the correlation of epigenetic factors with disease, is rapidly growing and becoming more relevant in the study of epigenetic diseases. In the last few years, there has been a massive technological development in epigenetic experimental procedures that will facilitate the work on the challenges ahead, like genome-wide association analyses, DNA methylation microarray platforms and the next sequencing generation [216, 262]. A deeper knowledge in this field will allow scientists to discover epigenetic modifications that could serve as biomarkers indicative of disease and will improve diagnosis, prognosis and therapies for the patients in the future.
References
Aapola U, Liiv I, Peterson P (2002) Imprinting regulator DNMT3L is a transcriptional repressor associated with histone deacetylase activity. Nucleic Acids Res 30:3602–3608
Abidi F, Schwartz CE, Carpenter NJ, Villard L, Fontes M, Curtis M (1999) Carpenter-Waziri syndrome results from a mutation in XNP. Am J Med Genet 85:249–251
Abidi FE, Cardoso C, Lossi AM, Lowry RB, Depetris D, Mattei MG, Lubs HA, Stevenson RE, Fontes M, Chudley AE, Schwartz CE (2005) Mutation in the 5′ alternatively spliced region of the XNP/ATR-X gene causes Chudley-Lowry syndrome. Eur J Hum Genet 13:176–183
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188
Anca MH, Gazit E, Loewenthal R, Ostrovsky O, Frydman M, Giladi N (2004) Different phenotypic expression in monozygotic twins with Huntington disease. Am J Med Genet A 124A:89–91
Anderson AN, Roncaroli F, Hodges A, Deprez M, Turkheimer FE (2008) Chromosomal profiles of gene expression in Huntington’s disease. Brain 131:381–388
Argentaro A, Yang JC, Chapman L, Kowalczyk MS, Gibbons RJ, Higgs DR, Neuhaus D, Rhodes D (2007) Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc Natl Acad Sci USA 104:11939–11944
Arrington CB, Cowley BC, Nightingale DR, Zhou H, Brothman AR, Viskochil DH (2005) Interstitial deletion 8q11.2-q13 with congenital anomalies of CHARGE association. Am J Med Genet A 133A:326–330
Augoustides-Savvopoulou P, Luka Z, Karyda S, Stabler SP, Allen RH, Patsiaoura K, Wagner C, Mudd SH (2003) Glycine N -methyltransferase deficiency: a new patient with a novel mutation. J Inherit Metab Dis 26:745–759
Aung PP, Matsumura S, Kuraoka K, Kunimitsu K, Yoshida K, Matsusaki K, Nakayama H, Yasui W (2005) No evidence of correlation between the single-nucleotide polymorphism of DNMT3B promoter and gastric cancer risk in a Japanese population. Oncol Rep 14:1151–1154
Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, Prieto J, Lu SC, Caballeria J, Rodes J, Mato JM (2000) Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol 33:907–914
Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, Wysocka J (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463:958–962
Balaghi M, Horne DW, Wagner C (1993) Hepatic one-carbon metabolism in early folate deficiency in rats. Biochem J 291(Pt 1):145–149
Balding DJ (2006) A tutorial on statistical methods for population association studies. Nat Rev Genet 7:781–791
Ballestar E, Esteller M, Richardson BC (2006) The epigenetic face of systemic lupus erythematosus. J Immunol 176:7143–7147
Bao Q, He B, Pan Y, Tang Z, Zhang Y, Qu L, Xu Y, Zhu C, Tian F, Wang S (2011) Genetic variation in the promoter of DNMT3B is associated with the risk of colorectal cancer. Int J Colorectal Dis 26:1107–1112
Baric I (2009) Inherited disorders in the conversion of methionine to homocysteine. J Inherit Metab Dis 32:459–471
Bashtrykov P, Jankevicius G, Smarandache A, Jurkowska RZ, Ragozin S, Jeltsch A (2012) Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem Biol 19:572–578
Baujat G, Cormier-Daire V (2007) Sotos syndrome. Orphanet J Rare Dis 2:36
Beaulieu N, Morin S, Chute IC, Robert MF, Nguyen H, MacLeod AR (2002) An essential role for DNA methyltransferase DNMT3B in cancer cell survival. J Biol Chem 277:28176–28181
Beerens N, Hoeijmakers JH, Kanaar R, Vermeulen W, Wyman C (2005) The CSB protein actively wraps DNA. J Biol Chem 280:4722–4729
Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM (2011) CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 48:334–342
Berube NG, Smeenk CA, Picketts DJ (2000) Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum Mol Genet 9:539–547
Bestor TH (2000) The DNA methyltransferases of mammals. Hum Mol Genet 9:2395–2402
Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59:74–79
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, Launer LJ, Eiriksdottir G, Leppert MF, Sapienza C, Gudnason V, Feinberg AP (2008) Intra-individual change over time in DNA methylation with familial clustering. JAMA 299:2877–2883
Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8:299–309
Bodega B, Ramirez GD, Grasser F, Cheli S, Brunelli S, Mora M, Meneveri R, Marozzi A, Mueller S, Battaglioli E, Ginelli E (2009) Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol 7:41
Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP (2005) Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet 14:3463–3476
Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294:2536–2539
Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D (2012) A long ncRNA links copy number variation to a Polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149:819–831
Cabianca DS, Gabellini D (2010) The cell biology of disease: FSHD: copy number variations on the theme of muscular dystrophy. J Cell Biol 191:1049–1060
Cacurri S, Piazzo N, Deidda G, Vigneti E, Galluzzi G, Colantoni L, Merico B, Ricci E, Felicetti L (1998) Sequence homology between 4qter and 10qter loci facilitates the instability of subtelomeric KpnI repeat units implicated in facioscapulohumeral muscular dystrophy. Am J Hum Genet 63:181–190
Cai J, Mao Z, Hwang JJ, Lu SC (1998) Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res 58:1444–1450
Cai J, Sun WM, Hwang JJ, Stain SC, Lu SC (1996) Changes in S-adenosylmethionine synthetase in human liver cancer: molecular characterization and significance. Hepatology 24:1090–1097
Cano A, Buqué X, Martínez-Uña M, Aurrekoetxea I, Menor A, García-Rodriguez JL, Lu SC, Martínez-Chantar M, Mato JM, Ochoa B, Aspichueta P (2011) Methionine adenosyltransferase 1A gene deletion disrupts hepatic VLDL assembly in mice. Hepatology doi:10.1002/hep.24607. (epub ahead of print)
Caramel J, Quignon F, Delattre O (2008) RhoA-dependent regulation of cell migration by the tumor suppressor hSNF5/INI1. Cancer Res 68:6154–6161
Cardoso C, Lutz Y, Mignon C, Compe E, Depetris D, Mattei MG, Fontes M, Colleaux L (2000) ATR-X mutations cause impaired nuclear location and altered DNA binding properties of the XNP/ATR-X protein. J Med Genet 37:746–751
Cardoso C, Timsit S, Villard L, Khrestchatisky M, Fontes M, Colleaux L (1998) Specific interaction between the XNP/ATR-X gene product and the SET domain of the human EZH2 protein. Hum Mol Genet 7:679–684
Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320:1224–1229
Chahrour M, Zoghbi HY (2007) The story of Rett syndrome: from clinic to neurobiology. Neuron 56:422–437
Chan HM, La Thangue NB (2001) p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114:2363–2373
Chang KP, Hao SP, Liu CT, Cheng MH, Chang YL, Lee YS, Wang TH, Tsai CN (2007) Promoter polymorphisms of DNMT3B and the risk of head and neck squamous cell carcinoma in Taiwan: a case-control study. Oral Oncol 43:345–351
Chang KP, Hao SP, Tsang NM, Chang YL, Cheng MH, Liu CT, Lee YS, Tsai CL, Lee TJ, Wang TH, Tsai CN (2008) Gene expression and promoter polymorphisms of DNA methyltransferase 3B in nasopharyngeal carcinomas in Taiwanese people: a case-control study. Oncol Rep 19:217–222
Chen CP, Lin SP, Chang TY, Chiu NC, Shih SL, Lin CJ, Wang W, Hsu HC (2002) Perinatal imaging findings of inherited Sotos syndrome. Prenat Diagn 22:887–892
Chen L, Zeng Y, Yang H, Lee TD, French SW, Corrales FJ, Garcia-Trevijano ER, Avila MA, Mato JM, Lu SC (2004) Impaired liver regeneration in mice lacking methionine adenosyltransferase 1A. FASEB J 18:914–916
Chen SY, Lin JR, Darbha R, Lin P, Liu TY, Chen YM (2004) Glycine N-methyltransferase tumor susceptibility gene in the benzo(a)pyrene-detoxification pathway. Cancer Res 64:3617–3623
Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME (2003) Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302:885–889
Chen YM, Chen LY, Wong FH, Lee CM, Chang TJ, Yang-Feng TL (2000) Genomic structure, expression, and chromosomal localization of the human glycine N-methyltransferase gene. Genomics 66:43–47
Chen YM, Liao YJ, Liu SP, Tsai TF (2009) Phenotypic differences between two Gnmt−/− mouse models for hepatocellular carcinoma. Hepatology 49:2130–2131 (author reply 2131)
Cho YY, Yao K, Pugliese A, Malakhova ML, Bode AM, Dong Z (2009) A regulatory mechanism for RSK2 NH(2)-terminal kinase activity. Cancer Res 69:4398–4406
Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, Vermeulen W (2000) ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol 20:7643–7653
Clark DE, Errington TM, Smith JA, Frierson HF Jr, Weber MJ, Lannigan DA (2005) The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res 65:3108–3116
Cohen DR, Matarazzo V, Palmer AM, Tu Y, Jeon OH, Pevsner J, Ronnett GV (2003) Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol Cell Neurosci 22:417–429
Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY (2004) Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13:2679–2689
Cook RJ, Wagner C (1984) Glycine N-methyltransferase is a folate binding protein of rat liver cytosol. Proc Natl Acad Sci USA 81:3631–3634
Couce ML, Boveda MD, Castineiras DE, Corrales FJ, Mora MI, Fraga JM, Mudd SH (2008) Hypermethioninaemia due to methionine adenosyltransferase I/III (MAT I/III) deficiency: diagnosis in an expanded neonatal screening programme. J Inherit Metab Dis 31(Suppl 2):233–239
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90:537–548
Daxinger L, Whitelaw E (2011) Transgenerational epigenetic inheritance: more questions than answers. Genome Res 20:1623–1628
De Cesare D, Jacquot S, Hanauer A, Sassone-Corsi P (1998) Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc Natl Acad Sci USA 95:12202–12207
de Greef JC, Lemmers RJ, van Engelen BG, Sacconi S, Venance SL, Frants RR, Tawil R, van der Maarel SM (2009) Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat 30:1449–1459
Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, Lahue RS (2012) Histone deacetylase complexes promote trinucleotide repeat expansions. Plos Biol 10:1001257
DelBove J, Kuwahara Y, Mora-Blanco EL, Godfrey V, Funkhouser WK, Fletcher CD, Van Dyke T, Roberts CW, Weissman BE (2009) Inactivation of SNF5 cooperates with p53 loss to accelerate tumor formation in Snf5(±);p53(±) mice. Mol Carcinog 48:1139–1148
Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, Hanash SM, Richardson BC (2001) Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum 44:397–407
Deng Z, Campbell AE, Lieberman PM (2010) TERRA, CpG methylation and telomere heterochromatin: lessons from ICF syndrome cells. Cell Cycle 9:69–74
Deng Z, Norseen J, Wiedmer A, Riethman H, Lieberman PM (2009) TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell 35:403–413
Denissenko MF, Chen JX, Tang MS, Pfeifer GP (1997) Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc Natl Acad Sci USA 94:3893–3898
Dhayalan A, Tamas R, Bock I, Tattermusch A, Dimitrova E, Kudithipudi S, Ragozin S, Jeltsch A (2011) The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum Mol Genet 20:2195–2203
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993
Dixit M, Ansseau E, Tassin A, Winokur S, Shi R, Qian H, Sauvage S, Matteotti C, van Acker AM, Leo O, Figlewicz D, Barro M, Laoudj-Chenivesse D, Belayew A, Coppee F, Chen YW (2007) DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA 104:18157–18162
Dolinoy DC (2008) The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr Rev 66(Suppl 1):S7–S11
Dugani CB, Paquin A, Kaplan DR, Miller FD (2010) Coffin-Lowry syndrome: a role for RSK2 in mammalian neurogenesis. Dev Biol 347:348–359
Duhl DM, Vrieling H, Miller KA, Wolff GL, Barsh GS (1994) Neomorphic agouti mutations in obese yellow mice. Nat Genet 8:59–65
Duthie SJ (1999) Folic acid deficiency and cancer: mechanisms of DNA instability. Br Med Bull 55:578–592
Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M et al (1993) Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet 4:387–392
Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S (2006) DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38:1378–1385
Ehrlich M (2003) The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin Immunol 109:17–28
Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10:2709–2721
Ehrlich M, Sanchez C, Shao C, Nishiyama R, Kehrl J, Kuick R, Kubota T, Hanash SM (2008) ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity 41:253–271
Ehrnhoefer DE, Sutton L, Hayden MR (2011) Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist 17:475–492
El-Maarri O, Becker T, Junen J, Manzoor SS, Diaz-Lacava A, Schwaab R, Wienker T, Oldenburg J (2007) Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum Genet 122:505–514
El-Maarri O, Kareta MS, Mikeska T, Becker T, Diaz-Lacava A, Junen J, Nusgen N, Behne F, Wienker T, Waha A, Oldenburg J, Chedin F (2009) A systematic search for DNA methyltransferase polymorphisms reveals a rare DNMT3L variant associated with subtelomeric hypomethylation. Hum Mol Genet 18:1755–1768
Ellis L, Atadja PW, Johnstone RW (2009) Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther 8:1409–1420
Esteller M (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8:286–298
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358:1148–1159
Ezzikouri S, El Feydi AE, Benazzouz M, Afifi R, El Kihal L, Hassar M, Akil A, Pineau P, Benjelloun S (2009) Single-nucleotide polymorphism in DNMT3B promoter and its association with hepatocellular carcinoma in a Moroccan population. Infect Genet Evol 9:877–881
Fan H, Zhang F, Hu J, Liu D, Zhao Z (2008) Promoter polymorphisms of DNMT3B and the risk of colorectal cancer in Chinese: a case-control study. J Exp Clin Cancer Res 27:24
Feinberg AP (2010) Genome-scale approaches to the epigenetics of common human disease. Virchows Arch 456:13–21
Feinberg AP, Tycko B (2004) The history of cancer epigenetics. Nat Rev Cancer 4:143–153
Feinberg AP, Vogelstein B (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301:89–92
Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 23:9418–9427
Fischer M, Pereira PM, Holtmann B, Simon CM, Hanauer A, Heisenberg M, Sendtner M (2009) P90 Ribosomal s6 kinase 2 negatively regulates axon growth in motoneurons. Mol Cell Neurosci 42:134–141
Flaus A, Martin DM, Barton GJ, Owen-Hughes T (2006) Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res 34:2887–2905
Fong CB, Thong MK, Sam CK, Mohamed Noor MN, Ariffin R (2009) MECP2 mutations in Malaysian Rett syndrome patients. Singapore Med J 50:529–533
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 102:10604–10609
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400
Fraga MF, Esteller M (2005) Towards the human cancer epigenome: a first draft of histone modifications. Cell Cycle 4:1377–1381
Friedman JH, Trieschmann ME, Myers RH, Fernandez HH (2005) Monozygotic twins discordant for Huntington disease after 7 years. Arch Neurol 62:995–997
Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, Miyazaki T, Ogura C, Okazaki Y, Jinno Y (2004) Age-related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet 68:196–204
Gabellini D, D’Antona G, Moggio M, Prelle A, Zecca C, Adami R, Angeletti B, Ciscato P, Pellegrino MA, Bottinelli R, Green MR, Tupler R (2006) Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature 439:973–977
Gabellini D, Green MR, Tupler R (2002) Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 110:339–348
Gallagher A, Hallahan B (2011) Fragile X-associated disorders: a clinical overview. J Neurol 259:401–413
Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF (2005) Neuroprotective effects of phenylbutyrate in the N171–82Q transgenic mouse model of Huntington’s disease. J Biol Chem 280:556–563
Garrick D, Sharpe JA, Arkell R, Dobbie L, Smith AJ, Wood WG, Higgs DR, Gibbons RJ (2006) Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet 2:e58
Gatto S, Della Ragione F, Cimmino A, Strazzullo M, Fabbri M, Mutarelli M, Ferraro L, Weisz A, D’Esposito M, Matarazzo MR (2010) Epigenetic alteration of microRNAs in DNMT3B-mutated patients of ICF syndrome. Epigenetics 5:427–443
Gaull GE, Tallan HH (1974) Methionine adenosyltransferase deficiency: new enzymatic defect associated with hypermethioninemia. Science 186:59–60
Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC (2003) Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J Biol Chem 278:32181–32188
Georgiou N, Bradshaw JL, Chiu E, Tudor A, O’Gorman L, Phillips JG (1999) Differential clinical and motor control function in a pair of monozygotic twins with Huntington’s disease. Mov Disord 14:320–325
Gervasini C, Castronovo P, Bentivegna A, Mottadelli F, Faravelli F, Giovannucci-Uzielli ML, Pessagno A, Lucci-Cordisco E, Pinto AM, Salviati L, Selicorni A, Tenconi R, Neri G, Larizza L (2007) High frequency of mosaic CREBBP deletions in Rubinstein-Taybi syndrome patients and mapping of somatic and germ-line breakpoints. Genomics 90:567–573
Gervasini C, Mottadelli F, Ciccone R, Castronovo P, Milani D, Scarano G, Bedeschi MF, Belli S, Pilotta A, Selicorni A, Zuffardi O, Larizza L (2010) High frequency of copy number imbalances in Rubinstein-Taybi patients negative to CREBBP mutational analysis. Eur J Hum Genet 18:768–775
Ghosh RP, Horowitz-Scherer RA, Nikitina T, Gierasch LM, Woodcock CL (2008) Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J Biol Chem 283:20523–20534
Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL (2010) MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol Cell Biol 30:4656–4670
Gibbons R (2006) Alpha thalassaemia-mental retardation. X linked. Orphanet J Rare Dis 1:15
Gibbons RJ, McDowell TL, Raman S, O’Rourke DM, Garrick D, Ayyub H, Higgs DR (2000) Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet 24:368–371
Gibbons RJ, Picketts DJ, Villard L, Higgs DR (1995) Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 80:837–845
Gibbons RJ, Suthers GK, Wilkie AO, Buckle VJ, Higgs DR (1992) X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: localization to Xq12-q21.31 by X inactivation and linkage analysis. Am J Hum Genet 51:1136–1149
Gibbons RJ, Wada T, Fisher CA, Malik N, Mitson MJ, Steensma DP, Fryer A, Goudie DR, Krantz ID, Traeger-Synodinos J (2008) Mutations in the chromatin-associated protein ATRX. Hum Mutat 29:796–802
Gibson JH, Slobedman B, NH K, Williamson SL, Minchenko D, El-Osta A, Stern JL, Christodoulou J (2010) Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci 11:53
Gil B, Casado M, Pajares MA, Bosca L, Mato JM, Martin-Sanz P, Alvarez L (1996) Differential expression pattern of S-adenosylmethionine synthetase isoenzymes during rat liver development. Hepatology 24:876–881
Giordano A, Avantaggiati ML (1999) p300 and CBP: partners for life and death. J Cell Physiol 181:218–230
Gomez-Esteban JC, Lezcano E, Zarranz JJ, Velasco F, Garamendi I, Perez T, Tijero B (2007) Monozygotic twins suffering from Huntington’s disease show different cognitive and behavioural symptoms. Eur Neurol 57:26–30
Gonzalgo ML, Jones PA (1997) Mutagenic and epigenetic effects of DNA methylation. Mutat Res 386:107–118
Gourley M, Miller FW (2007) Mechanisms of disease: Environmental factors in the pathogenesis of rheumatic disease. Nat Clin Pract Rheumatol 3:172–180
Gowher H, Liebert K, Hermann A, Xu G, Jeltsch A (2005) Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol Chem 280:13341–13348
Goyal R, Reinhardt R, Jeltsch A (2006) Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res 34:1182–1188
Group THsDCR (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72:971–983
Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, Smith TW, Imbalzano AN, Jones SN (2001) Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol 21:3598–3603
Guo X, Zhang L, Wu M, Wang N, Liu Y, Er L, Wang S, Gao Y, Yu W, Xue H, Xu Z (2010) Association of the DNMT3B polymorphism with colorectal adenomatous polyps and adenocarcinoma. Mol Biol Rep 37:219–225
Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science 315:1143–1147
Hagerman RJ, Hagerman PJ (2002) The fragile X premutation: into the phenotypic fold. Curr Opin Genet Dev 12:278–283
Hagleitner MM, Lankester A, Maraschio P, Hulten M, Fryns JP, Schuetz C, Gimelli G, Davies EG, Gennery A, Belohradsky BH, de Groot R, Gerritsen EJ, Mattina T, Howard PJ, Fasth A, Reisli I, Furthner D, Slatter MA, Cant AJ, Cazzola G, van Dijken PJ, van Deuren M, de Greef JC, van der Maarel SM, Weemaes CM (2008) Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICF syndrome). J Med Genet 45:93–99
Hanel ML, Sun CY, Jones TI, Long SW, Zanotti S, Milner D, Jones PL (2011) Facioscapulohumeral muscular dystrophy (FSHD) region gene 1 (FRG1) is a dynamic nuclear and sarcomeric protein. Differentiation 81:107–118
Hansen FJ, Friis B (1976) Familial occurrence of cerebral gigantism, Sotos’ syndrome. Acta Paediatr Scand 65:387–389
Hansen JC, Ghosh RP, Woodcock CL (2010) Binding of the Rett syndrome protein, MeCP2, to methylated and unmethylated DNA and chromatin. IUBMB Life 62:732–738
Hansen RS, Stoger R, Wijmenga C, Stanek AM, Canfield TK, Luo P, Matarazzo MR, D’Esposito M, Feil R, Gimelli G, Weemaes CM, Laird CD, Gartler SM (2000) Escape from gene silencing in ICF syndrome: evidence for advanced replication time as a major determinant. Hum Mol Genet 9:2575–2587
Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM (1999) The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA 96:14412–14417
Hendrich B, Bird A (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 18:6538–6547
Hennekam RC (2006) Rubinstein-Taybi syndrome. Eur J Hum Genet 14:981–985
Herman JG (1999) Hypermethylation of tumor suppressor genes in cancer. Semin Cancer Biol 9:359–367
Hess JL (2004) MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med 10:500–507
Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP (2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA 100:2041–2046
Hong YS, Lee HJ, You CH, Roh MS, Kwak JY, Lee MJ, Kim JY (2007) DNMT3b 39179GT polymorphism and the risk of adenocarcinoma of the colon in Koreans. Biochem Genet 45:155–163
Horibata K, Saijo M, Bay MN, Lan L, Kuraoka I, Brooks PJ, Honma M, Nohmi T, Yasui A, Tanaka K (2011) Mutant Cockayne syndrome group B protein inhibits repair of DNA topoisomerase I-DNA covalent complex. Genes Cells 16:101–114
Horikawa S, Tsukada K (1992) Molecular cloning and developmental expression of a human kidney S-adenosylmethionine synthetase. FEBS Lett 312:37–41
Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T (2005) Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet 37:31–40
Howard PJ, Lewis IJ, Harris F, Walker S (1985) Centromeric instability of chromosomes 1 and 16 with variable immune deficiency: a new syndrome. Clin Genet 27:501–505
Hu J, Fan H, Liu D, Zhang S, Zhang F, Xu H (2010) DNMT3B promoter polymorphism and risk of gastric cancer. Dig Dis Sci 55:1011–1016
Huang N, vom Baur E, Garnier JM, Lerouge T, Vonesch JL, Lutz Y, Chambon P, Losson R (1998) Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J 17:3398–3412
Hulten M (1978) Selective somatic pairing and fragility at 1q12 in a boy with common variable immunodeficiency: a new syndrome. Clin Genet 14:294
Huyhn K, Renfree MB, Graves JA, Pask AJ (2011) ATRX has a critical and conserved role in mammalian sexual differentiation. BMC Dev Biol 11:39
Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM, Wang X, Biegel JA, Pomeroy SL, Mesirov JP, Roberts CW (2005) Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci USA 102:17745–17750
Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, Cochrane JC, Allis CD, Picketts DJ, Patel DJ, Li H, Shi Y (2011) ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat Struct Mol Biol 18:769–776
Jacquot S, Zeniou M, Touraine R, Hanauer A (2002) X-linked Coffin-Lowry syndrome (CLS, MIM 303600, RPS6KA3 gene, protein product known under various names: pp 90(rsk2), RSK2, ISPK, MAPKAP1). Eur J Hum Genet 10:2–5
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–254
Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, Klekota J, Tamayo P, Nguyen PT, Tolstorukov M, Park PJ, Cho YJ, Hsiao K, Buonamici S, Pomeroy SL, Mesirov JP, Ruffner H, Bouwmeester T, Luchansky SJ, Murtie J, Kelleher JF, Warmuth M, Sellers WR, Roberts CW, Dorsch M (2010) Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med 16:1429–1433
Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M, Fraga MF, O’Hanlon TP, Rider LG, Jacinto FV, Lopez-Longo FJ, Dopazo J, Forn M, Peinado MA, Carreno L, Sawalha AH, Harley JB, Siebert R, Esteller M, Miller FW, Ballestar E (2010) Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 20:170–179
Jeanpierre M, Turleau C, Aurias A, Prieur M, Ledeist F, Fischer A, Viegas-Pequignot E (1993) An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum Mol Genet 2:731–735
Jefferson A, Colella S, Moralli D, Wilson N, Yusuf M, Gimelli G, Ragoussis J, Volpi EV (2010) Altered intra-nuclear organisation of heterochromatin and genes in ICF syndrome. PLoS One 5:e11364
Jiang YL, Rigolet M, Bourc’his D, Nigon F, Bokesoy I, Fryns JP, Hulten M, Jonveaux P, Maraschio P, Megarbane A, Moncla A, Viegas-Pequignot E (2005) DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum Mutat 25:56–63
Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, Fields CR, Delmas AL, Liu X, Qiu J, Robertson KD (2008) DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet 17:690–709
Jin F, Dowdy SC, Xiong Y, Eberhardt NL, Podratz KC, Jiang SW (2005) Up-regulation of DNA methyltransferase 3B expression in endometrial cancers. Gynecol Oncol 96:531–538
Johnson D, Morrison N, Grant L, Turner T, Fantes J, Connor JM, Murday V (2006) Confirmation of CHD7 as a cause of CHARGE association identified by mapping a balanced chromosome translocation in affected monozygotic twins. J Med Genet 43:280–284
Jones JS, Amos CI, Pande M, Gu X, Chen J, Campos IM, Wei Q, Rodriguez-Bigas M, Lynch PM, Frazier ML (2006) DNMT3b polymorphism and hereditary nonpolyposis colorectal cancer age of onset. Cancer Epidemiol Biomarkers Prev 15:886–891
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3:415–428
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19:187–191
Jones RS (2007) Epigenetics: reversing the ‘irreversible’. Nature 450:357–359
Judd BH (1955) Direct proof of a variegated-type position effect at the white locus in Drosophila melanogaster. Genetics 40:739–744
Kalkhoven E (2004) CBP and p300: HATs for different occasions. Biochem Pharmacol 68:1145–1155
Kalkhoven E, Roelfsema JH, Teunissen H, den Boer A, Ariyurek Y, Zantema A, Breuning MH, Hennekam RC, Peters DJ (2003) Loss of CBP acetyltransferase activity by PHD finger mutations in Rubinstein-Taybi syndrome. Hum Mol Genet 12:441–450
Kang S, Elf S, Lythgoe K, Hitosugi T, Taunton J, Zhou W, Xiong L, Wang D, Muller S, Fan S, Sun SY, Marcus AI, Gu TL, Polakiewicz RD, Chen ZG, Khuri FR, Shin DM, Chen J (2010) p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J Clin Invest 120:1165–1177
Karpf AR, Matsui S (2005) Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 65:8635–8639
Kernohan KD, Jiang Y, Tremblay DC, Bonvissuto AC, Eubanks JH, Mann MR, Berube NG (2010) ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev Cell 18:191–202
Kerr SJ (1972) Competing methyltransferase systems. J Biol Chem 247:4248–4252
Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC (2008) Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 83:511–519
Kim SZ, Santamaria E, Jeong TE, Levy HL, Mato JM, Corrales FJ, Mudd SH (2002) Methionine adenosyltransferase I/III deficiency: two Korean compound heterozygous siblings with a novel mutation. J Inherit Metab Dis 25:661–671
Kimura H, Shiota K (2003) Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem 278:4806–4812
Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M (2000) The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep 1:500–506
Klochendler-Yeivin A, Picarsky E, Yaniv M (2006) Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini1 subunit of the SWI/SNF chromatin remodeling complex. Mol Cell Biol 26:2661–2674
Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP (2005) DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell 19:667–678
Kohashi K, Oda Y, Yamamoto H, Tamiya S, Oshiro Y, Izumi T, Taguchi T, Tsuneyoshi M (2008) SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: a special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am J Surg Pathol 32:1168–1174
Kotb M, Mudd SH, Mato JM, Geller AM, Kredich NM, Chou JY, Cantoni GL (1997) Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet 13:51–52
Kourmouli N, Sun YM, van der Sar S, Singh PB, Brown JP (2005) Epigenetic regulation of mammalian pericentric heterochromatin in vivo by HP1. Biochem Biophys Res Commun 337:901–907
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Kreiger PA, Judkins AR, Russo PA, Biegel JA, Lestini BJ, Assanasen C, Pawel BR (2009) Loss of INI1 expression defines a unique subset of pediatric undifferentiated soft tissue sarcomas. Mod Pathol 22:142–150
Krueger DD, Bear MF (2011) Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu Rev Med 62:411–429
Kurotaki N, Harada N, Yoshiura K, Sugano S, Niikawa N, Matsumoto N (2001) Molecular characterization of NSD1, a human homologue of the mouse Nsd1 gene. Gene 279:197–204
Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, Sugimoto T, Sonoda T, Hasegawa T, Chinen Y, Tomita Ha HA, Kinoshita A, Mizuguchi T, Yoshiura Ki K, Ohta T, Kishino T, Fukushima Y, Niikawa N, Matsumoto N (2002) Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 30:365–366
Kuwahara Y, Charboneau A, Knudsen ES, Weissman BE (2010) Reexpression of hSNF5 in malignant rhabdoid tumor cell lines causes cell cycle arrest through a p21(CIP1/WAF1)-dependent mechanism. Cancer Res 70:1854–1865
Lake RJ, Geyko A, Hemashettar G, Zhao Y, Fan HY (2010) UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol Cell 37:235–246
Lee S, Cimica V, Ramachandra N, Zagzag D, Kalpana GV (2011) Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res 71:3225–3235
Lee SJ, Jeon HS, Jang JS, Park SH, Lee GY, Lee BH, Kim CH, Kang YM, Lee WK, Kam S, Park RW, Kim IS, Cho YL, Jung TH, Park JY (2005) DNMT3B polymorphisms and risk of primary lung cancer. Carcinogenesis 26:403–409
Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM (2010) A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329:1650–1653
Lemmers RJ, Wohlgemuth M, van der Gaag KJ, van der Vliet PJ, van Teijlingen CM, de Knijff P, Padberg GW, Frants RR, van der Maarel SM (2007) Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 81:884–894
Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A (1992) Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69:905–914
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128:707–719
Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3:662–673
Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926
Li J, Ramani K, Sun Z, Zee C, Grant EG, Yang H, Xia M, Oh P, Ko K, Mato JM, Lu SC (2010) Forced expression of methionine adenosyltransferase 1A in human hepatoma cells suppresses in vivo tumorigenicity in mice. Am J Pathol 176:2456–2466
Li Y, Dai Y, Wu SL, Pei P, Cao XH, Pu DF (2005) The C46359T polymorphism of DNMT3B promoter gene and pathogenesis of acute leukemia. Zhonghua Nei Ke Za Zhi 44:588–591
Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ 3rd, Qiao Q, Neubert TA, Xu RM, Gozani O, Reinberg D (2009) The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem 284:34283–34295
Liao YJ, Chen KH, Huang SF, Chen TL, Wang CK, Chien CH, Tsai TF, Liu SP, Chen YM (2010) Deficiency of glycine N-methyltransferase results in deterioration of cellular defense to stress in mouse liver. Proteomics Clin Appl 4:394–406
Liao YJ, Liu SP, Lee CM, Yen CH, Chuang PC, Chen CY, Tsai TF, Huang SF, Lee YH, Chen YM (2009) Characterization of a glycine N-methyltransferase gene knockout mouse model for hepatocellular carcinoma: implications of the gender disparity in liver cancer susceptibility. Int J Cancer 124:816–826
Lin CH, Hsieh SY, Sheen IS, Lee WC, Chen TC, Shyu WC, Liaw YF (2001) Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res 61:4238–4243
Lin H, Wong RP, Martinka M, Li G (2009) Loss of SNF5 expression correlates with poor patient survival in melanoma. Clin Cancer Res 15:6404–6411
Lin H, Yamada Y, Nguyen S, Linhart H, Jackson-Grusby L, Meissner A, Meletis K, Lo G, Jaenisch R (2006) Suppression of intestinal neoplasia by deletion of Dnmt3b. Mol Cell Biol 26:2976–2983
Liu SP, Li YS, Chen YJ, Chiang EP, Li AF, Lee YH, Tsai TF, Hsiao M, Huang SF, Chen YM (2007) Glycine N-methyltransferase-/- mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology 46:1413–1425
Liu Z, Wang L, Wang LE, Sturgis EM, Wei Q (2008) Polymorphisms of the DNMT3B gene and risk of squamous cell carcinoma of the head and neck: a case-control study. Cancer Lett 268:158–165
Loenen WA (2006) S-adenosylmethionine: jack of all trades and master of everything? Biochem Soc Trans 34:330–333
Lu SC, Alvarez L, Huang ZZ, Chen L, An W, Corrales FJ, Avila MA, Kanel G, Mato JM (2001) Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci USA 98:5560–5565
Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, Gudkov AV, Stark GR (2010) Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci USA 107:46–51
Lucio-Eterovic AK, Singh MM, Gardner JE, Veerappan CS, Rice JC, Carpenter PB (2010) Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc Natl Acad Sci USA 107:16952–16957
Luikenhuis S, Giacometti E, Beard CF, Jaenisch R (2004) Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci USA 101:6033–6038
Luka Z, Capdevila A, Mato JM, Wagner C (2006) A glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res 15:393–397
Luka Z, Cerone R, Phillips JA 3rd, Mudd HS, Wagner C (2002) Mutations in human glycine N-methyltransferase give insights into its role in methionine metabolism. Hum Genet 110:68–74
Martin-Subero JI, Esteller M (2011) Profiling epigenetic alterations in disease. Adv Exp Med Biol 711:162–177
Martinez-Chantar ML, Corrales FJ, Martinez-Cruz LA, Garcia-Trevijano ER, Huang ZZ, Chen L, Kanel G, Avila MA, Mato JM, Lu SC (2002) Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J 16:1292–1294
Martinez-Chantar ML, Latasa MU, Varela-Rey M, Lu SC, Garcia-Trevijano ER, Mato JM, Avila MA (2003) l-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells: role of S-adenosylmethionine. J Biol Chem 278:19885–19890
Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, Martinez N, Varela M, Luka Z, Capdevila A, Rodriguez J, Aransay AM, Matthiesen R, Yang H, Calvisi DF, Esteller M, Fraga M, Lu SC, Wagner C, Mato JM (2008) Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 47:1191–1199
Mato JM, Alvarez L, Ortiz P, Pajares MA (1997) S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther 73:265–280
Mato JM, Corrales FJ, Lu SC, Avila MA (2002) S-adenosylmethionine: a control switch that regulates liver function. FASEB J 16:15–26
Mato JM, Lu SC (2007) Role of S-adenosyl-l-methionine in liver health and injury. Hepatology 45:1306–1312
Mayne LV, Lehmann AR (1982) Failure of RNA synthesis to recover after UV irradiation: an early defect in cells from individuals with Cockayne’s syndrome and xeroderma pigmentosum. Cancer Res 42:1473–1478
McDowell TL, Gibbons RJ, Sutherland H, O’Rourke DM, Bickmore WA, Pombo A, Turley H, Gatter K, Picketts DJ, Buckle VJ, Chapman L, Rhodes D, Higgs DR (1999) Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and the short arms of acrocentric chromosomes. Proc Natl Acad Sci USA 96:13983–13988
McKenna ES, Sansam CG, Cho YJ, Greulich H, Evans JA, Thom CS, Moreau LA, Biegel JA, Pomeroy SL, Roberts CW (2008) Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol Cell Biol 28:6223–6233
Meda F, Folci M, Baccarelli A, Selmi C (2011) The epigenetics of autoimmunity. Cell Mol Immunol 8:226–236
Mehmood T, Schneider A, Sibille J, Marques Pereira P, Pannetier S, Ammar MR, Dembele D, Thibault-Carpentier C, Rouach N, Hanauer A (2011) Transcriptome profile reveals AMPA receptor dysfunction in the hippocampus of the Rsk2-knockout mice, an animal model of Coffin-Lowry syndrome. Hum Genet 129:255–269
Merienne K, Pannetier S, Harel-Bellan A, Sassone-Corsi P (2001) Mitogen-regulated RSK2-CBP interaction controls their kinase and acetylase activities. Mol Cell Biol 21:7089–7096
Mila M, Castellvi-Bel S, Sanchez A, Lazaro C, Villa M, Estivill X (1996) Mosaicism for the fragile X syndrome full mutation and deletions within the CGG repeat of the FMR1 gene. J Med Genet 33:338–340
Miller RW, Rubinstein JH (1995) Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 56:112–115
Miyake K, Hirasawa T, Soutome M, Itoh M, Goto Y, Endoh K, Takahashi K, Kudo S, Nakagawa T, Yokoi S, Taira T, Inazawa J, Kubota T (2011) The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: implication for pathogenesis of Rett syndrome. BMC Neurosci 12:81
Moarefi AH, Chedin F (2011) ICF syndrome mutations cause a broad spectrum of biochemical defects in DNMT3B-mediated de novo DNA methylation. J Mol Biol 409:758–772
Modena P, Lualdi E, Facchinetti F, Galli L, Teixeira MR, Pilotti S, Sozzi G (2005) SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 65:4012–4019
Momparler RL, Bovenzi V (2000) DNA methylation and cancer. J Cell Physiol 183:145–154
Montgomery KG, Liu MC, Eccles DM, Campbell IG (2004) The DNMT3B C– > T promoter polymorphism and risk of breast cancer in a British population: a case-control study. Breast Cancer Res 6:R390–R394
Morgan HD, Sutherland HG, Martin DI, Whitelaw E (1999) Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 23:314–318
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA (2011) Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476:298–303
Mudd SH, Cerone R, Schiaffino MC, Fantasia AR, Minniti G, Caruso U, Lorini R, Watkins D, Matiaszuk N, Rosenblatt DS, Schwahn B, Rozen R, LeGros L, Kotb M, Capdevila A, Luka Z, Finkelstein JD, Tangerman A, Stabler SP, Allen RH, Wagner C (2001) Glycine N-methyltransferase deficiency: a novel inborn error causing persistent isolated hypermethioninaemia. J Inherit Metab Dis 24:448–464
Muers MR, Sharpe JA, Garrick D, Sloane-Stanley J, Nolan PM, Hacker T, Wood WG, Higgs DR, Gibbons RJ (2007) Defining the cause of skewed X-chromosome inactivation in X-linked mental retardation by use of a mouse model. Am J Hum Genet 80:1138–1149
Muller H (1930) Types of visible variations induced by X-rays in Drosophila. J Genet 22:299
Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446
Murata T, Kurokawa R, Krones A, Tatsumi K, Ishii M, Taki T, Masuno M, Ohashi H, Yanagisawa M, Rosenfeld MG, Glass CK, Hayashi Y (2001) Defect of histone acetyltransferase activity of the nuclear transcriptional coactivator CBP in Rubinstein-Taybi syndrome. Hum Mol Genet 10:1071–1076
Nan X, Hou J, Maclean A, Nasir J, Lafuente MJ, Shu X, Kriaucionis S, Bird A (2007) Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci USA 104:2709–2714
Nan X, Meehan RR, Bird A (1993) Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res 21:4886–4892
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389
Nance MA, Berry SA (1992) Cockayne syndrome: review of 140 cases. Am J Med Genet 42:68–84
Newman JC, Bailey AD, Fan HY, Pavelitz T, Weiner AM (2008) An abundant evolutionarily conserved CSB-PiggyBac fusion protein expressed in Cockayne syndrome. PLoS Genet 4:e1000031
Newman JC, Bailey AD, Weiner AM (2006) Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci USA 103:9613–9618
Nikitina T, Shi X, Ghosh RP, Horowitz-Scherer RA, Hansen JC, Woodcock CL (2007) Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol Cell Biol 27:864–877
Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD (2009) Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet 41:465–472
Oelke K, Lu Q, Richardson D, Wu A, Deng C, Hanash S, Richardson B (2004) Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum 50:1850–1860
Ogawa H, Gomi T, Takusagawa F, Fujioka M (1998) Structure, function and physiological role of glycine N-methyltransferase. Int J Biochem Cell Biol 30:13–26
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19:219–220
Ostler KR, Davis EM, Payne SL, Gosalia BB, Exposito-Cespedes J, Le Beau MM, Godley LA (2007) Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene 26:5553–5563
Ozdag H, Teschendorff AE, Ahmed AA, Hyland SJ, Blenkiron C, Bobrow L, Veerakumarasivam A, Burtt G, Subkhankulova T, Arends MJ, Collins VP, Bowtell D, Kouzarides T, Brenton JD, Caldas C (2006) Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics 7:90
Pagon RA, Graham JM Jr, Zonana J, Yong SL (1981) Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 99:223–227
Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL (2008) Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet 17:3767–3775
Pampal A (2010) CHARGE: an association or a syndrome? Int J Pediatr Otorhinolaryngol 74:719–722
Park BL, Kim LH, Shin HD, Park YW, Uhm WS, Bae SC (2004) Association analyses of DNA methyltransferase-1 (DNMT1) polymorphisms with systemic lupus erythematosus. J Hum Genet 49:642–646
Park HJ, Yu E, Shim YH (2006) DNA methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma. Cancer Lett 233:271–278
Park YJ, Claus R, Weichenhan D, Plass C (2011) Genome-wide epigenetic modifications in cancer. Prog Drug Res 67:25–49
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, Nikolsky Y, Hartigan J, Smith DR, Gerhard DS, Fults DW, VandenBerg S, Berger MS, Marie SK, Shinjo SM, Clara C, Phillips PC, Minturn JE, Biegel JA, Judkins AR, Resnick AC, Storm PB, Curran T, He Y, Rasheed BA, Friedman HS, Keir ST, McLendon R, Northcott PA, Taylor MD, Burger PC, Riggins GJ, Karchin R, Parmigiani G, Bigner DD, Yan H, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE (2011) The genetic landscape of the childhood cancer medulloblastoma. Science 331:435–439
Pauli S, von Velsen N, Burfeind P, Steckel M, Mänz J, Buchholz A, Borozdin W, Kohlhase J (2011) CHD7 mutations causing CHARGE syndrome are predominantly of paternal origin. Clin Genet 81:234
Paulson HL, Fischbeck KH (1996) Trinucleotide repeats in neurogenetic disorders. Annu Rev Neurosci 19:79–107
Petri M (2002) Epidemiology of systemic lupus erythematosus. Best Pract Res Clin Rheumatol 16:847–858
Petrij F, Dauwerse HG, Blough RI, Giles RH, van der Smagt JJ, Wallerstein R, Maaswinkel-Mooy PD, van Karnebeek CD, van Ommen GJ, van Haeringen A, Rubinstein JH, Saal HM, Hennekam RC, Peters DJ, Breuning MH (2000) Diagnostic analysis of the Rubinstein-Taybi syndrome: five cosmids should be used for microdeletion detection and low number of protein truncating mutations. J Med Genet 37:168–176
Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ et al (1995) Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376:348–351
Petronis A (2010) Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465:721–727
Petrov A, Pirozhkova I, Carnac G, Laoudj D, Lipinski M, Vassetzky YS (2006) Chromatin loop domain organization within the 4q35 locus in facioscapulohumeral dystrophy patients versus normal human myoblasts. Proc Natl Acad Sci USA 103:6982–6987
Pradhan S, Bacolla A, Wells RD, Roberts RJ (1999) Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem 274:33002–33010
Rajender S, Avery K, Agarwal A (2011) Epigenetics, spermatogenesis and male infertility. Mutat Res 727:62–71
Rakyan VK, Blewitt ME, Druker R, Preis JI, Whitelaw E (2002) Metastable epialleles in mammals. Trends Genet 18:348–351
Rakyan VK, Chong S, Champ ME, Cuthbert PC, Morgan HD, Luu KV, Whitelaw E (2003) Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci USA 100:2538–2543
Ramocki MB, Tavyev YJ, Peters SU (2010) The MECP2 duplication syndrome. Am J Med Genet A 152A:1079–1088
Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH (2000) Cockayne syndrome and xeroderma pigmentosum. Neurology 55:1442–1449
Rayasam GV, Wendling O, Angrand PO, Mark M, Niederreither K, Song L, Lerouge T, Hager GL, Chambon P, Losson R (2003) NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J 22:3153–3163
Reed SC (1937) The Inheritance and Expression of Fused, a New Mutation in the House Mouse. Genetics 22:1–13
Reik W (1988) Genomic imprinting: a possible mechanism for the parental origin effect in Huntington’s chorea. J Med Genet 25:805–808
Richardson B (1986) Effect of an inhibitor of DNA methylation on T cells. II. 5-Azacytidine induces self-reactivity in antigen-specific T4+ cells. Hum Immunol 17:456–470
Richardson B, Powers D, Hooper F, Yung RL, O’Rourke K (1994) Lymphocyte function-associated antigen 1 overexpression and T cell autoreactivity. Arthritis Rheum 37:1363–1372
Richardson BC, Strahler JR, Pivirotto TS, Quddus J, Bayliss GE, Gross LA, O’Rourke KS, Powers D, Hanash SM, Johnson MA (1992) Phenotypic and functional similarities between 5-azacytidine-treated T cells and a T cell subset in patients with active systemic lupus erythematosus. Arthritis Rheum 35:647–662
Ridley RM, Frith CD, Crow TJ, Conneally PM (1988) Anticipation in Huntington’s disease is inherited through the male line but may originate in the female. J Med Genet 25:589–595
Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH (2000) Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA 97:13796–13800
Robertson KD (2001) DNA methylation, methyltransferases, and cancer. Oncogene 20:3139–3155
Robertson KD (2005) DNA methylation and human disease. Nat Rev Genet 6:597–610
Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA (1999) The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 27:2291–2298
Roelfsema JH, Peters DJ (2007) Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med 9:1–16
Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, Papadia F, Bacino CA, den Dunnen JT, van Ommen GJ, Breuning MH, Hennekam RC, Peters DJ (2005) Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet 76:572–580
Rubinstein JH, Taybi H (1963) Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child 105:588–608
Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ, Chotai K, Connarty M, Crauford D, Curtis A, Curtis D, Davidson MJ, Differ AM, Dode C, Dodge A, Frontali M, Ranen NG, Stine OC, Sherr M, Abbott MH, Franz ML, Graham CA, Harper PS, Hedreen JC, Hayden MR et al (1996) Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am J Hum Genet 59:16–22
Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ (2006) ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc Natl Acad Sci USA 103:19176–19181
Sabl JF, Laird CD (1992) Epigene conversion: a proposal with implications for gene mapping in humans. Am J Hum Genet 50:1171–1177
Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S (2002) Overexpression of a splice variant of DNA methyltransferase 3b, DNMT3b4, associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc Natl Acad Sci USA 99:10060–10065
Sanka M, Tangsinmankong N, Loscalzo M, Sleasman JW, Dorsey MJ (2007) Complete DiGeorge syndrome associated with CHD7 mutation. J Allergy Clin Immunol 120:952–954
Sasai N, Defossez PA (2009) Many paths to one goal? The proteins that recognize methylated DNA in eukaryotes. Int J Dev Biol 53:323–334
Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI (2000) Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci USA 97:5750–5755
Sawalha AH, Richardson B (2005) DNA Methylation in the Pathogenesis of Systemic Lupus Erythematosus Current Pharmacogenomics 3:73–78
Sawalha AH, Webb R, Han S, Kelly JA, Kaufman KM, Kimberly RP, Alarcon-Riquelme ME, James JA, Vyse TJ, Gilkeson GS, Choi CB, Scofield RH, Bae SC, Nath SK, Harley JB (2008) Common variants within MECP2 confer risk of systemic lupus erythematosus. PLoS One 3:e1727
Scarpa P, Faggioli R, Voghenzi A (1994) Familial Sotos syndrome: longitudinal study of two additional cases. Genet Couns 5:155–159
Schmickel RD, Chu EH, Trosko JE, Chang CC (1977) Cockayne syndrome: a cellular sensitivity to ultraviolet light. Pediatrics 60:135–139
Schmid M, Haaf T, Grunert D (1984) 5-Azacytidine-induced undercondensations in human chromosomes. Hum Genet 67:257–263
Schmidt WM, Sedivy R, Forstner B, Steger GG, Zochbauer-Muller S, Mader RM (2007) Progressive up-regulation of genes encoding DNA methyltransferases in the colorectal adenoma-carcinoma sequence. Mol Carcinog 46:766–772
Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, Zhang X, Song L, Wang Z, Laframboise T, Crawford GE, Scacheri PC (2009) Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res 19:590–601
Scofield RH, Bruner GR, Namjou B, Kimberly RP, Ramsey-Goldman R, Petri M, Reveille JD, Alarcon GS, Vila LM, Reid J, Harris B, Li S, Kelly JA, Harley JB (2008) Klinefelter’s syndrome (47, XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum 58:2511–2517
Shen H, Wang L, Spitz MR, Hong WK, Mao L, Wei Q (2002) A novel polymorphism in human cytosine DNA-methyltransferase-3B promoter is associated with an increased risk of lung cancer. Cancer Res 62:4992–4995
Shibayama A, Cook EH Jr, Feng J, Glanzmann C, Yan J, Craddock N, Jones IR, Goldman D, Heston LL, Sommer SS (2004) MECP2 structural and 3′-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am J Med Genet B Neuropsychiatr Genet 128B:50–53
Simon JA, Kingston RE (2009) Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10:697–708
Singal R, Das PM, Manoharan M, Reis IM, Schlesselman JJ (2005) Polymorphisms in the DNA methyltransferase 3b gene and prostate cancer risk. Oncol Rep 14:569–573
Smeets DF, Moog U, Weemaes CM, Vaes-Peeters G, Merkx GF, Niehof JP, Hamers G (1994) ICF syndrome: a new case and review of the literature. Hum Genet 94:240–246
Smith A, Farrar JR, Silink M, Judzewitsch R (1981) Investigations in dominant Sotos syndrome. Ann Genet 24:226–228
Smith CL, Peterson CL (2005) ATP-dependent chromatin remodeling. Curr Top Dev Biol 65:115–148
Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, Miller DG (2010) Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 6:e1001181
Song J, Rechkoblit O, Bestor TH, Patel DJ (2011) Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331:1036–1040
Sotos JF, Dodge PR, Muirhead D, Crawford JD, Talbot NB (1964) Cerebral Gigantism in Childhood. A Syndrome of Excessively Rapid Growth and Acromegalic Features and a Nonprogressive Neurologic Disorder. N Engl J Med 271:109–116
Srivastava K, Srivastava A, Mittal B (2010) DNMT3B -579 G > T promoter polymorphism and risk of gallbladder carcinoma in North Indian population. J Gastrointest Cancer 41:248–253
Statland JM, Tawil R (2011) Facioscapulohumeral muscular dystrophy: molecular pathological advances and future directions. Curr Opin Neurol 24:423–428
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413:739–743
Stevenson RE, Abidi F, Schwartz CE, Lubs HA, Holmes LB (2000) Holmes-Gang syndrome is allelic with XLMR-hypotonic face syndrome. Am J Med Genet 94:383–385
Suetake I, Shinozaki F, Miyagawa J, Takeshima H, Tajima S (2004) DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem 279:27816–27823
Sun CY, van Koningsbruggen S, Long SW, Straasheijm K, Klooster R, Jones TI, Bellini M, Levesque L, Brieher WM, van der Maarel SM, Jones PL (2011) Facioscapulohumeral muscular dystrophy region gene 1 is a dynamic RNA-associated and actin-bundling protein. J Mol Biol 411:397–416
Takata Y, Huang Y, Komoto J, Yamada T, Konishi K, Ogawa H, Gomi T, Fujioka M, Takusagawa F (2003) Catalytic mechanism of glycine N-methyltransferase. Biochemistry 42:8394–8402
Tang P, Frankenberg S, Argentaro A, Graves JM, Familari M (2011) Comparative analysis of the ATRX promoter and 5′ regulatory region reveals conserved regulatory elements which are linked to roles in neurodevelopment, alpha-globin regulation and testicular function. BMC Res Notes 4:200
Tatton-Brown K, Rahman N (2007) Sotos syndrome. Eur J Hum Genet 15:264–271
Tawil R, Figlewicz DA, Griggs RC, Weiffenbach B (1998) Facioscapulohumeral dystrophy: a distinct regional myopathy with a novel molecular pathogenesis. FSH Consortium. Ann Neurol 43:279–282
Tawil R, Van Der Maarel SM (2006) Facioscapulohumeral muscular dystrophy. Muscle Nerve 34:1–15
Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT (2000) Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet 66:1403–1406
Telenius H, Kremer HP, Theilmann J, Andrew SE, Almqvist E, Anvret M, Greenberg C, Greenberg J, Lucotte G, Squitieri F et al (1993) Molecular analysis of juvenile Huntington disease: the major influence on (CAG)n repeat length is the sex of the affected parent. Hum Mol Genet 2:1535–1540
Thienpont B, de Ravel T, Van Esch H, Van Schoubroeck D, Moerman P, Vermeesch JR, Fryns JP, Froyen G, Lacoste C, Badens C, Devriendt K (2007) Partial duplications of the ATRX gene cause the ATR-X syndrome. Eur J Hum Genet 15:1094–1097
Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM (2008) The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci USA 105:15564–15569
Tiepolo (1979) Multibranched chromosomes 1, 9, and 16 in a patient with combined IgA and IgE deficiency. Hum Genet 51:127–137
Trivier E, De Cesare D, Jacquot S, Pannetier S, Zackai E, Young I, Mandel JL, Sassone-Corsi P, Hanauer A (1996) Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature 384:567–570
Tseng TL, Shih YP, Huang YC, Wang CK, Chen PH, Chang JG, Yeh KT, Chen YM, Buetow KH (2003) Genotypic and phenotypic characterization of a putative tumor susceptibility gene, GNMT, in liver cancer. Cancer Res 63:647–654
Tuck-Muller CM, Narayan A, Tsien F, Smeets DF, Sawyer J, Fiala ES, Sohn OS, Ehrlich M (2000) DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet Cell Genet 89:121–128
Tupler R, Berardinelli A, Barbierato L, Frants R, Hewitt JE, Lanzi G, Maraschio P, Tiepolo L (1996) Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet 33:366–370
Turner BM (2002) Cellular memory and the histone code. Cell 111:285–291
Ueda Y, Okano M, Williams C, Chen T, Georgopoulos K, Li E (2006) Roles for Dnmt3b in mammalian development: a mouse model for the ICF syndrome. Development 133:1183–1192
Urdinguio RG, Sanchez-Mut JV, Esteller M (2009) Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 8:1056–1072
van Deutekom JC, Wijmenga C, van Tienhoven EA, Gruter AM, Hewitt JE, Padberg GW, van Ommen GJ, Hofker MH, Frants RR (1993) FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 2:2037–2042
van Overveld PG, Enthoven L, Ricci E, Rossi M, Felicetti L, Jeanpierre M, Winokur ST, Frants RR, Padberg GW, van der Maarel SM (2005) Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann Neurol 58:569–576
Varela-Rey M, Fernandez-Ramos D, Martinez-Lopez N, Embade N, Gomez-Santos L, Beraza N, Vazquez-Chantada M, Rodriguez J, Luka Z, Wagner C, Lu SC, Martinez-Chantar ML, Mato JM (2009) Impaired liver regeneration in mice lacking glycine N-methyltransferase. Hepatology 50:443–452
Varela-Rey M, Martinez-Lopez N, Fernandez-Ramos D, Embade N, Calvisi DF, Woodhoo A, Rodriguez J, Fraga MF, Julve J, Rodriguez-Millan E, Frades I, Torres L, Luka Z, Wagner C, Esteller M, Lu SC, Martinez-Chantar ML, Mato JM (2010) Fatty liver and fibrosis in glycine N-methyltransferase knockout mice is prevented by nicotinamide. Hepatology 52:105–114
Vasicek TJ, Zeng L, Guan XJ, Zhang T, Costantini F, Tilghman SM (1997) Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics 147:777–786
Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206
Viegas-Pequignot E, Dutrillaux B (1976) Segmentation of human chromosomes induced by 5-ACR (5-azacytidine). Hum Genet 34:247–254
Vilkaitis G, Suetake I, Klimasauskas S, Tajima S (2005) Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J Biol Chem 280:64–72
Villard L, Fontes M, Ades LC, Gecz J (2000) Identification of a mutation in the XNP/ATR-X gene in a family reported as Smith-Fineman-Myers syndrome. Am J Med Genet 91:83–85
Villard L, Gecz J, Mattei JF, Fontes M, Saugier-Veber P, Munnich A, Lyonnet S (1996) XNP mutation in a large family with Juberg-Marsidi syndrome. Nat Genet 12:359–360
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 36:955–957
Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384
Wagner C, Briggs WT, Cook RJ (1985) Inhibition of glycine N-methyltransferase activity by folate derivatives: implications for regulation of methyl group metabolism. Biochem Biophys Res Commun 127:746–752
Wakeland EK, Liu K, Graham RR, Behrens TW (2001) Delineating the genetic basis of systemic lupus erythematosus. Immunity 15:397–408
Wallace LM, Garwick-Coppens SE, Tupler R, Harper SQ (2011) RNA Interference improves myopathic phenotypes in mice over-expressing FSHD region gene 1 (FRG1). Mol Ther 19:2048
Wang J, Walsh G, Liu DD, Lee JJ, Mao L (2006) Expression of Delta DNMT3B variants and its association with promoter methylation of p16 and RASSF1A in primary non-small cell lung cancer. Cancer Res 66:8361–8366
Wang L, Rodriguez M, Kim ES, Xu Y, Bekele N, El-Naggar AK, Hong WK, Mao L, Oh YW (2004) A novel C/T polymorphism in the core promoter of human de novo cytosine DNA methyltransferase 3B6 is associated with prognosis in head and neck cancer. Int J Oncol 25:993–999
Wang L, Wang J, Sun S, Rodriguez M, Yue P, Jang SJ, Mao L (2006) A novel DNMT3B subfamily, DeltaDNMT3B, is the predominant form of DNMT3B in non-small cell lung cancer. Int J Oncol 29:201–207
Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, Workman JL, Crabtree GR (1996) Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J 15:5370–5382
Wang YM, Wang R, Wen DG, Li Y, Guo W, Wang N, Wei LZ, He YT, Chen ZF, Zhang XF, Zhang JH (2005) Single-nucleotide polymorphism in DNA methyltransferase 3B promoter and its association with gastric cardiac adenocarcinoma in North China. World J Gastroenterol 11:3623–3627
Waterland RA, Dolinoy DC, Lin JR, Smith CA, Shi X, Tahiliani KG (2006) Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis 44:401–406
Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ (2004) Epigenetic programming by maternal behavior. Nat Neurosci 7:847–854
Whitelaw NC, Whitelaw E (2008) Transgenerational epigenetic inheritance in health and disease. Curr Opin Genet Dev 18:273–279
Wijmenga C, Hansen RS, Gimelli G, Bjorck EJ, Davies EG, Valentine D, Belohradsky BH, van Dongen JJ, Smeets DF, van den Heuvel LP, Luyten JA, Strengman E, Weemaes C, Pearson PL (2000) Genetic variation in ICF syndrome: evidence for genetic heterogeneity. Hum Mutat 16:509–517
Winship IM (1985) Sotos syndrome–autosomal dominant inheritance substantiated. Clin Genet 28:243–246
Wu Y, Lin JS (2007) DNA methyltransferase 3B promoter polymorphism and its susceptibility to primary hepatocellular carcinoma in the Chinese Han nationality population: a case-control study. World J Gastroenterol 13:6082–6086
Xie ZH, Huang YN, Chen ZX, Riggs AD, Ding JP, Gowher H, Jeltsch A, Sasaki H, Hata K, Xu GL (2006) Mutations in DNA methyltransferase DNMT3B in ICF syndrome affect its regulation by DNMT3L. Hum Mol Genet 15:1375–1385
Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E (1999) Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402:187–191
Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W (2003) The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA 100:10635–10640
Yanagisawa Y, Ito E, Yuasa Y, Maruyama K (2002) The human DNA methyltransferases DNMT3A and DNMT3B have two types of promoters with different CpG contents. Biochim Biophys Acta 1577:457–465
Yang H, Huang ZZ, Zeng Z, Chen C, Selby RR, Lu SC (2001) Role of promoter methylation in increased methionine adenosyltransferase 2A expression in human liver cancer. Am J Physiol Gastrointest Liver Physiol 280:G184–G190
Yao TP, Oh SP, Fuchs M, Zhou ND, Ch’ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R (1998) Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93:361–372
Ye C, Beeghly-Fadiel A, Lu W, Long J, Shu XO, Gao YT, Zheng W, Cai Q (2010) Two-stage case-control study of DNMT-1 and DNMT-3B gene variants and breast cancer risk. Breast Cancer Res Treat 121:765–769
Yen TT, Gill AM, Frigeri LG, Barsh GS, Wolff GL (1994) Obesity, diabetes, and neoplasia in yellow A(vy)/- mice: ectopic expression of the agouti gene. FASEB J 8:479–488
Yeo EJ, Wagner C (1994) Tissue distribution of glycine N-methyltransferase, a major folate-binding protein of liver. Proc Natl Acad Sci USA 91:210–214
Yoon JH, Smith LE, Feng Z, Tang M, Lee CS, Pfeifer GP (2001) Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res 61:7110–7117
Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, Zoghbi HY (2005) Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci USA 102:17551–17558
Yung RL, Quddus J, Chrisp CE, Johnson KJ, Richardson BC (1995) Mechanism of drug-induced lupus. I. Cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol 154:3025–3035
Yung RL, Richardson BC (1994) Drug-induced lupus. Rheum Dis Clin North Am 20:61–86
Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL 3rd, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F (1997) The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90:181–192
Zeng W, de Greef JC, Chen YY, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, Kimonis VE, Balog J, Frants RR, Ball AR Jr, Lock LF, Donovan PJ, van der Maarel SM, Yokomori K (2009) Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 5:e1000559
Zeniou-Meyer M, Begle A, Bader MF, Vitale N (2009) The Coffin-Lowry syndrome-associated protein RSK2 controls neuroendocrine secretion through the regulation of phospholipase D1 at the exocytotic sites. Ann N Y Acad Sci 1152:201–208
Zentner GE, Hurd EA, Schnetz MP, Handoko L, Wang C, Wang Z, Wei C, Tesar PJ, Hatzoglou M, Martin DM, Scacheri PC (2010) CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum Mol Genet 19:3491–3501
Zentner GE, Layman WS, Martin DM, Scacheri PC (2010) Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A 152A:674–686
Zhao M, Wu X, Zhang Q, Luo S, Liang G, Su Y, Tan Y, Lu Q (2010) RFX1 regulates CD70 and CD11a expression in lupus T cells by recruiting the histone methyltransferase SUV39H1. Arthritis Res Ther 12:R227
Zimmermann N, Acosta AM, Kohlhase J, Bartsch O (2007) Confirmation of EP300 gene mutations as a rare cause of Rubinstein-Taybi syndrome. Eur J Hum Genet 15:837–842
Zonana J, Sotos JF, Romshe CA, Fisher DA, Elders MJ, Rimoin DL (1977) Dominant inheritance of cerebral gigantism. J Pediatr 91:251–256
Zraly CB, Dingwall AK (2012) The chromatin remodeling and mRNA splicing functions of the Brahma (SWI/SNF) complex are mediated by the SNR1/SNF5 regulatory subunit. Nucleic Acids Res 40:5975–5987
Acknowledgments
We thank OIB (FYCIT) for editorial assistance. This work has been financially supported by FIS (FI07/00380 to C·H.); the FIS/FEDER (PI11/01728) and the ISCIII (CP11/00131) (to A.F·F.); the Spanish Ministry of Health (PI061267; PS09/02454 to M.F·F.); the Spanish National Research Council (CSIC; 200820I172 to M.F·F.); the Community of Asturias (FYCIT IB09-106 to M.F·F.). The IUOPA is supported by the Obra Social Cajastur, Spain.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huidobro, C., Fernandez, A.F. & Fraga, M.F. The role of genetics in the establishment and maintenance of the epigenome. Cell. Mol. Life Sci. 70, 1543–1573 (2013). https://doi.org/10.1007/s00018-013-1296-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-013-1296-2