
Abstract
Among the pathogenic mechanisms underlying central nervous system (CNS) diseases, oxidative stress is almost invariably described. For this reason, numerous attempts have been made to decrease reactive oxygen species (ROS) with the administration of antioxidants as potential therapies for CNS disorders. However, such treatments have always failed in clinical trials. Targeting specific sources of reactive oxygen species in the CNS (e.g. NOX enzymes) represents an alternative promising option. Indeed, NOX enzymes are major generators of ROS, which regulate progression of CNS disorders as diverse as amyotrophic lateral sclerosis, schizophrenia, Alzheimer disease, Parkinson disease, and stroke. On the other hand, in autoimmune demyelinating diseases, ROS generated by NOX enzymes are protective, presumably by dampening the specific immune response. In this review, we discuss the possibility of developing therapeutics targeting NADPH oxidase (NOX) enzymes for the treatment of different CNS pathologies. Specific compounds able to modulate the activation of NOX enzymes, and the consequent production of ROS, could fill the need for disease-modifying drugs for many incurable CNS pathologies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: NOX NADPH oxidases and their emerging role in CNS diseases
Central nervous system (CNS) diseases are heterogeneous and have numerous etiologies. However, CNS pathologies as diverse as progressive neurodegenerative diseases, neuropsychopathological disorders, and stroke share many pathogenic mechanisms, such as inflammation, microglia activation, impaired neurotransmission, glutamate-mediated excitotoxicity, mitochondrial dysfunction, apoptosis, and increase of oxidative stress [78, 120]. Among these features of CNS diseases, NOX enzymes are emerging as an important source of oxidants in the CNS and key regulators of neurological pathologies [112, 136, 142].
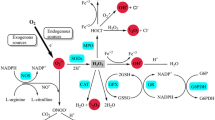
The NOX family consists of seven members (NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2), each with a specific tissue distribution. The isoforms present in the CNS are mostly NOX1, NOX2, and NOX4. Despite their similar core structures, NOX isoforms have different mechanisms of activation. NOX1, NOX2, and NOX3 require association with cytosolic components [p47phox, p67phox, NOXO1 (NOX organizer type 1), NOXA1 (NOX activator type 1)], NOX4 is constitutively active, and NOX5 and DUOXes are activated by intracellular Ca2+ elevation and direct phosphorylation [8, 42, 84].
NOX enzymes function to transfer electrons across membranes, which then react with oxygen generating the superoxide anion O2 −. Superoxide dismutates very rapidly to generate hydrogen peroxide and other reactive oxygen species (ROS).
In physiological conditions, ROS generated by NOX enzymes work as defense mechanisms against pathogens and signaling molecules. NOX enzymes are increasingly recognized as key regulators of pathological situations where oxidative stress is involved and leads to tissue oxidative damage, such as pulmonary fibrosis, diabetic nephropathy, and vascular and CNS pathologies [81]. The NOX2 isoform (previously known as gp91phox or the phagocyte oxidase) is responsible for the respiratory oxidative burst in neutrophils. However, NOX2 is also expressed in the CNS, where it controls key neuronal functions and neuroinflammatory processes. NOX2 is strongly upregulated in different CNS disorders where it generates large amounts of ROS. NOX2 activation is thought to regulate microglia activation, a hallmark of inflammatory gliosis observed in neuroinflammatory degenerative disorders [17, 85]. As a major source of ROS, NOX2 can induce direct neuronal damage [128] and maintain microglial cells in an activated stage where they produce neurotoxic molecules like peroxynitrite and other inflammatory molecules [94]. A protective role of NOX2 deficiency has been demonstrated in animal models of Parkinson disease (PD), Alzheimer disease (AD), amyotrophic lateral sclerosis (ALS), and stroke (reviewed in [142]). The fact that decreased NOX2-generated ROS is protective in diverse CNS diseases implies that it must regulate a key aspect of the pathological CNS. NOX2 in the microglia is not the only target to be pursued. In fact, NOX2 inhibition also prevents development of psychotic disorders without the microglia-mediated neuroinflammatory components by controlling key neuronal aspects such as neurotransmitter release and GABAergic interneurons function [13]. Although NOX2 deletion leads to decreased signs of oxidative stress, the exact role of NOX enzymes in CNS physiology and pathology is still very incomplete. Recent studies have involved NOX2 in the pathways related to NMDA receptor activation, opening new perspectives of research [20, 143].
Another striking aspect of the complexity of the role of NOX2 is the fact that NOX2 plays an anti-inflammatory role in autoimmune-mediated diseases including autoimmune neuroinflammation. Indeed, an insufficient ROS production by systemic antigen presenting cells, such as mononuclear phagocytes, will favor autoimmune neuroinflammation. Genetic studies identified a polymorphism in the Ncf1 gene coding for p47phox, a regulatory cytosolic factor of NOX-dependent oxidant production, which is associated with susceptibility in rheumatoid arthritis models and other autoimmune disorders [116], including experimental allergic encephalomyelitis, an animal model of multiple sclerosis (MS) [62], and experimental allergic neuritis, a model of Guillain Barré syndrome [58]. The link between Ncf1 and disease susceptibility was further confirmed in humans because circulating leukocytes of patients affected by MS [110] and other autoimmune neuropathies [109] have a lower capacity for ROS generation. These unexpected findings appear to be due to down-regulation of T cells by NOX2-dependent ROS generation in response to autoantigen presentation [63].
To summarize, there are two possible therapeutic options using NOX enzymes as pharmacological targets (Fig. 1): (1) in CNS diseases like stroke, PD, AD, ALS, with a strong glutamate excitotoxicity component, the inhibition of NOX enzymes is expected to be beneficial, while (2) when the neuroinflammatory process is mediated by peripheral immune cells, activation of NOX2 in antigen presenting cells of the immune system is predicted to bring a therapeutic effect.

Schematic overview of possible therapeutic indications for NOX targeting compounds. a In diseases characterized by neurodegeneration and neuroinflammation such as Alzheimer, Parkinson, Amyotrophic lateral sclerosis, and stroke, excessive NOX2 activation in injured neurons and activated microglia cells leads to production of ROS and oxidative damage. b In neurological autoimmune diseases, such as multiple sclerosis and Guillain Barré syndrome, NOX2 activation inhibits the excessive T cell response and contributes to remyelination and neuronal survival
Although it has been known for a long time that mitochondria are an important source of ROS in CNS disease [113, 2], the role of NOX enzymes in CNS pathologies is rapidly emerging. However, because of the presence of at least three NOX isoforms in the CNS and the absence of studies using specific NOX inhibitors, we focus here on disease models where proof-of-principle studies using knock-out or mutant mice are available. We discuss current therapies and the potential for NOX-based therapeutics either decreasing NOX activity or, on the contrary, enhancing low NOX activity in CNS autoimmune disorders. Finally, we discuss past and emerging small molecules targeting NOX enzymes and their possible advantages and risks in CNS-based pathologies.
Beneficial effect of NOX inhibition: potential CNS indications
Amyotrophic lateral sclerosis (ALS)
ALS is a neurodegenerative disease of the motoneurons that leads to complete paralysis and death. Its prevalence is around 2 per 100,000. Several therapeutic approaches have been proposed for the treatment of this disease with varying efficacy. Today, there are no treatments that can arrest, or even substantially delay, the progression of ALS. The only drug approved to treat ALS patients is Riluzole™, which is thought to inhibit the presynaptic release of glutamate. However, its therapeutic benefit is modest as in randomized controlled trials, it prolonged survival by approximately 4 months [15, 79], a statistical benefit which is subjectively not perceived by patients, family members, or physicians [50]. ALS has a strong inflammatory component with, for instance, increased expression of COX-2, but the benefit of COX-2 inhibitors, such as rofecoxib and celecoxib for the treatment of ALS, remains uncertain. A protective effect of numerous compounds has been described in animal models of ALS (anti-inflammatory drugs, anti-glutamate agents, neurotrophic factors, antioxidant, anti-apoptotic, gene inductors, autophagy inducers), but none of them significantly prolonged survival or improved quality of life when translated to ALS patients. Signs of oxidative stress are observed in ALS rodent models and in ALS patients, such as 8-oxodeoxyguanosine (8-oxodG), urinary 15-F(2t)-isoprostane, protein carbonylation, and markers of lipid peroxidation [5, 7, 104, 106]. The most studied form of ALS is a familial ALS (FALS) in which the antioxidant enzyme SOD1 is mutated (SOD1G93A). However, surprisingly, the increase of oxidative stress is not due to the lack of function of SOD1, but rather to aggregation of mutant protein [124], leading to a toxic gain of function of the mutated SOD1 protein, which in turn has been proposed to activate NOX enzymes [54]. NOX2 and subunits are strongly upregulated in both ALS mice and patients colocalizing with microglial markers [164]. Arguments in favor of a beneficial effect of NOX inhibition comes from experiments where transgenic mice expressing human SOD1G93A were bred with mice deficient for NOX1 or NOX2, which showed lifespan increases of 33 and 97 days, respectively [97]. Another study using a similar approach has shown that NOX2-deficiency in SOD1G93A overexpressing mice decreased the production of microglial-derived ROS, delayed neurodegeneration and prolonged survival, although more modestly than the other study (13 days) [164]. Because of absence of treatment and the inexorable development of ALS, there is a real need for testing new targets for the pharmacological approach of ALS. In spite of important differences in the protective potential of NOX inhibition in ALS mice, these preliminary proof-of-principle experiments converge at identifying NOX enzymes as primary target for a potential treatment of ALS.
Alzheimer disease (AD)
Alzheimer disease (AD) represents the most common single cause of age-associated dementia worldwide. Although AD etiology is unclear, neurodegeneration in AD patient brain is characterized by protein misfolding and accumulation of Aβ (derived from amyloid precursor protein APP) and tau (a microtubule-associated protein) in plaques and intraneuronal neurofibrillary tangles, respectively. Formation of these abnormal protein aggregates leads to severe neuronal death and synaptic loss, associated with microglial and astrocytic activation. Excessive production of inflammatory mediators and ROS by activated glial cells and damaged neurons contribute to reduced neuronal survival and disease progression.
Due to the decreased levels of neurotransmitters, and especially of acetylcholine, in AD patient brain, the main therapy currently used for AD consists in the administration of acetylcholinesterase inhibitors, such as tacrine, donepezil, rivastigmine, and galantamine [103]. Another drug also commonly used for AD treatment is memantine [103], which acts as a non-competitive antagonist of the NMDA receptor, thereby reducing excitotoxicity due to the excessive glutamate release associated with neurodegeneration [132]. Although these treatments can reduce AD symptoms and improve cognitive functions, they do not contribute to the resolution of disease or arrest its progression [103]. Current approaches to develop better therapies include inhibition of Aβ aggregation and/or production, as well as inhibition of Aβ/tau-dependent neurotoxic effects [24]. Indeed, preventing the protein misfolding or its direct consequences would represent a more efficacious way to reduce or even reverse the progression of the disease.
The involvement of NOX activation in the pathological mechanisms of AD has been described in several studies based on in vitro experiments with cultures of microglia, astrocytes, and neurons (reviewed in [142]). Moreover, disease progression was prevented in a mouse model of AD (overexpressing the Swedish mutation of APP, leading to Aβ fragment accumulations) after breeding with NOX2-deficient mice. Deletion of NOX2 gene reduced the pathologic effects of Aβ amyloid fragments, rather than the spontaneous accumulation of plaques in the mouse brain [119]. There is also evidence that NOX enzymes are expressed and activated in AD patients as compared to healthy controls: translocation of NOX2-associated subunits (p47phox, p67phox) to the membrane [140], as well as increased NOX2 activation in the frontal and temporal cortex of AD patients with mild cognitive impairment [3]. Interestingly, in this recent study, only the expression of NOX2 regulatory cytosolic subunits (p47phox, p67phox, and p40phox) were increased in AD patients, while the expression of membrane-associated proteins (p22phox and NOX2) remained stable [3]. In addition, increased levels of NOX1 and NOX3 mRNA were found in early stage AD patients [33], suggesting that other isoforms might contribute to this pathology. Nevertheless, in spite of increasingly recognised importance of NOX in AD, free radicals of specific mitochondrial origin and uncoupling of endothelial nitric oxide synthase are likely to be important mediators of the general oxidant status observed in AD [102].
Parkinson disease (PD)
Among neurodegenerative diseases, Parkinson disease is the disorder for which the best options for symptomatic treatment exist, at least for the initial phases. Neurodegeneration occurs in the substantia nigra, leading to loss of dopaminergic neurons, which is associated with motor dysfunctions. Typical Parkinson-related symptoms include shaking, rigidity, or slowness, and they can be diagnosed early in the course of the disease [135]. Treatment consists of the administration of levodopa, a precursor of dopamine, to compensate for the dopamine reduction. Although levodopa alleviates early movement dysfunctions, it does not provide a real cure to prevent the progressive degeneration of dopaminergic neurons. Similarly, other available treatments include dopamine receptor agonists, or monoamine oxidase (MAO-B) and catechol-O-methyl transferase (COMT) inhibitors, which reduce the dopamine metabolism and increase its availability. These drugs are often used alone at the first appearance of symptoms, while, at later stage, they are used in combination with levodopa, which remains more efficacious, but is associated with more undesirable side effects, such as dyskinesia (Table 1). Both genetic and environmental factors have been implicated in the etiology of Parkinson [135]. However, misfolding and deposition of toxic α-synuclein aggregates seem to be an initiating pathogenic event of the disease, whereas the progressive degeneration is due to glial activation [53]. Oxidative damage is thought to contribute to these mechanisms, and mitochondrial dysfunction has been proposed to be a major source of ROS [55]. Yet, there is experimental evidence supporting a role for NOX enzymes. Most of the data derive from in vitro studies, indicating a major involvement of NOX2 in microglia-dependent dopaminergic neurotoxicity (reviewed in [142]). In addition, a direct expression of NOX enzymes in dopaminergic neurons might play a role, while microglia serve to amplify the neurotoxic stimuli [17]. In line with these findings, decreased death of dopaminergic neurons in NOX2-deficient mice as compared to wild-type (WT) was detected following LPS-injections in the substantia nigra [129] and systemic administration of MPTP [170] or the herbicide paraquat [127]. A recent study identified a possible mechanism by which NOX2 could be involved in paraquat toxicity. In its native form, paraquat is a divalent cation and is not a substrate of dopaminergic transporters. It therefore requires reduction into a monovalent cation by microglial NOX2 in order to be a substrate for dopamine transporters leading to its accumulation in dopaminergic neurons [130]. Whether oxidative modification of α-synuclein or other Parkinson causative agents increase their toxicity to dopamine neurons remains unknown. Two recent studies show evidence of a role of neuronal NOX1 in Parkinson disease [28, 31]. At this stage, the role of different NOX isoforms is not clear, but one could speculate that a cross-talk exists between neuronal NOX (probably NOX1) and microglial NOX2, which would act by amplifying the neuronal damage and regulating a neuroinflammatory response. In order to evaluate the exact effect of different isoforms, backcross of NOX-deficient animals with genetic models of PD should be performed [35].
Schizophrenia
Schizophrenia is a severe psychiatric disorder characterized by three main symptoms: positive, such as hallucinations and delusions, negative, such as loss of motivation and blunted emotions, and cognitive impairment mainly due to deficits in working memory and attention [22]. Although symptoms of schizophrenia usually appear in the late second or third decade of life, it is considered to be a neurodevelopmental mental disorder. Both genetic and environmental factors contribute to the development of schizophrenia, but a complete picture of pathogenic events is still not clear. Alterations in glutamate and dopamine neurotransmission seem to be the main cause of the symptoms, but it is not known how these abnormalities develop [87]. Treatments for schizophrenia are limited and, in general, consist of drugs that act mainly on positive symptoms. The introduction of chlorpromazine in 1954 as a first compound for the treatment of schizophrenia opened the era of psychopharmacology. Similar drugs were developed in the 1950s and 1960s (typical antipsychotics), finally giving the possibility of controlling symptoms in schizophrenic patients and limiting behavioral abnormalities. Although these molecules are known to have different pharmacological actions [146], their antipsychotic effect is thought to be due to the inhibition of the D2 dopamine receptor. However, severe side effects are associated with these treatments, such as a strong sedative action or extra-pyramidal motor control disabilities, leading to a syndrome per se. The subsequent development of other antipsychotic therapies led to the discovery of different compounds (atypical antipsychotics), but with similar mechanisms of action. These drugs prevent positive symptoms, and, even if attenuated, they cause important side effects, especially related to metabolic syndromes and extrapyramidal symptoms [71]. Due to severe side effects, the treatment with antipsychotics (typical or atypical) is often associated with low compliance of the patient and non-adherence to the therapy. In addition, patients develop drug resistance. For these reasons, there is a real need for new treatments involving novel targets with increased efficacy and better tolerability. New therapies should also aim at targeting negative symptoms and cognitive deficits, and reducing the functional and social impairment, which prevent schizophrenic patients from living a normal life [71].
The role of NOX enzymes in the pathogenesis of schizophrenia has been shown in two different experimental animal models. By inducing schizophrenia-like symptoms in mice with subchronic administration of subanesthetic doses of ketamine, Behrens and colleagues first demonstrated that NOX2-dependent ROS production induces a loss of parvalbumin in interneurons [11, 12]. Parvalbumin is a Ca2+ binding protein, which normally regulates activity of GABAergic interneurons, thereby modulating the glutamatergic transmission. Decrease in parvalbumin expression has been detected in the brain of schizophrenic patients, suggesting an alteration in the control of excitatory glutamatergic neurotransmission, possibly related to schizophrenia symptoms [86]. By analyzing the acute effects of ketamine on mouse behavior and neurotransmission, it has been found that an increased production of ROS by NOX2 can be determinant to initially trigger the increase in glutamate and dopamine release [52, 143]. In contrast, the repetitive ketamine exposure leads to adaptive alteration of the post-synaptic NMDA receptor [143], possibly as a consequence of parvalbumin decrease [76]. The beneficial effect of NOX2 inhibition is not limited to the psychosis induced by ketamine, as similar observations were made in a model of social isolation in the rat. In rats grown in social isolation for 7 weeks after weaning, NOX2 is up-regulated concomitantly with behavioral alterations, and signs of oxidative stress and loss of parvalbumin. Treatment with apocynin, a non-specific NOX inhibitor, was able to prevent all these effects [136]. Since the social isolation of young rats after weaning induces a prolonged stress during the development of the CNS [83], these findings show in two unrelated models using two different species that NOX2-dependent generation of ROS can play a role in the pathogenesis of schizophrenia. However, at this stage, although signs of oxidative stress are known in schizophrenic patients, NOX2 expression pattern in post mortem specimens or even increased NOX2 activity in peripheral leukocytes has so far not been documented. NOX inhibition in animal models of psychosis does not only completely prevent behavioral changes but also blunts signs of oxidative stress as well as the histopathological and neurochemical alterations observed in these models. Development of CNS-targeted NOX inhibitors therefore represents an extremely promising alternative approach to existing therapies currently used in the treatment of psychotic disorders.
Stroke
Stroke is a leading cause of death and permanent disability worldwide. The majority of strokes are ischemic, due to the occlusion of a vessel in the brain. Intervention in these cases is mainly based on enzymatic or mechanical removal of the occlusion to restore blood flow. Even if beneficial, this treatment can be performed in patients only 3–4 h after the occurrence of stroke, because of the associated severe risk of inducing a hemorrhage [99]. Since it is difficult to rescue the infarcted area, possible therapies are designed to reduce further development of tissue damage and cognitive impairment associated with the ischemic insult. Another approach is to prevent the occurrence of stroke in subjects at risk [134].
Considerable variation in the outcome of ischemic stroke using transient middle cerebral artery occlusion (tMCAO) in NOX-deficient animals was observed. In an early study, Walder et al. [157] described that the infarct volume was reduced by approximately 50 % in NOX2-deficient mice after 2 h transient ischemia followed by 22 h reperfusion. These observations were further confirmed in different studies using similar ischemia–reperfusion approaches ranging from 30 to 75 to 120 min ischemia followed by 22–24 h reperfusion [27, 66, 70]. A recent study repeated exactly the study by Walder et al. [157] confirming the protective effect of NOX2 deficiency [165]. For NOX1-deficient mice, the time of ischemia appears to be critical for the switch between a protective or a deleterious effect of NOX1 activity. NOX1 deficiency decreased infarct size when 60 min ischemia was applied, while no differences were observed with occlusion time was 2 h or more [70]. In a model of 30 min ischemia followed by 24 h reperfusion, NOX1-deficient mice showed no difference in neurological score, total or subcortical cerebral infarct volume, or edema volume as compared to WT. However, cortical infarct volume was approximately fourfold greater in brains of NOX1-KO versus WT mice [66]. Moreover, a recent study using 60 min ischemia followed by 24 h reperfusion showed impressive protection in NOX4-deficient mice, but no difference in either NOX1- or NOX2-deficient mice. Substantial decrease of infarct size was also observed in NOX4-deficient mice when no reperfusion was applied 24 h after ischemia, while no data are available for NOX1- or NOX2-deficient mice for this model [77]. Such discrepancies are unfortunately not rare in rodent models of ischemia–reperfusion, as experimental details can affect the lesion in either beneficial or detrimental way, because tiny differences in the cerebral vasculature between mouse strains and surgical technique can account for important changes in infarct severity, such as body temperature control, blood pressure and blood monitoring, anesthetic used, and surgery time [36, 88]. In order to reconcile and understand these divergent data, some approaches can be proposed: (1) genotypic homogeneity of the strains could be verified, (2) a single operator could perform blindly both transient and permanent ischemia, (3) different times of ischemia could be tested for different knock-outs, and (4) permanent occlusion models could be used and neuronal cell death followed up at different time points as a relevant model of stroke. Although a direct neurotoxic role of ROS is possible, the possible mechanism played by NOX in stroke is still unclear, and a role in the BBB integrity and cerebrovascular permeability is emerging [41].
Obstructive sleep apnea syndrome (OSAS)
Obstructive sleep apnea is a respiratory disturbance characterized by recurrent occlusions of the upper airways and reduction in oxygen availability during sleep followed by sudden awakening and reoxygenation. The underlying neuropathological events of obstructive sleep apnea are still unclear, but it is known that intermittent hypoxia episodes cause behavioral alterations and cognitive impairments [9].
OSAS often affects obese patients, due to fat deposition in the parapharyngeal space, in the tongue, and under the mandible, reducing the upper airway caliber and predisposing them to breathing disorders during sleep [51].
The main remedy for OSAS consists of the application of devices to induce a nocturnal continuous positive airway pressure (CPAP), but this is poorly accepted by patients. Surgery can also be considered in certain cases to definitively remove the obstruction of upper airways. Otherwise, weight loss in obese patients or avoidance of risk factors, e.g., alcohol consumption, can be helpful to reduce the occurrence of this respiratory disturbance [51].
The animal model of OSAS is called long-term intermittent hypoxia (LTIH). It consists of the use of a particular chamber with an oxygen/nitrogen delivery system which automatically decreases the content of oxygen from 21 to 10 % at certain intervals for few seconds, inducing arterial oxyhemoglobin saturation [169]. Excessive production of ROS, neuronal death, and tissue damage are associated with the cognitive dysfunctions in this model [161].
In animals submitted to LTIH, genetic depletion of NOX2 (knock-out mice) or pharmacological (apocynin) inhibition of NOX2 reduced hypersomnolence and prevented oxidative damage in the wake-active regions of the brain [169, 173]. A recent report similarly shows that cognitive deficits induced by recurrent hypoxia events during sleep are mediated by excessive NOX2 activity [112].
In patients affected by OSAS, markers of inflammation (interleukin-6) and oxidative stress (8-isoprostane) can be detected in exhaled breath condensate [23]. Interestingly, neutrophils (which mainly express NOX2) of OSAS patients also show an enhanced production of superoxide [137]. The presence of these signs of oxidative stress has been mainly associated with the cardiovascular consequences of the disease. However, in light of the results obtained from animal models, it is possible that increase of NOX2 activity also contributes to the cognitive decline. Increased mRNA expression of p22phox was detected in peripheral blood mononuclear cells of 107 subjects affected by OSAS. In addition, in these patients, a significantly higher frequency of the C242T polymorphism in CYBB, the gene coding for p22phox, a NOX subunit necessary for the function of 4 NOX isoforms (NOX1-4), was detected as compared to 69 healthy subjects [90]. Altogether, these findings suggest a potential for a NOX2-based therapy to treat the neurological consequences of sleep-associated breathing disorders.
Beneficial effect of NOX enhancement: potential CNS indications
Neurological autoimmune diseases
Multiple sclerosis (MS) is an inflammatory disease leading to myelin damage, which progresses to physical and cognitive disabilities. Disease onset usually occurs in young adults and affects 3 times more females than males. MS prevalence ranges between 2 and 150 per 100,000, but MS is much more common in northern Europe. MS takes several forms, with new symptoms occurring either in discrete attacks (relapsing forms) or slowly accumulating over time (progressive forms). There is no known cure for MS. Available treatments attempt to return function after an attack, prevent new attacks, and prevent disability. Administration of β-interferon 1a (Rebif®) shows some beneficial effects in a subset of patients, but, despite claims to the contrary, their ability to modify disease course has not been clearly established [39]. Antibodies to α4-integrin (Natalizumab®) suppress the extravasation of lymphocytes into the CNS, but may trigger progressive multifocal leukoencephalopathy. Inhibition of the sphingosine-1-phosphate receptor (Fingolimod®) results in the sequestration of lymphocytes in lymph nodes. Use of drugs which non-specifically suppress the immune system (glucocorticoids and the antineoplastic agent mitoxantrone) slows the progression of the disease but are associated with harmful side effects.
Although autoimmunity is a primary trigger in MS lesion formation, it is now widely accepted that immune-mediated inflammation contributes to MS pathogenesis. Thus, the role of inflammatory auto-reactive CD4-positive T helper (Th) cells has been extensively proven in animal models of MS [57, 168]. However, although oxidative damage is a known feature of MS [153], ROS produced by NOX2 have been shown to be anti-inflammatory in autoimmune diseases. Indeed, low ROS generation by NOX2 appears to prevent autoimmune responses in the chronic EAE model of MS [62]. NOX2-dependent ROS of antigen presenting cells are a key regulator of T cell activation. Interestingly, leukocyte ROS production correlates inversely with disease severity in MS [110] and recurrent Guillain Barré syndrome (GBS) [108, 109]. Recurrent GBS is also mediated by autoimmunity and is caused by damage to the myelin sheet of the peripheral nerves. Current treatments of recurrent GBS consist of corticosteroids, plasmapheresis, and intravenous immunoglobulins.
Due to the critical role played by NOX enzymes in CNS pathological states, it is almost certain that other pathologies where oxidative stress is known to regulate disease progression will show beneficial effects of NOX inhibition (e.g., epilepsy, HIV-mediated dementia, or Huntington among others) or NOX enhancement in autoimmune diseases, such as leukodystrophies or progressive multifocal leukoencephalopathy.
Therapeutics targeting ROS and NOX enzymes
Antioxidants
In the case of schizophrenia, several clinical trials have recently been made with Vitamin C, E, omega-3 fatty acids, or N-acetyl cysteine, suggesting a certain efficacy of improving the antioxidant defence as adjunctive to a primary antipsychotic treatment [131]. Of particular note, parallel administration of N-acetyl cysteine could also moderately diminish negative symptoms, such as akathisia [16]. However, as for cardiovascular diseases, the use of antioxidant therapies has led to contradictory and mostly disappointing outcomes in clinical trials for CNS diseases [82], in spite of promising results obtained in in vitro studies and in animal models for AD [141], and other neuropathologies, such as stroke [134] or ALS [10]. Failure of antioxidant treatments should not necessarily preclude the search of therapies targeting oxidative stress to treat neuropathologies because there are numerous possible reasons for the apparent failure of antioxidant therapies, including lack of specificity, potency, and bioavailability of antioxidant drugs, poor trial design, or lack of relevant biomarkers of oxidation. Such issues are discussed in details elsewhere [19, 47]. Nevertheless, as of today, the medical use of such an approach still awaits solid evidence of therapeutic benefit. On the other hand, the concerted search and discovery of NOX inhibitors is only emerging. Neuroprotective action of several compounds have been described as acting on the NOX pathway, but they are probably acting upstream of NOX and, therefore, are blocking other pharmacological targets [29]. However, recently, systematic screenings of chemical libraries were performed and have identified new chemical entities targeting NOX enzymes [18, 46, 80]. Although peptidic and siRNAs have been designed to target NOX enzymes, we will only describe small molecules that are currently described as NOX inhibitors.
Natural compounds
Apocynin
Apocynin or acetovanillone (MW 166.174) is a natural organic compound widely used as a NOX inhibitor in models involving NOX enzymes [144], and has been shown to be beneficial in numerous models of CNS diseases at different doses and types of administration (summarized in Table 2). However, the use of this molecule as a NOX inhibitor remains controversial as its mode of action is thought to be mostly through oxidant scavenging activity, although formation of an apocynin dimer (diapocynin) accounts for NOX2 inhibition through the activity of a peroxidase, such as the myeloperoxidase of leukocytes [145, 56]. Surprisingly, for such a compound which seems to represent a panacea for a large panel of diseases, few studies of its bioavailability in vivo are available. One study showed that, following single intraperitoneal injection (5 mg/kg), apocynin was detected in the brain, but in a glycosylated form, while no diapocynin could be detected [159]. However, another study showed a contrary result following chronic oral treatment (150 and 300 mg/kg/day for above 100 days): no glycosylation, but conversion of apocynin into diapocynin was detected in the brain and spinal cord [152]. In almost all studies described in Table 2, the compound has been administered (in concentrations ranging from 0.4 to 300 mg/kg) as preventive treatment ranging from more than 2 months before disease onset in the case of ALS [54] to a few minutes before ischemic stroke [27]. It is clear that a curative approach would be more relevant, although the outcome of such studies is sometimes disappointing, as for hemorrhagic stroke [151]. In the case of ALS, chronic treatment with apocynin in the drinking water led to extremely variable results: a first study described an increase of survival (113 days) in SODG93A mice, a real hope for a potential treatment in patients with ALS [54]. However, this impressive outcome could not be repeated. A study following exactly the same treatment protocol showed no benefit at all [152], while another study using a similar protocol showed a modest increase of survival (5 days) [89] (see Table 2). Interestingly, direct administration of diapocynin (150 mg/kg/day) after disease onset showed an 8-day increase in mean survival of SODG93A mice [152]. As diapocynin is considered the active form of apocynin [56], the rationale for NOX inhibition as a treatment for ALS remains valid.
Discrepancies have also been described in studies with chronic administration of apocynin (over 4 months) in transgenic models of AD (see Table 2) [34, 95]. Differences in the dose, mouse model, and the age of the mice at the beginning of the treatment may account for these contrasting results. Future comprehensive PK/PD studies for apocynin would greatly help researchers in the choice of a dose and mode of administration. Due to such discrepancies, lack of clear mechanism of action, lack of specificity, high metabolism, previous failure of antioxidant drugs, low potency on NOX enzymes, and extensive patenting status, apocynin has a low potential for development by pharmaceutical companies [1]. Nevertheless, apocynin shows several advantages, which could potentially make it a therapeutic agent for CNS disorders: low toxicity, oral bioavailability, high potency in neuropathologies, and impact on surrogate markers of oxidative stress in phase I clinical studies following aerosol administration [123, 144]. Therefore, future studies associating apocynin bioavailability (PK/PD) with real therapeutic benefit in controlled preclinical trials after disease onset would provide proof of concept for a possible clinical development as therapeutics for intractable CNS diseases.
Celastrol
Celastrol (MW 450.6) is a natural compound extracted from the medicinal plant Tripterygium wilfordii, which has recently been shown to be a bone fide NOX inhibitor as it blocks within minutes both the increase of superoxide and hydrogen peroxide (the product of the reaction calatysed by NOX enzymes) and the decrease of its substrate, i.e. oxygen [68]. Although celastrol has only recently been identified as a NOX inhibitor, its neuroprotective properties have been established several years ago. Injection of celastrol (2 and 8 mg/kg/day) improved survival of SODG93A mice by 9.4 and 13 %, respectively [74], improved dopaminergic neuron survival in the MPTP model of PD (3 mg/kg, i.p before and after MPTP injection) and with a dose of 3 mg/kg twice a day for 5 days significantly decreased the striatal lesion volume induced by 3-nitropropionic acid, a neurotoxin used to model Huntington disease in rats [30]. In a transgenic model of AD (Tg PS1/APPsw), celastrol (1 mg/kg i.p. for 4 days) and chronic treatment (32 days) with celastrol (2.5 mg/kg/day s.c. in a matrix-driven delivery pellet system) reduced the levels of both soluble and insoluble amyloid beta peptides, microglial activation, and amyloid beta plaque deposition [118]. Recently, intraperitoneal administration of celastrol improved the cognitive decline following major surgery in old mice and reduced β-amyloid accumulation and τ phosphorylation in the brain [158]. However, celastrol is a complex molecule with numerous targets [72], and its use as therapeutic awaits further study about its tolerability and possible efficacy for CNS pathologies.
Phytol
In contrast to previous examples, phytol (3,7,11,15-tetramethyl-2-hexadecene-1-ol, MW 296.53) is a compound which enhances NOX activity. Phytol has impressive effects in vivo as it completely blunts autoimmune inflammation. In the case of autoimmune disorders, phytol (i.p., but mostly s.c.) therapeutic effect was first described in arthritis-prone Ncf1 (DA) rats, which have a decreased NOX2-dependent oxidative burst. This compound has been shown to act by increasing the phagocyte oxidative burst in vivo [61]. In an acute model of Guillain Barré, phytol treatment led to a strong reduction in experimental allergic neuritis disease severity and a lower number of IFN-γ-secreting cells in late disease stage [58]. The fact that phytol is an oil makes it an unlikely candidate for development by the pharmaceutical industry because of challenging SAR, pharmacokinetics, and metabolism. Nevertheless, it represents a proof of concept for therapies aiming at enhancing NOX activity.
Chemically synthesized molecules
Triazolopyrimidines
Several compounds developed by Vasopharm have been described as NOX inhibitors (for review, see [69, 75, 162]. Recently, VAS2870l, a low-molecular-weight pharmacological NADPH oxidase inhibitor was shown to inhibit NOX1, NOX2, and NOX4, but this may be through an indirect mode of action, at least for NOX2, because it does not block NOX2 activity in a semi-recombinant membrane assay [44]. When 2 mg VAS2870l solubilized in 10 % DMSO was injected intrathecally 2 and 12 h after ischemia, brain infarct volumes were reduced by 75 % compared to vehicle-treated mice thereby improving neurological outcome and mice viability [77]. However, this study failed to demonstrate efficacy of apocynin (100 μg, i.v. 1 h before occlusion), thereby challenging other studies (Table 2). It was also the first report that provides evidence for a potential use of NOX inhibitors in the clinic for CNS diseases and suggests that investigation of other compounds targeting NOX should be similarly performed. Nevertheless, low solubility of this compound and mode of administration (intrathecal) makes it an unlikely candidate CNS drug for use in humans.
2-Acetylphenothiazine
Athough they have not yet been used in CNS disorders, it can be expected that the compound 2-acetylphenothiazine (ML171), which shows potent inhibitory activity (0.25 μM in a cellular assay) on NOX1, but also NOX2, NOX3, and NOX4 in the low micromolar range [46], can be active in the CNS because it belongs to the phenothiazine family of compounds, which are known to cross the BBB and to act as antipsychotics, but also act on other targets and are potent antioxidants [114].
3-Pyrazolopyridines
Recently, orally bioavailable small molecules developed by Genkyotex show high potency against NOX1 and NOX4 [43, 80] have shown high efficacy in animal models [21, 138, 154]. However, at this stage, no information about CNS permeability and efficacy in CNS disorders has been documented.
4-Perhexiline
Perhexiline is primarily considered as a carnitine palmitoyltransferase inhibitor, and is used in patients to treat angina pectoris [4]. Perhexiline has been known for a long time to have NOX inhibitory activity [69], but a direct action on the NOX2 isoform (IC50 = 13.2 μM in a semi-recombinant assay) was recently demonstrated [44]. Because of oral bioavailability, perhexiline might be recommended as a NOX inhibitor to treat CNS diseases. However, such an indication should, unfortunately, not be recommended for this compound because in many cases it induces drug-induced neuropathies [49, 133].
NOX inhibitors are currently developed by both academic [18] and industrial laboratories, such as Shionogi or Mitsubishi, for which only patents have been published (for review, see [69, 75], but, unfortunately, BBB penetration or efficacy in CNS indications has to our knowledge not been published.
Discussion and conclusion
Research and development of drugs for CNS disorders are particularly difficult and characterized by several challenges: (1) in the majority of the cases, the etiology of the disease is unknown, (2) mimicking CNS disorders in animal models offers a limited predictive value, (3) direct analysis of human brain samples can in most cases only be performed post mortem, (4) most CNS pathologies have a slow progression and can be diagnosed only when the disease is already at an advanced stage of development, making difficult both the identification of pathogenic events and the possible therapeutic intervention, (5) the absence or low availability of validated biomarkers to study disease progression, and (6) the presence of the blood–brain barrier, which limits access of compounds to the CNS, and requires complex additional assays to measure the concentration of drug that can effectively reach the target [117].
In this review, we have summarized the present knowledge showing that NOX enzymes represent new therapeutic targets with high potential for treatment of a large panel of CNS disorders. However, today, most of our knowledge is based on the protective effect of congenital absence of NOX2 (stable knock-out mice) or pretreatment with the antioxidant/NOX inhibitor apocynin. Therefore, alterations or adaptive responses due to the lack of NOX2 activity during the development of the brain cannot be excluded. As for future directions, use of conditional knock-outs as well as patient studies should be performed to measure NOX expression and activation. It is still unclear, for example, whether increased activity of NOX2 in neurons or microglia is responsible for diseases such as schizophrenia (with low or no inflammation) or ALS (with a strong neuroinflammation). In addition, the role of other NOX isoforms especially NOX1 (Parkinson) and NOX4 (stroke) is only just emerging. Nevertheless, it is striking how consistently a key role of NOX activity is found in CNS pathologies and may in the future represent either a therapeutic target or a biomarker of CNS disease progression. However, many more studies are required to understand the exact impact of NOX enzymes in the different human CNS pathologies.
What would be the advantages of developing compounds to target NOX2?
If antioxidant treatments have shown no efficacy in clinical trials, why should it be worthwhile developing compounds to target NOX enzymes in the CNS? There are several reasons to sustain such an effort:
-
1.
Inhibition of ROS production at the source: antioxidants act as scavengers and they are not able to prevent the actual generation of ROS. In contrast, a molecule designed to target NOX enzymes will be able to block directly the source, with the possibility of having a curative effect. Indeed, blocking NOX2-dependent ROS production, for example in microglia, would also allow for diminishing its activation and subsequent production of neuroinflammatory mediators [17].
-
2.
Specificity: antioxidant molecules do not have a specific target and, as a consequence, their molecular structure cannot be systematically improved. In contrast, developing NOX targeting molecules allow for studying the effects of structural modifications on potency and isoform selectivity. Structure–activity relationships can be developed and molecules can be improved to target the desired protein.
-
3.
Modulation: decreasing ROS is not always the therapeutic solution. It has been demonstrated that in certain pathological situations diminishing ROS can even be deleterious, such as in autoimmune CNS diseases [63]. In those cases, targeting the NOX enzyme can allow the design of compounds able to increase or restore NOX activity.
How to measure NOX activity in vivo? Use of oxidation biomarkers
Although NOX genetic deletion shows efficacy in preclinical models, a key requirement for a successful development of NOX targeted approach in humans would be the in vivo demonstration that the targeted NOX isoforms are blocked. This can only be achieved by the identification of specific biomarkers as molecular signatures of excessive NOX activity. Such biomarkers would provide extremely useful information for in vivo demonstration and for the organization of clinical trials. Increased concentration of oxidized molecules in biological fluids as biomarkers of neuropathologies is extensively documented. As an example, increased markers of lipid peroxidation (isoprostanes) and nucleic acid oxidation (8-oxodeoxyguanosine) were detected in ALS sporadic patients [106] and PD patients [139]. However, the reliability of the correlation between the presence of oxidized molecules and disease has been questioned because of numerous unresolved experimental and technical flaws (for a detailed discussion of these specific issues, see the excellent critical review in [47]). The causes involved are the fact that biomarkers of oxidative stress are often chemically unstable molecules, which require complicated and expensive detection methods for reliable quantification, such as mass spectrometry and radioimmunoassay, but also that the source of these oxidized molecules remains unresolved.
However, levels of isoprostane, a marker of lipid peroxidation, and 8-hydroxy-2’-deoxyguanosine, a marker of nucleic acid oxidation, were markedly decreased in urine of patients with hereditary deficiency in NOX2 (CGD patients) [155, 156]. NOX2 in CNS pathologies show a positive relationship of isoprostanes with AD [126] and oxidized nucleic acids in psychosis [136, 143] as well as other CNS pathologies [112]. Such biomarkers could prove invaluable for CNS pathologies, such as Alzheimer disease: currently the only way to tell whether a patient is affected by AD in a conclusive manner is post mortem histological analysis of amyloid plaques deposition. Whether NOX enzymes are a main cause of the formation of oxidized biomarkers in CNS disease awaits further confirmation. This could be done by correlating the presence of isoprostanes and oxidized nucleic acids during the progression of different neuropathological animal models using available NOX knock-outs (i.e. NOX1, NOX2, and NOX4). Validated oxidized biomarkers for NOX activity would be extremely useful in drug development, such as to assess the effect of a NOX inhibitor on disease progression, help select drug candidates, define dose effects, and facilitate the selection of populations for clinical trials, and therefore optimize chances of the successful development of a drug, and drastically decrease development costs. It would also help in comparing therapies (NOX inhibitors versus antioxidant or drugs with another mode of action).
For which pathologies would it be useful to develop NOX targeting drugs?
As mentioned above, the involvement of the NOX2 enzyme has emerged from experimental animal models of several incurable CNS diseases (Fig. 1). Because of the absence of treatment, severity, and strong rational for NOX involvement, ALS could be a primary clinical indication for NOX inhibitors. Although ALS is quite rare, it is still possible to recruit a sufficient number of patients to organize placebo versus treatment clinical trials, while for recurrent Guillain Barré, for example, its incidence is so low that only a few patients can be included, generating data of poor statistical significance. In the case of therapeutic benefit, this would pave the way to other intractable CNS pathologies, such as Huntington disease or other more frequent neurodegenerative diseases, including AD and PD. A large effort is being put into the identification of novel therapeutic options for AD with promising targets [24]. The therapeutic benefit of NOX inhibition requires more proof of concept animal models. Similarly to AD, the potential for NOX inhibitors for PD and stroke as well as activators for MS are high, but therapeutic options are available and new drugs are emerging, so more studies will be necessary to demonstrate an added benefit compared to existing drugs. However, the future of drug development for neurodegenerative disease probably relies on complex therapies with several drugs that target various pharmaceutical targets, such as other anti-inflammatory drugs, glutamate excitotoxicity, and drugs targeting neurotransmitters [92, 107].
Since the discovery of chlorpromazine in 1952, little progress has been made for the therapy of schizophrenia. Indeed, available drugs to treat schizophrenia are not curative and cause considerable side effects. They all target the same pathway (dopaminergic neurotransmission), and they primarily decrease positive symptoms, thereby inhibiting behaviors not well tolerated by the society (Fig. 2a). Excessive dopaminergic release may not be the primary event, but rather the result of alteration of GABAergic-glutamatergic neurotransmission [38]. However, these findings have not provided new opportunities for treatment. Generation of ROS by NOX2 is emerging as a possible novel mechanism, which (1) can first elicit the abnormal release of glutamate and dopamine and (2) promote neurochemical adaptive responses [11, 12, 136, 143]. NOX2 would represent an upstream target to this cascade of events, leading to the manifestation of behavioral alterations (Fig. 2b). If the complete protection seen with NOX2 inhibition in animal models translates into schizophrenic patients, the potential for the design of novel treatment for schizophrenia is huge and would provide a new paradigm in the fields of psychiatric diseases.

Schematic representation of possible use of NOX2 inhibitors in schizophrenia. a Available antipsychotic compounds inhibit excessive dopaminergic transmission, but not curative and are associated with considerable side effects. b From experimental data, it emerges that NOX2 activation could be the primary source of neurotransmission alterations, which lead to psychotic symptoms. Therefore, blocking NOX2 activation could represent a novel therapeutic approach
Possible side effects of NOX therapeutics
As most CNS diseases are generally chronic diseases, intake of NOX-based therapeutics would possibly last for years. Therefore, side effects represent a serious concern. Although off-targets effects are difficult to predict and can be determined by systemic assessment of toxicity, possible on-target side effects may result in clinical manifestations similar to what is known from animals and humans carrying genetic mutations in NOX genes [63, 67, 115]. Safety concerns regarding NOX inhibition include (1) NOX2 inhibition on microglial killing and development of hyperinflammatory states, (2) inhibition of NOX3 could lead to balance disorders as mice affected by mutations in NOX3 genes and its regulatory subunits show impaired otoconia formation and balance disorders, and (3) mutations affecting DUOX2 function lead to impaired thyroid hormone synthesis and congenital hypothyroidism. The effect of enhancing NOX2 activity, although potentially beneficial in autoimmune diseases, might result in phagocyte-mediated tissue damage. Therefore, to ensure proper monitoring of those effects, preclinical toxicity studies should include evaluation of balance disorders, measurements of circulating thyroid hormones, and inflammatory autoimmune manifestations.
Perspectives
Today, most treatments for CNS diseases are palliative or symptomatic, rather than curative or disease-modifying, and hence provide only slight relief instead of a cure. Also, they are generally associated with numerous side effects. With the increasing understanding of the biological basis of CNS diseases, common pathological mediators have been identified [65]. These include glutamate, ROS, aggregated misfolded proteins, and inflammation, and, now, NOX enzymes are emerging as key upstream regulators of at least some of the above-mentioned features. NOX enzymes represent promising CNS therapeutic targets as (1) they are major ROS generators in the CNS, (2) they regulate microglia and/or astrocyte activation, and (3) they are key modulators of T lymphocyte activation in autoimmune diseases.
These remarkable features show that, although NOX enzymes are not likely to be responsible for the etiology of CNS diseases, they represent a novel and extremely promising therapeutic area for CNS pathologies.
References
Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D (2008) Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab 9:686–696
Aliev G, Palacios HH, Walrafen B, Lipsitt AE, Obrenovich ME, Morales L (2009) Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int J Biochem Cell Biol 41:1989–2004
Ansari MA, Scheff SW (2011) NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic Biol Med 51:171–178
Ashrafian H, Horowitz JD, Frenneaux MP (2007) Perhexiline. Cardiovasc Drug Rev 25:76–97
Barber SC, Shaw PJ (2010) Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med 48:629–641
Barten LJ, Allington DR, Procacci KA, Rivey MP (2010) New approaches in the management of multiple sclerosis. Drug Des Dev Ther 4:343–366
Beal MF (2002) Oxidatively modified proteins in aging and disease. Free Radic Biol Med 32:797–803
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Beebe DW, Gozal D (2002) Obstructive sleep apnea and the prefrontal cortex: towards a comprehensive model linking nocturnal upper airway obstruction to daytime cognitive and behavioral deficits. J Sleep Res 11:1–16
Beghi E, Chio A, Couratier P, Esteban J, Hardiman O, Logroscino G, Millul A, Mitchell D, Preux PM, Pupillo E, Stevic Z, Swingler R, Traynor BJ, Van den Berg LH, Veldink JH, Zoccolella S (2011) The epidemiology and treatment of ALS: focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph Later Scler 12:1–10
Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL (2007) Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318:1645–1647
Behrens MM, Ali SS, Dugan LL (2008) Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci 28:13957–13966
Behrens MM, Sejnowski TJ (2009) Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex? Neuropharmacology 57:193–200
Bendtzen K (2010) Critical review: assessment of interferon-beta immunogenicity in multiple sclerosis. J Interfer Cytokine Res 30:759–766
Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D (1994) Alignment and sensitive detection of DNA by a moving interface. Science 265:2096–2098
Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Judd F, Katz F, Katz P, Ording-Jespersen S, Little J, Conus P, Cuenod M, Do KQ, Bush AI (2008) N-acetyl cysteine as a glutathione precursor for schizophrenia–a double-blind, randomized, placebo-controlled trial. Biol Psychiatry 64:361–368
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Borbely G, Szabadkai I, Horvath Z, Marko P, Varga Z, Breza N, Baska F, Vantus T, Huszar M, Geiszt M, Hunyady L, Buday L, Orfi L, Keri G (2010) Small-molecule inhibitors of NADPH oxidase 4. J Med Chem 53:6758–6762
Braunersreuther V, Jaquet V (2011) Reactive Oxygen Species in Myocardial Reperfusion Injury: From Physiopathology to Therapeutic Approaches. Curr Pharm Biotechnol 13:97–114
Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA (2009) NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci 12:857–863
Briones AM, Tabet F, Callera GE, Montezano AC, Yogi A, He Y, Quinn MT, Salaices M, Touyz RM (2011) Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertens 5:137–153
Bromet EJ, Fennig S (1999) Epidemiology and natural history of schizophrenia. Biol Psychiatry 46:871–881
Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ (2002) Increased 8-isoprostane and interleukin-6 in breath condensate of obstructive sleep apnea patients. Chest 122:1162–1167
Carter MD, Simms GA, Weaver DF (2010) The development of new therapeutics for Alzheimer’s disease. Clin Pharmacol Ther 88:475–486
Carter NJ, Keating GM (2010) Glatiramer acetate: a review of its use in relapsing-remitting multiple sclerosis and in delaying the onset of clinically definite multiple sclerosis. Drugs 70:1545–1577
Caslake R, Macleod A, Ives N, Stowe R, Counsell C (2009) Monoamine oxidase B inhibitors versus other dopaminergic agents in early Parkinson’s disease. Cochrane Database Syst Rev: CD006661
Chen H, Song YS, Chan PH (2009) Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab 29:1262–1272
Choi DH, Cristovao AC, Guhathakurta S, Joh T, Beal F, Kim YS (2011) NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid Redox Signal 16:1033–1045
Choi DK, Koppula S, Choi M, Suk K (2010) Recent developments in the inhibitors of neuroinflammation and neurodegeneration: inflammatory oxidative enzymes as a drug target. Expert Opin Ther Pat 20:1531–1546
Cleren C, Calingasan NY, Chen J, Beal MF (2005) Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J Neurochem 94:995–1004
Cristovao AC, Choi DH, Baltazar G, Beal MF, Kim YS (2009) The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signal 11:2105–2118
Cui C, Chen AF, Jiang Z, Wu Q, Lin J, Wen H, Zeng J (2007) Inhibition of NAD(P)H oxidase reduces fibronectin expression in stroke-prone renovascular hypertensive rat brain. Clin Exp Pharmacol Physiol 34:304–309
de la Monte SM, Wands JR (2006) Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis 9:167–181
Dumont M, Stack C, Elipenhali C, Calingasan NY, Wille E, Beal MF (2011) Apocynin administration does not improve behavioral and neuropathological deficits in a transgenic mouse model of Alzheimer’s disease. Neurosci Lett 492:150–154
Duty S, Jenner P (2011) Animal models of Parkinson’s disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 164:1357–1391
Engel O, Kolodziej S, Dirnagl U, Prinz V (2011) Modeling stroke in mice—middle cerebral artery occlusion with the filament model. J Vis Exp. doi: 10.3791/2423
Fabian RH, Perez-Polo JR, Kent TA (2008) Perivascular nitric oxide and superoxide in neonatal cerebral hypoxia-ischemia. Am J Physiol Heart Circ Physiol 295:H1809–H1814
Farber NB (2003) The NMDA receptor hypofunction model of psychosis. Ann NY Acad Sci 1003:119–130
Filippini G, Munari L, Incorvaia B, Ebers GC, Polman C, D’Amico R, Rice GP (2003) Interferons in relapsing remitting multiple sclerosis: a systematic review. Lancet 361:545–552
Fragoso YD, Frota ER, Lopes JS, Noal JS, Giacomo MC, Gomes S, Goncalves MV, da Gama PD, Finkelsztejn A (2010) Severe depression, suicide attempts, and ideation during the use of interferon beta by patients with multiple sclerosis. Clin Neuropharmacol 33:312–316
Fraser PA (2011) The role of free radical generation in increasing cerebrovascular permeability. Free Radic Biol Med 51:967–977
Fulton DJ (2009) Nox5 and the regulation of cellular function. Antioxid Redox Signal 11:2443–2452
Gaggini F, Laleu B, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P (2011) Design, synthesis and biological activity of original pyrazolo-pyrido-diazepine, -pyrazine and -oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg Med Chem 19:6989–6999
Gatto GJ, Ao Z, Kearse MG, Zhou M, Morales CR, Daniels E, Bradley BT, Goserud MT, Goodman KB, Douglas SA, Harpel MR, Johns DG (2012) NADPH oxidase-dependent and -independent mechanisms of reported inhibitors of reactive oxygen generation. J Enzyme Inhib Med Chem (in press)
Genovese T, Mazzon E, Paterniti I, Esposito E, Bramanti P, Cuzzocrea S (2011) Modulation of NADPH oxidase activation in cerebral ischemia/reperfusion injury in rats. Brain Res 1372:92–102
Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM, Rosen H (2010) A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol 5:981–993
Giustarini D, Dalle-Donne I, Tsikas D, Rossi R (2009) Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit Rev Clin Lab Sci 46:241–281
Gold R (2011) Oral therapies for multiple sclerosis: a review of agents in phase III development or recently approved. CNS Drugs 25:37–52
Gordon M, Gordon AS (1981) Perhexiline maleate as a cause of reversible parkinsonism and peripheral neuropathy. J Am Geriatr Soc 29:259–262
Gordon PH (2011) Amyotrophic lateral sclerosis: pathophysiology, diagnosis and management. CNS Drugs 25:1–15
Gupta RK, Chandra A, Verm AK, Kumar S (2010) Obstructive sleep apnoea: a clinical review. J Assoc Physicians India 58:438–441
Hall DJ, Han SH, Chepetan A, Inui EG, Rogers M, Dugan LL (2011) Dynamic optical imaging of metabolic and NADPH oxidase-derived superoxide in live mouse brain using fluorescence lifetime unmixing. J Cereb Blood Flow Metab 32:23–32
Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord 26:6–17
Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schoneich C, Engelhardt JF (2008) SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest 118:659–670
Henchcliffe C, Beal MF (2008) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol 4:600–609
Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP (2008) Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51:211–217
Hofstetter HH, Grau C, Buttmann M, Forsthuber TG, Gaupp S, Toyka KV, Gold R (2007) The PLPp-specific T-cell population promoted by pertussis toxin is characterized by high frequencies of IL-17-producing cells. Cytokine 40:35–43
Huberle A, Beyeen AD, Ockinger J, Ayturan M, Jagodic M, de Graaf KL, Fissolo N, Marta M, Olofsson P, Hultqvist M, Holmdahl R, Olsson T, Weissert R (2009) Advanced intercross line mapping suggests that ncf1 (ean6) regulates severity in an animal model of guillain-barre syndrome. J Immunol 182:4432–4438
Hughes RA, Pritchard J, Hadden RD (2011) Pharmacological treatment other than corticosteroids, intravenous immunoglobulin and plasma exchange for Guillain Barre syndrome. Cochrane Database Syst Rev: CD008630
Hui-guo L, Kui L, Yan-ning Z, Yong-jian X (2010) Apocynin attenuate spatial learning deficits and oxidative responses to intermittent hypoxia. Sleep Med 11:205–212
Hultqvist M, Olofsson P, Gelderman KA, Holmberg J, Holmdahl R (2006) A new arthritis therapy with oxidative burst inducers. PLoS Med 3:e348
Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, Holmdahl R (2004) Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci USA 101:12646–12651
Hultqvist M, Olsson LM, Gelderman KA, Holmdahl R (2009) The protective role of ROS in autoimmune disease. Trends Immunol 30:201–208
Hutchinson M (2010) Natalizumab therapy of multiple sclerosis. J Interfer Cytokine Res 30:787–789
Infante-Duarte C, Waiczies S, Wuerfel J, Zipp F (2008) New developments in understanding and treating neuroinflammation. J Mol Med 86:975–985
Jackman KA, Miller AA, De Silva TM, Crack PJ, Drummond GR, Sobey CG (2009) Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br J Pharmacol 156:680–688
Jaquet V, Bedard K (2009) Editorial: Genetic mapping–the path of discovery for novel functions of the NOX NADPH oxidases. J Leukoc Biol 86:461–463
Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, Westermaier Y, Perozzo R, Plastre O, Fioraso-Cartier L, Diebold B, Scapozza L, Nauseef WM, Fieschi F, Krause KH, Bedard K (2011) NOX NADPH oxidase isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol 164:507–520
Jaquet V, Scapozza L, Clark R, Krause KH, Lambeth JD (2009) Small Molecule NOX Inhibitors: ROS-generating NADPH Oxidases as Therapeutic Targets. Antioxid Redox Signal 11:2535–2552
Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP (2007) NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke 38:3000–3006
Kane JM, Correll CU (2010) Past and present progress in the pharmacologic treatment of schizophrenia. J Clin Psychiatry 71:1115–1124
Kannaiyan R, Shanmugam MK, Sethi G (2011) Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett 303:9–20
Kelly KA, Li X, Tan Z, VanGilder RL, Rosen CL, Huber JD (2009) NOX2 inhibition with apocynin worsens stroke outcome in aged rats. Brain Res 1292:165–172
Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF (2005) Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis 2:246–254
Kim JA, Neupane GP, Lee ES, Jeong BS, Park BC, Thapa P (2011) NADPH oxidase inhibitors: a patent review. Expert Opin Ther Pat 21:1148–1158
Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM (2006) A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci 26:1604–1615
Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH (2010) Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8(9). (pii: e1000479)
Kurz A, Perneczky R (2009) Neurobiology of cognitive disorders. Curr Opin Psychiatry 22:546–551
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V (1996) Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 347:1425–1431
Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P (2010) First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem 53:7715–7730
Lambeth JD, Krause KH, Clark RA (2008) NOX enzymes as novel targets for drug development. Semin Immunopathol 30:339–363
Lee HP, Zhu X, Casadesus G, Castellani RJ, Nunomura A, Smith MA, Lee HG, Perry G (2010) Antioxidant approaches for the treatment of Alzheimer’s disease. Expert Rev Neurother 10:1201–1208
Leng A, Feldon J, Ferger B (2004) Long-term social isolation and medial prefrontal cortex: dopaminergic and cholinergic neurotransmission. Pharmacol Biochem Behav 77:371–379
Leto TL, Morand S, Hurt D, Ueyama T (2009) Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal 11:2607–2619
Levesque S, Wilson B, Gregoria V, Thorpe LB, Dallas S, Polikov VS, Hong JS, Block ML (2010) Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain 133:808–821
Lewis DA, Hashimoto T, Volk DW (2005) Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6:312–324
Lewis DA, Lieberman JA (2000) Catching up on schizophrenia: natural history and neurobiology. Neuron 28:325–334
Liebeskind DS, Kasner SE (2001) Neuroprotection for ischaemic stroke: an unattainable goal? CNS Drugs 15:165–174
Lincecum JM, Vieira FG, Wang MZ, Thompson K, De Zutter GS, Kidd J, Moreno A, Sanchez R, Carrion IJ, Levine BA, Al-Nakhala BM, Sullivan SM, Gill A, Perrin S (2010) From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat Genet 42:392–399
Liu HG, Liu K, Zhou YN, Xu YJ (2009) Relationship between reduced nicotinamide adenine dinucleotide phosphate oxidase subunit p22phox gene polymorphism and obstructive sleep apnea-hypopnea syndrome in the Chinese Han population. Chin Med J (Engl) 122:1369–1374
Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, Miyake M, Liu KJ (2008) Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem 107:1196–1205
Lo A (2008) Advancement of therapies for neuroprotection in multiple sclerosis. Expert Rev Neurother 8:1355–1366
Lopate G, Pestronk A (2011) Inflammatory demyelinating neuropathies. Curr Treat Options Neurol 13:131–142
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7:354–365
Lull ME, Levesque S, Surace MJ, Block ML (2011) Chronic apocynin treatment attenuates beta amyloid plaque size and microglial number in hAPP(751)(SL) mice. PLoS ONE 6:e20153
Maldonado PD, Molina-Jijon E, Villeda-Hernandez J, Galvan-Arzate S, Santamaria A, Pedraza-Chaverri J (2010) NAD(P)H oxidase contributes to neurotoxicity in an excitotoxic/prooxidant model of Huntington’s disease in rats: protective role of apocynin. J Neurosci Res 88:620–629
Marden JJ, Harraz MM, Williams AJ, Nelson K, Luo M, Paulson H, Engelhardt JF (2007) Redox modifier genes in amyotrophic lateral sclerosis in mice. J Clin Invest 117:2913–2919
Marriott JJ, Miyasaki JM, Gronseth G, O’Connor PW (2010) Evidence Report: the efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 74:1463–1470
Marsh JD, Keyrouz SG (2010) Stroke prevention and treatment. J Am Coll Cardiol 56:683–691
Martinelli V, Radaelli M, Straffi L, Rodegher M, Comi G (2009) Mitoxantrone: benefits and risks in multiple sclerosis patients. Neurol Sci 30(Suppl 2):S167–S170
Masood A, Nadeem A, Mustafa SJ, O’Donnell JM (2008) Reversal of oxidative stress-induced anxiety by inhibition of phosphodiesterase-2 in mice. J Pharmacol Exp Ther 326:369–379
Massaad CA, Amin SK, Hu L, Mei Y, Klann E, Pautler RG (2010) Mitochondrial superoxide contributes to blood flow and axonal transport deficits in the Tg2576 mouse model of Alzheimer’s disease. PLoS ONE 5:e10561
Melnikova I (2007) Therapies for Alzheimer’s disease. Nat Rev Drug Discov 6:341–342
Miana-Mena FJ, Piedrafita E, Gonzalez-Mingot C, Larrode P, Munoz MJ, Martinez-Ballarin E, Reiter RJ, Osta R, Garcia JJ (2011) Levels of membrane fluidity in the spinal cord and the brain in an animal model of amyotrophic lateral sclerosis. J Bioenerg Biomembr 43:181–186
Miller RG, Mitchell JD, Lyon M, Moore DH (2003) Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph Lateral Scler Other Motor Neuron Disord 4:191–206
Mitsumoto H, Santella RM, Liu X, Bogdanov M, Zipprich J, Wu HC, Mahata J, Kilty M, Bednarz K, Bell D, Gordon PH, Hornig M, Mehrazin M, Naini A, Flint Beal M, Factor-Litvak P (2008) Oxidative stress biomarkers in sporadic ALS. Amyotroph Lateral Scler 9:177–183
Mohamed T, Rao PN (2011) Alzheimer’s disease: emerging trends in small molecule therapies. Curr Med Chem 18:4299–4320
Mossberg N, Andersen O, Nilsson S, Dahlgren C, Hellstrand K, Lindh M, Svedhem A, Bergstrom T, Movitz C (2007) Oxygen radical production and severity of the Guillain–Barre syndrome. J Neuroimmunol 192:186–191
Mossberg N, Andersen O, Nordin M, Nilsson S, Svedhem A, Bergstrom T, Hellstrand K, Movitz C (2010) Leukocyte oxygen radical production determines disease severity in the recurrent Guillain–Barre syndrome. J Inflamm (Lond) 7:40
Mossberg N, Movitz C, Hellstrand K, Bergstrom T, Nilsson S, Andersen O (2009) Oxygen radical production in leukocytes and disease severity in multiple sclerosis. J Neuroimmunol 213:131–134
Murotomi K, Takagi N, Takeo S, Tanonaka K (2011) NADPH oxidase-mediated oxidative damage to proteins in the postsynaptic density after transient cerebral ischemia and reperfusion. Mol Cell Neurosci 46:681–688
Nair D, Dayyat EA, Zhang SX, Wang Y, Gozal D (2011) Intermittent hypoxia-induced cognitive deficits are mediated by NADPH oxidase activity in a murine model of sleep apnea. PLoS ONE 6:e19847
Nakanishi N, Tu S, Shin Y, Cui J, Kurokawa T, Zhang D, Vincent Chen H-S, Tong G, Lipton SA (2009) Neuroprotection by the NR3A subunit of the NMDA receptor. J Neurosci 29:5260–526
Ohlow MJ, Moosmann B (2011) Phenothiazine: the seven lives of pharmacology’s first lead structure. Drug Discov Today 16:119–131
Ohye H, Sugawara M (2010) Dual oxidase, hydrogen peroxide and thyroid diseases. Exp Biol Med (Maywood) 235:424–433
Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, Holmdahl R (2003) Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet 33:25–32
Palmer AM, Stephenson FA (2005) CNS drug discovery: challenges and solutions. Drug News Perspect 18:51–57
Paris D, Ganey NJ, Laporte V, Patel NS, Beaulieu-Abdelahad D, Bachmeier C, March A, Ait-Ghezala G, Mullan MJ (2010) Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J Neuroinflamm 7:17
Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C (2008) Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA 105:1347–1352
Patten DA, Germain M, Kelly MA, Slack RS (2010) Reactive oxygen species: stuck in the middle of neurodegeneration. J Alzheimers Dis 20(Suppl 2):S357–S367
Perez-Lloret S, Rascol O (2010) Dopamine receptor agonists for the treatment of early or advanced Parkinson’s disease. CNS Drugs 24:941–968
Pestana RR, Kinjo ER, Hernandes MS, Britto LR (2010) Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci Lett 484:187–191
Peters EA, Hiltermann JT, Stolk J (2001) Effect of apocynin on ozone-induced airway hyperresponsiveness to methacholine in asthmatics. Free Radic Biol Med 31:1442–1447
Polymenidou M, Cleveland DW (2011) The seeds of neurodegeneration: prion-like spreading in ALS. Cell 147:498–508
Pozzilli C, Prosperini L, Borriello G (2010) Treating multiple sclerosis with fingolimod or intramuscular interferon. Expert Opin Pharmacother 11:1957–1960
Pratico D (2010) The neurobiology of isoprostanes and Alzheimer’s disease. Biochim Biophys Acta 1801:930–933
Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA (2007) Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol Dis 25:392–400
Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH (2006) A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging 27:1577–1587
Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS (2004) NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279:1415–1421
Rappold PM, Cui M, Chesser AS, Tibbett J, Grima JC, Duan L, Sen N, Javitch JA, Tieu K (2011) Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc Natl Acad Sci USA 108:20766–20771
Reddy R (2011) Antioxidant Therapeutics for Schizophrenia. Antioxid Redox Signal 15:2047–2055
Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ (2003) Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348:1333–1341
Said G (1978) Perhexiline neuropathy: a clinicopathological study. Ann Neurol 3:259–266
Sanchez-Moreno C, Jimenez-Escrig A, Martin A (2009) Stroke: roles of B vitamins, homocysteine and antioxidants. Nutr Res Rev 22:49–67
Savitt JM, Dawson VL, Dawson TM (2006) Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest 116:1744–1754
Schiavone S, Sorce S, Dubois-Dauphin M, Jaquet V, Colaianna M, Zotti M, Cuomo V, Trabace L, Krause KH (2009) Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry 66:384–392
Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, Seeger W, Grimminger F (2000) Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 162:566–570
Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM, Hebert RL (2010) Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol 299:F1348–F1358
Seet RC, Lee CY, Lim EC, Tan JJ, Quek AM, Chong WL, Looi WF, Huang SH, Wang H, Chan YH, Halliwell B (2010) Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radic Biol Med 48:560–566
Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S (2000) Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun 273:5–9
Simonyi A, He Y, Sheng W, Sun AY, Wood WG, Weisman GA, Sun GY (2010) Targeting NADPH oxidase and phospholipases A2 in Alzheimer’s disease. Mol Neurobiol 41:73–86
Sorce S, Krause KH (2009) NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal 11:2481–2504
Sorce S, Schiavone S, Tucci P, Colaianna M, Jaquet V, Cuomo V, Dubois-Dauphin M, Trabace L, Krause KH (2010) The NADPH oxidase NOX2 controls glutamate release: a novel mechanism involved in psychosis-like ketamine responses. J Neurosci 30:11317–11325
Stefanska J, Pawliczak R (2008) Apocynin: molecular aptitudes. Mediat Inflamm 2008:106507
Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ (1994) Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11:95–102
Sudeshna G, Parimal K (2010) Multiple non-psychiatric effects of phenothiazines: a review. Eur J Pharmacol 648:6–14
Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA (2007) Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest 117:910–918
Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA (2008) Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol 64:654–663
Tang LL, Ye K, Yang XF, Zheng JS (2007) Apocynin attenuates cerebral infarction after transient focal ischaemia in rats. J Int Med Res 35:517–522
Tang XN, Cairns B, Cairns N, Yenari MA (2008) Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience 154:556–562
Titova E, Ostrowski RP, Sowers LC, Zhang JH, Tang J (2007) Effects of apocynin and ethanol on intracerebral haemorrhage-induced brain injury in rats. Clin Exp Pharmacol Physiol 34:845–850
Trumbull KA, McAllister D, Gandelman MM, Fung WY, Lew T, Brennan L, Lopez N, Morre J, Kalyanaraman B, Beckman JS (2012) Diapocynin and apocynin administration fails to significantly extend survival in G93A SOD1 ALS mice. Neurobiol Dis 45:137–144
van Horssen J, Witte ME, Schreibelt G, de Vries HE (2011) Radical changes in multiple sclerosis pathogenesis. Biochim Biophys Acta 1812:141–150
Vendrov AE, Madamanchi NR, Niu XL, Molnar KC, Runge M, Szyndralewiez C, Page P, Runge MS (2010) NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J Biol Chem 285:26545–26557
Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De Mattia D, Martire B, Pietrogrande MC, Martino S, Gambineri E, Soresina AR, Pignatelli P, Martino F, Basili S, Loffredo L (2009) Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation 120:1616–1622
Violi F, Sanguigni V, Loffredo L, Carnevale R, Buchetti B, Finocchi A, Tesauro M, Rossi P, Pignatelli P (2006) Nox2 is determinant for ischemia-induced oxidative stress and arterial vasodilatation: a pilot study in patients with hereditary Nox2 deficiency. Arterioscler Thromb Vasc Biol 26:e131–e132
Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR (1997) Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke 28:2252–2258
Wan Y, Xu J, Meng F, Bao Y, Ge Y, Lobo N, Vizcaychipi MP, Zhang D, Gentleman SM, Maze M, Ma D (2010) Cognitive decline following major surgery is associated with gliosis, beta-amyloid accumulation, and tau phosphorylation in old mice. Crit Care Med 38:2190–2198
Wang Q, Smith RE, Luchtefeld R, Sun AY, Simonyi A, Luo R, Sun GY (2008) Bioavailability of apocynin through its conversion to glycoconjugate but not to diapocynin. Phytomedicine 15:496–503
Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY (2006) Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res 1090:182–189
Wang Y, Zhang SX, Gozal D (2010) Reactive oxygen species and the brain in sleep apnea. Respir Physiol Neurobiol 174:307–316
Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, Wingler K (2010) Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 161:885–898
Wipfler P, Harrer A, Pilz G, Oppermann K, Trinka E, Kraus J (2011) Recent developments in approved and oral multiple sclerosis treatment and an update on future treatment options. Drug Discov Today 16:8–21
Wu DC, Re DB, Nagai M, Ischiropoulos H, Przedborski S (2006) The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci USA 103:12132–12137
Xian NT, Zhen Z, Rona GG, Midori AY (2011) Significance of marrow derived NADPH oxidase in experimental ischemic stroke. Ann Neurol 70:606–615
Yamamoto E, Tamamaki N, Nakamura T, Kataoka K, Tokutomi Y, Dong YF, Fukuda M, Matsuba S, Ogawa H, Kim-Mitsuyama S (2008) Excess salt causes cerebral neuronal apoptosis and inflammation in stroke-prone hypertensive rats through angiotensin II-induced NADPH oxidase activation. Stroke 39:3049–3056
Yoshioka H, Niizuma K, Katsu M, Okami N, Sakata H, Kim GS, Narasimhan P, Chan PH (2011) NADPH oxidase mediates striatal neuronal injury after transient global cerebral ischemia. J Cereb Blood Flow Metab 31:868–880
Zamvil SS, Steinman L (1990) The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol 8:579–621
Zhan G, Serrano F, Fenik P, Hsu R, Kong L, Pratico D, Klann E, Veasey SC (2005) NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med 172:921–929
Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, Hong JS, Liu B (2004) Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: role of NADPH oxidase. FASEB J 18:589–591
Zhao H, Mayhan WG, Arrick DM, Xiong W, Sun H (2010) Alcohol-induced exacerbation of ischemic brain injury: role of NAD(P)H oxidase. Alcohol Clin Exp Res 34:1948–1955
Zheng JS, Zhan RY, Zheng SS, Zhou YQ, Tong Y, Wan S (2005) Inhibition of NADPH oxidase attenuates vasospasm after experimental subarachnoid hemorrhage in rats. Stroke 36:1059–1064
Zhu Y, Fenik P, Zhan G, Mazza E, Kelz M, Aston-Jones G, Veasey SC (2007) Selective loss of catecholaminergic wake active neurons in a murine sleep apnea model. J Neurosci 27:10060–10071
Zia MT, Csiszar A, Labinskyy N, Hu F, Vinukonda G, LaGamma EF, Ungvari Z, Ballabh P (2009) Oxidative-nitrosative stress in a rabbit pup model of germinal matrix hemorrhage: role of NAD(P)H oxidase. Stroke 40:2191–2198
Acknowledgments
We are grateful to Dr Karen Bedard and Dr Freddy Heitz for critical reading of the manuscript and to all the members of the NEURINOX consortium for their input in the elaboration of the concepts described in this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sorce, S., Krause, KH. & Jaquet, V. Targeting NOX enzymes in the central nervous system: therapeutic opportunities. Cell. Mol. Life Sci. 69, 2387–2407 (2012). https://doi.org/10.1007/s00018-012-1014-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1014-5