
Abstract
Alzheimer’s disease (AD) is considered the sixth leading cause of death in elderly patients and is characterized by progressive neuronal degeneration and impairment in memory, language, etc. AD is characterized by the deposition of senile plaque, accumulation of fibrils, and neurofibrillary tangles (NFTs) which are responsible for neuronal degeneration. Amyloid-β (Aβ) plays a key role in the process of neuronal degeneration in the case of AD. It has been reported that Aβ is responsible for the production of reactive oxygen species (ROS), depletion of endogenous antioxidants, increase in intracellular Ca2+ which further increases mitochondria dysfunctions, oxidative stress, release of pro-apoptotic factors, neuronal apoptosis, etc. Thus, oxidative stress plays a key role in the pathogenesis of AD. Antioxidants are compounds that have the ability to counteract the oxidative damage conferred by ROS. Therefore, the antioxidant therapy may provide benefits and halt the progress of AD to advance stages by counteracting neuronal degeneration. However, despite the beneficial effects imposed by the antioxidants, the findings from the clinical studies suggested inconsistent results which might be due to poor study design, selection of the wrong antioxidant, inability of the molecule to cross the blood–brain barrier (BBB), treatment in the advanced state of disease, etc. The present review insights into the neuroprotective effects and limitations of the antioxidant therapy for the treatment of AD by targeting mitochondrial-derived ROS. This particular article will certainly help the researchers to search new avenues for the treatment of AD by utilizing mitochondrial-derived ROS-targeted antioxidant therapies.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
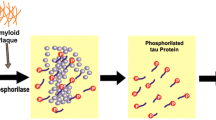
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive neuronal degeneration in the brain resulting in the impairment of behavior, cognition, memory, language, etc. AD mainly affects individuals in the later years of life and is considered the sixth leading cause of death in elderly patients. In AD, the individual initially shows the loss of short-term episodic and the retention of the long-term memory. However, in the later stages, psychiatric impairment occurs in the patients [1,2,3]. AD can be categorized into two types: familial AD (FAD) presents the minority of AD cases (approx. 1%) and sporadic AD (SAD) or late-onset AD (LOAD) presents the majority of the cases of the AD and aging is considered the main cause of this form of AD [4, 5]. Pathologically, in AD, there is the deposition of senile plaque, accumulation of fibrils, and neurofibrillary tangles (NFTs that ultimately leads to neuronal degeneration in the brain [6, 7]. Amyloid beta (Aβ is the major component of the senile plaques [8], whereas tau protein is the main component of neurofibrillary tangles observed in the brain of AD patients [9]. Aβ is generated as a heterogeneous pool of peptides, and it occurs mainly in two isoforms i.e., Aβ1-40 and Aβ1-42 of which Aβ1-42 peptide is considered more amyloidogenic than Aβ1-40 [10]. Aβ is generated by the sequential cleavage of amyloid precursor protein (APP) by the enzymes, i.e., α and β-secretase [11]. The production of the Aβ is catalyzed by the enzyme γ-secretase [12]. Aβ so produced is mainly non-toxic in its monomeric form but when aggregates to form oligomers followed by the production of senile plaques (fibrils) confers neurotoxicity [11]. Aβ is also responsible for the formation of NFTs by causing the hyperphosphorylation of tau protein [13], inhibition of the binding of the tau with the microtubules [14], leading to their destabilization resulting in the neuronal injury and death [15]. It is suggested that the aggregated tau is more toxic while the soluble and prefibrillar tau is more likely to cause neurodegeneration [16]. It has been reported that oxidative stress may increase the phosphorylation of tau and thus may accelerate the neurodegeneration in the patients of AD [17,18,19]. Further higher levels of phosphorylated tau have been observed in AD patients [20, 21]. In AD, the neurofibrillary degeneration starts in the allocortex and spreads to the associative isocortex [22]. However, the neuronal loss often exceeds NFTs [23]. Therefore, the therapies that inhibit the phosphorylation of tau and its oligomerization may confer therapeutic benefits in patients of AD [12]. However, besides promoting the phosphorylation of tau protein, Aβ also increases the intracellular levels of Ca2+ by opening the calcium channels [24] which further promote the influx of Ca2+ [25] resulting in the Ca2+ overload responsible for the increase production of Aβ (by increasing the expression of β-secretase enzyme), aggregation of Aβ [26, 27], and Aβ-induced membrane damage, fragmentation, and lipid loss [28]. Ca2+ overload also promotes the influx of Ca2+ inside the mitochondria resulting in the mitochondrial outer membrane permeabilization (MOMP), release of pro-apoptotic factors, and subsequent neuronal apoptosis [29]. These facts suggested that reducing the Ca2+ influx may counteract the neurotoxicity confers by the Aβ oligomers in the hippocampus of AD transgenic mice [30]. It has been reported that ROS produced during oxidative stress also utilize the signaling pathways involving Ca2 + to confer mitochondrial and neuronal damage. Further, mitochondrial dysfunction also promotes the production of ROS [31] and the increase production of ROS has been implicated in aging and AD [32]. Antioxidants are the chemical compounds that counteract the oxidative damage conferred by ROS [33, 34]. Preclinical studies suggested that the antioxidant supplementation improves the learning ability [35] and memory functions in rodents [36] by providing protection against extracellular and intracellular ROS [37, 38]. Besides this, the antioxidants inhibit the aggregation of Aβ [39, 40], improve cognition and Mini-Mental State Exam (MMSE) score in AD patients [41, 42], and modulate the therapeutic effect of vitamin B in AD patients [43]. Mitochondria is the cell organelle that is affected mainly by oxidative stress thus the treatment with the mitochondriotropic antioxidants having low cytotoxic effects may improve the outcomes by protecting the mitochondria against the deleterious effects of oxidative stress [44]. However, despite these positive findings, several studies suggested no clear significant effect of antioxidants in AD patients [45,46,47] and convincing evidence regarding their therapeutic efficacy in AD is still lacking [48, 49]. In the present review, the authors describe the production of ROS, ROS-mediated damage in AD, the role of antioxidants, and the therapeutic efficacy of antioxidants in the treatment of AD.
Mitochondrial ROS Formation and Oxidative Stress
Mitochondria are considered the powerhouse of cells and play an important role in the production of energy, thus the mitochondrial impairment results in the impaired energy metabolism, increased production of ROS, and impaired Ca2+ buffering [50]. Mitochondrial formation utilizes the electron transport chain (ETC) and its several complexes for the production of ATP. These complexes are located on the inner mitochondrial membrane together with complex-V [51]. Besides this, the ETC required cytochrome c and co-enzyme Q10. Electrons are transported through a series of complexes and ultimately transferred to the molecular oxygen (O2) resulting in the production of water. The simultaneous translocation of protons from the matrix to intermembrane space creates a proton gradient responsible for the production of ATP, and this reaction is catalyzed by the enzyme ATP synthase [52, 53]. The transfer of the electron across the complexes is a very efficient process,however, when the electron gets leaks from the ETC or its complexes, the electron is captured by the molecular oxygen resulting in the production of ROS. The capturing of the electron by the oxygen molecule is responsible for the production of superoxide radical (O2−), which is then converted into hydrogen peroxides (H2O2) (by the enzyme superoxide dismutase) and the later is converted into hydroxyl radical (OH−) (by the enzyme catalase) [54,55,56,57], and these ROS confer oxidative damage [58, 59] (Fig. 1). Besides increasing the production of ROS, Aβ also depletes endogenous antioxidants of the body [60] resulting in lipoperoxidation and neuronal dysfunctions [61]. Aβ aggregates also promote phosphorylation of tau proteins [62,63,64] which then interferes in the activity of complex-I of ETC, contributing to mitochondrial dysfunction and ROS production [65]. Aβ oligomers mediated ROS formation resulting in the NMDA activation and increased influx of Ca2+ ions through the calcium ion channel responsible for neuronal excitotoxicity and cell death [66,67,68,69]. Besides this, the Aβ itself induces the increase in intracellular Ca2+ concentrations in neuronal cells [70, 71]. The Ca2+ overload causes the impairment in the normal functioning of the mitochondria resulting in the excessive production of ROS responsible for the MOMP and the release of pro-apoptotic factors in the cytoplasm responsible for the neuronal apoptosis [29]. ROS may trigger the opening of mitochondrial permeability transition pore (mPTP) which is involved in the regulation of mitochondrial Ca2+ homeostasis [72] (Fig. 2). ROS induces the opening of mPTP resulting in further release of ROS, and this phenomenon is referred to as “ROS-induced ROS release (RIRR)” [73]. It has been reported that the short and transient opening of mPTP is important for the passage of protons, Ca2+, and other substances [74, 75], but the prolonged opening of mPTP causes mitochondrial disruption and deposition of Ca2+ in the mitochondrial matrix [76]. Besides this, Ca2+ ions also participate in the Aβ-promoted membrane damage and lipid loss [28]. Aging is characterized by the increased production of ROS [77, 78]. ROS further participates in the glycation processes and the formation of advanced glycation end products (AGEs) that has been observed in the plaques of AD patients [79, 80], causing mitochondrial dysfunction [81,82,83,84] and subsequent neuronal degeneration [85,86,87,88,89].

Production of reactiveoxygen species

ROS-mediated mitochondrial impairment and cell death
Aβ-Mediated Oxidative Stress and Damage in AD
AD is characterized by the accumulation of Aβ plaques and NFT formation [90] both of which are linked with the production of ROS responsible for the neuronal, synaptic, and mitochondrial dysfunctions in AD patients [50, 91,92,93,94,95]. Oxidative stress facilitates the expression of protein, i.e., amyloid precursor protein (APP) and beta-secretase (BACE1) involved in the increased generation of Aβ [96]. Oxidative stress also induces a pathogenic PS1 conformational change in neurons, resulting in the increased ratio of Aβ42/40 [97]. Oxidative stress may also produce interference in the clearance of Aβ by oxidizing the LRP1. LRP1 is a multifunctional protein that is responsible for the efflux of Aβ across the BBB [98, 99]. Aβ is responsible for the disruption of the normal functioning of the LRP1 protein and thus restricts its clearance and favors its accumulation in the brain [100]. Also, the reduced activity of LRP1 protein has been observed in the brain of AD patients [101]. Aβ activates the JNK/c-jun pathway and induces apoptosis in in vitro models [102,103,104]. JNK has been shown to promote the phosphorylation of tau protein in various cells and animal models [105]. Tau plays a key role in the maintenance of microtubule dynamics and the protection of DNA and RNA from ROS-mediated damage. Thus, the alteration in the tau protein is responsible for the increased oxidation of DNA and RNA [106] and mitochondrial dysfunction in an animal model of AD. Aβ-mediated oxidative stress is responsible for the modification and the alterations in the normal conformation of tau protein resulting in the formation of NFTs [105]. ROS formations facilitate the self-assembly of tau protein into fibrillary polymers [107]. The conformational changes in the tau protein and the formation of the NFTs may further evoke neuronal degeneration [108]. However, it has been reported that the formation of neurofibrillary lesions counteracts the damaging effects of oxidative damage on neuronal cells suggesting neurofibrillary formation is a neuronal adaptation to chronic oxidative stress and functions as the antioxidant to protect the neurons [109]. Besides this, Aβ also induces the rise in the intracellular Ca2+ levels by the activations of calcium channels, glutamate receptors, etc. [110,111,112,113,114]. The increased conductance through the Ca2+ channels resulted in the mitochondrial Ca2+ overload and ROS overproduction resulting in the in mitochondrial outer membrane permeabilization (MOMP) and release of pro-apoptotic responsible for neuronal apoptosis [29].
This evidence suggested that oxidative and nitrosative stress play a key role in the pathogenesis and damage in AD [115, 116]. Oxidative stress is imposed by the overproduction and the increased levels of the Aβ which is responsible for the oxidation of proteins, lipids, and nucleic acids in the hippocampus and cortex region of the brain [117]. This is further confirmed by the finding suggesting the increased oxidation of protein and lipid in the brain regions rich in Aβ [118, 119]. The increased levels of the carbonylated proteins in the hippocampus and parietal cortex suggested protein oxidation in the AD brain [120]. Further, the membrane proteins are damaged more as compared to the cytoplasmic proteins [121]. Protein modification resulting from the reaction of the proteins with 4-hydroxynonenal (4-HNE) and peroxynitrite (ONOO–) increases the proteosomal degradation of brain proteins [122]. Further, the evidence suggesting the lipid oxidation is suggested by the increased concentrations of 4-HNE in the brain hippocampus of AD patients [123]. 4-HNE is also responsible for the oxidative modification of lipoic acid [124] “apparently” evident in hippocampal regions of the brain [125]. Oxidative stress also confers the oxidative damage to the nuclear and mitochondrial DNA in AD in frontal, parietal, and temporal cortex [126, 127]. It is further confirmed by the studies suggesting the increased levels of 8-hydroxyguanine in the hippocampus of AD patients in the preclinical stage of disease [128]. The oxidative DNA damage has also been observed in the peripheral blood cells of AD patients [129]. RNA oxidation especially mRNA oxidation has been reported in the frontal cortex region of the brain [130]. Oxidative stress is also responsible for the reduced production of ATP and brain energy metabolism both of which have been considered the key event in AD pathogenesis [131]. The reduction of the ATP levels causes the leakage of the electron from the ETC resulting in the overproduction of ROS [132]. Oxidative stress reduces the expression or inactivates the enzyme ATP synthase in the AD brain [133,134,135]. It has been reported that the accumulated Aβ in the AD brain directly interact with the ATP synthase, inhibit it [136], and thus impaired the ATP production in AD [137] further suggesting the link between AD and diabetes [118, 119].
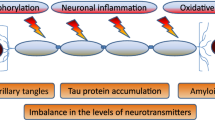
Oxidative stress also influences the expression of ApoE4 [138] which is involved in the regulation of aggregation and clearance of Aβ [139, 140], mitochondrial dysfunction, calcium homeostasis [141, 142], synaptic architecture, and functioning [143,144,145]. It has been reported that the impaired ApoE expression is linked to oxidative stress implicated in AD [146,147,148,149]. The role of ROS in the emergence of AD and the consequences of Aβ deposition and its role in AD-related neuronal degeneration are presented in Fig. 3a, b.

a Factors contributingto the ROS Production and Alzheimer’s disease. b Amyloid beta-mediated neuronal dysfunction and neuronal cell death in AD
Oxidative Stress Induced by AD Inducing Agents in the Preclinical Models of AD
AlCl3 is mainly used to induce Alzheimer-like disease in laboratory animals. Al also binds with the Aβ promoting its precipitation in a β-sheet conformation responsible for its retention and accumulation in brain tissue [150]. Al also promotes the oligomerization of Aβ and promotes the Ca2+ overload, inducing ER stress and apoptotic pathway governed by specific caspase-12 [151]. It has been reported that the administration of AlCl3 enhances ROS production and oxidative stress, LPO, and NO levels but reduces the GSH levels as compared to control [152]. Further, the administration of AlCl3 led to the deposition of aluminum in the hippocampus and cortex. This is responsible for the induction of memory impairment novel object recognition test and passive avoidance test. It has been reported that the transferrin favors the transport of aluminum across the BBB responsible for its entry into the brain. The deposition of aluminum in the brain is responsible for neuronal structural damage, neuronal cell death, and memory deterioration [153]. This is further in line with the evidence that the AlCl3 significantly reduces contextual memory in the passive avoidance test and spatial memory in the Morris water maze test. AlCl3 is known to upregulate the expression of proapoptotic protein and downregulate the expression of an anti-apoptotic protein. AlCl3 also promotes the translocation of cyt c and increases the expression of enzymes capsase-9 and capsase-3 to cause neuronal cell death [154].
STZ is a glucosamine-nitrosourea derivative, used frequently for the induction of AD-like pathology in experimental animals. STZ administration resulted in the impaired metabolism of glucose in the brain responsible for neuronal degeneration in AD. Administration of STZ (icv route) resulted in acute memory impairment, partial recovery of memory, and then chronic slow memory impairment. However, the administration of higher doses of STZ resulted in the severe and faster degeneration of neurons resulting in more cognitive deficits. It is in line with the evidence that the administration of STZ (≤ 1 mg/kg, icv) promotes slower neuronal degeneration, STZ (2 mg/kg, icv) administration induces cognitive impairment and neurodegeneration while STZ (≥ 3 mg/kg, icv) produces interference in locomotor activity, neurotoxicity, and faster severe neurodegeneration [155]. The findings from the behavioral studies suggested that the administration of STZ resulted in the impairment in learning and memory in the passive avoidance test and Morris water maze (MWM) test [156]. The further administration of STZ resulted in the emergence of oxidative stress evident by the increased LPO, nitrite levels and decreased GSH and SOD levels [156]. The pathological investigation suggested that the administration of STZ increases the hippocampal Aβ level by increasing the expression of APP [156] and increases the total cerebral Aβ fragments, tau protein, and Aβ deposits [157]. Insulin is known to negatively regulate the metabolism of Aβ and tau proteins [157]. STZ (3 mg/kg, icv) administration does not interfere with the peripheral blood glucose level, induces significant cognitive impairment and resulted in the insulin-resistance state which is analogous to AD [158]. STZ (3 mg/kg, icv) further decreases the mitochondrial transmembrane potential, ATP content, and calcium-induced mitochondrial permeability transition responsible for neuronal cell death [159].
Iron plays a key role in the pathogenesis of AD [160] by increasing the production of ROS by participating in the Fenton reaction [161, 162]. It has been reported that Fe3+ reacts with water to form hydroxyl radicals in the presence of H2O2. The hydroxyl radicals so formed causes the conversion of soluble fibrinogen into insoluble fibrin which is resistant to hydrolytic actions of enzymes and is thus responsible for the neurological consequences [163, 164]. The iron itself is also responsible for the oxidation of the cellular molecules [165, 166] and causes neuronal cell damage in the AD and related conditions [167]. The iron-mediated neuronal cell death is known as ferroptosis, a recent mechanism that is targeted by the researchers in the context of AD [168, 169]. Besides this, iron itself increases the production of and aggregation of Aβ [160, 170,171,172,173], the later causes the conversion of Fe3+ form to Fe2+ further responsible for the excessive generation of ROS and subsequent neuronal cell death [174, 175]. Further studies have suggested that the excess of iron induces cognitive deficits, and interacting with Aβ increases ROS production and contributes to neuronal cell death [176, 177].
Endogenous Antioxidant Defense and the Emergence of AD
Brain is the organ that is most susceptible to the damage confer by the ROS, and it is due to the increased production of ROS, low levels of antioxidants, low repair capacity, and non-replicating natures of neurons [178]. Antioxidants are the chemical compounds that have the ability to counteract the oxidative damage confer by ROS [33, 34]. Thus, the antioxidant either endogenous or exogenous mitigates the oxidative damage by maintaining the balance between pro-oxidant and antioxidant defense [179]. The antioxidants may be enzymatic and non-enzymatic, but they perform the same function that is the mitigation of oxidative damage [180, 181]. The antioxidants confer the antioxidant effect either by trapping the oxygen, by chelation with the prooxidative metals [182,183,184], or by inhibiting the production of ROS or by converting the ROS into stable compounds [183, 185, 186]. Thus, the treatment of neurodegenerative disorders by antioxidants may provide benefits and halt the progression of the disease to advance stages by counteracting the process of neuronal degeneration [187]. However, despite of the beneficial effects imposed by the antioxidants, the findings from the clinical studies suggested inconsistent results which might be due to poor study design, selection of the wrong antioxidant, inability of the molecule to cross the BBB, treatment in the advanced state of disease, etc.[188].
Endogenous Antioxidants
Glutathione
Glutathione is the most prevalent antioxidantof the cell existing in two isoforms, thiol-reduced (GSH) and disulfide-oxidized form (GSSG). In the reduced form, the glutathione reacts with ROS, and in this process, it is converted into the oxidized form. The conversion of the reduced form to the oxidized form is catalyzed by the enzyme glutathione peroxidase (GPx) whereas the conversion of oxidized form into reduced form is catalyzed by the enzyme glutathione reductase (GR). Thus, GSH/GSSG ratio is frequently used as a frequent indicator of oxidative stress in the cells [189, 190]. The results from the biochemical analysis of the AD brain suggested the reduced level of GSH and increased levels of GSSG in the frontal cortex region and the hippocampus region of the brain as compared to control [191,192,193]. Further, the extent of the changes in the levels of the oxidized and reduced form is closely related to the severity of the disease [191, 193].
Superoxide Dismutase
Superoxide dismutase (SOD) is an enzyme that catalyzes the conversion of the superoxide radicals into hydrogen peroxide [194,195,196]. Superoxide dismutase exists in three forms (includes copper/zinc-superoxide dismutase, manganese-superoxide dismutase, and extracellular superoxide dismutase) in the body (mainly in the cytoplasm and mitochondria) [196,197,198]. It has been reported that AD is characterized by the reduced expression and the activity of enzyme SOD in the hippocampus region of the brain [199, 200].
Catalase
Catalase is an enzyme present in the peroxisomes [201] responsible for the conversion of approx. 6 million molecules of H2O2 into H2O and O2 every minute [202]. The findings from the preclinical studies suggested the reduced activity of catalase in the STZ-induced AD in rats [203]. Further, the findings from the clinical studies suggested the reductions in CAT activity in several regions of the brain of AD patients [204]. It has been reported that the Aβ in AD promotes the accumulation of H2O2 in the hippocampal neurons [79, 205] by inhibiting the enzyme catalase [206]. It may happen that Aβ directly inhibits the enzyme catalase or it may inhibit the catalase by promoting oxidative stress [207].
Methionine Sulfoxide Reductase
Methionine sulfoxide reductase (MsrA) is responsible for the conversion of methionine sulfoxide into methionine, and the reduced expression of the MsrA activity has been reported in several regions of the brain AD [208].
Nuclear Factor E2-Related Factor 2
E2-related factor 2 (Nrf2) regulates the endogenous antioxidant response in response to oxidative stress. The interaction of Nrf2 with the kelch-like ECH-associated protein (Keap1) sequesters it into the cytoplasm, leading to ubiquitination and subsequent degradation. Nrf2 in its free form stabilizes and translocates inside the nucleus to carry out the gene transcription for the antioxidant enzymes and to confer protection against oxidative damage. However, when the antioxidant response is not required, Nrf2 is removed from the nucleus by a nuclear export sequence near its nuclear localization signal [209]. It has been reported that the Nrf2 increases the expression of the various endogenous enzyme (heme oxygenase-1 (HMOX1), glutathione S-transferase (GST), superoxide dismutase (SOD), catalase (CAT), NAD(P)H dehydrogenase (quinone) 1 (NQO1)) to confer the protection against the oxidative stress [210,211,212,213,214]. NRF2 also regulates the expression of anti-inflammatory mediators such as interleukin (IL)-17D, CD36, etc. [215,216,217] and reduces the expression of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, etc. [218, 219]. Nrf2 signaling impairment has been implicated in the pathogenesis of AD [209]. It has been reported that in AD, Nrf2 is predominantly localized to the cytoplasm in hippocampal neurons and failed to get translocated inside the nucleus. It is further accompanied by the reduced levels of nuclear Nrf2 in AD. Further, in aged individuals, Nrf2 often fails to perform its normal functions responsible for the emergence of oxidative damage and further neuronal degeneration in later stages [209].
Exogenous Antioxidants in the Treatment of AD
Antioxidants provide protection against ROS and counteract the deleterious effects of oxidative stress [37]. Antioxidant, e.g., luteolin has been shown to significantly inhibit the production of ROS and increases the neuronal cell survival by improving the antioxidant status [220]. Further, luteolin and epigallocatechin-3-gallate (EGCG) have the mitochondrial restorative capability and suppress the burden of Aβ in the brain to confer the therapeutic benefits in the AD [221]. Numerous antioxidants have been screened for their anti-AD potential, but the antioxidants which are used mainly in the clinical studies are given below:
Curcumin
Curcumin is a polyphenolic natural compound that has shown its beneficial therapeutic effect in AD and related conditions in various preclinical and clinical studies. A metanalysis of six clinical trials with a total of 289 subjects suggested the beneficial effect of curcumin on cognitive function [222]. The findings from the preclinical studies suggested that the curcumin treatment counteracts the deleterious effects of oxidative stress and reverses the memory impairment [38]. The results from a study in which the participants were administered with curcumin (180 mg/day for 12 weeks) suggested that the curcumin treatment reduces GSK-3β activity, IAPP, and insulin resistance significantly as compared to the placebo group [223]. In a study of 34 AD patients receiving curcumin (1 g/day or 4 g/day for 6 months), curcumin treatment did not show any significant effect as compared to placebo [41]. Another study suggested that the daily administration of curcumin 2–4 g/day for 24 weeks did not produce any significant alterations in plasma and CFS levels of Aβ and tau levels [42]. Further studies have reported that the curcumin inhibits the aggregation of Aβ [39, 40], reduces the plaque burden [224, 225] and improve cognition and MMSE score [41, 42].
Omega-3 Fatty Acids
Omega-3 fatty acids (OFA) is one of the key molecules with diverse antioxidant properties and has been shown to reduce the risk of several neurological disorders. Docosahexaenoic acid (DHA) is the most important component of OFA. In the brain, the enzyme phospholipase A2 and lipoxygenase convert DHA into neuroprotection 1 (NPD1) which exerts neuroprotective effects [226]. DHA also regulate neurogenesis, neuronal survival, and normal cognitive functions [227]. DHA supplementation thus improves learning ability [35] and memory functions in aged rats [36]. Besides this, DHA also confers protection against the deleterious effects of ROS [228, 229], enhances the endogenous antioxidant system [230], provide neuroprotection against Aβ-induced neurotoxicity [231, 232], limit the production and accumulation of Aβ [233, 234], and increases the cerebral acetylcholine levels [235, 236]. Several studies have reported the therapeutic benefits of OFA and its component DHA in AD patients. In a study on 44 individuals, OFA supplementation has been shown to improve the recall of objects as compared with placebo [237]. A study on 204 AD patients on acetylcholine esterase inhibitor treatment having a MMSE score of 15 points were supplemented with OFA for 6 months. It was observed that OFA supplementation did not delay the rate of cognitive decline but improve MMSE score in a smaller group of patients further suggesting the beneficial effect of OFA [238].A study on 174 AD patients receiving OFA for 6 months suggested that the OFA supplementation lowers the rate of deterioration and preserves the cognition in the AD patients [239]. In a study on 39 AD patients with OFA supplementation for 12 months, it was observed that the supplementation slowed down the cognitive and functional decline in the AD patients [240]. A double study on 204 AD patients suggested that the OFA supplementation for 6 months positively improve weight and appetite AD patients [241]. Oral supplementation with OFA results in the reduction of Ab burden, total and phosphorylated tau levels in CSF, inflammatory biomarkers and neuronal loss in AD patients [242,243,244]. Phillips et al. [245] reported no benefit of OFA supplementation for 4 months in AD patients. OFA is known to modulate the therapeutic effect of vitamin B such that when the levels of OFA are low, then vitamin B produces no effect on cognition,however, when the levels of OFA are higher then the vitamin B, treatment may slow down the cognitive dysfunctioning in the AD patients [43]. Further, it has been reported that vitamin B in the presence of OFA confers beneficial effects on brain atrophy [246].
Vitamin E
Vitamin E is a well-known antioxidant that is known to confer neuroprotection in AD [247, 248]. The antioxidant potential of vitamin E is due to the presence of the hydroxyl group in the phenolic ring structure [249]. Further, the studies have suggested the reduced level of vitamin E in the patients of AD [46, 47, 250]. Various clinical studies suggested the beneficial effect of vitamin E in AD. However, some of the studies present either no effect of vitamin E or the worsening of the condition upon vitamin E treatment. A recent meta-analysis suggested that vitamin E exhibit the most pronounced protective effects [251]. A study on 613 AD patients receiving vitamin E 2000 IU/day (n = 152) for 2 years suggested that the vitamin E delays clinical progression, functional decline, and caregiver burden [252]. Another study on 341 patients also suggested that vitamin E at 2000 IU a day for 2 years slows the progression of AD [253]. In another study on 57 patients receiving vitamin E 800 at IU per day for 6 months suggested the beneficial effect of vitamin E treatment on the cognitive functions of AD patients [254]. However, some of the studies suggested that the vitamin E treatment did not affect the progression of AD. For example, a study on 516 AD patients receiving 1000 IU of alpha-tocopherol twice daily for 3 years suggested no effect on the progression from MCI to Alzheimer’s dementia over 3 years [255]. Similar results have been reported by another study on 756 AD patients receiving donepezil and vitamin E. The results suggested that the treatment with vitamin E did not delay progression to AD whereas treatment with donepezil delayed the progression [256]. Similarly, in another double-blind study on 769 patients receiving 2000 IU of vitamin E daily shows that 10 mg of donepezil daily for 3 years suggested no significant effect of vitamin E treatment on the progression to AD whereas the donepezil therapy reduces the rate of progression to AD [257]. One of the studies on 40 AD patients receiving vitamin E at 2000 IU/day suggested the worsening of neuropsychologic test scores upon vitamin E treatment [258]. Mechanistically, vitamin E treatment counteracts the lipid peroxidation by neutralizing the peroxyl radicals [259]; suppresses the Aβ-mediated production of ROS [260], Aβ-induced neurotoxicity, and cognitive impairment [261,262,263],manages acetylcholinesterase activity [262, 264,265,266],improves cognitive functions [267],and delays the progression of AD [236, 252, 261, 262, 268,269,270]. Thus,vitamin E supplementation may provide therapeutic benefits in AD patients [46, 47, 252], reduce the emergence of AD [267], and slow down the functional decline in AD patients [252].
Vitamin C
Vitamin C is a well-known antioxidant. It is suggested that the level of vitamin C decreases in the brain of the cognitively impaired and AD individuals [271]. It has been reported that the AD patients are characterized by increased level and activity of ROS [272] due to the deposition of Aβ [273], activation of microglia cells [274], neuronal mitochondrial dysfunctioning [275], disturbance in the redox metal metabolic pathway [63, 64, 276], and the deficiencies of neurotransmitters such as noradrenaline and serotonin [277]. Further, the observation from the studies suggested that the CSF:plasma ratio of vitamin C rises significantly in the AD patients compared to the controls, suggesting the increased consumption of antioxidants by the brain in AD patients [278]. Vitamin C acts as a first-line barrier to ROS [279] and thus might be an important therapeutic option in the pharmacotherapy of AD. Vitamin C has been shown to improve cognition, and various studies suggested the relationship between plasma concentration of vitamin C and cognitive performance [280,281,282]. Further, some of the studies suggested no relationship or failed to show the relationship between the plasma concentration of vitamin C and cognitive performance [283,284,285]. The findings from the clinical studies and trials suggested that the higher level of vitamin C improves cognition and MMSE score in AD patients [281, 286,287,288,289]. However, a study suggested that there is no correlation between vitamin C concentrations and MMSE cognitive function in cognitively impaired individuals [290].
Vitamin C and Vitamin E Combination Therapy
The effect of the combined treatment with vitamins C and E has been evaluated in various studies. A study on two groups of AD patients supplemented with vitamin E 400 IU and vitamin C 1000 daily for 1 month was conducted. It was observed that the supplementation increased the concentrations of both vitamins in plasma and CSF. The study suggested that the combination is superior over the alone treatment in AD on the basis of the biochemical findings of the study [291]. One of the observational studies on 4740 individuals receiving vitamin E 400 IU daily and vitamin C 500 mg daily for 3 years suggested that the combination reduces the prevalence and the incidence of AD [292].
Others
Selenium (Se)
It plays a main role in the maintenance of health and treatment of neurologic disorders including AD [293, 294]. The level of the Se decreases with the increase in age [295] and contributes to the emergence of AD [296]. The reduced levels of Se have been described in AD patients [297]. Further, the treatment with Se slows down the cognitive decline [298,299,300] and reduces the Aβ burden [301], tau pathology, and neurodegeneration in AD patients [302].
Melatonin or N-Acetyl-5-methoxytryptamine
Melatoninis are neurohormones secreted by the pineal gland and confer antioxidant effect [303,304,305,306]. It has been reported that aging is accompanied by the reduction of CSF melatonin level, and this may favor the emergence of AD and related conditions [307]. Melatonin inhibits the aggregation and deposition of Aβ [308] and Aβ-induced oxidative damage, mitochondria dysfunction, and caspase-3 activity [309, 310]. Melatonin also activates GABAA receptors to counteract excitotoxicity and corrects the circadian rhythm in AD patients [311]. Melatonin (6 mg/day, daily) treatment has been demonstrated to enhance sleep quality and mood in MCI patients [312].
Estrogens
Estrogens also confer neuroprotection against oxidative stress and excitotoxicity [313]. Estrogen may confer the beneficial effect in AD by enhancing the trafficking of APP in the trans-Golgi network [314], by reducing the expression of BACE1 [313], by enhancing the uptake of Aβ in cortical microglia [315], and by preventing the aggregation of Aβ [316]. Further, selective estrogen receptor modulators have been shown to confer neuroprotective effects in the neuronal cultures by preventing Aβ-induced neurotoxicity [317]. Estrogen treatment also improves memory function in AD women and sustains neuronal viability [318].
Coenzyme Q10
Coenzyme Q10 (CoQ10) is a potent antioxidant in mitochondrial membranes [319] and exerts a neuroprotective effect by increasing the stability of membranes, decreasing the DNA damage, and potentiating the endogenous antioxidants [320]. It has been reported that the levels of CoQ10 decrease with the increase in age, making the mitochondria more susceptible to damage [321]. CoQ10 further exerts anti-inflammatory effects by inhibiting NF-κβ [322], by reducing the cortical levels [323] and deposition of Aβ [324], and by improving the brain energetics [325].
Galantamine
Galantamine is an effective scavenger of ROS and confers neuroprotective effects by inhibiting the activation of P2X7 receptors, protects the mitochondria from the damaging effect of ROS, increases the levels of acetylcholine by inhibiting the expression of acetylcholinesterase, and also inhibits the enzymatic activity of γ-secretase responsible for the production of Aβ [326].
Berberine
Berberine is known to confer antioxidant effects, reduces the levels of ROS, potentiates the level and the signaling of BDNF, reduces the levels of apoptotic proteins Bax/Bcl-2, down-regulates the activities of caspases, and suppresses neuronal apoptosis [327].
Edaravone
Edaravone is a potent free radical scavenger, and it has been reported that the edaravone administration (5 mg/kg) prevented the deleterious effect of Ab on memory and the acetylcholine levels of rats [328]. Administration of edaravone also reduces the production of Aβ by inhibiting the expression of enzyme BACE1, reduces the deposition of Aβ and inhibits the Aβ-mediated hyperphosphorylation of Tau, inhibits neuronal loss and neuronal degeneration, and restores the cognition in aged APPswe/PS1dE9 (APP/PS1) mice [329]. Edaravone at 25 μM counteracted the ZnO NP-induced reduction of ATP level and the activity of complexes I and V of the mitochondria and lipid peroxidation and blocked the expression of NF-κβ to confer protective effects in SH-SY5Y cells [330].
Hyperoside
Hyperoside has been shown to inhibit the Aβ-induced ROS production, mitochondrial dysfunction cytotoxicity, apoptosis, etc. Hyperoside also increases the expression of PI3K/Akt pathway and inhibits the expression of Bad and Bcl(XL) and caspase to suppress the neuronal cell death, suggesting its potential in the treatment of AD in the preclinical and clinical settings [331].
Miscellaneous: Molecular Hydrogen Therapy
Molecular hydrogen is made up of two protons and two electrons, and its antioxidant potential was first studied in 2007 [332]. It has been reported that molecular hydrogen therapy may confer therapeutic benefits in neurodegenerative disorders [333]. It is suggested that less than 4% of hydrogen shows greater efficacy and a better safety profile [334]. The greatest advantage of molecular hydrogen therapy is that the hydrogen even at higher concentrations is still non-toxic [335] and the inhalation of the hydrogen gas has no prominent adverse effects [332, 336]. Further, hydrogen-rich water has been considered safe for administration by many researchers [337,338,339]. In comparison with the conventional antioxidants, the molecular hydrogen only reduces ·OH and therefore showed lesser side effects [332]. Treatment with the molecular hydrogen suppresses the proinflammatory cytokines such as IL-1, IL-6, IL-10, and TNF-a [143, 144, 340], neutralizes Aβ-induced ROS generation and mitochondrial dysfunction [341], modulates Ca2+ signal transduction [342], inhibits the activation of NLRP3 [343], stimulates energy metabolism by stimulating AMPK-Sirt1-FoxO3a pathway [341], maintains cell homeostasis [344], inhibits the translocation of BAX, and suppresses the phosphorylation of JNK responsible for neuronal cell apoptosis [345, 346]. Molecular hydrogen therapy further improves learning and memory ability [347], improves cognition [343], and confers anti-inflammatory and anti-oxidant effects [348, 349]. Besides this, the previous study has shown that hydrogen-rich water improves cognition in the mice model [350]. Further, the administration of hydrogen-rich saline for 14 days has been shown to counteract the cognitive decline induced by i.c.v. administration of Aβ and improve performance in MWM and preserved LTP by inhibiting the IL-1β, JNK, and NF-κβ [351, 352]. Molecular hydrogen suppresses the inflammatory responses and oxidative stress, which are more pronounced in female than in male AD mice [343]. Hippocampus plays a key role in the regulation of learning and memory-related activities [353, 354]. Hydrogen water has been shown to restore the proliferating cells in the dentate gyrus following restraint stress [350], suggesting the beneficial effect. Further, another study suggested that the administration of hydrogen-rich water for 30 days improves the performance in MWM and counteracts the hippocampal neuronal degeneration upon 18 weeks of treatment [355].
A study on 73 MCI subjects receiving 300 mL of H2-water (water infused with H2) for 1 year suggested that the improvement of ADAS-cog score after 1 year in the APOE4 carriers [356]. Another study on 20 adults receiving hydrogen-rich water for 4 weeks suggested that the administration of hydrogen-rich water increases the BAP in the individuals aged ≥ 30 years, suggesting the antioxidant effect [357]. These lines of evidence suggested that molecular hydrogen therapy offers a new direction for the treatment of AD. However, the therapy is in the initial phase and very few clinical studies have been conducted till now. Further, the molecular hydrogen modulates all the pathways which are modulated by the antioxidants, but the antioxidants are in clinical use for a very long period of time. So, the exact superiority between the molecular hydrogen therapy and the conventional antioxidants can be elucidated only after the suitable clinical studies on them.
Evidence from the Studies on Nutraceutical Formulations or Dietary Supplements in AD Patients
Oxidative stress performs major function in the pathogenesis of AD and thus the supplementation of the antioxidant may confer therapeutic benefits in AD patients. Further, the findings from the AD patients suggested that they often have some nutritional deficiency due to the impaired availability of those nutrients [358]. It has been found that the MCI is a risk factor that makes the patients prone to AD and the supplementation of antioxidant in the MCI lowers the risk of further development of AD and related dementia [286]. It has been reported that the correction of oxidative stress and the consumption of tocopherol, ascorbic acid, and β-carotene lowers the risk of AD [251]. Further, AD patients are characterized by the reduced plasma levels of tocopherol, ascorbic acid, and β-carotene, suggesting the importance of these interventions in the pharmacotherapy of AD patients [359]. A study in which several vitamins, antioxidants, amino acids, nutrients, etc. are administered in the form of a pill to the AD patients suggested the reduction in the oxidative stress parameters. The study also suggested that the above pill when used in combination with the donepezil improves the condition of AD patients significantly [360]. Another study on the nutraceutical formulation also claimed that the use of the formulation in AD patients improves cognitive functions [361]. In another study, the authors suggested that the use of the nutraceuticals in AD patients for 3–6 months improves cognition and mood, further suggesting therapeutic benefits of these formulations [362]. Another study by the same author on 24 patients of AD suggested that the daily supplementation with the nutraceutical for 1 year improves the cognitive performance [363]. However, a previous study in which the participants were supplemented with various vitamins and antioxidants for 5 years reported no statistically significant differences in the scores [364]. Another double-blind randomized clinical study on 3786 AD patients receiving vitamin E 400 IU, selenium 200 IU per day, or placebo suggested that neither supplement prevented dementia [365].
Limitations and Failure of Antioxidant Therapy in AD
Oxidative stress has been concerned in the pathogenesis of AD, but despite of this, the role of antioxidants in the pharmacotherapeutics of AD is still controversial. AD patients are characterized by enhanced lipid peroxidation, cognitive dysfunction, abnormal heavy metal metabolism, and a decrease in the total antioxidant capacity. However, the studies have revealed that there is no significant decrease in the levels of the major internal enzymatic and non-enzymatic antioxidants in the body of AD patients. Therefore, the antioxidant therapy should be initiated in the patients followed by the determination of the cognitive outcomes and oxidative parameters in the patients [366]. Theoretically, it is suggested that the antioxidants provide therapeutic benefit in AD patients by providing protection against the ROS and by preventing the accumulation of Aβ [367]. Various studies have described the beneficial effects of antioxidants, suggesting that the treatment with the antioxidants may improve the cognitive performance in AD patients [179, 368, 369]. However, some of the studies suggested no clear significant effect of antioxidants in AD patients [45,46,47]. Thus, despite of the beneficial effects of these compounds in in vitro and in vivo studies, convincing evidence regarding their therapeutic efficacy in AD is still lacking [48, 49]. The result is that only a few studies supported the therapeutic benefits of antioxidants in AD patients [48, 370, 371]. The lack of evidence regarding the therapeutic effects of antioxidant treatment might be due to several reasons, as follows:
First Is the Lack of a Suitable and Relevant Animal Model
The preclinical studies on some antioxidants show their excellent therapeutic potential, but the same compound did not show any significant or promising effect in the clinical studies. These types of incidence suggesting that relevant animal models of AD should be developed to screen the therapeutic potential of the compounds targeted for AD treatment. Further, the difference in the methodology used in the preclinical and clinical studies on the same compound further affects their clinical value or therapeutic efficacy [372]. Further, the dose of antioxidants used in the preclinical studies is very high, and it is impractical to extrapolate that dose to human studies [368]. It has been found that Aβ deposition is the characteristic of AD. However, the findings from the animal models suggested that oxidative stress is responsible for the overproduction and accumulation of Aβ in the brain and vice versa [373,374,375].
Second Is the Clinical Study Design, Protocol, and Their Conflicting Results
The therapeutic potential of the antioxidants remains of question because sometimes they are started at the stages where the disease already progressed to the last stages or sometimes they are started too early. Further, some of the antioxidants were studied at very high doses or in very low doses. Sometimes, antioxidants were used in the combinations where their true potential always remains hidden. These constraints may greatly affect the therapeutic effectiveness of antioxidants for the treatment of AD [368, 376].Various studies suggested that the antioxidants including vitamin C, vitamin E, melatonin, CoQ, etc. failed to inhibit the process of neuronal degeneration, deposition of Aβ, any significant improvement in cognitive functions, etc. [26, 27, 377]. A population-based investigation suggested a relationship between the GPx activity and cognitive decline [378], but the results were inconsistent when observed in AD patients [293]. Further, the confusing and conflicting results have been reported by the studies on SOD and catalase activities in AD patients [199, 200, 379].
Third Is the Nature of Antioxidants and Their Antioxidant Potential
Some of the antioxidants can also act as pro-oxidants under deleterious conditions, and thus, they may potentiate oxidative stress and thus their antioxidant activity remains questionable in these conditions [380]. Consistent with these findings, some of the agents (e.g., CoQ) which has been suggested and speculated to confer therapeutic benefits in AD patients cannot cross the BBB and failed to produce any change in the cognitive functions in AD patients [381, 382]. For CoQ, it has been reported that to exert its therapeutic effect, CoQ must be retained in the mitochondrial ETC. Currently available antioxidants also suffer from similar problems. For example, vitamin E has very slow and lesser bioavailability in the human brain to inhibit AD [383].
Future Directions
Antioxidant therapies have several limitations (as discussed above) for the treatment of AD. Therefore, it further demands in-depth studies to explore and delineate the efficacy of antioxidants for the treatment of AD. This particular review paper will certainly help the researchers/scientists to substantiate/explore the efficacy of antioxidants in the treatment of AD.
Data Availability
Not applicable.
Code Availability
Not applicable.
Change history
27 November 2021
A Correction to this paper has been published: https://doi.org/10.1007/s12035-021-02665-7
References
Maccioni RB, González A, Andrade V, Cortés N, Tapia JP, Guzmán-Martínez L (2018) Alzheimer s disease in the perspective of neuroimmunology. Open Neurol J 12:50
Tang Y, Lutz MW, Xing Y (2019) A systems-based model of Alzheimer’s disease. Alzheimers Dement 15(1):168–171
Zilberzwige-Tal S, Gazit E (2018) Go with the flow—microfluidics approaches for amyloid research. Chem Asian J 13(22):3437–3447
Gomez-Ramos A, Podlesniy P, Soriano E, Avila J (2015) Distinct X-chromosome SNVs from some sporadic AD samples. Sci Rep 5(1):1–11
Nacmias B, Piaceri I, Bagnoli S, Tedde A, Piacentini S, Sorbi S (2014) Genetics of Alzheimer’s disease and frontotemporal dementia. Curr Mol Med 14(8):993–1000
Katzman R (1986) Alzheimer’s disease. N Engl J Med 314(15):964–973
Smith MA (1998) Alzheimer disease. Int Rev Neurobiol 42:1–54
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci 82(12):4245–4249
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM (1986) Microtubule associated protein tau A component of Alzheimer paired helical filaments. J Biol Chem 261(13):6084–6089
Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, ... Tabaton M (2005) β‐siteAPP cleaving enzyme up‐regulation induced by 4‐hydroxynonenal is mediated by stress‐activated protein kinases pathways. J Neurochem 92(3): 628-636
Sáez-Orellana F, Godoy PA, Bastidas CY, Silva-Grecchi T, Guzmán L, Aguayo LG, Fuentealba J (2016) ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of β-amyloid peptide in hippocampal neurons. Neuropharmacology 100:116–123
Godyń J, Jończyk J, Panek D, Malawska B (2016) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacological Reports: PR 68(1):127–138
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Lee VMY, Trojanowski JQ (1992) The disordered neuronal cytoskeleton in Alzheimer’s disease. Curr Opin Neurobiol 2(5):653–656
Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, ... Trojanowski JQ (2003) Cerebrospinal fluid tau and β-amyloid: How well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol 60(12): 1696-1702
Spires-Jones TL, Kopeikina KJ, Koffie RM, de Calignon A, Hyman BT (2011) Are tangles as toxic as they look? J Mol Neurosci 45(3):438–444
Davis DR, Anderton BH, Brion J-P, Reynolds CH, Hanger DP (1997) Oxidative stress induces dephosphorylation of τ in rat brain primary neuronal cultures. J Neurochem 68(4):1590–1597
Galas MC, Dourlen P, Bégard S, Ando K, Blum D, Hamdane M, Buée L (2006) The peptidylprolyl cis/trans-isomerase Pin1 modulates stress-induced dephosphorylation of Tau in neurons: implication in a pathological mechanism related to Alzheimer disease. J Biol Chem 281(28):19296–19304
Zambrano CA, Egaña JT, Núñez MT, Maccioni RB, González-Billault C (2004) Oxidative stress promotes τ dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radical Biol Med 36(11):1393–1402
De Leon MJ, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Segal S, ... Davies P (2006) Longitudinal CSF and MRI biomarkers improve the diagnosis of mild cognitive impairment. Neurobiol Aging, 27(3), 394-401
Fagan AM, Holtzman DM (2010) Cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med 4(1):51–63
Garcia ML, Cleveland DW (2001) Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr Opin Cell Biol 13(1):41–48
Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41(1):17–24
Ranjan B, Chong KH, Zheng J (2018) Composite mathematical modeling of calcium signaling behind neuronal cell death in Alzheimer’s disease. BMC Syst Biol 12(1):10
Yang L, Wang Z, Wang B, Justice NJ, Zheng H (2009) Amyloid precursor protein regulates Cav1. 2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci 29(50):15660–15668
Wang YY, Zheng W, Ng CH, Ungvari GS, Wei W, Xiang YT (2017) Meta-analysis of randomized, double-blind, placebo-controlled trials of melatonin in Alzheimer’s disease. Int J Geriatr Psychiatry 32(1):50–57
Wang Y, Shi Y, Wei H (2017) Calcium dysregulation in Alzheimer’s disease: a target for new drug development. Journal of Alzheimer’s Disease & Parkinsonism 7(5)
Sciacca MF, Monaco I, La Rosa C, Milardi D (2018) The active role of Ca2+ ions in Aβ-mediated membrane damage. Chem Commun 54(29):3629–3631
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2(3):156–162
Samad N, Ishaq S, Bano S, Manzoor N (2017) Calcium regulation in Alzheimer’s disease: mechanistic understanding. J Coll Physicians Surg Pak 27(9):566–571
Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X (2010) A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J Alzheimers Dis 20(s2):S401–S412
Mancuso M, Coppedè F, Murri L, Siciliano G (2007) Mitochondrial cascade hypothesis of Alzheimer’s disease: myth or reality? Antioxid Redox Signal 9(10):1631–1646
Cui K, Luo X, Xu K, Ven Murthy MR (2004) Role of oxidative stress in neurodegeneration: recent developments in assay methods for oxidative stress and nutraceutical antioxidants. Prog Neuropsychopharmacol Biol Psychiatry 28(5):771–799
Teleanu RI, Chircov C, Grumezescu AM, Volceanov A, Teleanu DM (2019) Antioxidant therapies for neuroprotection-a review. J Clin Med 8(10):1659
Yokota A (1993) Relationship between polyunsaturated fatty acid (PUFA) and learning ability in the brain of rat fetus and newborn. Nihon Sanka Fujinka Gakkai Zasshi 45(1):15–22
Yamamoto N, Okaniwa Y, Mori S, Nomura M, Okuyama H (1991) Effects of a high-linoleate and a high-α-linolenate diet on the learning ability of aged rats evidence against an autoxidation-related lipid peroxide theory of aging. J Gerontol 46(1):B17–B22
Pagani L, Eckert A (2011) Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis 2011
Sanei M, Saberi-Demneh A (2019) Effect of curcumin on memory impairment: A systematic review. Phytomedicine 52:98–106
Ono K, Hasegawa K, Naiki H, Yamada M (2004) Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J Neurosci Res 75(6):742–750
Reddy ACP, Lokesh BR (1992) Studies on spice principles as antioxidants in the inhibition of lipid peroxidation of rat liver microsomes. Mol Cell Biochem 111(1):117–124
Baum L, Lam CWK, Cheung SKK, Kwok T, Lui V, Tsoh J, ... Mok V (2008) Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol 28(1): 110-113
Ringman JM, Frautschy SA, Teng E, Begum AN, Bardens J, Beigi M, Gylys KH, Badmaev V, Heath DD, Apostolova LG, Porter V, Vanek Z, Marshall GA, Hellemann G, Sugar C, Masterman DL, Montine TJ, Cummings JL, Cole GM (2012) Oral curcumin for Alzheimer’s disease: tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimer’s Res Ther 4(5):43
Oulhaj A, Jernerén F, Refsum H, Smith AD, de Jager CA (2016) Omega-3 fatty acid status enhances the prevention of cognitive decline by B vitamins in mild cognitive impairment. Journal of Alzheimer’s Disease: JAD 50(2):547–557
Benfeito S, Oliveira C, Fernandes C, Cagide F, Teixeira J, Amorim R, ... Borges F (2019) Fine-tuning the neuroprotective and blood-brain barrier permeability profile of multi-target agents designed to prevent progressive mitochondrial dysfunction. Eur J Med Chem 167: 525-545
Grimm MO, Mett J, Hartmann T (2016) The impact of vitamin E and other fat-soluble vitamins on Alzheimer’s disease. Int J Mol Sci 17(11)
La Fata G, Weber P, Mohajeri MH (2014) Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 6:5453–5472
La Fata G, Weber P, Mohajeri MH (2014) Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 6(12):5453–5472
Mazzanti G, Di Giacomo S (2016) Curcumin and resveratrol in the management of cognitive disorders: what is the clinical evidence? Molecules 21(9):1243
Sawda C, Moussa C, Turner RS (2017) Resveratrol for Alzheimer’s disease. Ann N Y Acad Sci 1403(1):142–149
Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58(4):495–505
Di Mauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348(26):2656–2668
Koopman WJ, Distelmaier F, Smeitink JA, Willems PH (2013) OXPHOS mutations and neurodegeneration. EMBO J 32(1):9–29
Noji H, Yoshida M (2001) The rotary machine in the cell, ATP synthase. J Biol Chem 276(3):1665–1668
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408(6809):239–247
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84
Zhu X, Perry G, Smith MA, Wang X (2013) Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 33(s1):S253–S262
Ávila F, Schmeda-Hirschmann G, Silva E (2017) The major chromophore arising from glucose degradation and oxidative stress occurrence during lens proteins glycation induced by glucose. Molecules (Basel, Switzerland) 23(1):6. https://doi.org/10.3390/molecules23010006
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120(4):483–495
Van Houten B, Woshner V, Santos JH (2006) Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 5(2):145–152
Crouch PJ, Harding SME, White AR, Camakaris J, Bush AI, Masters CL (2008) Mechanisms of Aβ mediated neurodegeneration in Alzheimer’s disease. Int J Biochem Cell Biol 40(2):181–198
Morris G, Walder K, Puri BK, Berk M, Maes M (2016) The deleterious effects of oxidative and nitrosative stress on palmitoylation, membrane lipid rafts and lipid-based cellular signalling: new drug targets in neuroimmune disorders. Mol Neurobiol 53(7):4638–4658
Boom A, Authelet M, Dedecker R, Frédérick C, Van Heurck R, Daubie V, ... Brion JP (2009) Bimodal modulation of tau protein phosphorylation and conformation by extracellular Zn2+ in human-tau transfected cells. Biochim Biophys Acta Mol Cell Res 1793(6), 1058–1067
Sayre LM, Perry G, Atwood CS, Smith MA (2000) The role of metals in neurodegenerative diseases. Cell Mol Biol (Noisy-le-Grand) 46(4):731–741
Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA (2000) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J Neurochem 74(1):270–279
Mondragón-Rodríguez S, Perry G, Zhu X, Moreira PI, Acevedo-Aquino MC, Williams S (2013) Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease. Oxidative Medicine and Cellular Longevity, 2013
De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL (2007) Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 282(15):11590–11601
Ferreira ST, Vieira MN, De Felice FG (2007) Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. Int Union Biochem Mol Biol Life 59(4–5):332–345
Mattson MP, Chan SL (2003) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 34(4–5):385–397
Rai S, Kamat PK, Nath C, Shukla R (2013) A study on neuroinflammation and NMDA receptor function in STZ (ICV) induced memory impaired rats. J Neuroimmunol 254(1–2):1–9
Kelly BL, Ferreira A (2006) β-amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem 281(38):28079–28089
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31(18):6627–6638
Briston T, Roberts M, Lewis S, Powney B, Staddon JM, Szabadkai G, Duchen MR (2017) Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci Rep 7(1):1–13
Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ (2000) Reactive oxygen species (Ros-Induced) Ros release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192(7):1001–1014
Ichas F, Jouaville LS, Mazat JP (1997) Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89(7):1145–1153
Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F (1999) Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J 76(2):725–734
Duchen MR (2000) Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium 28(5–6):339–348
Gerschman R, Gilbert DL, Nye SW, Dwyer P, Fenn WO (1954) Oxygen poisoning and x-irradiation: a mechanism in common. Science 119(3097):623–626
Harman E (1956) Protein oxidation in aging and age-related diseases. Gerontology 11:298–300
Mattson MP, Lovell MA, Furukawa K, Markesbery WR (1995) Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem 65(4):1740–1751
Münch G, Lüth HJ, Wong A, Arendt T, Hirsch E, Ravid RA, Riederer P (2000) Crosslinking of α-synuclein by advanced glycation endproducts—an early pathophysiological step in Lewy body formation? J Chem Neuroanat 20(3–4):253–257
Floyd RA, Hensley K (2002) Oxidative stress in brain aging: implications for therapeutics of neurodegenerative diseases. Neurobiol Aging 23(5):795–807
Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, ... Moraes CT (2005) Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci 25(1): 164-172
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G (2002) Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem 277(33):29626–29633
Baloyannis SJ (2006) Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis 9(2):119–126
Exner N, Lutz AK, Haass C, Winklhofer KF (2012) Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J 31(14):3038–3062
Harman D (1972) The biologic clock: The mitochondria? J Am Geriatr Soc 20(4):145–147
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, ... Shimohama S (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21(9), 3017-3023
Schon EA, Przedborski S (2011) Mitochondria: the next (neurode) generation. Neuron 70(6):1033–1053
Christen Y (2000) Oxidative stress and Alzheimer disease. Am J Clin Nutr 71(2):621S-629S
Baloyannis SJ, Costa V, Michmizos D (2004) Mitochondrial alterations Alzheimer’s disease. Am J Alzheimers Dis Other Demen 19(2):89–93
García-Matas S, de Vera N, Aznar AO, Marimon JM, Adell A, Planas AM, ... Sanfeliu C (2010) In vitro and in vivo activation of astrocytes by amyloid-β is potentiated by pro-oxidant agents. J Alzheimers Dis 20(1), 229-245
Ichimura H, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J (2003) Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Investig 111(5):691–699
Shah SB, Nolan R, Davis E, Stokin GB, Niesman I, Canto I, ... Goldstein LS (2009) Examination of potential mechanisms of amyloid-induced defects in neuronal transport. Neurobiol Dis 36(1), 11–25
Szabados T, Dul C, Majtényi K, Hargitai J, Pénzes Z, Urbanics R (2004) A chronic Alzheimer’s model evoked by mitochondrial poison sodium azide for pharmacological investigations. Behav Brain Res 154(1):31–40
Xiong K, Cai H, Luo XG, Struble RG, Clough RW, Yan XX (2007) Mitochondrial respiratory inhibition and oxidative stress elevate β-secretase (BACE1) proteins and activity in vivo in the rat retina. Exp Brain Res 181(3):435–446
Wahlster L, Arimon M, Nasser-Ghodsi N, Post KL, Serrano-Pozo A, Uemura K, Berezovska O (2013) Presenilin-1 adopts pathogenic conformation in normal aging and in sporadic Alzheimer’s disease. Acta Neuropathol 125(2):187–199
Ito S, Ohtsuki S, Kamiie J, Nezu Y, Terasaki T (2007) Cerebral clearance of human amyloid-β peptide (1–40) across the blood–brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J Neurochem 103(6):2482–2490
Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, ... Zlokovic BV (2007). Clearance of amyloid-β by circulating lipoprotein receptors. Nat Med, 13(9): 1029-1031
Owen JB, Sultana R, Aluise CD, Erickson MA, Price TO, Bu G, …& Butterfield, D. A. (2010) Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Aβ accumulation in AD brain. Free Radical Biol Med 49(11):1798–1803
Jeynes B, Provias J (2008) Evidence for altered LRP/RAGE expression in Alzheimer lesion pathogenesis. Curr Alzheimer Res 5(5):432–437
Bozyczko-Coyne D, O’Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL, Scott RW (2001) CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Aβ-induced cortical neuron apoptosis. J Neurochem 77(3):849–863
Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, ... Greenberg ME (2001) β-Amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci 21(19), 7551-7560
Wei W, Wang X, Kusiak JW (2002) Signaling events in amyloid β-peptide-induced neuronal death and insulin-like growth factor I protection. J Biol Chem 277(20):17649–17656
Liu Q, Smith MA, Avilá J, DeBernardis J, Kansal M, Takeda A, ... Perry G (2005) Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radical Biology and Medicine 38(6), 746-754
Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, ... Galas MC (2014) A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci 8:84
Santa-María I, Hernández F, Martín CP, Avila J, Moreno FJ (2004) Quinones facilitate the self-assembly of the phosphorylated tubulin binding region of tau into fibrillar polymers. Biochemistry 43(10):2888–2897
Su JH, Deng G, Cotman CW (1997) Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer’s disease brain. Brain Res 774(1–2):193–199
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, ... Smith MA (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60(8): 759-767
Ferreira IL, Ferreiro E, Schmidt J, Cardoso JM, Pereira CM, Carvalho AL, Oliveira CR, Rego AC (2015) Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol Aging 36(2):680–692
Hermann D, Mezler M, Müller MK, Wicke K, Gross G, Draguhn A, ... Nimmrich V (2013) Synthetic Aβ oligomers (Aβ1–42 globulomer) modulate presynaptic calcium currents: prevention of Aβ-induced synaptic deficits by calcium channel blockers. Eur J Pharmacol 702(1-3): 44-55
Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K (1997) Amyloid β protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem 68(1):265–271
Woods NK, Padmanabhan J (2012) Neuronal calcium signaling and Alzheimer’s disease. Calcium signaling 1193–1217
Yagami T, Ueda K, Sakaeda T, Itoh N, Sakaguchi G, Okamura N, ... Fujimoto M (2004) Protective effects of a selective L-type voltage-sensitive calcium channel blocker, S-312-d, on neuronal cell death. Biochem Pharmacol 67(6), 1153-1165
Butterfield DA, Lange MLB, Sultana R (2010) Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim Biophys Acta Mol Cell Biol L 1801(8):924–929
Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis 1842(8):1240–1247
Butterfield DA, Lauderback CM (2002) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radical Biol Med 32(11):1050–1060
Butterfield DA (2014) The 2013 SFRBM discovery award: selected discoveries from the butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Radical Biol Med 74:157–174
Butterfield DA, Di Domenico F (1842) Barone E (2014) Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim Biophys Acta Mol Basis Dis 9:1693–1706
Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, ... Butterfield DA (1995) Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem 65(5): 2146-2156
Granold M, Moosmann B, Staib-Lasarzik I, Arendt T, Del Rey A, Engelhard K, Behl C, Hajieva P (2015) High membrane protein oxidation in the human cerebral cortex. Redox Biol 4:200–207
Gow AJ, Duran D, Malcolm S, Ischiropoulos H (1996) Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett 385(1–2):63–66
Markesbery WR, Lovell MA (1998) Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging 19(1):33–36
Hardas SS, Sultana R, Clark AM, Beckett TL, Szweda LI, Murphy MP, Butterfield DA (2013) Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol 1(1):80–85
Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA (1997) 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem 68(5):2092–2097
Gabbita SP, Lovell MA, Markesbery WR (1998) Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem 71(5):2034–2040
Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36(5):747–751
Lovell MA, Soman S, Bradley MA (2011) Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev 132(8–9):443–448
Coppedè F, Migliore L (2015) DNA damage in neurodegenerative diseases. Mutat Res 776:84–97. https://doi.org/10.1016/j.mrfmmm.2014.11.010
Shan X, Lin CLG (2006) Quantification of oxidized RNAs in Alzheimer’s disease. Neurobiol Aging 27(5):657–662
Tramutola A, Lanzillotta C, Perluigi M, Butterfield DA (2017) Oxidative stress, protein modification and Alzheimer disease. Brain Res Bull 133:88–96
Aluise CD, Robinson RA, Cai J, Pierce WM, Markesbery WR, Butterfield DA (2011) Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: insights into memory loss in MCI. J Alzheimers Dis 23(2):257–269
Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, ... Butterfield DA (2008) Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis 30(1):107-120
Schagger H, Ohm TG (1995) Human diseases with defects in oxidative phosphorylation: 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur J Biochem 227(3):916–921
Terni B, Boada J, Portero-Otin M, Pamplona R, Ferrer I (2010) Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol 20(1):222–233
Cha MY, Cho HJ, Kim C, Jung YO, Kang MJ, Murray ME, ... Mook-Jung I (2015) Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum Mol Genet 24(22): 6492-6504
Chen Z, Zhong C (2013) Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog Neurobiol 108:21–43
Lauderback CM, Kanski J, Hackett JM, Maeda N, Kindy MS, Butterfield DA (2002) Apolipoprotein E modulates Alzheimer’s Aβ (1–42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res 924(1):90–97
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, ... Zlokovic BV (2008) apoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J Clin Investig 118(12): 4002-4013
Ma J, Yee A, Brewer HB, Das S, Potter H (1994) Amyloid-associated proteins α 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372(6501):92–94
Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y (2005) Lipid-and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci 102(51):18694–18699
Veinbergs I, Everson A, Sagara Y, Masliah E (2002) Neurotoxic effects of apolipoprotein E4 are mediated via dysregulation of calcium homeostasis. J Neurosci Res 67(3):379–387
Chen CH, Manaenko A, Zhan Y, Liu WW, Ostrowki RP, Tang JIPING, Zhang JH (2010) Hydrogen gas reduced acute hyperglycemia-enhanced hemorrhagic transformation in a focal ischemia rat model. Neuroscience 169(1):402–414
Chen Y, Durakoglugil MS, Xian X, Herz J (2010) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci 107(26):12011–12016
Hamanaka H, Katoh-Fukui Y, Suzuki K, Kobayashi M, Suzuki R, Motegi Y, …& Fujita, S. C. (2000) Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet 9(3):353–361
Di Domenico F, Pupo G, Giraldo E, Badìa MC, Monllor P, Lloret A, ... Perluigi M (2016) Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic Biol Med 91: 1-9
Kharrazi H, Vaisi-Raygani A, Rahimi Z, Tavilani H, Aminian M, Pourmotabbed T (2008) Association between enzymatic and non-enzymatic antioxidant defense mechanism with apolipoprotein E genotypes in Alzheimer disease. Clin Biochem 41(12):932–936
Ramassamy C, Krzywkowski P, Averill D, Lussier-Cacan S, Theroux L, Christen Y, ... Poirier J (2001) Impact of apoE deficiency on oxidative insults and antioxidant levels in the brain. Mol Brain Res 86(1-2): 76-83
Tamaoka A, Miyatake F, Matsuno S, Ishii K, Nagase S, Sahara N, …& Shoji, S. (2000) Apolipoprotein E allele–dependent antioxidant activity in brains with Alzheimer’s disease. Neurology 54(12):2319–2321
Mirza A, King A, Troakes C, Exley C (2017) Aluminium in brain tissue in familial Alzheimer’s disease. J Trace Elem Med Biol 40:30–36
Mustafa Rizvi SH, Parveen A, Verma AK, Ahmad I, Arshad M, Mahdi AA (2014) Aluminium induced endoplasmic reticulum stress mediated cell death in SH-SY5Y neuroblastoma cell line is independent of p53. PloS One 9(5):e98409
Al-Olayan EM, El-Khadragy MF, Abdel Moneim AE (2015) The protective properties of melatonin against aluminium-induced neuronal injury. Int J Exp Pathol 96(3):196–202
Hosseini-Sharifabad A, Rabbani M, Seyed-Yousefi Y, Safavi M (2020) Magnesium increases the protective effect of citicoline on aluminum chloride-induced cognitive impairment. Clin Psychopharmacol Neurosci 18(2):241–248
Prema A, Thenmozhi AJ, Manivasagam T, Essa MM, Akbar MD, Akbar M (2016) Fenugreek seed powder nullified aluminium chloride induced memory loss, biochemical changes, Aβ burden and apoptosis via regulating Akt/GSK3β signaling pathway. PloS One 11(11):e0165955
Moreira-Silva D, Vizin R, Martins T, Ferreira TL, Almeida MC, Carrettiero DC (2019) Intracerebral injection of streptozotocin to model Alzheimer disease in rats. Bio-protocol 9(20):e3397
Alluri R, Ambati SR, Routhu K, Kopalli SR, Koppula S (2020) Phosphoinositide 3-kinase inhibitor AS605240 ameliorates streptozotocin-induced Alzheimer’s disease like sporadic dementia in experimental rats. EXCLI J 19:71–85
Kamat PK (2015) Streptozotocin induced Alzheimer’s disease like changes and the underlying neural degeneration and regeneration mechanism. Neural Regen Res 10(7):1050–1052
Retinasamy T, Shaikh MF, Kumari Y, Abidin S, Othman I (2020) Orthosiphon stamineus standardized extract reverses streptozotocin-induced Alzheimer’s disease-like condition in a rat model. Biomedicines 8(5):104
Correia SC, Santos RX, Santos MS, Casadesus G, Lamanna JC, Perry G, Smith MA, Moreira PI (2013) Mitochondrial abnormalities in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. Curr Alzheimer Res 10(4):406–419
Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L (2014) The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 13(10):1045–1060
Casadesus G, Smith MA, Zhu X, Aliev G, Cash AD, Honda K, ... Perry G (2004) Alzheimer disease: evidence for a central pathogenic role of iron-mediated reactive oxygen species. J Alzheimers Dis 6(2): 165-169
Castellani RJ, Moreira PI, Perry G, Zhu X (2012) The role of iron as a mediator of oxidative stress in Alzheimer disease. BioFactors 38:133–138
Lipinski B, Sajdel-Sulkowska EM (2006) New insight into Alzheimer disease: demonstration of fibrin (ogen)-serum albumin insoluble deposits in brain tissue. Alzheimer Dis Assoc Disord 20(4):323–326
Lipinski B, Pretorius E (2012) Novel pathway of iron-induced blood coagulation: implications for diabetes mellitus and its complications. Polskie Archiwum Medycyny Wewnetrznej 122(3):115–122
Cao JY, Dixon SJ (2016) Mechanisms of ferroptosis. Cell Mol Life Sci 73(11):2195–2209
Fanzani A, Poli M (2017) Iron, oxidative damage and ferroptosis in rhabdomyosarcoma. Int J Mol Sci 18(8):1718
Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C (2019) Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radical Biol Med 133:221–233
Nikseresht S, Bush AI, Ayton S (2019) Treating Alzheimer’s disease by targeting iron. Br J Pharmacol 176(18):3622–3635
Weiland A, Wang Y, Wu W, Lan X, Han X, Li Q, Wang J (2019) Ferroptosis and its role in diverse brain diseases. Mol Neurobiol 56(7):4880–4893
Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI (2004) Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Aβ peptides. J Biol Inorg Chem 9(8):954–960
Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, …& Crowther, D. C. (2011) Iron promotes the toxicity of amyloid β peptide by impeding its ordered aggregation. J Biol Chem 286(6):4248–4256
Mantyh PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER, Maggio JE (1993) Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem 61:1171–1174
Schubert D, Chevion M (1995) The role of iron in beta amyloid toxicity. Biochem Biophys Res Commun 216(2):702–707
Everett J, Collingwood JF, Tjendana-Tjhin V, Brooks J, Lermyte F, Plascencia-Villa G, ... Telling ND (2018) Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale 10(25), 11782-11796
Liu JL, Fan YG, Yang ZS, Wang ZY, Guo C (2018) Iron and Alzheimer’s disease: from pathogenesis to therapeutic implications. Front Neurosci 12:632
Derry PJ, Hegde ML, Jackson GR, Kayed R, Tour JM, Tsai AL, Kent TA (2020) Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog Neurobiol 184:101716
de Lima PDL, Yamada ES, da Costa ET, do O Pessoa C, Rabenhorst SHB, de Oliveira Bahia M, Cardoso PC, Santos RA, de Arruda Cardoso Smith M, Burbano RR (2005) Genotoxic effects of rotenone on cultured lymphocytes. Genetics and Molecular Research : GMR 4(4):822–831
Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, Filip M (2016) Oxidative stress in neurodegenerative diseases. Mol Neurobiol 53(6):4094–4125
Jiang T, Sun Q, Chen S (2016) Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog Neurobiol 147:1–19
Pisoschi AM, Pop A (2015) The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem 97:55–74
Poljsak B, Šuput D (2013) an, Milisav I (2013). ROS and antioxidants: Achieving the balance between when to use the synthetic antioxidants. Oxidative Medicine and Cellular Longevity, 956792
Al-Oud SS (2003) Heavy metal contents in tea and herb leaves. Pakistan Journal of Biological Sciences (Pakistan) 6:208–212
El-Sayed IH, Lotfy M, El-Khawaga OA, Nasif WA, El-Shahat M (2006) Prominent free radicals scavenging activity of tannic acid in lead-induced oxidative stress in experimental mice. Toxicol Ind Health 22(4):157–163
Lopes GK, Schulman HM, Hermes-Lima M (1999) Polyphenol tannic acid inhibits hydroxyl radical formation from Fenton reaction by complexing ferrous ions. Biochim Biophys Acta Gen Subj 1472(1–2):142–152
Leopoldini M, Russo N, Toscano M (2011) The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem 125(2):288–306
Marković Z (2016) Study of the mechanisms of antioxidative action of different antioxidants. J Serb Soc Comput Mech 10(1):135–150
Golden TR, Patel M (2009) Catalytic antioxidants and neurodegeneration. Antioxid Redox Signal 11(3):555–570
Amato A, Terzo S, Mulè F (2019) Natural compounds as beneficial antioxidant agents in neurodegenerative disorders: a focus on Alzheimer’s disease. Antioxidants (Basel, Switzerland) 8(12):608. https://doi.org/10.3390/antiox8120608
Forman HJ, Zhang H, Rinna A (2009) Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 30(1–2):1–12
Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta Gen Subj 1830(5):3143–3153
Ansari MA, Scheff SW (2010) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69(2):155–167
Karelson E, Bogdanovic N, Garlind A, Winblad B, Zilmer K, Kullisaar T, …& Zilmer, M. (2001) The cerebrocortical areas in normal brain aging and in Alzheimer’s disease: noticeable differences in the lipid peroxidation level and in antioxidant defense. Neurochem Res 26(4):353–361
Mandal PK, Saharan S, Tripathi M, Murari G (2015) Brain glutathione levels–a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol Psychiat 78(10):702–710
Docampo R, Moreno SNJ (2017) 17-biochemistry of trypanosoma cruzi. In: Telleria J, Tibayrenc M (eds) American Trypanosomiasis Chagas Disease, 2nd edn. Elsevier; London, p 371–400
Nabi S (2014) Endogenous antioxidants. In: Toxic effects of mercury. Springer, New Delhi, p 117–120
Undeland I (2016). Oxidative stability of seafood. In: Oxidative stability and shelf life of foods containing oils and fats (pp. 391–460). AOCS Press
Asakura H, Kitahora T (2018) Antioxidants and polyphenols in inflammatory bowel disease: ulcerative colitis and Crohn disease. In: Polyphenols: prevention and treatment of human disease (pp. 279–292). Academic Press
Homma T, Fujii J (2019) Chapter 5-oxidative stress and dysfunction of the intracellular proteolytic machinery: A pathological hallmark of nonalcoholic fatty liver disease. In: Watson RR, Preedy VR (eds) Dietary Interventions in Liver Disease. Academic Press, Boston, pp 59–70
Padurariu M, Ciobica A, Hritcu L, Stoica B, Bild W, Stefanescu C (2010) Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neurosci Lett 469(1):6–10
Sultana R, Piroddi M, Galli F, Butterfield DA (2008) Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem Res 33(12):2540–2546
Thimraj TA, George L, Asrafuzzaman S, Upadhyay S, Ganguly K (2018) Chapter 7-oxidative signaling in chronic obstructive airway diseases. In: Chatterjee S, Jungraithmayr W, Bagchi D (eds) Immunity and Inflammation in Health and Disease. Academic Press, Boston, pp 79–98
Matés JM, Pérez-Gómez C, De Castro IN (1999) Antioxidant enzymes and human diseases. Clin Biochem 32(8):595–603
Sofic E, Salkovic-Petrisic M, Tahirovic I, Sapcanin A, Mandel S, Youdim M, Riederer P (2015) Brain catalase in the streptozotocin-rat model of sporadic Alzheimer’s disease treated with the iron chelator–monoamine oxidase inhibitor, M30. J Neural Transm 122(4):559–564
Gsell W, Conrad R, Hickethier M, Sofic E, Frölich L, Wichart I, Jellinger K, Moll G, Ransmayr G, Beckmann H (1995) Decreased catalase activity but unchanged superoxide dismutase activity in brains of patients with dementia of Alzheimer type. J Neurochem 64(3):1216–1223
Behl C (1994) Davis JB, Lesley R, and Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77:817–827
Habib LK, Lee MT, Yang J (2010) Inhibitors of catalase-amyloid interactions protect cells from β-amyloid-induced oxidative stress and toxicity. J Biol Chem 285(50):38933–38943
Hane F, Tran G, Attwood SJ, Leonenko Z (2013) Cu 2+ affects amyloid-β (1–42) aggregation by increasing peptide-peptide binding forces. PloS One 8(3):e59005
Gabbita SP, Aksenov MY, Lovell MA, Markesbery WR (1999) Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. J Neurochem 73(4):1660–1666
Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, ... Jordan-Sciutto KL (2007) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66(1): 75-85
Chen K, Gunter K, Maines MD (2000) Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J Neurochem 75(1):304–313
Giordano G, White CC, Mohar I, Kavanagh TJ, Costa LG (2007) Glutathione levels modulate domoic acid induced apoptosis in mouse cerebellar granule cells. Toxicol Sci 100(2):433–444
Lim JH, Kim KM, Kim SW, Hwang O, Choi HJ (2008) Bromocriptine activates NQO1 via Nrf2-PI3K/Akt signaling: novel cytoprotective mechanism against oxidative damage. Pharmacol Res 57(5):325–331
Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, ... Lipton SA (2006) Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophillic phase II inducers. Proc Natl Acad Sci 103(3), 768-773
Tanito M, Agbaga MP, Anderson RE (2007) Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radical Biol Med 42(12):1838–1850
Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, Mann GE (2004) Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res 94(5):609–616
Saddawi-Konefka R, Seelige R, Gross ET, Levy E, Searles SC, Washington Jr A, ... Bui JD (2016) Nrf2 induces IL-17D to mediate tumor and virus surveillance. Cell Rep 16(9): 2348-2358
Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S (2002) Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Can Res 62(18):5196–5203
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, ... Yamamoto M (2016) Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 7(1): 1-14
Quinti L, Naidu SD, Träger U, Chen X, Kegel-Gleason K, Llères D, ... Kazantsev AG (2017) KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc Natl Acad Sci 114(23): E4676-E4685
Zhao G, Yao-Yue C, Qin GW, Guo LH (2012) Luteolin from Purple Perilla mitigates ROS insult particularly in primary neurons. Neurobiol Aging 33(1):176–186
Dragicevic N, Smith A, Lin X, Yuan F, Copes N, Delic V, ... Bradshaw PC (2011) Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J Alzheimers Dis 26(3), 507-521
Zhu LN, Mei X, Zhang ZG, Xie YP, Lang F (2019) Curcumin intervention for cognitive function in different types of people: a systematic review and meta-analysis. Phytother Res 33(3):524–533
Thota RN, Rosato JI, Dias CB, Burrows TL, Martins RN, Garg ML (2020) Dietary supplementation with curcumin reduce circulating levels of glycogen synthase kinase-3β and islet amyloid polypeptide in adults with high risk of type 2 diabetes and Alzheimer’s disease. Nutrients 12(4):1032
Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, ... Frautschy SA (2008) Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther 326(1), 196-208
Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, …& Cole, G. M. (2009) β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci 29(28):9078–9089
Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG (2004) Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci 101(22):8491–8496
Cole GM, Lim GP, Yang F, Teter B, Begum A, Ma Q, ... Frautschy SA (2005) Prevention of Alzheimer’s disease: omega-3 fatty acid and phenolic antioxidantinterventions. Neurobiol Aging 26(1): 133-136
Green P, Glozman S, Yavin E (2001) Ethyl docosahexaenoate-associated decrease in fetal brain lipid peroxide production is mediated by activation of prostanoid and nitric oxide pathways. Biochem Biophys Acta 1531(1–2):156–164
Yavin E, Brand A, Green P (2002) Docosahexaenoic acid abundance in the brain: a biodevice to combat oxidative stress. Nutr Neurosci 5(3):149–157
Hossain MS, Hashimoto M, Gamoh S, Masumura S (1999) Antioxidative effects of docosahexaenoic acid in the cerebrum versus cerebellum and brainstem of aged hypercholesterolemic rats. J Neurochem 72(3):1133–1138
Grimm MO, Kuchenbecker J, Grösgen S, Burg VK, Hundsdörfer B, Rothhaar TL, Friess P, de Wilde MC, Broersen LM, Penke B, Péter M, Vígh L, Grimm HS, Hartmann T (2011) Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J Biol Chem 286(16):14028–14039
Hashimoto M, Hossain S (2011) Neuroprotective and ameliorative actions of polyunsaturated fatty acids against neuronal diseases: beneficial effect of docosahexaenoic acid on cognitive decline in Alzheimer’s disease. J Pharmacol Sci 116(2):150–162
Cole GM, Ma QL, Frautschy SA (2009) Omega-3 fatty acids and dementia. Prostaglandins Leukot Essent Fatty Acids 81(2–3):213–221
Green KN, Martinez-Coria H, Khashwji H, Hall EB, Yurko-Mauro KA, Ellis L, LaFerla FM (2007) Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci 27(16):4385–4395
Lim SY, Suzuki H (2000) Intakes of dietary docosahexaenoic acid ethyl ester and egg phosphatidylcholine improve maze-learning ability in young and old mice. J Nutr 130(6):1629–1632
Minami M, Kimura S, Endo T, Hamaue N, Hirafuji M, Togashi H, …& Okuyama, H. (1997) Dietary docosahexaenoic acid increases cerebral acetylcholine levels and improves passive avoidance performance in stroke-prone spontaneously hypertensive rats. Pharmacol Biochem Behav 58(4):1123–1129
Külzow N, Witte AV, Kerti L, Grittner U, Schuchardt JP, Hahn A, Flöel A (2016) Impact of omega-3 fatty acid supplementation on memory functions in healthy older adults. J Alzheimers Dis 51(3):713–725
Freund-Levi Y, Eriksdotter-Jönhagen M, Cederholm T, Basun H, Faxén-Irving G, Garlind A, Vedin I, Vessby B, Wahlund LO, Palmblad J (2006) Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch Neurol 63(10):1402–1408
Eriksdotter M, Vedin I, Falahati F, Freund-Levi Y, Hjorth E, Faxen-Irving G, Wahlund LO, Schultzberg M, Basun H, Cederholm T, Palmblad J (2015) Plasma fatty acid profiles in relation to cognition and gender in Alzheimer’s disease patients during oral omega-3 fatty acid supplementation: the omega D study. J Alzheimers Dis 48(3):805–812
Shinto L, Quinn J, Montine T, Dodge HH, Woodward W, Baldauf-Wagner S, Waichunas D, Bumgarner L, Bourdette D, Silbert L, Kaye J (2014) A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J Alzheimers Dis 38(1):111–120
Irving GF, Freund-Levi Y, Eriksdotter-Jönhagen M, Basun H, Brismar K, Hjorth E, Palmblad J, Vessby B, Vedin I, Wahlund LO, Cederholm T (2009) Omega-3 fatty acid supplementation effects on weight and appetite in patients with Alzheimer’s disease: the omega-3 Alzheimer’s disease study. J Am Geriatr Soc 57(1):11–17
Freund Levi Y, Vedin I, Cederholm T, Basun H, Faxén Irving G, Eriksdotter M, Hjorth E, Schultzberg M, Vessby B, Wahlund LO, Salem N Jr, Palmblad J (2014) Transfer of omega-3 fatty acids across the blood-brain barrier after dietary supplementation with a docosahexaenoic acid-rich omega-3 fatty acid preparation in patients with Alzheimer’s disease: the OmegAD study. J Intern Med 275(4):428–436
Hooijmans CR, Pasker-de Jong PC, de Vries RB, Ritskes-Hoitinga M (2012) The effects of long-term omega-3 fatty acid supplementation on cognition and Alzheimer’s pathology in animal models of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis 28(1):191–209
Vedin I, Cederholm T, Freund-Levi Y, Basun H, Hjorth E, Irving GF, Eriksdotter-Jönhagen M, Schultzberg M, Wahlund LO, Palmblad J (2010) Reduced prostaglandin F2 alpha release from blood mononuclear leukocytes after oral supplementation of omega3 fatty acids: the OmegAD study. J Lipid Res 51(5):1179–1185
Phillips MA, Childs CE, Calder PC, Rogers PJ (2015) No effect of omega-3 fatty acid supplementation on cognition and mood in individuals with cognitive impairment and probable Alzheimer’s disease: a randomised controlled trial. Int J Mol Sci 16(10):24600–24613
Jernerén F, Elshorbagy AK, Oulhaj A, Smith SM, Refsum H, Smith AD (2015) Brain atrophy in cognitively impaired elderly: the importance of long-chain ω-3 fatty acids and B vitamin status in a randomized controlled trial. Am J Clin Nutr 102(1):215–221
Guan JZ, Guan W-P, Maeda T, Makino N (2011) Effect of vitamin E administration on the elevated oxygen stress and the telomeric and subtelomeric status in Alzheimer’s disease. Gerontology 58:62–69
Kaneai N, Arai M, Takatsu H, Fukui K, Urano S (2012) Vitamin E inhibits oxidative stress-induced denaturation of nerve terminal proteins involved in neurotransmission. J Alzheimers Dis 28(1):183–189
Wang X, Quinn PJ (1999) Vitamin E and its function in membranes. Prog Lipid Res 38(4):309–336
Dong S, Huang X, Zhen J, Van Halm-Lutterodt N, Wang J, Zhou C, Yuan L (2018) Dietary vitamin E status dictates oxidative stress outcomes by modulating effects of fish oil supplementation in Alzheimer disease model APP swe/PS1 dE9 mice. Mol Neurobiol 55(12):9204–9219
Li FJ, Shen L, Ji HF (2012) Dietary intakes of vitamin E, vitamin C, and β-carotene and risk of Alzheimer’s disease: a meta-analysis. J Alzheimers Dis 31(2):253–258
Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD, McCarten JR, Malphurs J, Prieto S, Chen P, Loreck DJ, Trapp G, Bakshi RS, Mintzer JE, Heidebrink JL, Vidal-Cardona A, Arroyo LM, Cruz AR, … Guarino PD (2014) Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 311(1), 33–44
Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ (1997) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med 336(17):1216–1222
Lloret A, Badía MC, Mora NJ, Pallardó FV, Alonso MD, Viña J (2009) Vitamin E paradox in Alzheimer’s disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis 17(1):143–149
McCleery J, Abraham RP, Denton DA, Rutjes AW, Chong LY, Al-Assaf AS, Griffith DJ, Rafeeq S, Yaman H, Malik MA, Di Nisio M, Martínez G, Vernooij RW, Tabet N (2018) Vitamin and mineral supplementation for preventing dementia or delaying cognitive decline in people with mild cognitive impairment. Cochrane Database Syst Rev 11(11):CD011905
Lu PH, Edland SD, Teng E, Tingus K, Petersen RC, Cummings JL, Alzheimer’s Disease Cooperative Study Group (2009) Donepezil delays progression to AD in MCI subjects with depressive symptoms. Neurology 72(24):2115–2121
Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ, Alzheimer’s Disease Cooperative Study Group (2005) Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 352(23):2379–2388
Thomas A, Iacono D, Bonanni L, D’Andreamatteo G, Onofrj M (2001) Donepezil, rivastigmine, and vitamin E in Alzheimer disease: a combined P300 event-related potentials/neuropsychologic evaluation over 6 months. Clin Neuropharmacol 24(1):31–42
Brigelius-Flohé R (2009) Vitamin E: the shrew waiting to be tamed. Free Radical Biol Med 46(5):543–554
Yatin SM, Varadarajan S, Butterfield DA (2000) Vitamin E prevents Alzheimer’s amyloid ß-Peptide (1–42)-induced neuronal protein oxidation and reactive oxygen species production. Journal of Alzheimer’s Disease 2(2):123–131
Devore EE, Grodstein F, van Rooij FJ, Hofman A, Stampfer MJ, Witteman JC, Breteler MM (2010) Dietary antioxidants and long-term risk of dementia. Arch Neurol 67(7):819–825
Giraldo E, Lloret A, Fuchsberger T, Viña J (2014) Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol 2:873–877
Montiel T, Quiroz-Baez R, Massieu L, Arias C (2006) Role of oxidative stress on β-amyloid neurotoxicity elicited during impairment of energy metabolism in the hippocampus: protection by antioxidants. Exp Neurol 200(2):496–508
Bittner DM (2009) Combination therapy of acetylcholinesterase inhibitor and vitamin E in Alzheimer disease. J Clin Psychopharmacol 29(5):511–513
Kim HS, Lee BM (2001) Protective effects of antioxidant supplementation on plasma lipid peroxidation in smokers. J Toxicol Environ Health A 63(8):583–598
Vogiatzoglou A, Refsum H, Johnston C, Smith SM, Bradley KM, De Jager C, ... Smith AD (2008) Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology 71(11): 826-832.
Morris MC, Evans DA, Tangney CC, Bienias JL, Wilson RS, Aggarwal NT, Scherr PA (2005) Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am J Clin Nutr 81(2):508–514
Ahmed HH (2012) Modulatory effects of vitamin E, acetyl-l-carnitine and α-lipoic acid on new potential biomarkers for Alzheimer’s disease in rat model. Exp Toxicol Pathol 64(6):549–556
Arlt S, Müller-Thomsen T, Beisiegel U, Kontush A (2012) Effect of one-year vitamin C-and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem Res 37(12):2706–2714
Pavlik VN, Doody RS, Rountree SD, Darby EJ (2009) Vitamin E use is associated with improved survival in an Alzheimer’s disease cohort. Dement Geriatr Cogn Disord 28(6):536–540
Rivière S, Birlouez-Aragon I, Nourhashémi F, Vellas B (1998) Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry 13(11):749–754
Richardson JS (1993) Free radicals in the genesis of Alzheimer’s disease a. Ann N Y Acad Sci 695(1):73–76
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, ... Relkin NR (2000) Intraneuronal Aβ42 accumulation in human brain. Am J Pathol 156(1): 15-20
Markesbery WR, Carney JM (1999) Oxidative alterations in Alzheimer’s disease. Brain Pathol 9(1):133–146
Beal MF (1995) Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 38(3):357–366
Connor JR, Menzies SL, St. Martin SM, Mufson EJ (1992) A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J Neurosci Res 31(1):75–83
Thomas T, Thomas G, McLendon C, Sutton T, Mullan M (1996) β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380(6570):168–171
Quinn J, Suh J, Moore MM, Kaye J, Frei B (2003) Antioxidants in Alzheimer’s disease-vitamin C delivery to a demanding brain. J Alzheimers Dis 5(4):309–313
Frei B, Stocker R, Ames BN (1988) Antioxidant defenses and lipid peroxidation in human blood plasma. Proc Natl Acad Sci 85(24):9748–9752
Gale CR, Martyn CN, Cooper C (1996) Cognitive impairment and mortality in a cohort of elderly people. BMJ 312(7031):608–611
Lindeman RD, Romero LJ, Koehler KM, Liang HC, LaRue A, Baumgartner RN, Garry PJ (2000) Serum vitamin B12, C and folate concentrations in the New Mexico elder health survey: correlations with cognitive and affective functions. J Am Coll Nutr 19(1):68–76
Perrig WJ, Perrig P, Stähelin HB (1997) The relation between antioxidants and memory performance in the old and very old. J Am Geriatr Soc 45(6):718–724
Perkins AJ, Hendrie HC, Callahan CM, Gao S, Unverzagt FW, Xu Y, ... Hui SL (1999) Association of antioxidants with memory in a multiethnic elderly sample using the Third National Health and Nutrition Examination Survey. Am J Epidemiol 150(1), 37-44
Schmidt R, Hayn M, Reinhart B, Roob G, Schmidt H, Schumacher M, ... Launer LJ (1998) Plasma antioxidants and cognitive performance in middle‐aged and older adults: results of the Austrian stroke prevention study. J Am Geriatr Soc 46(11):1407-1410
Whalley LJ, Fox HC, Lemmon HA, Duthie SJ, Collins AR, Peace H, ... Deary IJ (2003) Dietary supplement use in old age: associations with childhood IQ, current cognition and health. Int J Geriatr Psychiatry 18(9): 769-776
Rinaldi P, Polidori MC, Metastasio A, Mariani E, Mattioli P, Cherubini A, ... Mecocci P (2003) Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol Aging 24(7): 915-919
Sato R, Helzlsouer KJ, Comstock GW, Hoffman SC (2006) A cross-sectional study of vitamin C and cognitive function in older adults: the differential effects of gender. J Nutr Health Aging 10(1):37
Rinaldi CA, Bucknall CA, Gill JS (2002) Beneficial effects of biventricular pacing in a patient with hypertrophic cardiomyopathy and intraventricular conduction delay. Heart (British Cardiac Society), 87(6), e6. https://doi.org/10.1136/heart.87.6.e6
Chandra S (2001) Studies of cell division (mitosis and cytokinesis) by dynamic secondary ion mass spectrometry ion microscopy: LLC-PK1 epithelial cells as a model for subcellular isotopic imaging. J Microsc 204(Pt 2):150–165. https://doi.org/10.1046/j.1365-2818.2001.00944.x
Travica N, Ried K, Sali A, Scholey A, Hudson I, Pipingas A (2017) Vitamin C status and cognitive function: a systematic review. Nutrients 9(9):960
Kontush A, Mann U, Arlt S, Ujeyl A, Lührs C, Müller-Thomsen T, Beisiegel U (2001) Influence of vitamin E and C supplementation on lipoprotein oxidation in patients with Alzheimer’s disease. Free Radical Biol Med 31(3):345–354
Boothby LA, Doering PL (2005) Vitamin C and vitamin E for Alzheimer’s disease. Ann Pharmacother 39(12):2073–2080
Loef M, Schrauzer GN, Walach H (2011) Selenium and Alzheimer’s disease: a systematic review. J Alzheimers Dis 26(1):81–104
Pinton S, Brüning CA, Oliveira CES, Prigol M, Nogueira CW (2013) Therapeutic effect of organoselenium dietary supplementation in a sporadic dementia of Alzheimer’s type model in rats. J Nutr Biochem 24(1):311–317
Savarino L, Granchi D, Ciapetti G, Cenni E, Ravaglia G, Forti P, ... Mattioli R (2001) Serum concentrations of zinc and selenium in elderly people: results in healthy nonagenarians/centenarians. Exp Gerontol 36(2):327-339
Akbaraly TN, Hininger-Favier I, Carriere I, Arnaud J, Gourlet V, Roussel AM, Berr C (2007) Plasma selenium over time and cognitive decline in the elderly. Epidemiology 52–58
Cardoso BR, Ong TP, Jacob-Filho W, Jaluul O, Freitas MIDÁ, Cozzolino SMF (2010) Nutritional status of selenium in Alzheimer’s disease patients. Br J Nutr 103(6):803–806
Ishrat T, Parveen K, Khan MM, Khuwaja G, Khan MB, Yousuf S, ... Islam F (2009) Selenium prevents cognitive decline and oxidative damage in rat model of streptozotocin-induced experimental dementia of Alzheimer’s type. Brain Res 1281L: 117-127
Song G, Zhang Z, Wen L, Chen C, Shi Q, Zhang Y, ... Liu Q (2014) Selenomethionine ameliorates cognitive decline, reduces tau hyperphosphorylation, and reverses synaptic deficit in the triple transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 41(1): 85-99
Tolonen M, Halme M, Sarna S (1985) Vitamin E and selenium supplementation in geriatric patients. Biol Trace Elem Res 7(3):161–168
Lovell MA, Xiong S, Lyubartseva G, Markesbery WR (2009) Organoselenium (Sel-Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radical Biol Med 46(11):1527–1533
Van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Götz J, Ittner LM (2010) Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci 107(31):13888–13893
Bucana CD, Nadakavukaren MJ, Frehn JL (1974) Novel features of hamster pinealocyte ultrastructure. Tissue Cell 6(1):85–93. https://doi.org/10.1016/0040-8166(74)90024-x
Buendia I, Navarro E, Michalska P, Gameiro I, Egea J, Abril S, ... León R (2015) New melatonin–cinnamate hybrids as multi-target drugs for neurodegenerative diseases: Nrf2-induction, antioxidant effect and neuroprotection. Future Med Chem 7(15): 1961-1969
Calvo J, Boya J (1984) Ultrastructure of the pineal gland in the adult rat. J Anat 138(Pt 3):405
Hardeland R (2005) Atioxidative protection by melatonin. Endocrine 27(2):119–130
Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, Swaab DF (1999) Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-ε4/4 genotype. J Clin Endocrinol Metab 84(1):323–327
Lin L, Huang QX, Yang SS, Chu J, Wang JZ, Tian Q (2013) Melatonin in Alzheimer’s disease. Int J Mol Sci 14(7):14575–14593
Abdel Moneim AE, Ortiz F, Leonardo-Mendonça RC, Vergano-Villodres R, Guerrero-Martínez JA, López LC, Acuña-Castroviejo D, Escames G (2015) Protective effects of melatonin against oxidative damage induced by Egyptian cobra (Naja haje) crude venom in rats. Acta Trop 143:58–65
Feng Y, Wang X (2012) Antioxidant therapies for Alzheimer’s disease. Oxidative Medicine and Cellular Longevity 2012
Cardinali DP, Furio AM, Brusco LI (2010) Clinical aspects of melatonin intervention in Alzheimer’s disease progression. Curr Neuropharmacol 8(3):218–227
Jean-Louis G, von Gizycki H, Zizi F (1998) Melatonin effects on sleep, mood, and cognition in elderly with mild cognitive impairment. J Pineal Res 25(3):177–183
Simpkins JW, Perez E, Wang X, Yang S, Wen Y, Singh M (2009) The potential for estrogens in preventing Alzheimer’s disease and vascular dementia. Ther Adv Neurol Disord 2(1):31–49
Greenfield JP, Leung LW, Cai D, Kaasik K, Gross RS, Rodriguez-Boulan E, Greengard P, Xu H (2002) Estrogen lowers Alzheimer beta-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J Biol Chem 277(14):12128–12136
Li R, Shen Y, Yang LB, Lue LF, Finch C, Rogers J (2000) Estrogen enhances uptake of amyloid β-protein by microglia derived from the human cortex. J Neurochem 75(4):1447–1454
Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE (1994) Estrogen regulates metabolism of Alzheimer amyloid beta precursor protein. J Biol Chem 269(18):13065–13068
Benvenuti S, Luciani P, Vannelli GB, Gelmini S, Franceschi E, Serio M, Peri A (2005) Estrogen and selective estrogen receptor modulators exert neuroprotective effects and stimulate the expression of selective Alzheimer’s disease indicator-1, a recently discovered antiapoptotic gene, in human neuroblast long-term cell cultures. J Clin Endocrinol Metab 90(3):1775–1782
Asthana S, Craft S, Baker LD, Raskind MA, Birnbaum RS, Lofgreen CP, … Plymate SR (1999) Cognitive and neuroendocrine response to transdermal estrogen in postmenopausal women with Alzheimer’s disease: results of a placebo-controlled, double-blind, pilot study. Psychoneuroendocrinology 24(6): 657-678
Forsmark-Andrée P, Lee CP, Dallner G, Ernster L (1997) Lipid peroxidation and changes in the ubiquinone content and the respiratory chain enzymes of submitochondrial particles. Free Radical Biol Med 22(3):391–400
Crane FL (2001) Biochemical functions of coenzyme Q10. J Am Coll Nutr 20(6):591–598
Ernster L, Dallner G (1995) Biochemical, physiological and medical aspects of ubiquinone function. Biochimica etBiophysica Acta 1271(1):195–204
Sharma SK, El ReFaey H, Ebadi M (2006) Complex-1 activity and 18F-DOPA uptake in genetically engineered mouse model of Parkinson’s disease and the neuroprotective role of coenzyme Q10. Brain Res Bull 70(1):22–32
Dumont M, Kipiani K, Yu F, Wille E, Katz M, Calingasan NY, ... Beal MF (2011) Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 27(1), 211-223
Yang X, Yang Y, Li G, Wang J, Yang ES (2008) Coenzyme Q10 attenuates β-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J Mol Neurosci 34(2):165–171
Horecky J, Gvozdjáková A, Kucharská J, Obrenovich ME, Palacios HH, Li Y, ... Aliev G (2011) Effects of coenzyme Q and creatine supplementation on brain energy metabolism in rats exposed to chronic cerebral hypoperfusion. Curr Alzheimer Res 8(8): 868-875
Tsvetkova D, Obreshkova D, Zheleva-Dimitrova D, Saso L (2013) Antioxidant activity of galantamine and some of its derivatives. Curr Med Chem 20(36):4595–4608
Wen C, Huang C, Yang M, Fan C, Li Q, Zhao J, ... Lu D (2020) The secretion from bone marrow Mesenchymal stem cells pretreated with Berberine rescues neurons with oxidative damage through activation of the Keap1-Nrf2-HO-1 signaling pathway. Neurotoxicity Research, 38(1): 59-73
Yang R, Wang Q, Li F, Li J, Liu X (2015) Edaravone injection ameliorates cognitive deficits in rat model of Alzheimer’s disease. Neurol Sci 36(11):2067–2072
Jiao SS, Yao XQ, Liu YH, Wang QH, Zeng F, Lu JJ, Liu J, Zhu C, Shen LL, Liu CH, Wang YR, Zeng GH, Parikh A, Chen J, Liang CR, Xiang Y, Bu XL, Deng J, Li J, Xu J, … Wang YJ (2015) Edaravone alleviates Alzheimer’s disease-type pathologies and cognitive deficits. Proc Natl Acad Sci USA 112(16): 5225-5230
Singh S, Gautam U, Manvi FV (2020) Protective impact of edaravone against ZnO NPs-induced oxidative stress in the human neuroblastoma SH-SY5Y cell line. Cell Mol Neurobiol, 1–22
Zeng KW, Wang XM, Ko H, Kwon HC, Cha JW, Yang HO (2011) Hyperoside protects primary rat cortical neurons from neurotoxicity induced by amyloid β-protein via the PI3K/Akt/Bad/BclXL-regulated mitochondrial apoptotic pathway. Eur J Pharmacol 672(1–3):45–55
Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, ... Ohta S (2007) Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med 13(6): 688-694
Yoshii Y, Inoue T, Uemura Y, Iwasaki Y, Yada T, Nakabeppu Y, Noda M (2017) Complexity of stomach–brain interaction induced by molecular hydrogen in Parkinson’s disease model mice. Neurochem Res 42(9):2658–2665
Hayashida K, Sano M, Ohsawa I, Shinmura K, Tamaki K, Kimura K, ... Fukuda K (2008) Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia–reperfusion injury. Biochem Biophys Res Commun 373(1): 30-35
Abraini JH, Gardette-Chauffour MC, Martinez E, Rostain JC, Lemaire C (1994) Psychophysiological reactions in humans during an open sea dive to 500 m with a hydrogen-helium-oxygen mixture. J Appl Physiol 76(3):1113–1118
Ono H, Nishijima Y, Adachi N, Sakamoto M, Kudo Y, Kaneko K, ... Imaoka T (2012) A basic study on molecular hydrogen (H 2) inhalation in acute cerebral ischemia patients for safety check with physiological parameters and measurement of blood H 2 level. Med Gas Res 2(1):1-7
Ara J, Fadriquela A, Ahmed MF, Bajgai J, Sajo MEJ, Lee SP, ... Lee KJ (2018) Hydrogen water drinking exerts antifatigue effects in chronic forced swimming mice via antioxidative and anti-inflammatory activities. BioMed Research International 2018
Jin L, Yu SQ, Zhang X, Ge Q, Zhang XL, Wang Y, Qin ML (2018) Clinical study of hydrogen-rich saline in the treatment of moderate to severe allergic rhinitis. Lin Chuang er bi yan hou tou Jing wai ke za zhi= Journal of Clinical Otorhinolaryngology, Head, and Neck Surgery 32(7): 493-496
Koyama Y, Taura K, Hatano E, Tanabe K, Yamamoto G, Nakamura K, ... Uemoto S (2014) Effects of oral intake of hydrogen water on liver fibrogenesis in mice. Hepatol Res 44(6):663-677
Ohta S (2014) Molecular hydrogen as a preventive and therapeutic medical gas: initiation, development and potential of hydrogen medicine. Pharmacol Ther 144(1):1–11
Lin CL, Huang WN, Li HH, Huang CN, Hsieh S, Lai C, Lu FJ (2015) Hydrogen-rich water attenuates amyloid β-induced cytotoxicity through upregulation of Sirt1-FoxO3a by stimulation of AMP-activated protein kinase in SK-N-MC cells. Chem Biol Interact 240:12–21
Iuchi K, Imoto A, Kamimura N, Nishimaki K, Ichimiya H, Yokota T, Ohta S (2016) Molecular hydrogen regulates gene expression by modifying the free radical chain reaction-dependent generation of oxidized phospholipid mediators. Sci Rep 6:18971
Hou C, Peng Y, Qin C, Fan F, Liu J, Long J (2018) Hydrogen-rich water improves cognitive impairment gender-dependently in APP/PS1 mice without affecting Aβ clearance. Free Radical Res 52(11–12):1311–1322
Riahi Y, Wikstrom JD, Bachar-Wikstrom E, Polin N, Zucker H, Lee MS, ... Leibowitz G (2016) Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 59(7):1480-1491
Liu X, Chen Z, Mao N, Xie Y (2012) The protective of hydrogen on stress-induced gastric ulceration. Int Immunopharmacol 13(2):197–203
Zhai Y, Zhou X, Dai Q, Fan Y, Huang X (2015) Hydrogen-rich saline ameliorates lung injury associated with cecal ligation and puncture-induced sepsis in rats. Exp Mol Pathol 98(2):268–276
Fukuda M, Takatori A, Nakamura Y, Suganami A, Hoshino T, Tamura Y, Nakagawara A (2016) Effects of novel small compounds targeting TrkB on neuronal cell survival and depression-like behavior. Neurochem Int 97:42–48
Cui Y, Zhang H, Ji M, Jia M, Chen H, Yang J, Duan M (2014) Hydrogen-rich saline attenuates neuronal ischemia–reperfusion injury by protecting mitochondrial function in rats. J Surg Res 192(2):564–572
Mano Y, Kotani T, Ito M, Nagai T, Ichinohashi Y, Yamada K, ... Toyokuni S (2014) Maternal molecular hydrogen administration ameliorates rat fetal hippocampal damage caused by in utero ischemia–reperfusion. Free Radic Biol Med 69: 324-330
Nagata K, Nakashima-Kamimura N, Mikami T, Ohsawa I, Ohta S (2009) Consumption of molecular hydrogen prevents the stress-induced impairments in hippocampus-dependent learning tasks during chronic physical restraint in mice. Neuropsychopharmacology 34(2):501–508
Li J, Wang C, Zhang JH, Cai JM, Cao YP, Sun XJ (2010) Hydrogen-rich saline improves memory function in a rat model of amyloid-beta-induced Alzheimer’s disease by reduction of oxidative stress. Brain Res 1328:152–161
Wang C, Li J, Liu Q, Yang R, Zhang JH, Cao YP, Sun XJ (2011) Hydrogen-rich saline reduces oxidative stress and inflammation by inhibit of JNK and NF-κB activation in a rat model of amyloid-beta-induced Alzheimer’s disease. Neurosci Lett 491(2):127–132
Hollands C, Tobin MK, Hsu M, Musaraca K, Yu TS, Mishra R, ... Lazarov O (2017) Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol Neurodegener 12(1):1-13
Radad K, Moldzio R, Al-Shraim M, Kranner B, Krewenka C, Rausch WD (2017) Recent advances on the role of neurogenesis in the adult brain: therapeutic potential in Parkinson’s and Alzheimer’s diseases. CNS Neurol Disord Drug Targets 16(7):740–748
Gu Y, Huang CS, Inoue T, Yamashita T, Ishida T, Kang KM, Nakao A (2010) Drinking hydrogen water ameliorated cognitive impairment in senescence-accelerated mice. J Clin Biochem Nutr 46(3):269–276
Nishimaki K, Asada T, Ohsawa I, Nakajima E, Ikejima C, Yokota T, Kamimura N, Ohta S (2018) Effects of molecular hydrogen assessed by an animal model and a randomized clinical study on mild cognitive impairment. Curr Alzheimer Res 15(5):482–492
Sim M, Kim CS, Shon WJ, Lee YK, Choi EY, Shin DM (2020) Hydrogen-rich water reduces inflammatory responses and prevents apoptosis of peripheral blood cells in healthy adults: A randomized, double-blind, controlled trial. Sci Rep 10(1):1–10
Butler M, Nelson VA, Davila H, Ratner E, Fink HA, Hemmy LS, McCarten JR, Barclay TR, Brasure M, Kane RL (2018) Over-the-counter supplement interventions to prevent cognitive decline, mild cognitive impairment, and clinical alzheimer-type dementia: a systematic review. Ann Intern Med 168(1):52–62
Mullan K, Cardwell CR, McGuinness B, Woodside JV, McKay GJ (2018) Plasma antioxidant status in patients with Alzheimer’s disease and cognitively intact elderly: a meta-analysis of case-control studies. J Alzheimers Dis 62(1):305–317
Cornelli U (2010) Treatment of Alzheimer’s disease with a cholinesterase inhibitor combined with antioxidants. Neurodegener Dis 7(1–3):193–202
Chan A, Remington R, Kotyla E, Lepore A, Zemianek J, Shea TB (2010) A vitamin/nutriceutical formulation improves memory and cognitive performance in community-dwelling adults without dementia. J Nutr Health Aging 14(3):224–230
Remington R, Bechtel C, Larsen D, Samar A, Doshanjh L, Fishman P, Luo Y, Smyers K, Page R, Morrell C, Shea TB (2015) A phase II randomized clinical trial of a nutritional formulation for cognition and mood in Alzheimer’s disease. J Alzheimers Dis 45(2):395–405
Remington R, Bechtel C, Larsen D, Samar A, Page R, Morrell C, Shea TB (2016) Maintenance of cognitive performance and mood for individuals with Alzheimer’s disease following consumption of a nutraceutical formulation: a one-year, open-label study. J Alzheimers Dis 51(4):991–995
Chew EY, Clemons TE, Agrón E, Launer LJ, Grodstein F, Bernstein PS, age-related eye disease study 2 (AREDS2) research group (2015) Effect of omega-3 fatty acids, lutein/zeaxanthin, or other nutrient supplementation on cognitive function: the areds2 randomized clinical trial. JAMA 314(8):791–801
Kryscio RJ, Abner EL, Caban-Holt A, Lovell M, Goodman P, Darke AK, Yee M, Crowley J, Schmitt FA (2017) Association of antioxidant supplement use and dementia in the prevention of Alzheimer’s disease by vitamin E and selenium trial (PREADViSE). JAMA Neurol 74(5):567–573
Schrag M, Mueller C, Zabel M, Crofton A, Kirsch WM, Ghribi O, Squitti R, Perry G (2013) Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: a meta-analysis. Neurobiol Dis 59:100–110
Ahmed T, Javed S, Javed S, Tariq A, Šamec D, Tejada S, ... Nabavi SM (2017) Resveratrol and Alzheimer’s disease: mechanistic insights. Mol Neurobiol 54(4): 2622-2635
Adair JC, Knoefel JE, Morgan N (2001) Controlled trial of N-acetylcysteine for patients with probable Alzheimer’s disease. Neurology 57(8):1515–1517
Di Domenico F, Barone E, Perluigi M, Butterfield DA (2015) Strategy to reduce free radical species in Alzheimer’s disease: an update of selected antioxidants. Expert Rev Neurother 15(1):19–40
Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM (2002) Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 287(24):3223–3229
Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, ... Scherr PA (2002) Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA 287(24): 3230-3237
Hsu CH, Cheng AL (2007) Clinical studies with curcumin. In: The molecular targets and therapeutic uses of curcumin in health and disease, 471–480
Misonou H, Morishima-Kawashima M, Ihara Y (2000) Oxidative stress induces intracellular accumulation of amyloid β-protein (Aβ) in human neuroblastoma cells. Biochemistry 39(23):6951–6959
Paola D, Domenicotti C, Nitti M, Vitali A, Borghi R, Cottalasso D, ... Pronzato MA (2000) Oxidative stress induces increase in intracellular amyloid β-protein production and selective activation of βI and βII PKCs in NT2 cells. Biochem Biophys Res Commun 268(2): 642-646
Shen C, Chen Y, Liu H, Zhang K, Zhang T, Lin A, Jing N (2008) Hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-secretase. J Biol Chem 283(25):17721–17730
Persson T, Popescu BO, Cedazo-Minguez A (2014) Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxid Med Cell Longev 2014:427318
Galasko DR, Peskind E, Clark CM, Quinn JF, Ringman JM, Jicha GA, ... Aisen P (2012) Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 69(7):836-841
Berr C, Richard MJ, Gourlet V, Garrel C, Favier A (2004) Enzymatic antioxidant balance and cognitive decline in aging—the EVA study. Eur J Epidemiol 19(2):133–138
Torres LL, Quaglio NB, de Souza GT, Garcia RT, Dati LMM, Moreira WL, ... Marcourakis T (2011) Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 26(1):59-68
Terada A, Yoshida M, Seko Y, Kobayashi T, Yoshida K, Nakada M, …& Rikihisa, T. (1999) Active oxygen species generation and cellular damage by additives of parenteral preparations: selenium and sulfhydryl compounds. Nutrition 15(9):651–655
Kwong LK, Kamzalov S, Rebrin I, Bayne ACV, Jana CK, Morris P, …& Sohal, R. S. (2002) Effects of coenzyme Q10 administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radical Biol Med 33(5):627–638
Thal LJ, Grundman M, Berg J, Ernstrom K, Margolin R, Pfeiffer E, ... Thomas RG (2003) Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology 61(11):1498-1502
Sung S, Yao Y, Uryu K, Yang H, Lee VMY, Trojanowski JQ, Praticò D (2004) Early vitamin E supplementation in young but not aged mice reduces Aβ levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J 18(2):323–325
Avila J (2010) Common mechanisms in neurodegeneration. Nat Med 16(12):1372–1372
Butterfield SM, Lashuel HA (2010) Amyloidogenic protein–membrane interactions: mechanistic insight from model systems. Angew Chem Int Ed 49(33):5628–5654
Di Domenico F, Barone E, Perluigi M, Butterfield DA (2017) The triangle of death in Alzheimer’s disease brain: the aberrant cross-talk among energy metabolism, mammalian target of rapamycin signaling, and protein homeostasis revealed by redox proteomics. Antioxid Redox Signal 26(8):364–387
Fillenbaum GG, Kuchibhatla MN, Hanlon JT, Artz MB, Pieper CF, Schmader KE, Dysken MW, Gray SL (2005) Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann Pharmacother 39(12):2009–2014
Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P (1997) Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett 409(1):57–62
González-Muñoz GC, Arce MP, Pérez C, Romero A, Villarroya M, López MG, ... Rodríguez-Franco MI (2014) Dibenzo [1, 4, 5] thiadiazepine: a hardly-known heterocyclic system with neuroprotective properties of potential usefulness in the treatment of neurodegenerative diseases. Eur J Med Chem 81: 350-358
Isaac MGEKN, Quinn R, Tabet N (2008) Vitamin E for Alzheimer’s disease and mild cognitive impairment. Cochrane Database of Systematic Reviews, (3)
Rafieipour F, Hadipour E, Emami SA, Asili J, Tayarani-Najaran Z (2019) Safranal protects against beta-amyloid peptide-induced cell toxicity in PC12 cells via MAPK and PI3 K pathways. Metab Brain Dis 34(1):165–172
Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH (2000) Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3β. J Neurochem 74(4):1587–1595
Reynolds CH, Nebreda AR, Gibb GM, Utton MA, Anderton BH (1997) Reactivating kinase/p38 phosphorylates τ protein in vitro. J Neurochem 69(1):191–198
Richardson TIL, Ball L, Rosenfeld T (2002) Will an orange a day keep the doctor away? Postgrad Med J 78(919):292–294
Schweizer U, Streckfuß F, Pelt P, Carlson BA, Hatfield DL, Köhrle J, Schomburg L (2005) Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem J 386(2):221–226
Tahirbegi IB, Pardo WA, Alvira M, Mir M, Samitier J (2016) Amyloid Aβ 42, a promoter of magnetite nanoparticle formation in Alzheimer’s disease. Nanotechnology 27(46):465102
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270(5240):1326–1331
Funding
This work was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, Saudi Arabia under grant no. (KEP-3-140-42). The authors, therefore, acknowledge with thanks DSR technical and financial support.
Author information
Authors and Affiliations
Contributions
Vaibhav Walia: writing—original draft preparation. Deepak Kaushik: conceptualization and supervision. Vineet Mittal: writing—review and editing. Kuldeep Kumar: writing—review and editing. Ravinder Verma: writing—review and editing. Jatin Parashar: software. Rokeya Akter: review and editing. Md. Habibur Rahman: conceptualization. Saurabh Bhatia: writing—review and editing. Ahmed Al-Harrasi: writing—review and editing. Chenmala Karthika: writing—review and editing. Tanima Bhattacharya: writing—review and editing. Hitesh Chopra: writing—review and editing. Ghulam Md Ashraf: writing—review and editing.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Antioxidant therapy prevents the emergence and the progression of AD to advanced stages
• Antioxidant therapy counteracts the neuronal degeneration in various region of brain
• Antioxidant therapy improves learning and memory, cognition, and MMSE scores
• Antioxidant activity of antioxidant(s) is largely affected by the antioxidant, patients, and clinical study-related parameters
• The failure of antioxidant therapy fails in clinical settings in AD patients does not mean the loss of effectiveness of antioxidants in AD patients or clinical settings
• The major reasons for the failure of antioxidant therapy involve wrong selection of the dose or duration and the stage of disease in which they are used
Rights and permissions
About this article

Cite this article
Walia, V., Kaushik, D., Mittal, V. et al. Delineation of Neuroprotective Effects and Possible Benefits of AntioxidantsTherapy for the Treatment of Alzheimer’s Diseases by Targeting Mitochondrial-Derived Reactive Oxygen Species: Bench to Bedside. Mol Neurobiol 59, 657–680 (2022). https://doi.org/10.1007/s12035-021-02617-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02617-1