
Abstract
The primary cilium protrudes from the cell surface and acts as a sensor for chemical and mechanical growth cues, with receptors for a number of growth factors (PDGFα, Hedgehog, Wnt, Notch) concentrated within the ciliary membrane. In normal tissues, the cilium assembles after cells exit mitosis and is resorbed as part of cell cycle re-entry. Although regulation of the cilium by cell cycle transitions has been appreciated for over 100 years, only recently have data emerged to indicate the cilium also exerts influence on the cell cycle. The resorption/protrusion cycle, regulated by proteins including Aurora-A, VHL, and GSK-3β, influences cell responsiveness to growth cues involving cilia-linked receptors; further, resorption liberates the ciliary basal body to differentiate into the centrosome, which performs discrete functions in S-, G2-, and M-phase. Besides these roles, the cilium provides a positional cue that regulates polarity of cell division, and thus directs cells towards fates of differentiation versus proliferation. In this review, we summarize the specific mechanisms mediating the cilia-cell cycle dialog. We then emphasize the examples of polycystic kidney disease (PKD), nephronopthisis (NPHP), and VHL-linked renal cysts as cases in which defects of ciliary function influence disease pathology, and may also condition response to treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The primary cilium is a microtubule-based organelle that extends from a modified centriolar anchor, termed the basal body, which is located below the surface of the plasma membrane. Although related to the motile flagella found in lower eukaryotes and specialized cell types such as sperm, in mammals most cilia are non-motile. Non-motile or “primary cilia” are organized around a central cytoskeletal core termed the axoneme, composed of nine microtubule doublets arranged in a ring (a 9+0 configuration) [1]. The axoneme is coated with a lipid bilayer and typically protrudes 3–10 μm from the cell surface [2]. Ultrastructural features of the cilium have recently been reviewed in detail by Ishikawa and Marshall [3]. In humans, cells in the majority of tissues have a single non-motile cilium protruding; lymphocytes have none, while a small number of specialized cell types, such as the lung epithelial lining and the reproductive tract, have motile cilia [4, 5].
In tissue culture, the primary cilium is readily observed in non-transformed cells that are serum-starved and confluent, in the stationary (G0) phase of the cell cycle. A number of early studies noted the consistent absence of cilia in mitotic cells, with resorption occurring around the G2/M boundary [6–8]. More recent work, including investigation of the ciliary cycle in many cell types, indicates that ciliary resorption sometimes occurs at earlier stages in the cell cycle, prior to S phase [9–12]. Subsequent studies demonstrated that ciliary resorption occurs in two waves in quiescent cells stimulated to reenter cycle, with a first wave occurring within 1–3 h during G1, and a second wave occurring after ~18 h, as cells enter mitosis [10, 13]. This dynamic profile of ciliary resorption and protrusion led to early debate as to whether the ciliary cycle passively reflected or actively contributed to cell cycle [14]. This debate was given fuel by the observations that oncogenically transformed cancer cell lines and cells in primary tumors, which have deregulated cell growth, commonly lack cilia [15–20], and that mutations eliminating proteins such as IFT88 both eliminate ciliary assembly and are associated with deregulated cell growth [21, 22]. Defects in cilia are now appreciated as contributing to numerous pathological conditions involving cell growth deficiencies, with these including not only cystic kidney diseases but also retinal degeneration, obesity, mental retardation, and developmental malformation [23–28]. For these reasons, understanding the function of cilia is now appreciated to have high medical relevance.
The past decade has been marked by enormous growth in studies of cilia, with the net benefit of elucidating multiple discrete mechanisms for bidirectional signaling between cilia and cell cycle. The field has become too large to summarize in a single review; recent relevant reviews addressing different aspects of ciliary formation and function include Refs. [3, 23]. In this article, we first briefly summarize mechanisms by which cell cycle cues regulate the ciliary cycle, and, reciprocally, mechanisms by which the presence or absence of cilia may influence the cell cycle and oriented cell division. As our main focus, we then describe the relevance of these mechanisms to pathological conditions, focusing in particular on the numerous diseases of the kidney in which a ciliary role is well documented [29–32]. We then summarize the current status of clinical management for these diseases, and explore the hypothesis that defects arising from compromised ciliary function may specifically influence response to treatment.
Regulation of ciliary protrusion and resorption during cell cycle
Cilia extend from the cell surface soon after completion of cytokinesis. As protrusion commences, the centrioles undergo specialized differentiation to become basal bodies. There is a fundamental asymmetry in the age of centrioles in individual post-mitotic cells, arising from the mother–daughter relationship in centriolar duplication [33]. Former mother centrioles are morphologically distinct, containing distal and subdistal appendages, while the daughter does not. After cytokinesis, cilia arise more rapidly in cells inheriting a former mother centriole, and these cells are initially more responsive to Sonic Hedgehog (Shh), which binds receptors displayed on the cilia [34], although this asynchrony is lost over time. Distal appendages contain CEP164, a protein that accumulates at the centrosome at G2/M and has been functionally defined as required for ciliary formation [35], suggesting a role for marks placed on the centriole prior to M phase as contributing to ciliary emergence in G0/G1. The process of protrusion involves the interaction of the interflagellar transport (IFT) machinery with polarized protein secretion, coupled with modification of the ciliary axonome by acetylation [36], and has been thoroughly reviewed [37–39]. As a number of the proteins that have been shown to trigger ciliary resorption (e.g., AURKA, NDE1, discussed below) are abundant in mitosis but inactivated or degraded at cytokinesis, and removal of these proteins is a likely prerequisite of effective ciliogenesis.
Quiescent (G0) cells maintained in culture or organized in tissues, and cells transitioning through G1, retain cilia. Depending on cell type, cilia are preferentially resorbed either at G1/S transition, or prior to mitotic entry, with the latter pattern more commonly reported [9–13] (Fig. 1). In studies with deciliation induced in G0-synchronized hTERT1-RPE1 cells, resorption occurred in two waves, with the loss of ~20 % of cell cilia in late G1, and the remainder during G2/M transition [13]. While serum is commonly used to induce cell cycle reentry, a number of studies have parsed out serum components that are sufficient to trigger ciliary resorption. Among those tested to date, the most critical growth factor for induction of resorption is platelet-derived growth factor (PDGF) [40], which can independently trigger ciliary resorption, and which is required for the process. Insulin growth factor (IGF), epidermal growth factor (EGF), and fibroblast growth factor (FGF) lack this capacity when applied independently, but augment PDGF activity, as can treatment of cells with calcium ionophores to increase cytoplasmic Ca2+ levels [40].

Primary cilia disassembly prior to mitosis. The primary cilium present in G0/G1 phase starts to disassemble at G1-S transition, and is completely resorbed at early prophase. In some descriptions of quiescent cultured cells stimulated to reenter cell cycle by administration of serum, the primary cilium reassembles after disassembly at G1-S and is present during DNA and centriole duplication at S phase, and centriole maturation at G2 phase. Other reports describe two waves of deciliation, with some cells losing cilia prior to G1/S transition and the remainder losing cilia at the end of G2 phase
The action of a number of proteins has been more proximally linked to the regulation of ciliary resorption (Fig. 2). Tctex-1 is a light chain of cytoplasmic dynein complex, which participates in intraflagellar transport (IFT) [41] but also has dynein-independent functions [42]. Two hours following serum-treatment of quiescent, ciliated cells, Tctex-1 is phosphorylated and targeted to the ciliary base, at the transition zone [12]. Although Tctex-1-deficient cells are able to generate primary cilia, these cells are unable to resorb cilia. Tctex-1 is phosphorylated on T94 during the resorption process; a non-phosphorylatable mutant cannot resorb cilia, while overexpression of a T94E phosphomimic accelerates ciliary disassembly. Although Tctex-1 action is not yet well understood, at least part of its activity involves integrity of the actin cytoskeleton, as cytochalasin-D, which inhibits actin polymerization, blocked Tctex-1-dependent disassembly.

Cilia disassembly at the G1-S transition. Activation of Aurora A, NIMA kinases and phosphorylation of Tectex-1 induces cilia resorption, whereas activity of Nde1 and INPPE5 blocks this process. The disassembly of cilia triggers phosphorylation and inactivation of pRb leading to cell cycle progression. Aurora A can be activated by PIFO and by inactivation of VHL to regulate cilia disassembly
Aurora-A (AURKA) in mammals and CALK, an AURKA-like kinase in Chlamydomonas, are each activated at the ciliary basal body immediately prior to ciliary (flagellar) resorption [13, 43]. Experiments involving measurement of ciliary resorption following treatment of mammalian cells with small molecule inhibitors or siRNAs to deplete AURKA, or microinjection of inactive or constitutively active mutants of AURKA, demonstrated that AURKA activity was necessary and sufficient to induce ciliary resorption, paralleling genetic experiments performed in Chlamydomonas. AURKA activity in resorption was reported to involve the phosphorylation and activation of HDAC6, leading to deacetylation and destabilization of the ciliary axoneme. AURKA expression and activity in ciliary resorption are based on interactions with NEDD9, a multifunctional scaffolding protein [44, 45]. AURKA was recently defined to function in some contexts as a Ca2+-regulated kinase [46], although the relationship of this activation mechanism to ciliary resorption has not yet been addressed. More recently, Pitchfork (PIFO) was identified as expressed in the embryonic node, and found to accumulate at the basal body during the disassembly process. Haploinsufficiency of PIFO causes left–right asymmetry and other developmental defects associated with ciliary anomalies. Providing one mechanistic explanation for these defects, a naturally occurring pathogenic R80K mutation of PIFO was found to be unable to activate AURKA, and cells expressing this mutation failed to resorb cilia during cell cycle [47].
Balancing the action of these proteins that promote resorption, other proteins act as brakes on the disassembly process. The tumor suppressor encoded by the von Hippel–Lindau (VHL) locus interacts with glycogen synthase kinase-3 beta (GSK-3β) to support ciliary maintenance [48]. Interestingly, mutation-induced loss of VHL induces the expression of the AURKA-NEDD9 complex [49], and destabilizes cilia. The lipid phosphatase INPP5E, mutated in MORM syndrome, also stabilizes cilia during the disassembly process. Pathogenic mutants of INPP5E both inhibit the ciliary localization of INPP5E and influence ciliary stability during disassembly [50, 51]. There is growing evidence that genes controlling the length of cilia thereby affect the timing of ciliary resorption, given a cell cycle cue. Proteins in this category include members of the NIMA kinase family [52, 53] and the centrosomal protein NDE1 [11].
Influence of ciliary dynamics on cell cycle progression
Whether the presence or absence of a primary cilium controls cell cycle entry, or instead passively reflects cell cycle progression, has long been a topic of discussion [54–57]. This issue is difficult to resolve, because many proteins shown to influence ciliary dynamics have complex functions that might independently explain concurrent changes in cell cycle.
Several studies suggest that the forced retention of cilia imposes a brake on cell cycle progression [11, 12, 58]. For example, the NDE1 phosphoprotein localizes to the mother centriole [11, 59]. The expression of NDE1 is high in M phase but low after cytokinesis, timing which coincides with the burst of primary cilium formation in G0/G1 phase. Following the depletion of NDE1 in hTERT-RPE1 cells, cells have longer cilia, and cell cycle re-entry is delayed. Importantly, abrogation of ciliogenesis by knocking down expression of IFT88 or IFT20 genes was sufficient to reverse this delay in cell cycle re-entry, suggesting the ciliary function of NDE1 was an essential cell cycle determinant [11]. On the other hand, NDE1 also functions cytoplasmically, with roles in mitosis, organellar positioning, and other processes; potentially, knockdown of the IFT genes counteracts a non-ciliary NDE1 function.
In a separate study, knockdown of Tctex-1 expression blocked ciliary resorption and cell cycle progression in G1 upstream of the phosphorylation and inactivation of pRb [12]. This block was seen in the ciliated NIH3T3 or RPE1 cell lines, but not in non-ciliated HeLa cells, and not in RPE1 cell lines with IFT20 or IFT88 knocked down. The authors of this study further showed that knockdown of AURKA or HDAC6 not only blocked Tctex-1-associated ciliary resorption but also blocked new DNA synthesis; as AURKA is not known to have any other essential functions in G1 phase, this suggested the role of AURKA in ciliary resorption was the critical limit on DNA synthesis. As with NDE1, these results may suggest cilia disassembly is a prerequisite for G1-S transition, or alternatively indicate a cytoplasmic action of Tctex-1. Certainly, a mechanistic explanation for how the presence of cilium would constraints activation of G1-S transition is not currently available. Potentially, cilia or the ciliary basal body have the capacity to sequester proteins or other factors that activate G1-S transition, and the resorption of cilia and differentiation of basal body to centrosome releases and/or activates these factors.
Indirect regulation of cell cycle progression through ciliary signaling: growth factor receptors and mechanosensation
Under normal conditions of organismal growth, the primary cilium serves as a unique platform for sensory functions in many organs, including the kidney, eye, nose, and brain. The signaling pathways mediated by cilia are summarized in Fig. 3. Stimulation of cilia-localized receptors by diffusible cues, and mechanical stimulation of the cilia by fluid flow, activate a number of effector pathways that independently or cooperatively contribute to cell cycle control. Some of the better studied of these pathways include receptor tyrosine kinases (RTKs) such as PDGFR, cAMP/mTOR, polycystin/Ca2+, Hedgehog, Wnt, and Notch [60–65].

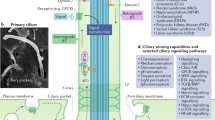
Ciliary signaling pathways implicated in control of cell proliferation. Mechanical sensation of cilia induced by fluid flow activates the LKb1-AMPK pathway in a calcium independent manner and inhibits mTOR1 pathway. Mechanical flow induces activation of PC1 and PC2 and results in elevation of internal calcium level, which suppresses cAMP signaling. In addition, calcium is required for PC1-CT targeting into the nucleus. PC1 also activates JAK2, which induces gene expression that promotes cell cycle progression. Ligand binding of PDGFRα in the cilia activates Raf-ERK pathway. Activation of cAMP signaling promotes PKA and AKAP activity, triggering cell cycle progression, whereas calcium inhibits this pathway. In addition, aberrant activation of wnt or hedgehog pathway stimulates cell cycle progression
PDGF signaling
PDGF (platelet-derived growth factor) regulates cell growth and proliferation for many cell types [66]. In NIH3T3 cells and in vitro culture of mouse embryo fibroblasts (MEFs), serum starvation concurrently induces primary cilium formation and expression of PDGFRα, the receptor for the PDGFαα ligand isoform in this signaling pathway, predominantly within the nascent primary cilium. Ligand binding to PDGFRα activates downstream ERK signaling within the cilium and at the basal body [67], and triggers cells to re-enter cell cycle, as demonstrated by protein phosphorylation of the G1-S checkpoint protein retinoblastoma (Rb). The evidence for the importance of ciliary location for this receptor–ligand interaction is robust. In serum-starved mutant MEF cells derived from the Tg737 mouse, which has no or stumpy cilia due to deficiency in the intraflagellar transport protein ift88, PDGFRα is instead localized in the basal body region. In these cells, addition of ligand does not induce protein phosphorylation of MEK, and cells remain in growth arrest. Ongoing work in normal MEFs and the ift88 model has shown PDGFRα also acts at the cilium to activate the Na+/H+ exchange protein NHE1 to control growth factor-induced chemotaxis [68, 69], implying an organization function for the cytoskeleton that may also support cell cycle signaling.
Polycystins, mechanosensation, and calcium signaling
In the kidney, the cilium serves as a flow sensor in the kidney tubules, with flow-induced ciliary bending causing a transient increase in intracellular calcium [70]. Polycystins (PC) 1 and 2, gene products of PKD1 and PKD2, are large multi-pass transmembrane proteins of the TRPC family of calcium transporters [71]. PC1 and PC2 heterodimerize in the primary cilium to mediate flow-induced calcium flux, with the large extracellular domain of PC1 receiving mechanical cues, then triggering the activity of the PC2 calcium ion channel [72, 73]. Inhibition of PC1 and PC2 activity either by gene mutation (as discussed below) or experimentally through use of antibodies interrupts the calcium flux generated by fluid flow [74]. Because mutations of PKD1 and PKD2 induce cyst formation in the kidney, and cyst formation in part arises because of de-restricted cell proliferation, PC1 and PC2 at least indirectly participate in cell cycle control. Signaling downstream of PC1 and PC2 is complicated. In normal cells, these proteins negatively regulate the cAMP and Raf-MEK-ERK signaling pathways [75], both of which are hyperactivated in renal epithelial cysts [76–78].
Interestingly, it was recently shown that loss of cilia is sufficient to increase intracellular cAMP levels [78]. This was observed when cilia were lost due to null mutation of the IFT motor protein KIF3a, due to maintenance of cells in non-confluent growth conditions, or following other stimuli, specifically linking the phenotype to ciliary loss. The mechanism involved requires further investigation. However, adenylyl cyclases (AC) 5 and 6, the A-kinase anchoring protein AKAP150, and phosphodiesterase (PDE) 4C, a negative regulator of cAMP, are all localized in the cilium and form a complex [78], implying cilia-dependent, spatially localized regulation of cAMP levels. Supporting the relevance of this pathway to disease, the transcription factor HNF-1b regulates PDE4C transcription, and mutation of HNF-1b results in kidney cysts [79]. It is known that calcium regulates AC5/6 activities [80]. Further, the PC2 calcium channel directly interacts with AC5/6, and regulates AC activities [78]. In PC2 mutant mice, the cAMP level in kidney cysts increases, and phosphorylation of the cAMP responsive transcription factor CREB is detected. AC5/6 is not detected in renal cilia expressing a mutant, inactive form of PC2, indicating that AC5/6 requires PC2 for ciliary localization [78]. Assembling this information, the primary cilium constrains cAMP signaling by tethering AC5/6 and regulating PDE4C in the cilium, with AC5/6 activation suppressed by calcium flux mediated by PC2 under fluid flow. The loss or dysfunction of cilia may abrogate calcium signaling to allow activation of cAMP pathway, which promotes cell cycle progression.
Actions of PC1 and PC2 at the cilium do not only involve controlling activity of cAMP. For example, under some conditions, PC1 interacts with PC2 and JAK2 to positively regulate the JAK-STAT pathway; this induces p21, which inhibits Cdk2, leading to cell cycle arrest [81]. PC2 interacts directly with AURKA, which governs ciliary resorption; the increase in cytoplasmic Ca2+ mediated by PC2 very transiently activates AURKA, while AURKA phosphorylation of PC2 limits its Ca2+ channel activity [46, 82]. PC2 may also prevent cell proliferation by physical interaction with the helix-loop-helix protein ID2, an inhibitor of cell differentiation, and blocking its translocation into the nucleus [83]. Interestingly, NEDD9, which supports the action of AURKA in ciliary resorption, has separately been shown to interact with ID2 [84]. In other work, PC1 was found to be cleaved in response to changes in mechanical stimuli, generating a C-terminal fragment that is translocated into the nucleus where it activates AP-1 signaling [85], but also inhibits Wnt/TCF signaling [86]. Apart from their well-described functions at the primary cilium, PC1 and PC2 also localize to the plasma membrane, with a large population of PC2 also found in the endoplasmic reticulum (reviewed in detail in [63]). Although it is clear that localization of a functional PC1/PC2 heterodimer to the cilium is critical for preventing cystogesis, it is possible that roles in other compartments are also important. Targeted experiments blocking traffic of PC1 or PC2 to the primary cilium may provide further insight [87, 88].
LKB1-AMPK-mTOR pathway
Although the mTOR (mammalian target of rapamycin) pathway is well recognized for its role in metabolism, cell cycle and size control, and cell polarity, it has only recently been recognized that mTOR signaling may have an important connection to cilia. A screen for yeast mutants that escaped the cell cycle arrest induced by rapamycin, an inhibitor of the yeast ortholog of mTOR [89, 90], led to the initial identification of the yeast TOR gene [91, 92]. mTOR forms two distinct complexes: the rapamycin-sensitive mTORC1 complex and the rapamycin-insensitive mTORC2 complex. mTORC1 predominantly regulates metabolism and cell growth while mTORC2 regulates cell polarity [89, 90]. Increased intracellular AMP levels activates AMPK to phosphorylate Raptor, which binds and inactivates mTORC1 signaling [93]. AMPK can also indirectly suppress the mTORC1 pathway by phosphorylating the tuberous sclerosis complex (TSC) proteins TSC1 and TSC2, which also negatively regulate mTORC1 [94, 95].
The link of the mTOR pathway to cilia, and the importance of this connection in ciliopathies, has been suggested by several studies. Mutation of the TSC2 gene induces a cystic kidney disease, among other symptoms [96]. Hyperactivation of mTORC1 signaling is observed in PKD-associated cysts, and treatment with rapamycin reduces cyst formation [97]. In a recent study, it was shown that the tumor suppressor LKB1 (also called STK11) kinase phosphorylates AMPK and negatively regulates the mTOR pathway [89]. Intriguingly, a genome-wide RNAi screen identified loss of LKB1 as stimulating ciliary disassembly [98]. Although a specific connection to mTOR or cilia has not yet been explored, it is suggestive that, in lung cancers associated with loss of LKB1, the AURKA activator NEDD9 is highly upregulated [99]. LKB1-deficient MEFs double more rapidly than control MEFs [98, 100], while activation [101] or overexpression [102] of LKB1 induces cell cycle arrest at G1.
The above data demonstrates the role of LKB1 in cell cycle arrest and that LKB1 affects cilia maintenance. Reciprocally, several studies suggest that changes in cilia affect LKB1 functionality. Boehlke et al. [64] have shown the LKB1-AMPK-mTOR pathway is regulated by cilia. First, LKB1 is localized in the primary cilium. Second, under fluid flow conditions, LKB1 activates AMPK at the basal body to suppress the mTOR pathway and downregulate cell size. Since fluid flow induces calcium signaling through PC1 and PC2, but a PC2 mutant does not abrogate activity of the mTORC1 pathway [64], which suggests that calcium signaling is not involved in LKB1 activation. How mechanical sensation by the primary cilium triggered by fluid flow activates LKB1 remains unclear. Collectively, the above data show that the LKB1-AMPK-mTOR pathway is mediated by the cilium and regulates both cell size and cell cycle arrest; more studies of these signaling relations are merited.
Hedgehog and Notch
Cilia control additional pathways with important roles in regulation of cell cycle, cell differentiation, and organogenesis. The obligate existence of cilia for cellular response to Hedgehog (Hh) family proteins was first established by Huangfu and Anderson in 2005, following their observation that mice with mutations in the IFT proteins IFT172 or IFT88 lacked cilia and failed to respond to Hh [103]. Hh is a soluble ligand for the transmembrane receptor protein Patched (Ptc), which in its inactive state localizes to the cilia. Upon Hh binding to Ptc, Ptc moves out of the cilia and ceases to repress a third protein, Smoothened (Smo), which now enters the cilia. Ciliary Smo is then able to activate the Gli transcription factors, allowing their translocation to the nucleus [104]. The Gli transcription factors activate a suite of genes, including cyclins D and E, that contribute to cell cycle transition from G1 to S, and support cell survival [105]. Upregulation of Hh/Gli signaling is observed in a number of forms of cancer, and may be linked in part to the typical loss of cilia in transformed cells. This upregulation, together with the observation that Hh/Gli signaling collaborates with RTK signaling to promote the malignant state, have caused some to propose combination therapies targeting Hh and EGFR [106]. The majority of studies of Hh signaling at cilia have focused on clear developmental roles for these proteins at the neural tube and in the central nervous system (reviewed in [107, 108]), with the potential role of this pathway in cystic kidney disorders not well addressed. Interestingly, a recent RNA interference study identified LKB1 as essential both for Hedgehog and for Wnt signaling (discussed in “Oriented cell division and planar cell polarity”), emphasizing the coordinate regulation of these growth regulatory pathways at intact cilia [98].
A role for the developmental regulator Notch in supporting the differentiation of multiciliated cells was initially established in studies of the development of Xenopus skin and the zebrafish pronephros [109–111], and subsequently shown to be important for the development of the motile multiciliated cells in the mammalian lung [112, 113]. In one recent study, microRNA (miR)-dependent inhibition of Notch was necessary for the emergence of multi-ciliated cells [114]. Notch receptors and processing enzymes localize to the cilia during epidermal differentiation. Cells with mutated IFT genes that lacked cilia were deficient in Notch signaling, failed to differentiate, and were hyperproliferative, with these latter defects rescued by re-expression of pre-activated Notch [62]. Notch represses Multicilin, a recently identified nuclear coiled-coil protein that appears to be a key inducer of a group of genes essential for the formation of multiciliated airway epithelial cells [115]. While Notch is emerging as an important ciliary regulator of the cell proliferation versus differentiation in cell types relevant to ciliopathies, particularly kidney [116], studies of the Notch-cilia connection are at an early stage, with mechanistic details still to emerge. It is also important to note that the Notch studies cited above are predominantly performed in multiciliated cells of the lung epithelium, rather than in cells with non-motile primary cilia: the signaling in these two cell types is not necessarily equivalent.
Oriented cell division and planar cell polarity
During cytokinesis, the cleavage furrow forms in perpendicular to the orientation of the spindle, determining the outcome of the spatial arrangement of two daughter cells. In multicellular organisms, appropriate orientation of cell division is an essential determinant in the morphogenetic process leading to the final architecture for many organized tissues. The kidney, an organ often affected in ciliopathies, is composed of a repeated unit in which a glomerulus (the site of blood filtration) is connected by an extended tubular structure (the proximal convoluted tubule, the loop of Henle, the distal convoluted tubule) to the collecting duct. During tubular development, the proliferation of tubular cells must be precisely controlled to allow tubular lengthening while maintaining a constant tube diameter. A number of studies have suggested the idea that the enforced orientation of cell division parallel to the tubular axis supports normal morphogenesis, while misoriented cell division may cause enlargement rather than lengthening of the tubule [31]. Examination of kidney tubular development in PKD models in the mouse reveals that the orientation of mitotic spindles of tubular cells is parallel to the tubular axis in normal tissues, but significantly distorted in the context of mutations of Tcf2, associated with PKD. Such mis-oriented mitoses appear to occur at an early step in disease pathology, preceding tubular dilation and cyst formation [117].
Defects involving the cilium can influence the orientation of the cell division plane in several ways. The first is based on perturbation of the cilia-centrosome cycle. The resorption of the primary cilium prior to mitosis seems to be essential to release the centriole for maturation into a centrosome capable of nucleating the bipolar mitotic spindle [33]. Normally, the mother centriole that forms the basal body is constrained to the apical cell surface based on interactions of the ciliary distal appendages with the membrane, as well as polarization of secretory apparatus in support of the ciliary membranes [27]. These mechanical constraints are reduced during the ordered disassembly of the primary cilia as cells approach mitosis; nevertheless, some cues governing the localization of the centrosomes are maintained. The loss or structural defects in primary cilia arising from many cyst-associated mutations may result in uncontrolled centrosome positioning, thus increasing randomness in orientation of cell division.
One important recent study has shown that the intraflagellar transport protein IFT88 directly controls spindle orientation through regulation of astral microtubule formation [21]. Although IFT88 has long been known to localize to cilia and basal bodies, it has now been recognized as a constituent of spindle poles [21, 22]. Cells with mutated or RNAi depleted IFT88 are characterized by spindle misorientation, and spindle poles lacking the astral microtubule arrays that interact with the cell cortex to orient the spindle [118, 119]. The formation of astral microtubule arrays requires the movement of peripheral microtubule clusters, dependent upon formation of a large protein complex containing cytoplasmic dynein1 and IFT52. This complex is lost in the absence of IFT88. Thus, IFT88 mediates orientation of cell division by controlling formation of astral microtubules to ensure proper spindle orientation (Fig. 4).

Oriented cell division regulated by cilia. Ciliary protein IFT88 (red ball, semi-transparent) is localized at the basal body of the cilium and at the centriole during spindle formation. Formation of astral microtubules (green lines) requires IFT88, which is essential for proper spindle orientation. Dysfunction of IFT88 results in malformation of astral microtubules leading to misoriented cell division
In contrast, another recent study has addressed function of the IFT protein IFT140 [120]. This work found that, although mutation of IF140 causes defects in cilia formation and formation of renal cysts, in this case pre-cystic dilated tubules have normal orientation of cell division. IFT140 has been much less studied than IFT88; how and if it interacts with the mitotic spindle is not well understood. However, in contrast to IFT88, which functions as a component of IFT complex B, which controls anterograde transport, IFT140 is a component of the IFT complex A, which controls retrograde signaling [121, 122]. Other studies comparing defects linked to mutation or depletion of IFT complex members have indicated weaker phenotypes associated with the targeting of complex A than with the loss of complex B. For example, Tsujikawa and Malicki found degeneration of sensory cells following targeting of the IFT complex B proteins IFT88, IFT52, or IFT57, but not after targeting IFT140 [123]. Earlier work studying C. elegans formation of amphid cilia following mutations of orthologs of IFT88 (osm-5), IFT52 (osm-6), IFT57 (che-13) versus IFT140 (che-11) or IFT122 (daf-10) also indicated greater ciliary defects among the complex B mutants [124]. In this context, the difference between the phenotypes associated with IFT88 and IFT140 mutation for cystogenesis either implies less severe consequences of IFT140, or, alternatively, suggests that defects in cell division orientation are sufficient, but not necessary, for cyst formation.
Tissue architecture is also influenced by planar cell polarity (PCP) controls, which provide an additional means of regulating cell distribution relative to an axis within a plane of growing cells. One mechanism by which this regulation is accomplished is by influencing the migration of cells during development, such that controlled, oriented interdigitation of cells leads to elongation of a tissue along a single axis. This process, termed convergent extension, is important in the formation of multiple tissue types, and has only recently been documented as important in kidney morphogenesis, with defects in this process suggested as a cause of cystogenesis [125–129]. A growing body of evidence suggests that cilia and some cilia-associated proteins are intimately involved in control of PCP, while some PCP regulatory proteins reciprocally influence ciliogenesis and ciliary orientation. This topic has recently been comprehensively reviewed by Wallingford and Mitchell [130].
Canonical [131] and non-canonical [132] signaling downstream of the soluble factor Wnt has an essential role in conditioning many aspects of cell growth and differentiation. PCP signaling involves the activity of the non-canonical Wnt/Frizzled (Fz)/Disheveled (Dvl1) signaling pathway [133, 134]. Inversin, also known as NPHP2, localizes to the cilia and basal body, and is mutated in the cystic kidney disease nephronophthisis. Simons et al. [125] first showed that a direct interaction between inversin and Dvl1 promotes non-canonical Wnt signaling, with defects in this pathway proposed to support the abnormal cell growth that characterizes cyst formation. In contrast, cilia downregulate the amplitude of canonical Wnt signaling, in part through the action of the ciliopathy-associated protein Jouberin in blocking nuclear entry of the canonical Wnt effector β-catenin [135]. BBS-associated proteins are associated with control of cilia; some BBS proteins interact directly with and regulate the function of the PCP signaling effector Vangl2 [136]. Conversely, PCP signaling proteins can influence ciliary dynamics. For example, knockdown of the PCP effector Fritz in Xenopus embryos brings about defects in PCP-mediated convergent extension as well as defects in ciliogenesis [137]. However, as discussed in depth in Ref. [130], the interactions between PCP signaling proteins and cilia are complicated, and in some cases do not support crosstalk as a direct cause of cystogenesis.
Alternatively, defects in cilia may affect PCP downstream of non-canonical Wnt signaling, at the level of control of the polarization of the cytoskeleton to support directional movement. The centrosome has many functions that impinge upon cell polarization in migration (reviewed in [138]). A number of proteins that affect ciliary formation and function have also been shown to affect placement of the centrosome. Besides regulating Dvl1, loss of Inversin also induces randomly oriented cell division prior to the detection of increased rates of proliferation in nascent kidney cysts [139]. Analysis of precystic tubules in Kif3a mutant mice reveals that cells lacking primary cilia have an abnormality in PCP that was suggested to initiate cyst formation [140]. IFT20 [141] and IFT88 [142] have both been shown to directly impact cell polarization in some tissues. MKS1 and meckelin are essential for ciliogenesis, but also influence centrosomal migration based on coordinated interactions with the actin cytoskeleton [143]. Given the various roles of basal body/centrosomal structures in polarizing the mitotic plane, organizing polarized cell migration, and providing a platform to integrate the function of proteins that signal to indirectly influence these processes, it is hard to prioritize which if any functions are dominant.
We note that, in sum, the polarity controls discussed in this section would seem to address the qualitative features of cell organization rather than control of cell proliferation. However, these qualitative changes result in dilated tubules in which cells are exposed to different fluid pressures, and have altered their ability to respond to chemotactic cues because of defects in orientation of the internal cytoskeleton to the cilia, or in orientation of the cilia to the direction of fluid flow. Hence, they ultimately have the potential to support the abnormal proliferation that occurs in cystic growth.
The contribution of ciliary defects to human renal diseases
Given the dense web of connections between cilia and control of cell growth described above, it is not surprising that defects in cilia have been implicated in multiple clinically significant pathologies, involving developmental defects and later onset syndromes. Established ciliopathies include Bardet–Biedl Syndrome (BBS), Joubert syndrome (JBTS), Meckel–Gruber syndrome (MKS), polycystic kidney disease (PKD), and nephronophthisis (NPHP) [32]. Many of these rare autosomal recessive diseases present with phenotypes affecting multiple organs, with symptoms that include renal cysts, neural tube defects, retinal degeneration, obesity, and situs inversus. A recent proteomic analysis of proteins co-purifying with 9 proteins known to be mutated in NPHP, JBTS, or MKS syndrome identified 850 interactive partners, with dense physical connections to proteins functioning in cell polarity (apical positioning) and centrosome control, and Hedgehog signaling [144]. A targeted search for mutations affecting these genes in a first set of 250 patients with symptoms associated with NPHP, JBTS, or MKS identified several novel causative lesions [144]; it is likely that more will be identified in the future. We here take cystic kidney diseases as an example of how ciliary defects translate to pathological response, and discuss how current approaches to treatment align with what is known about ciliary biology.
PKD and NPHP
Cystic kidney diseases represent a significant group of genetically inherited renal disorders [145]. A kidney cyst is a sac-like structure comprised of a liquid-filled monolayer of cells. Formation of a cyst represents the integrated endpoint of multiple stimuli leading to abnormal cell division among the epithelial cells lining the collecting ducts and convoluted tubules of the kidney. The development of cysts eventually leads to end-stage renal disease (ESRD) [146], at which point patients afflicted with these conditions must either undergo dialysis or receive kidney transplants. The majority of cystic kidney diseases arise from inheritance of mutations asociated with autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD), or NPHP, with more sporadic linkage to BBS [145, 147].
Although the genetic deficiencies leading to formation of kidney cysts are diverse, most mutated genes have a close connection to control of ciliary structure and function [29, 145, 148, 149], and localization to the cilia or basal body [29, 31, 145, 150–153], although many also function at other cellular locations (e.g., PC1 at cell junctions; [154]). Mutations in the PKD1 or PKD2 genes cause ADPKD, which is the most common cystic kidney disorder, affecting approximately 1 in 500. The PKD1 and PKD2-encoded protein products (polycystins 1 and 2) heterodimerize on cilia to integrate growth-limiting signals initiating from fluid pressure; defects in cilia impair this function [74]. Reciprocally, mutations in these genes have recently been found to cause defects in formation of cilia and/or centrosomes [155–157]. ARPKD and NPHP are much less common than ADPKD, with incidence estimated at around 1 in 5,000 for ARPKD and less than 1 in 50,000 for NPHP. ARPKD arises from mutations in the PKHD1 gene, which encodes the ciliary protein fibrocystin [158]. Mutations in at least 11 independent genes of the NPHP group have been found to lead to NPHP. Many of these proteins have already been shown to localize to cilia or centrioles, and to contribute to signaling at these structures [159].
von Hippel–Lindau (VHL) disease
VHL disease, caused by germline mutation in the VHL tumor suppressor gene, is an autosomal dominant genetic disease that has in the past been thought of predominantly in terms of hereditary cancer. The most clinically significant manifestation of the disease includes tumors in the central nervous system, retina, and kidney, including hemangioblastomas, pheochromocytomas, and renal cell carcinomas [160]. Interestingly, around 70 % of VHL patients also develop renal cysts [161, 162], a fact that has more recently led to targeted investigations of whether VHL might have a specific function at cilia, as with other cyst-associated proteins. Indeed, pVHL localizes to cilia [48, 163]. Further, both the kidney cysts found in VHL patients, as well as renal clear cell carcinoma cell lines lacking pVHL, have either no cilia or sparse, rudimentary cilia [164]. Importantly, ectopic expression of VHL gene in renal clear cell carcinoma cell lines restored cilia formation [163–165], implying that pVHL might directly support ciliogenesis.
In one elegant study, combined inactivation of VHL and GSK-3β was required to allow loss of cilia, based on cooperative function of these proteins in ciliary maintenance [48]. Although the mechanism by which these proteins interact is not yet clear, VHL normally promotes the proteasomal degradation of HIF-1α, a protein that activates transcription in hypoxic cells. Maxwell and colleagues showed that suppression of HIF-1a in VHL-deficient cell lines restored cilia [164]; separately, GSK-3β triggers HIF-1α degradation [166]. The absence of cilia mediated by pVHL may be due to loss of cilia maintenance or defects in ciliogenesis. Benzing and colleagues found that pVHL regulates microtubule orientation during ciliogenesis and also interacts with the Par3-Par6-atypical PKC complex, which supports ciliogenesis [163, 167]. pVHL also associates with kinesin 2, allowing pVHL to influence microtubule dynamics in support of cilia [168, 169]. Some evidence suggests that pVHL also negatively regulates ciliary disassembly. AURKA and its homologue CALK in Chlamydomonas are necessary and sufficient for cilia disassembly [13, 43]. Loss of VHL activates HIF signaling to increase the expression of AURKA and its activating partner NEDD9 [170]; suppression of this pathway improves formation of cilia [49].
Cilia and cysts
Cementing the requirement for intact ciliary signaling in cystic disease, cyst formation can be experimentally induced by mutation of genes that are essential for cilia assembly and maintenance, such as IFT88 [22] and the cilia-associated kinesin II, KIF3A [171]. Subsequent investigation of the consequences of the inactivation of KIF3A in young versus adult mice emphasized the requirement of kidney injury as a co-factor, and suggested an important consequence of ciliary inactivation was loss of PCP controls rather than a change in proliferation [140]. As noted above, one consequence of mutations relevant to clinically significant human cystic diseases is to impair ciliary function, and by extension to influence all of the cilia-associated, cell division regulating processes summarized throughout this review. In spite of the complexity this already invokes, it is important to note that proteins such as the polycystins, targeted in PKD, also have well-documented functions in cellular compartments other than cilia, acting at the endoplasmic reticulum, the plasma membrane, and at focal adhesions [63, 172]. The inevitably pleiotropic consequences of mutations eliminating polycystins makes it essential to move cautiously in assigning proliferation defects solely to ciliary deficiency, when considering the clinical management of “ciliary” diseases. Certainly, much more careful study is required to dissect relevant functions.
Targeted treatments
With the above cautions in mind, it is instructive to consider how the rapidly increasing knowledge of the root causes of diseases such as renal cysts impacts their management in the clinic, taking PKD as primary example. For additional perspective, recent reviews addressing this topic at length include Refs. [173–175]. For many years, the standard of care has been primarily management of the symptoms associated with the disease, rather than addressing the root causes. As of 2012, the patient-directed description of treatment options assembled by the National Kidney and Urologic Disease Information Clearinghouse (NKUDIC), managed by the US National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), suggested to patients that they manage pain associated with growing cysts by using aspirin or acetaminophen; control associated urinary tract infections with antibiotics; control high blood pressure by lifestyle changes or standard medications; and prepare for ultimate ESRD, which will require dialysis or kidney transplant (http://www.kidney.niddk.nih.gov/kudiseases/pubs/polycystic/#dominant).
In contrast, Table 1 summarizes clinical and preclinical evaluation that have either recently been completed or are currently in progress, and Table 2 summarizes rodent models used for preclinical work. Beginning with the more advanced clinical studies, a first theme has been to continue to address the symptoms, but with a more advanced underpinning of molecular biology. For example, the HALT-PKD study evaluates efficacy of the angiotensin-converting enzyme (ACE) inhibitor lisinopril alone or in conjunction with the angiotensin II receptor antagonist Telmisartan, to more efficiently lower blood pressure (described in [176]).
Other studies have addressed signaling effects classically associated with PKD. Deficient activity of the PC2 Ca2+ channel at the cilia reduces intracellular Ca2+ levels in PKD. Reduced Ca2+-dependent inhibition of adenylate cyclase (AC) helps elevate intracellular levels of cAMP; see also direct activation of AC through interaction with the AKAP150 complex and phosphodiesterase, discussed in [78] and above. In turn, elevation of cAMP contributes to activation of PKA signaling, which supports SRC and other RTK-dependent signaling effectors, and stimulates activity of the Cl− efflux channel CFTR, among other consequences [177]. A number of studies experimentally limit AC activity by targeting proteins associated with the AC-activating complex, using therapies such as somatostatin [178, 179] and vasopressin V2R antagonists [180–183]. For the trials with somatostatin, a significant decrease in the rate of kidney growth was observed, but only limited efficacy in improving kidney function. PKA effector limitation is also the theme of more recent pre-clinical studies, in which the point of intervention is inhibition of CFTR [184]. Triptolide, the active ingredient of an ancient Chinese medicine known as Lei Gong Teng, enhances PC2 channel activity, potentially restoring normal homeostasis in cases of PKD not directly associated with inactivating mutations of PC2 [185]; a clinical trial of triptolide is in progress.
An alternative approach in designing trials has been to target signaling pathways canonically associated with the proliferative state of the cell, which are active in cystogenesis, and for which the degree of connection to cilia has been discussed above. Bosutinib inhibits multiple tyrosine kinases, including SRC [186]; a trial is ongoing. Sirolimus and everolimus inhibit mTOR, which is essential for protein synthesis, and known to be regulated by PC1 and TSC, which have cilia-linked functions, making it natural to evaluate these compounds in PKD [187–189]. Disappointingly, although some activity was seen in reducing kidney growth, there was no significant change in kidney function; one study found an increased level of proteinuria and albuminuria in sirolimus-treated patients, and increased adverse events were reported with treatment. It has been suggested that a longer time of treatment would be beneficial; alternatively, this may increase the side effects of mTOR inhibitors in non-renal cells. mTOR-directed therapies are discussed at length in Refs. [190, 191]. For all the advanced or completed trials, an emergent theme is the non-congruence between results in reducing the rate of kidney or cystic growth, which can often be partially achieved, and results in improving renal function, which is clearly harder to achieve. Identifying the reasons for these differences requires more study.
As is clear from Table 1, a number of other pathway-directed therapies are now being pre-clinically evaluated for action in PKD, and may lead to trials in coming years. These generally follow the same approach as more advanced studies. Targets of therapies include: metformin, targeting AMPK [192]; roscovitine, targeting CDK kinases [193]; STAT6 [194]; the PPAR-γ receptor [195–197]; HDACs [198, 199]; MEK [200]; Raf [201]; Cdc25A [202]; and other agents. A number of these agents were previously developed for other disease indications, such as treatment of cancer, and are being explored based on their ability to inhibit cell proliferation. Given the discussion above as to whether it is proliferation per se, or qualitatively altered cell growth associated with ciliary defects (e.g., PCP changes), that is the predominant cause of PKD, it is currently an open question as to how well these compounds will work.
Another pragmatic issue is how patients will respond to long-term treatment with powerful agents that interfere with cell growth controls. Much care will be necessary to prevent compensatory changes that lead to severe adverse events, such as induction of cancer. Use of therapies targeted at proteins that specifically act in the kidney would reduce this last concern; as proteomic studies of cilia-associated proteins [144, 203–205] and tissue-specific protein expression and activation [206] advance, these data may help guide target selection. On the other hand, because PKD arises from an autosomally inherited dominant mutation, the mutant allele for PKD is found in every cell of the body, and some recent studies have documented striking cell cycle defects in non-renal cells associated with PKD lesions. For instance, endothelial cells from humans and mice with PKD were characterized by abnormal cilia and deficient cytokinesis [174, 207]. Based on these recent findings, it is plausible that the hypertension associated with PKD may not just be a secondary consequence of aberrant cell growth and signaling in the kidney but may arise directly from cilia-associated defects in the vasculature. If so, a kidney-focused therapeutic strategy would be inadequate. A potentially promising approach would be to try to correct the deficient function of PC2 specifically at the cilium. For instance, the observed AURKA activation in cysts is predicted to help suppress PC2 calcium channel activity [82]; inhibition of AURKA might be expected to enhance PC2 activity. Alternatively, as discussed at length above, cilia coordinate signaling of PDGFα, Wnt, Hedgehog, Notch, and other pathways; to date, there has been little effort to explore the consequences of manipulating these pathways for therapeutic gain in PKD. There is clearly much room for investigation of potential strategies.
References
Sorokin S (1962) Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol 15:363–377
Wheatley DN, Bowser SS (2000) Length control of primary cilia: analysis of monociliate and multiciliate PtK1 cells. Biol Cell 92:573–582
Ishikawa H, Marshall WF (2011) Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol 12:222–234
Wheatley DN (1995) Primary cilia in normal and pathological tissues. Pathobiology 63:222–238
Eley L, Yates LM, Goodship JA (2005) Cilia and disease. Curr Opin Genet Dev 15:308–314
Fonte VG, Searls RL, Hilfer SR (1971) The relationship of cilia with cell division and differentiation. J Cell Biol 49:226–229
Archer FL, Wheatley DN (1971) Cilia in cell-cultured fibroblasts. II. Incidence in mitotic and post-mitotic BHK 21–C13 fibroblasts. J Anat 109:277–292
Henneguy LF (1898) Sur le rapports des cils vibratiles avec les centrosomes. Arch Anat Microsc Morphol Exp 1:481
Rieder CL, Jensen CG, Jensen LC (1979) The resorption of primary cilia during mitosis in a vertebrate (PtK1) cell line. J Ultrastruct Res 68:173–185
Tucker RW, Pardee AB, Fujiwara K (1979) Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell 17:527–535
Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L (2011) Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol 13:351–360
Li A, Saito M, Chuang JZ, Tseng YY, Dedesma C, Tomizawa K, Kaitsuka T, Sung CH (2011) Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol 13:402–411
Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA (2007) HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129:1351–1363
Bloodgood RA (2009) From central to rudimentary to primary: the history of an underappreciated organelle whose time has come. The primary cilium. Methods Cell Biol 94:3–52
Barakat MT, Scott MP (2009) Tail wags dog: primary cilia and tumorigenesis. Cancer Cell 16:276–277
Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15:1062–1065
Moser JJ, Fritzler MJ, Rattner JB (2009) Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer 9:448
Schraml P, Frew IJ, Thoma CR, Boysen G, Struckmann K, Krek W, Moch H (2009) Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod Pathol 22:31–36
Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M (2009) Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res 69:422–430
Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr, Dlugosz AA, Reiter JF (2009) Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med 15:1055–1061
Delaval B, Bright A, Lawson ND, Doxsey S (2011) The cilia protein IFT88 is required for spindle orientation in mitosis. Nat Cell Biol 13:461–468
Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 151:709–718
Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11:331–344
Gerdes JM, Davis EE, Katsanis N (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137:32–45
Pan J (2008) Cilia and ciliopathies: from Chlamydomonas and beyond. Sci China C Life Sci 51:479–486
Fliegauf M, Benzing T, Omran H (2007) When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 8:880–893
Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139:663–678
Pazour GJ, Rosenbaum JL (2002) Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol 12:551–555
Pazour GJ (2004) Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol 15:2528–2536
Zhang Q, Taulman PD, Yoder BK (2004) Cystic kidney diseases: all roads lead to the cilium. Physiology (Bethesda) 19:225–230
Simons M, Walz G (2006) Polycystic kidney disease: cell division without a c(l)ue? Kidney Int 70:854–864
Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. N Engl J Med 364:1533–1543
Nigg EA, Stearns T (2011) The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13:1154–1160
Anderson CT, Stearns T (2009) Centriole age underlies asynchronous primary cilium growth in mammalian cells. Curr Biol 19:1498–1502
Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA (2007) Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol 179:321–330
Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV (2010) The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci USA 107:21517–21522
Pedersen LB, Rosenbaum JL (2008) Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol 85:23–61
Avasthi P, Marshall WF (2011) Stages of ciliogenesis and regulation of ciliary length. Differentiation 83:S30–S42
Azimzadeh J, Marshall WF (2011) Building the centriole. Curr Biol 20:R816–R825
Tucker RW, Scher CD, Stiles CD (1979) Centriole deciliation associated with the early response of 3T3 cells to growth factors but not to SV40. Cell 18:1065–1072
Pazour GJ, Wilkerson CG, Witman GB (1998) A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT). J Cell Biol 141:979–992
Chuang JZ, Yeh TY, Bollati F, Conde C, Canavosio F, Caceres A, Sung CH (2005) The dynein light chain Tctex-1 has a dynein-independent role in actin remodeling during neurite outgrowth. Dev Cell 9:75–86
Pan J, Wang Q, Snell WJ (2004) An aurora kinase is essential for flagellar disassembly in Chlamydomonas. Dev Cell 6:445–451
Singh M, Cowell L, Seo S, O’Neill G, Golemis E (2007) Molecular basis for HEF1/NEDD9/Cas-L action as a multifunctional co-ordinator of invasion, apoptosis and cell cycle. Cell Biochem Biophys 48:54–72
Tikhmyanova N, Tulin AV, Roegiers F, Golemis EA (2010) Dcas supports cell polarization and cell–cell adhesion complexes in development. PLoS One 5:e12369
Plotnikova OV, Pugacheva EN, Dunbrack RL, Golemis EA (2010) Rapid calcium-dependent activation of Aurora-A kinase. Nat Commun 1:64
Kinzel D, Boldt K, Davis EE, Burtscher I, Trumbach D, Diplas B, Attie-Bitach T, Wurst W, Katsanis N, Ueffing M, Lickert H (2010) Pitchfork regulates primary cilia disassembly and left-right asymmetry. Dev Cell 19:66–77
Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W (2007) pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol 9:588–595
Xu J, Li H, Wang B, Xu Y, Yang J, Zhang X, Harten SK, Shukla D, Maxwell PH, Pei D, Esteban MA (2010) VHL inactivation induces HEF1 and Aurora kinase A. J Am Soc Nephrol 21:2041–2046
Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compere P, Schiffmann SN, Gergely F, Riley JH, Perez-Morga D, Woods CG, Schurmans S (2009) INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 41:1027–1031
Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, Kayserili H, Swistun D, Scott LC, Bertini E, Boltshauser E, Fazzi E, Travaglini L, Field SJ, Gayral S, Jacoby M, Schurmans S, Dallapiccola B, Majerus PW, Valente EM, Gleeson JG (2009) Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41:1032–1036
Quarmby LM, Mahjoub MR (2005) Caught Nek-ing: cilia and centrioles. J Cell Sci 118:5161–5169
Wloga D, Camba A, Rogowski K, Manning G, Jerka-Dziadosz M, Gaertig J (2006) Members of the NIMA-related kinase family promote disassembly of cilia by multiple mechanisms. Mol Biol Cell 17:2799–2810
Quarmby LM, Parker JD (2005) Cilia and the cell cycle? J Cell Biol 169:707–710
Plotnikova OV, Pugacheva EN, Golemis EA (2009) Primary cilia and the cell cycle. Methods Cell Biol 94:137–160
Kim S, Tsiokas L (2011) Cilia and cell cycle re-entry: more than a coincidence. Cell Cycle 10:2683–2690
Pan J, Snell W (2007) The primary cilium: keeper of the key to cell division. Cell 129:1255–1257
Jackson PK (2011) Do cilia put brakes on the cell cycle? Nat Cell Biol 13:340–342
Hirohashi Y, Wang Q, Liu Q, Li B, Du X, Zhang H, Furuuchi K, Masuda K, Sato N, Greene MI (2006) Centrosomal proteins Nde1 and Su48 form a complex regulated by phosphorylation. Oncogene 25:6048–6055
Wong SY, Reiter JF (2008) The primary cilium at the crossroads of mammalian hedgehog signaling. Curr Top Dev Biol 85:225–260
Christensen ST, Clement CA, Satir P, Pedersen LB (2012) Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J Pathol 226:172–184
Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E (2011) A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145:1129–1141
Gallagher AR, Germino GG, Somlo S (2010) Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 17:118–130
Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Godel M, Muller K, Herbst M, Hornung M, Doerken M, Kottgen M, Nitschke R, Igarashi P, Walz G, Kuehn EW (2010) Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol 12:1115–1122
Ajima R, Hamada H (2011) Wnt signalling escapes to cilia. Nat Cell Biol 13:636–637
Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79:1283–1316
Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST (2005) PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15:1861–1866
Schneider L, Stock CM, Dieterich P, Jensen BH, Pedersen LB, Satir P, Schwab A, Christensen ST, Pedersen SF (2009) The Na+/H+ exchanger NHE1 is required for directional migration stimulated via PDGFR-alpha in the primary cilium. J Cell Biol 185:163–176
Schneider L, Cammer M, Lehman J, Nielsen SK, Guerra CF, Veland IR, Stock C, Hoffmann EK, Yoder BK, Schwab A, Satir P, Christensen ST (2010) Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cell Physiol Biochem 25:279–292
Praetorius HA, Spring KR (2001) Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 184:71–79
Delmas P (2004) Polycystins: from mechanosensation to gene regulation. Cell 118:145–148
Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG (2000) Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408:990–994
Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S (2002) Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 4:191–197
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33:129–137
Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP (2004) Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279:40419–40430
Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ (2003) Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int 63:1983–1994
Calvet JP (2008) Strategies to inhibit cyst formation in ADPKD. Clin J Am Soc Nephrol 3:1205–1211
Choi YH, Suzuki A, Hajarnis S, Ma Z, Chapin HC, Caplan MJ, Pontoglio M, Somlo S, Igarashi P (2011) Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc Natl Acad Sci USA 108:10679–10684
Igarashi P, Shao X, McNally BT, Hiesberger T (2005) Roles of HNF-1beta in kidney development and congenital cystic diseases. Kidney Int 68:1944–1947
Hanoune J, Defer N (2001) Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol 41:145–174
Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu PN, Germino FJ, Germino GG (2002) PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 109:157–168
Plotnikova OV, Pugacheva EN, Golemis EA (2011) Aurora A kinase activity influences calcium signaling in kidney cells. J Cell Biol 193:1021–1032
Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J (2005) Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol 7:1202–1212
Law SF, Zhang YZ, Fashena SJ, Toby G, Estojak J, Golemis EA (1999) Dimerization of the docking/adaptor protein HEF1 via a carboxy-terminal helix-loop-helix domain. Exp Cell Res 252:224–235
Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, Caplan MJ (2004) Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest 114:1433–1443
Lal M, Song X, Pluznick JL, Di Giovanni V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ (2008) Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet 17:3105–3117
Geng L, Okuhara D, Yu Z, Tian X, Cai Y, Shibazaki S, Somlo S (2006) Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci 119:1383–1395
Hoffmeister H, Babinger K, Gurster S, Cedzich A, Meese C, Schadendorf K, Osten L, de Vries U, Rascle A, Witzgall R (2011) Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J Cell Biol 192:631–645
Yang Q, Guan KL (2007) Expanding mTOR signaling. Cell Res 17:666–681
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124:471–484
Heitman J, Movva NR, Hall MN (1991) Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253:905–909
Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN (1993) Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 73:585–596
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226
Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL (2004) Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 18:1533–1538
Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC (2004) The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6:91–99
Brook-Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, Nellist M, Gamble V, Harris PC, Sampson JR (1994) Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease–a contiguous gene syndrome. Nat Genet 8:328–332
Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T (2006) The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 103:5466–5471
Jacob LS, Wu X, Dodge ME, Fan CW, Kulak O, Chen B, Tang W, Wang B, Amatruda JF,Lum L (2011) Genome-wide RNAi screen reveals disease-associated genes that are common to Hedgehog and Wnt signaling. Sci Signal, 4, ra4
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, Bao L, Contreras CM, Li D, Chen L, Krishnamurthy J, Koivunen J, Chirieac LR, Padera RF, Bronson RT, Lindeman NI, Christiani DC, Lin X, Shapiro GI, Janne PA, Johnson BE, Meyerson M, Kwiatkowski DJ, Castrillon DH, Bardeesy N, Sharpless NE, Wong KK (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature 448:807–810
Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, Loda M, Carrasco DR, DePinho RA (2002) Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature 419:162–167
Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18:283–293
Tiainen M, Ylikorkala A, Makela TP (1999) Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA 96:9248–9251
Huangfu D, Anderson KV (2005) Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA 102:11325–11330
Rohatgi R, Milenkovic L, Scott MP (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317:372–376
Hui CC, Angers S (2011) Gli proteins in development and disease. Annu Rev Cell Dev Biol 27:513–537
Mangelberger D, Kern D, Loipetzberger A, Eberl M, Aberger F (2012) Cooperative Hedgehog-EGFR signaling. Front Biosci 17:90–99
Louvi A, Grove EA (2011) Cilia in the CNS: the quiet organelle claims center stage. Neuron 69:1046–1060
Drummond IA (2012) Cilia functions in development. Curr Opin Cell Biol 24:24–30
Stubbs JL, Davidson L, Keller R, Kintner C (2006) Radial intercalation of ciliated cells during Xenopus skin development. Development 133:2507–2515
Ma M, Jiang YJ (2007) Jagged2a-notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genet 3:e18
Liu Y, Pathak N, Kramer-Zucker A, Drummond IA (2007) Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development 134:1111–1122
Morimoto M, Liu Z, Cheng HT, Winters N, Bader D, Kopan R (2010) Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci 123:213–224
Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV (2009) Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development 136:2297–2307
Marcet B, Chevalier B, Luxardi G, Coraux C, Zaragosi LE, Cibois M, Robbe-Sermesant K, Jolly T, Cardinaud B, Moreilhon C, Giovannini-Chami L, Nawrocki-Raby B, Birembaut P, Waldmann R, Kodjabachian L, Barbry P (2011) Control of vertebrate multiciliogenesis by miR-449 through direct repression of the Delta/Notch pathway. Nat Cell Biol 13:693–699
Stubbs JL, Vladar EK, Axelrod JD, Kintner C (2011) Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat Cell Biol 14:140–147
Sirin Y, Susztak K (2012) Notch in the kidney: development and disease. J Pathol 226:394–403
Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M (2006) Defective planar cell polarity in polycystic kidney disease. Nat Genet 38:21–23
Palmer RE, Sullivan DS, Huffaker T, Koshland D (1992) Role of astral microtubules and actin in spindle orientation and migration in the budding yeast, Saccharomyces cerevisiae. J Cell Biol 119:583–593
Toyoshima F, Nishida E (2007) Integrin-mediated adhesion orients the spindle parallel to the substratum in an EB1- and myosin X-dependent manner. EMBO J 26:1487–1498
Jonassen JA, Sanagustin J, Baker SP, Pazour GJ (2012) Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J Am Soc Nephrol 23(4):641–651
Pedersen LB, Geimer S, Sloboda RD, Rosenbaum JL (2003) The Microtubule plus end-tracking protein EB1 is localized to the flagellar tip and basal bodies in Chlamydomonas reinhardtii. Curr Biol 13:1969–1974
Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL (1998) Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol 141:993–1008
Tsujikawa M, Malicki J (2004) Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron 42:703–716
Perkins LA, Hedgecock EM, Thomson JN, Culotti JG (1986) Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev Biol 117:456–487
Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37:537–543
Yates LL, Papakrivopoulou J, Long DA, Goggolidou P, Connolly JO, Woolf AS, Dean CH (2010) The planar cell polarity gene Vangl2 is required for mammalian kidney-branching morphogenesis and glomerular maturation. Hum Mol Genet 19:4663–4676
Lienkamp S, Ganner A, Boehlke C, Schmidt T, Arnold SJ, Schafer T, Romaker D, Schuler J, Hoff S, Powelske C, Eifler A, Kronig C, Bullerkotte A, Nitschke R, Kuehn EW, Kim E, Burkhardt H, Brox T, Ronneberger O, Gloy J, Walz G (2010) Inversin relays Frizzled-8 signals to promote proximal pronephros development. Proc Natl Acad Sci USA 107:20388–20393
Luyten A, Su X, Gondela S, Chen Y, Rompani S, Takakura A, Zhou J (2010) Aberrant regulation of planar cell polarity in polycystic kidney disease. J Am Soc Nephrol 21:1521–1532
Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ (2009) Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet 41:793–799
Wallingford JB, Mitchell B (2011) Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev 25:201–213
MacDonald BT, Tamai K, He X (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17:9–26
Gao C, Chen YG (2010) Dishevelled: the hub of Wnt signaling. Cell Signal 22:717–727
Segalen M, Bellaiche Y (2009) Cell division orientation and planar cell polarity pathways. Semin Cell Dev Biol 20:972–977
Morin X, Bellaiche Y (2011) Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev Cell 21:102–119
Lancaster MA, Schroth J, Gleeson JG (2011) Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol 13:700–707
Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S, Tan PL, Phillips HM, Leroux MR, Henderson DJ, Murdoch JN, Copp AJ, Eliot MM, Lupski JR, Kemp DT, Dollfus H, Tada M, Katsanis N, Forge A, Beales PL (2005) Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 37:1135–1140
Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie-Bittach T, Katsanis N, Wallingford JB (2010) Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science 329:1337–1340
Pugacheva EN, Roegiers F, Golemis EA (2006) Interdependence of cell attachment and cell cycle signaling. Curr Opin Cell Biol 18:507–515
Sugiyama N, Tsukiyama T, Yamaguchi TP, Yokoyama T (2011) The canonical Wnt signaling pathway is not involved in renal cyst development in the kidneys of inv mutant mice. Kidney Int 79:957–965
Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P (2008) Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet 17:1578–1590
Jonassen JA, San Agustin J, Follit JA, Pazour GJ (2008) Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol 183:377–384
Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P (2008) Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 40:69–77
Dawe HR, Adams M, Wheway G, Szymanska K, Logan CV, Noegel AA, Gull K, Johnson CA (2009) Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J Cell Sci 122:2716–2726
Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, Huntzicker EG, Sfakianos MK, Sandoval W, Bazan JF, Kulkarni P, Garcia-Gonzalo FR, Seol AD, O’Toole JF, Held S, Reutter HM, Lane WS, Rafiq MA, Noor A, Ansar M, Devi AR, Sheffield VC, Slusarski DC, Vincent JB, Doherty DA, Hildebrandt F, Reiter JF, Jackson PK (2011) Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145:513–528
Guay-Woodford LM (2006) Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. Pediatr Nephrol 21:1369–1376
Grantham JJ (1993) Homer Smith Award: fluid secretion, cellular proliferation, and the pathogenesis of renal epithelial cysts. J Am Soc Nephrol 3:1841–1857
Witzgall R (2005) New developments in the field of cystic kidney diseases. Curr Mol Med 5:455–465
Calvet JP (2002) Cilia in PKD–letting it all hang out. J Am Soc Nephrol 13:2614–2616
Menezes LF, Germino GG (2009) Polycystic kidney disease, cilia, and planar polarity. Methods Cell Biol 94:273–297
Badano JL, Teslovich TM, Katsanis N (2005) The centrosome in human genetic disease. Nat Rev Genet 6:194–205
Hildebrandt F, Otto E (2005) Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet 6:928–940
Yoder BK (2007) Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol 18:1381–1388
Watnick T, Germino G (2003) From cilia to cyst. Nat Genet 34:355–356
Wilson PD (2011) Apico-basal polarity in polycystic kidney disease epithelia. Biochim Biophys Acta 1812:1239–1248
Steigelman KA, Lelli A, Wu X, Gao J, Lin S, Piontek K, Wodarczyk C, Boletta A, Kim H, Qian F, Germino G, Geleoc GS, Holt JR, Zuo J (2011) Polycystin-1 is required for stereocilia structure but not for mechanotransduction in inner ear hair cells. J Neurosci 31:12241–12250
Bataille S, Demoulin N, Devuyst O, Audrezet MP, Dahan K, Godin M, Fontes M, Pirson Y, Burtey S (2011) Association of PKD2 (polycystin 2) mutations with left-right laterality defects. Am J Kidney Dis 58:456–460
Battini L, Macip S, Fedorova E, Dikman S, Somlo S, Montagna C, Gusella GL (2008) Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum Mol Genet 17:2819–2833
Bakeberg JL, Tammachote R, Woollard JR, Hogan MC, Tuan HF, Li M, van Deursen JM, Wu Y, Huang BQ, Torres VE, Harris PC, Ward CJ (2011) Epitope-tagged Pkhd1 tracks the processing, secretion, and localization of fibrocystin. J Am Soc Nephrol 22:2266–2277
Hurd TW, Hildebrandt F (2011) Mechanisms of nephronophthisis and related ciliopathies. Nephron Exp Nephrol 118:e9–e14
Maher ER, Neumann HP, Richard S (2011) von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 19:617–623
Glenn GM, Linehan WM, Hosoe S, Latif F, Yao M, Choyke P, Gorin MB, Chew E, Olfield E, Manolatos C et al (1992) Screening for von Hippel-Lindau disease by DNA polymorphism analysis. JAMA 267:1226–1231
Van Poppel H, Nilsson S, Algaba F, Bergerheim U, Dal Cin P, Fleming S, Hellsten S, Kirkali Z, Klotz L, Lindblad P, Ljungberg B, Mulders P, Roskams T, Ross RK, Walker C, Wersall P (2000) Precancerous lesions in the kidney. Scand J Urol Nephrol Suppl 205:136–165
Schermer B, Ghenoiu C, Bartram M, Muller RU, Kotsis F, Hohne M, Kuhn W, Rapka M, Nitschke R, Zentgraf H, Fliegauf M, Omran H, Walz G, Benzing T (2006) The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol 175:547–554
Esteban MA, Harten SK, Tran MG, Maxwell PH (2006) Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J Am Soc Nephrol 17:1801–1806
Lutz MS, Burk RD (2006) Primary cilium formation requires von hippel-lindau gene function in renal-derived cells. Cancer Res 66:6903–6907
Flugel D, Gorlach A, Michiels C, Kietzmann T (2007) Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol 27:3253–3265
Fan S, Hurd TW, Liu CJ, Straight SW, Weimbs T, Hurd EA, Domino SE, Margolis B (2004) Polarity proteins control ciliogenesis via kinesin motor interactions. Curr Biol 14:1451–1461
Thoma CR, Matov A, Gutbrodt KL, Hoerner CR, Smole Z, Krek W, Danuser G (2010) Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J Cell Biol 190:991–1003
Lolkema MP, Mans DA, Snijckers CM, van Noort M, van Beest M, Voest EE, Giles RH (2007) The von Hippel-Lindau tumour suppressor interacts with microtubules through kinesin-2. FEBS Lett 581:4571–4576
Pugacheva EN, Golemis EA (2005) The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol 7:937–946
Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 100:5286–5291
Wilson PD (2004) Polycystic kidney disease: new understanding in the pathogenesis. Int J Biochem Cell Biol 36:1868–1873
Takiar V, Caplan MJ (2011) Polycystic kidney disease: pathogenesis and potential therapies. Biochim Biophys Acta 1812:1337–1343
Abdul-Majeed S, Moloney BC, Nauli SM (2011) Mechanisms regulating cilia growth and cilia function in endothelial cells. Cell Mol Life Sci 69:165–173
Harris PC, Torres VE (2009) Polycystic kidney disease. Annu Rev Med 60:321–337
Chapman AB (2008) Approaches to testing new treatments in autosomal dominant polycystic kidney disease: insights from the CRISP and HALT-PKD studies. Clin J Am Soc Nephrol 3:1197–1204
Wallace DP (2011) Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta 1812:1291–1300
Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR 3rd, Rossetti S, Harris PC, LaRusso NF, Torres VE (2010) Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 21:1052–1061
van Keimpema L, Hockerstedt K (2009) Treatment of polycystic liver disease. Br J Surg 96:1379–1380
Gattone VH 2nd, Wang X, Harris PC, Torres VE (2003) Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9:1323–1326
Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH 2nd (2004) Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 10:363–364
Wang X, Wu Y, Ward CJ, Harris PC, Torres VE (2008) Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 19:102–108
Meijer E, Gansevoort RT, de Jong PE, van der Wal AM, Leonhard WN, de Krey SR, van den Born J, Mulder GM, van Goor H, Struck J, de Heer E, Peters DJ (2011) Therapeutic potential of vasopressin V2 receptor antagonist in a mouse model for autosomal dominant polycystic kidney disease: optimal timing and dosing of the drug. Nephrol Dial Transplant 26:2445–2453
Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS (2008) Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol 19:1300–1310
Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, Crews CM (2007) Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci USA 104:4389–4394
Sweeney WE Jr, von Vigier RO, Frost P, Avner ED (2008) Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol 19:1331–1341
Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, Wuthrich RP (2010) Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 363:820–829
Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Horl WH, Obermuller N, Arns W, Pavenstadt H, Gaedeke J, Buchert M, May C, Gschaidmeier H, Kramer S, Eckardt KU (2010) Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 363:830–840
Perico N, Antiga L, Caroli A, Ruggenenti P, Fasolini G, Cafaro M, Ondei P, Rubis N, Diadei O, Gherardi G, Prandini S, Panozo A, Bravo RF, Carminati S, De Leon FR, Gaspari F, Cortinovis M, Motterlini N, Ene-Iordache B, Remuzzi A, Remuzzi G (2010) Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol 21:1031–1040
Huber TB, Walz G, Kuehn EW (2011) mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int 79:502–511
Ibraghimov-Beskrovnaya O, Natoli TA (2011) mTOR signaling in polycystic kidney disease. Trends Mol Med 17:625–633
Takiar V, Nishio S, Seo-Mayer P, King JD Jr, Li H, Zhang L, Karihaloo A, Hallows KR, Somlo S, Caplan MJ (2011) Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci USA 108:2462–2467
Ibraghimov-Beskrovnaya O, Bukanov N (2008) Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell Mol Life Sci 65:605–619
Olsan EE, Mukherjee S, Wulkersdorfer B, Shillingford JM, Giovannone AJ, Todorov G, Song X, Pei Y, Weimbs T (2011) Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proc Natl Acad Sci USA 108:18067–18072
Nofziger C, Blazer-Yost BL (2009) PPARgamma agonists, modulation of ion transporters, and fluid retention. J Am Soc Nephrol 20:2481–2483
Yoshihara D, Kurahashi H, Morita M, Kugita M, Hiki Y, Aukema HM, Yamaguchi T, Calvet JP, Wallace DP, Nagao S (2011) PPAR-gamma agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol 300:F465–F474
Dai B, Liu Y, Mei C, Fu L, Xiong X, Zhang Y, Shen X, Hua Z (2010) Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han: SPRD rats. Clin Sci (Lond) 119:323–333
Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R, Sun Z (2009) Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci USA 106:21819–21824
Xia S, Li X, Johnson T, Seidel C, Wallace DP, Li R (2010) Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts. Development 137:1075–1084
Omori S, Kitagawa H, Koike J, Fujita H, Hida M, Pringle KC, Awazu M (2008) Activated extracellular signal-regulated kinase correlates with cyst formation and transforming growth factor-beta expression in fetal obstructive uropathy. Kidney Int 73:1031–1037
Yamaguchi T, Reif GA, Calvet JP, Wallace DP (2010) Sorafenib inhibits cAMP-dependent ERK activation, cell proliferation, and in vitro cyst growth of human ADPKD cyst epithelial cells. Am J Physiol Renal Physiol 299:F944–F951
Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Masyuk AI, Gradilone SA, Gajdos GB, Chandok N, Bakeberg JL, Ward CJ, Ritman EL, Kiyokawa H, Larusso NF (2012) Inhibition of Cdc25A Suppresses Hepato-renal Cystogenesis in Rodent Models of Polycystic Kidney and Liver Disease. Gastroenterology 142(3):622–633
Gherman A, Davis EE, Katsanis N (2006) The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat Genet 38:961–962
Hogan MC, Manganelli L, Woollard JR, Masyuk AI, Masyuk TV, Tammachote R, Huang BQ, Leontovich AA, Beito TG, Madden BJ, Charlesworth MC, Torres VE, LaRusso NF, Harris PC, Ward CJ (2009) Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol 20:278–288
van Reeuwijk J, Arts HH, Roepman R (2011) Scrutinizing ciliopathies by unraveling ciliary interaction networks. Hum Mol Genet 20:R149–R157
Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143:1174–1189
AbouAlaiwi WA, Ratnam S, Booth RL, Shah JV, Nauli SM (2011) Endothelial cells from humans and mice with polycystic kidney disease are characterized by polyploidy and chromosome segregation defects through survivin down-regulation. Hum Mol Genet 20:354–367
Shillingford JM, Piontek KB, Germino GG, Weimbs T (2010) Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol 21:489–497
Zafar I, Ravichandran K, Belibi FA, Doctor RB, Edelstein CL (2010) Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int 78:754–761
Cadnapaphornchai MA, Masoumi A, Strain JD, McFann K, Schrier RW (2011) Magnetic resonance imaging of kidney and cyst volume in children with ADPKD. Clin J Am Soc Nephrol 6:369–376
Higashihara E, Torres VE, Chapman AB, Grantham JJ, Bae K, Watnick TJ, Horie S, Nutahara K, Ouyang J, Krasa HB, Czerwiec FS (2011) Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 6:2499–2507
Torres VE (2008) Vasopressin antagonists in polycystic kidney disease. Semin Nephrol 28:306–317
Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM (2008) Triptolide reduces cystogenesis in a model of ADPKD. J Am Soc Nephrol 19:1659–1662
Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM (2010) Triptolide reduces cyst formation in a neonatal to adult transition Pkd1 model of ADPKD. Nephrol Dial Transplant 25:2187–2194
Elliott J, Zheleznova NN, Wilson PD (2011) c-Src inactivation reduces renal epithelial cell-matrix adhesion, proliferation, and cyst formation. Am J Physiol Cell Physiol 301:C522–C529
Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O (2006) Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 444:949–952
Omori S, Hida M, Fujita H, Takahashi H, Tanimura S, Kohno M, Awazu M (2006) Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol 17:1604–1614
Leonhard WN, van der Wal A, Novalic Z, Kunnen SJ, Gansevoort RT, Breuning MH, de Heer E, Peters DJ (2011) Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways: in vivo evidence from a Pkd1-deletion model. Am J Physiol Renal Physiol 300:F1193–F1202
Natoli TA, Smith LA, Rogers KA, Wang B, Komarnitsky S, Budman Y, Belenky A, Bukanov NO, Dackowski WR, Husson H, Russo RJ, Shayman JA, Ledbetter SR, Leonard JP, Ibraghimov-Beskrovnaya O (2010) Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med 16:788–792
Blazer-Yost BL, Haydon J, Eggleston-Gulyas T, Chen JH, Wang X, Gattone V, Torres VE (2010) Pioglitazone attenuates cystic burden in the PCK rodent model of polycystic kidney disease. PPAR Res 2010:274376
Wang X, Harris PC, Somlo S, Batlle D, Torres VE (2009) Effect of calcium-sensing receptor activation in models of autosomal recessive or dominant polycystic kidney disease. Nephrol Dial Transplant 24:526–534
Gattone VH 2nd, Chen NX, Sinders RM, Seifert MF, Duan D, Martin D, Henley C, Moe SM (2009) Calcimimetic inhibits late-stage cyst growth in ADPKD. J Am Soc Nephrol 20:1527–1532
Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, Louvi A, Velazquez H, Ishibe S, Cantley LG, Igarashi P, Somlo S (2008) Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 17:1505–1516
Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, Germino GG (2004) A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol 15:3035–3043
Takakura A, Contrino L, Beck AW, Zhou J (2008) Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol 19:2351–2363
Lu W, Shen X, Pavlova A, Lakkis M, Ward CJ, Pritchard L, Harris PC, Genest DR, Perez-Atayde AR, Zhou J (2001) Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum Mol Genet 10:2385–2396
Lantinga-van Leeuwen IS, Leonhard WN, van de Wal A, Breuning MH, Verbeek S, de Heer E, Peters DJ (2006) Transgenic mice expressing tamoxifen-inducible Cre for somatic gene modification in renal epithelial cells. Genesis 44:225–232
Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJ (2007) Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet 16:3188–3196
Wu G, D’Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H Jr, Kucherlapati R, Edelmann W, Somlo S (1998) Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93:177–188
Cowley BD Jr, Gudapaty S, Kraybill AL, Barash BD, Harding MA, Calvet JP, Gattone VH 2nd (1993) Autosomal-dominant polycystic kidney disease in the rat. Kidney Int 43:522–534
Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, Gretz N, Walz G, Schermer B, Benzing T, Hildebrandt F, Omran H (2003) Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet 34:455–459
Takahashi H, Ueyama Y, Hibino T, Kuwahara Y, Suzuki S, Hioki K, Tamaoki N (1986) A new mouse model of genetically transmitted polycystic kidney disease. J Urol 135:1280–1283
Smith LA, Bukanov NO, Husson H, Russo RJ, Barry TC, Taylor AL, Beier DR, Ibraghimov-Beskrovnaya O (2006) Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. J Am Soc Nephrol 17:2821–2831
Atala A, Freeman MR, Mandell J, Beier DR (1993) Juvenile cystic kidneys (jck): a new mouse mutation which causes polycystic kidneys. Kidney Int 43:1081–1085
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC (2002) The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30:259–269
Nauta J, Ozawa Y, Sweeney WE Jr, Rutledge JC, Avner ED (1993) Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr Nephrol 7:163–172
Brown NE, Murcia NS (2003) Delayed cystogenesis and increased ciliogenesis associated with the re-expression of polaris in Tg737 mutant mice. Kidney Int 63:1220–1229
Hou X, Mrug M, Yoder BK, Lefkowitz EJ, Kremmidiotis G, D’Eustachio P, Beier DR, Guay-Woodford LM (2002) Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J Clin Invest 109:533–540
Ricker JL, Gattone VH 2nd, Calvet JP, Rankin CA (2000) Development of autosomal recessive polycystic kidney disease in BALB/c-cpk/cpk mice. J Am Soc Nephrol 11:1837–1847
Acknowledgments
This work was supported by National Basic Research Program (973 program) (Grant 2012CB945000) and National Natural Science Foundation of China (Grants 30830057, 30988004), and Tsinghua Research Program (to J.P.) and R01s CA63366 and CA113342 (to E.A.G.).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Pan, J., Seeger-Nukpezah, T. & Golemis, E.A. The role of the cilium in normal and abnormal cell cycles: emphasis on renal cystic pathologies. Cell. Mol. Life Sci. 70, 1849–1874 (2013). https://doi.org/10.1007/s00018-012-1052-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1052-z