
Abstract
Following the initial discovery that adipose tissue actively synthesizes and secretes cytokines, obesity-induced inflammation has been implicated in the etiology of a host of disease states related to obesity, including cardiovascular disease and type II diabetes. Interestingly, a growing body of evidence similarly implicates sphingolipids as prime instigators in these same diseases. From the recent discovery that obesity-related inflammatory pathways modulate sphingolipid metabolism comes a novel perspective—sphingolipids may act as the dominant mediators of deleterious events stemming from obesity-induced inflammation. This paradigm may identify sphingolipids as an effective target for future therapeutics aimed at ameliorating diseases associated with chronic inflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite an explosion in interventions aimed at holding back the tide of obesity and improving public health, the rising trends in its prevalence show little sign of slowing. The economic and personal toll of obesity is enormous. Medical costs associated with treating obesity and its complications are roughly $150 billion annually, and obese individuals can expect average annual medical expenses to be approximately $1,500 more than normal-weight persons [1]. The problem is not merely one of money, however, considering that obesity is estimated to result in up to 20 years of life lost when compared to the non-obese [2]. If interventions aimed at averting obesity continue to fail, trends can be expected to increase with an estimated 2.4 million more adults becoming obese annually, followed closely by an ever-greater financial burden. The observation by the ancient Greek physician Hippocrates that “sudden death is more common in those who are naturally fat than in the lean” astutely describes the increase in mortality associated with obesity.
Of course, the increased health-related expenses and risk of mortality are not simply due to the mechanical discomforts and limitations that accompany an expanding fat mass. Obesity not only adversely affects tissue function but also contributes to an elevated risk of developing several fatal diseases, such as hypertension, atherosclerosis, type 2 diabetes mellitus, and nonalcoholic fatty liver disease [3]. As such, it is not surprising that extensive efforts have been devoted to understanding the role of obesity in the etiology of these prominent diseases. In particular, two deleterious factors often associated with obesity are implicated in increasing disease risk—lipotoxicity and inflammation. Lipotoxicity refers to the ectopic deposition of fat in tissues not intended as lipid storage sites. Of the myriad of bioactive lipids in tissues, however, the sphingolipids warrant particular attention due not only due to being highly correlated with the degree of obesity, but also implicated in the etiology of various diseases [4–7]. In addition, inflammation, defined by elevated levels of proinflammatory cytokines and increased presence and activity of monocytes/macrophages, is also present in obesity [8–11]. Interestingly, recent efforts reveal that these two seemingly distinct characteristics of obesity, lipotoxicity and inflammation, share a degree of linearity that might alter our perspective on the mechanism of inflammation-induced complications in various diseases. Thus, it is the intention of this review to explore the role of sphingolipids in the pathophysiology of some of the prominent diseases associated with obesity-induced inflammation.
Obesity, inflammation, and sphingolipids
Obesity-induced inflammation
Gone are the days when adipose tissue was considered a passive organ whose exclusive functions are to accumulate triglycerides in hypertrophied adipocytes during caloric excess and release these lipids during caloric restriction. The exploration of obesity and inflammation began in adipose tissue. From the initial observation that adipose is capable of synthesizing and releasing tumor necrosis factor α (TNFα) [12], the total number of adipose-derived molecules (termed “adipokines”) has ballooned. Compared to adipose from lean individuals, adipose tissue from the obese has increased levels of several inflammatory proteins, such as interleukin-6 (IL-6), iNOS, C-reactive protein (CRP), monocyte chemotactic protein-1 (MCP-1), and plasminogen activator inhibitor type-1 (PAI-1), among others [13–16]. Transcriptional profiling reveals that inflammatory genes are among the most abundantly regulated in adipose tissue in obesity [17]. However, adipose tissue is not simply a homogenous collection of adipocytes. Expanding adipose tissue is accompanied by increased infiltration of activated macrophages, and, while adipocytes themselves appear to express the cellular machinery to enable cytokine synthesis and responsiveness [18], these resident macrophages play a dominant role in adipose-derived proinflammatory gene expression [19].
The adipokine profile secreted from adipose tissue is dynamic and is affected by fat mass status (Fig. 1). While proinflammatory cytokine expression (e.g., TNFα, MCP-1, IL-1β) is increased with weight gain by fat mass expansion [12], anti-inflammatory protein expression (e.g., adiponectin, IL-10) is elevated in adipose from lean or increased with weight loss in the obese [20, 21]. Additionally, circulating and tissue ceramide levels follow a trend similar to proinflammatory cytokines. Though once considered to be nothing more than correlative companions [22], we now know that sphingolipid biosynthesis requires activation of immune receptors and pathways.

Adipocyte expansion, leading to obesity, is associated with reduced adiponectin levels and elevated TNFα and ceramide
TLR4 and sphingolipids
Recent findings have refuted the old notion that production of ceramide, the backbone of all higher-order sphingolipids, is exclusively controlled by substrate availability. Rather, it appears hormonal signals have a powerful influence to alter ceramide metabolism, resulting in ceramide accumulation or degradation. Similar to inflammatory status in lean and obese, ceramide metabolism and accumulation is altered in the tissues of lean and obese. Both rodent and human models of obesity have shown an increase in ceramide levels in a variety of tissues, such as skeletal muscle, liver, and hypothalamus [7, 23–26]. Also, paralleling the shift in inflammatory status with weight loss [11], a reduction in fat mass is associated with a reduction in tissue and circulating ceramide levels [7, 27, 28]. For example, Huang et al. [27] found that substantial surgery-induced weight loss (~25% of body weight) in morbidly obese humans correlated with a reduction in plasma ceramides. However, Dube et al. [29] found that even modest weight loss (2% body weight) is associated with a significant drop in ceramide levels.
The parallel changes in inflammatory status and ceramide metabolism are not coincidental. We recently found that the activation of inflammatory pathways is a necessary event in ceramide biosynthesis, with both saturated fatty acids and bacterial endotoxins converging on the Toll-like receptor 4 (TLR4) pathway to induce ceramide accumulation (Fig. 2)[23]. Using mutant mice lacking a functional TLR4 (C.C2-Tlr4 Lps−d), we found that TLR4 is required for saturated fatty acid- and endotoxin-induced ceramide biosynthesis in skeletal muscle and liver. Interestingly, while saturated fatty acids are known to induce both an inflammatory response and de novo ceramide biosynthesis, unsaturated fatty acids, which do not activate TLR4, fail to do both [23]. In addition to saturated fats, lipopolysaccharide (LPS), a major bacterial membrane lipid and TLR4 ligand, similarly induced ceramide biosynthesis in a TLR4-dependent manner [23].

The TLR4-induced activation of ceramide biosynthesis represents a common pathway between lipotoxicity- and inflammation-induced insulin resistance. Saturated fatty acids (SFA); lipopolysaccharide (LPS); tumor necrosis factor (TNF) α; inhibitor of κB kinase (IKK) β; sphingomyelinase (SMAase)
TLR4 initiates signaling through the canonical IKKβ-NF-κB pathway. Briefly, upon activation, IKKβ phosphorylates and marks the NF-κB inhibitor, IκBα, for degradation, after which NF-κB is liberated and free to migrate into the nucleus to initiate transcription of various cytokines. IKKβ has been previously implicated in acting as a prime mediator of inflammation-induced metabolic disorders via its inhibitory effects on proximal insulin signaling [30, 31]. We found that IKKβ was a necessary downstream factor in TLR4-induced ceramide biosynthesis. Not only was IKKβ ablation associated with dramatically reduced ceramides in spite of TLR4 activation, it was necessary for the fatty acid- and inflammation-induced increase in transcription of several of the enzymes involved in de novo ceramide synthesis [23]. Additionally, diet-induced obese mice treated with the IKKβ inhibitor sodium salicylate fail to accrue ceramides [23].
As mentioned, both saturated fatty acids (SFA) and bacterial endotoxins appear to activate TLR4, though whether they activate the receptor via similar or disparate mechanisms is not clear. The findings that SFA, but not unsaturated FA, activate TLR4 are widely reported [23, 32–34]. In contrast, Erridge et al. [35] found conflicting results, reporting that SFA failed to activate TLR4 in multiple cell types and implicating LPS contamination in fatty acid preparations as the source of TLR4 activation. However, this fails to explain the contrasting effects of SFA compared to unsaturated counterparts, which should be similarly contaminated [23]. Another argument against SFA and endotoxins affecting TLR4 via a single mechanism is the different kinetics of TLR4 activation between the two purported ligands. In exploring the inflammatory effects of SFA and LPS, Shi et al. [36] observed that orders of magnitude more SFA are necessary to get a comparable response seen with LPS, indicating LPS is a far more potent TLR4 activator, regardless of the mechanism. Similarly, LPS elicited a significantly greater increase in NF-κB activation compared to SFA [36]. Additionally, non-TLR4 mechanisms may explain the ability of SFA to activate inflammatory pathways [37].
Cytokines and sphingolipids
However, the absence or presence of TLR4 signaling does not explain every alteration in sphingolipid metabolism in response to the myriad of proinflammatory or anti-inflammatory stimuli. While TNFα and other proinflammatory cytokines (e.g., IL-1β) can activate de novo ceramide synthesis [38], likely via increased IKKβ-NF-κB action, they can also exploit a ‘backdoor’. Several proinflammatory cytokines result in ceramide accrual by activating sphingomyelinase, which mediates the conversion of sphingomyelin to ceramide. Specifically, TNFα and IL-1β signaling activates acid and neutral sphingomyelinase activity, resulting in significant ceramide pooling [39–41]. Considering that sphingomyelin is the dominant membrane lipid, its role as a source of ceramide is potentially enormous.
In contrast, adiponectin has long been known to oppose proinflammatory cytokines. Whereas TNFα activates proinflammatory pathways and induces cell death, adiponectin inhibits proinflammatory pathways and promotes cell survival [42, 43]. Importantly, evidence suggests that the contrasting effects of the quintessential pro- and anti-inflammatory cytokines TNFα and adiponectin, respectively, can be explained by how they modulate ceramide metabolism (Fig. 3). Holland et al. [44] recently demonstrated that adiponectin receptors contain inherent ceramidase activity [45], resulting in the degradation of ceramide to form sphingosine [46]. Sphingosine, in turn, is phosphorylated by sphingosine kinase, forming sphingosine 1-phosphate (S1P). Of the several metabolic fates available to ceramide, its deacylation and phosphorylation (via ceramidase and sphingosine kinase, respectively) to form S1P is intriguing, inasmuch as ceramide and S1P exert completely different and even antagonistic actions. In stark contrast to ceramide, S1P has been repeatedly shown to activate Akt and promote cell survival and growth, even directly antagonizing ceramide [46–50]. Indeed, S1P has been shown to reduce ceramide synthesis by inhibiting de novo enzymatic activity [51], which gives rise to the theory that ceramide and S1P constitute a rheostat system [52]. Interestingly, S1P may be a critical mediator of adiponectin’s anti-inflammatory profile. S1P has been shown to inhibit actions of pro-inflammatory cytokines [53] and regulate inflammation-related gene expression [54].

TNFα induces ceramide biosynthesis and accrual through multiple mechanisms, whereas adiponectin activates ceramide degradation and eventual formation of S1P. Ceramide (Cer); sphingosine 1-phosphoate (S1P)
Sphingolipids in the pathophysiology of obesity-induced inflammation
The inflammatory state associated with obesity is implicated in several clinically important complications. Similarly, a growing body of literature suggests that ceramide and other sphingolipids are also present in these disease states and may play a prominent role in the etiology. In light of findings suggesting that sphingolipid metabolism is affected by inflammatory profile [23, 44], it seems likely that sphingolipids are downstream effectors of obesity-induced inflammation and are critical mediators of inflammation-associated diseases. This section will highlight the role of sphingolipids in the etiology of several diseases associated with obesity-induced inflammation (Fig. 4).

Ceramide has been shown to cause virtually all of the pathological states elicited by obesity-induced inflammation
Cardiovascular disease
A role for inflammation in the etiology of cardiovascular disease, a term used to collectively describe diseases that involve the heart or blood vessels, has become so well established over the past two decades that a number of inflammatory markers are now measured as potential predictors of prevalent or incident cardiovascular disease [55]. Additionally, anti-inflammatory medications are now used to reduce the risk of cardiovascular disease [55, 56]. Similarly, sphingolipids have been shown to play a role in the regulation of vascular growth and tone, thus impacting cardiovascular function. We will explore the impact of sphingolipids in three prominent cardiovascular disorders commonly associated with obesity-induced inflammation, namely hypertension, atherosclerosis, and cardiomyopathy.
Hypertension
Hypertension, identified as clinically elevated blood pressure, is a major risk factor for cardiac and cerebrovascular disease. Noting the vascular effects of sphingolipids, Spijkers et al. [57] sought to determine the role of sphingolipids in essential hypertension. They found that shifting the ceramide/S1P ratio towards ceramide dominance by administering a sphingosine kinase inhibitor or by exogenous sphingomyelinase induced pronounced endothelium-dependent contractions in isolated carotid arteries. Additionally, in vivo administration of a sphingosine kinase inhibitor resulted in a marked rise in blood pressure in spontaneous hypertensive rats, and, further implicating ceramide, hypertensive rats have significantly increased levels of total ceramides in arterial tissues. Moreover, both hypertensive rats and humans have elevated plasma ceramide levels [57]. Similarly, in an effort to decipher the genetics of hypertension, Fenger et al. [58] conducted a genomic analysis focusing on components of ceramide metabolism and concluded that the ceramide/S1P rheostat has a substantial influence on blood pressure regulation. Interestingly, they found that genes involved in de novo ceramide synthesis, rather than ceramide formation via sphingomyelinase, were the most important sources of ceramide in a hypertensive population.
Atherosclerosis
Several early reports have cited a consistent positive correlation between circulating sphingolipids and occurrence of cardiovascular complications [59–62]. In fact, the correlation is so consistently observed that sphingomyelin levels are considered an independent risk factor for coronary artery disease [63]. In support of these observations, recent findings have established that sphingolipids indeed play a causative role in the etiology of atherosclerosis. Park et al. [64] found that treating apoE KO mice, which develop advanced atherosclerosis and have elevated circulating sphingolipids [59], with an inhibitor of de novo ceramide synthesis, myriocin, not only reduced plasma ceramide and sphingomyelin, but also total plasma cholesterol and triglycerides. Further, inhibition of ceramide synthesis in the KO mice resulted in a substantial reduction in atherosclerotic lesion area in the aortic root and slowed the progression of atherosclerosis in the brachiocephalic artery. Macrophage content in the aortic root was similarly reduced in myriocin-treated mice [59]. Interestingly, inhibition of de novo ceramide synthesis also results in increased hepatic apoA-I synthesis and elevated circulating levels of favorable HDL cholesterol [65]. Similar to treatments using inhibitors of ceramide synthesis, the use of a myriocin-based drug FTY720, an S1P homologue, also inhibits atherosclerosis [66].
Underlining the connection between inflammation and sphingolipids, LDL receptor KO mice that lack the ability to generate sphingomyelin in macrophages, the archetypal immune cell, have decreased atherosclerotic lesions in the entire aorta. Moreover, plaque morphology analysis from brachiocephalic arteries of LDL receptor KO mice reveal reduced necrotic core area [67].
Cardiomyopathy
Like most cardiovascular complications, cardiomyopathy, or weakening of the heart, has strong ties to inflammation [68, 69]. However, inflammation per se may be a sufficient, but unnecessary factor in cardiomyopathy etiology. To tease out the role of inflammation versus the inflammation-related factor NF-κB, Kawamura et al. [70] blocked the activation of cardiac NF-κB by crossing transgenic mice harboring cardiac-specific TNFα overexpression with mutant mice carrying a disrupted NF-κB subunit. Interestingly, they found that while NF-κB blockade did not ameliorate myocardial inflammation as determined by inflammatory cell infiltration, it significantly improved cardiac function and survival, hinting that the classic inflammation-immune response is less important than the activation of NF-κB in cardiomyopathy. Combined with the separate observation that TLR4 deficiency protects against cardiomyopathy [63], and the fact that TLR4 and NF-κB are regulators of de novo sphingolipid synthesis [23], these findings may suggest that a product(s) of inflammation, rather than inflammation per se, is responsible for the inflammation-induced cardiomyopathy.
An oft-used rodent model of cardiomyopathy is the cardiomyocyte overexpression of a glycosylphatidylinositol membrane-anchored form of lipoprotein lipase (LpLGPI). Park et al. [63] explored the role of ceramide in this model by (a) crossing the LpLGPI mouse with a heterozygous deletion of LCB1, a SPT subunit, and (b) treating LpLGPI mice with myriocin. In addition to finding that the hearts from LpLGPI mice contained 45% more ceramide than control levels, they found that both genetic (LpLGPI-LCB1 cross) and pharmacological (myriocin) inhibition of ceramide synthesis improves cardiac function and reduces expression of heart failure markers [63].
Non-alcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) is defined as fat accumulation in excess of 5 to 10% of liver weight [71]. While seemingly benign at onset, NAFLD, or hepatic steatosis, can develop into considerably less benign conditions, such as steatohepatitis, cirrhosis, and even hepatocellular carcinoma [72, 73]. In fact, NALFD is the most common cause of liver dysfunction in the United States [72].
A large part of NALFD’s prevalence is its association with obesity. The hyperlipidemia and chronic inflammation associated with obesity impact NAFLD risk. Although lipid accumulation is the hallmark of early NAFLD, latter stages are marked by elevated inflammation [74]. Patients with NALFD have elevated TNFα levels compared to obese and non-obese controls [75], and TNFα receptor level and TNFα gene expression are increased in the livers of those with NAFLD when compared to healthy livers [76]. In contrast, adiponectin levels are reduced in patients with NAFLD [75]. Moreover, adiponectin transcript expression in higher-fat livers is negatively associated with hepatic ceramides and sphingomyelinase transcript levels [22]. By investigating adiponectin and its receptors, Peng et al. [77] found that circulating adiponectin was decreased in diet-induced obese (DIO) mice compared to lean controls, and that DIO mice have dramatically reduced hepatic expression of adipoR2, the predominant adiponectin receptor in the liver. Similar examinations in humans have revealed comparable results—a reduction in hepatic adiponectin and adipoR2 expression with advancing steatosis [78, 79].
Considering the contrasting presence of TNFα and adiponectin in NAFLD, and given that TLR4 has been implicated in the etiology of NAFLD complications [80], it is not surprising that modulation of sphingolipid metabolism impacts NAFLD. Memon et al. [81] observed that LPS treatments, a TLR4 activator, resulted in a twofold increase in hepatic SPT gene expression and enzyme activity, which was supported by a 75 and 200% increase in hepatic sphingomyelin and ceramide, respectively. Further, they found that IL-1β injection increased hepatic SPT transcription and activity in vivo and both IL-1β and TNFα induced SPT transcript levels in cultured hepatocytes [81]. Additionally, SPT inhibition with myriocin results in a significant reduction in hepatic triglycerides in DIO mice [82].
In addition to implicating the de novo ceramide synthesis pathway, several groups have explored the role of the salvage pathway in NAFLD, where ceramide is synthesized from sphingomyelin (SM) via sphingomyelinase (SMase). Deevska et al. [83] demonstrated that mice lacking acid SMase and functional LDL receptors are protected from diet-induced hepatic steatosis when compared to littermates with functional acid SMase. Moreover, inhibition of SMase in palmitic acid-treated hepatocytes exhibited significantly reduced triglyceride levels compared to untreated controls, suggesting a role for SMase in mediating steatosis in response to elevated fatty acids in vivo [83]. SMase is mediated by numerous stimuli, including TNFα [84–90], which induces SMase by binding the p55 TNFα receptor (also known as TNF type 1 receptor)[91]. This is noteworthy, given that p55 TNFR KO mice are resistant to diet-induced steatosis and liver injury [92].
Insulin resistance
The development of the concept that obesity-induced inflammation mediates disease onset started with a pivotal publication by Hotamisligil et al. [9], demonstrating that adipose-released cytokines inhibit insulin signaling. Insulin resistance mediates a surplus of diseases, including cardiovascular disease [93], type II diabetes, and even some cancers [94, 95]. Hence, understanding the etiology of insulin resistance with regards to inflammatory pathways is intensely pursued.
IKKβ
Following their novel observation that adipose-secreted TNFα inhibits insulin signaling [9], Hotamisligil et al. [96] found that the molecular mechanisms linking TNFα to insulin resistance involved serine phosphorylation of insulin receptor substrate (IRS)-1, inhibiting normal tyrosine phosphorylation and reducing IRS-1 action. This was later revealed to be through the actions of the serine kinase inhibitor of κB kinase-β (IKK-β) that phosphorylates IRS-1 and impairs the ability of IRS-1 to associate with the insulin receptor, which subsequently inhibits insulin-stimulated tyrosine phosphorylation and activation of IRS-1-associated PI3-kinase [11, 97].
The realization that IKKβ mediates insulin resistance elicited a rediscovery of insulin-sensitizing therapies. Over a century ago, Williamson et al. [98] showed that salicylate treatment reduced symptoms associated with diabetes. This was later further confirmed when Reid et al. [99] demonstrated that a regimen of aspirin improved glucose tolerance in diabetic patients. More recently, salicylate was found to inhibit IKKβ activity [100] and the Shoelson lab was instrumental in establishing that salicylates, via IKKβ disruption, were effective in preventing inflammation-induced insulin resistance [101].
Research has continued to support a role for IKKβ in mediating inflammation-induced insulin resistance, though the originally proposed mechanism, namely serine phosphorylation of IRS-1, is debated [102]. A novel perspective to IKKβ-induced insulin resistance is the observation that IKKβ regulates ceramide synthesis. In murine myotubes overexpressing a kinase dead IKKβ, ceramide levels are dramatically reduced compared to wild-type cells, and the lack of functional IKKβ prevents ceramide biosynthesis in response to common insults, like saturated fatty acids [23]. This was found to be a result of IKKβ-NF-κB-mediated transcription of enzymes involved in de novo ceramide biosynthesis, including SPT2, various ceramide synthase isoforms, and dihydroceramide desaturase 1. Additionally, further evidence for ceramide as a mediator of IKKβ-induced insulin resistance comes from treatment of DIO mice with sodium salicylate, an IKKβ inhibitor. Sodium salicylate-treated mice fail to accrue ceramide in skeletal muscle and liver and remain insulin sensitivity despite DIO [23].
Toll-like receptor 4
Obesity is the most important known risk factor contributing to insulin resistance [103]. As mentioned earlier, efforts to identify the molecular mechanisms to explain obesity-induced insulin resistance have revealed lipotoxicity and inflammation as two prominent explanations linking obesity to insulin resistance. A milestone was reached when the Flier laboratory [36] revealed that TLR4, a pattern-recognition receptor that plays a critical role in innate immunity by activating the canonical NF-κB inflammatory pathway, is a carrefour of lipid- and inflammation-induced insulin resistance. They found that saturated fatty acids, which are elevated in obesity, activate TLR4 and evoke a TLR4-dependent inflammatory response. Additionally, mice lacking TLR4 were protected from acute lipid- and chronic HFD-induced insulin resistance, despite weight gain [36]. Similar to earlier reports, IRS-1 serine phosphorylation was increased in insulin-resistant WT mice, but not in insulin-sensitive TLR4-deficient mice. However, as with IKKβ, accumulation of ceramide might provide an alternate explanation to TLR4-incuced insulin resistance.
An association between TLR4 and ceramides has long been known, showing that TLR4 agonists activate ceramide-generating pathways, including de novo synthesis and sphingomyelin salvage [104–106]. Similar to Flier’s observations [36], we found that mice lacking functional TLR4 were protected from a variety of insulin-desensitizing interventions, such as acute lipid infusions, diet-induced obesity, and LPS infusions. Additionally, we found that TLR4 mutant mice, in contrast to controls, failed to accrue ceramides in response to HFD in insulin-responsive tissues, like the muscle, liver, and hypothalamus. Therefore, we posit that TLR4 stimulates ceramide synthesis, which represents the common molecular mediator behind lipid- and inflammation-induced insulin resistance.
Adiponectin
In contrast to many of its fellow adipose-secreted hormones, adiponectin is known for its anti-inflammatory and insulin-sensitizing functions. Similarly, rather than increasing with obesity, like many adipokines, adiponectin secretion is reduced with fat mass expansion [107] and increased with weight loss [21]. It is therefore little surprise that adiponectin reverses insulin resistance in obesity. Yamauchi et al. [108] found that obese insulin-resistant mice experienced significant improvements in insulin sensitivity and glucose tolerance following adiponectin treatment.
For some time, the positive effects of adiponectin treatment were thought to be mediated almost entirely by its activation of AMP-activated protein kinase (AMPK)[109], though this is not necessarily the case [110]. Adiponectin research took a significant step forward when it was found that adiponectin receptors contain inherent ceramidase activity, degrading ceramide and forming sphingosine and S1P [45, 111]. Indeed, it appears that many of adiponectin’s beneficial effects are mediated by accumulation of S1P, including the activation of AMPK [44]. Unsurprisingly, where its proinflammatory counterparts inhibit insulin sensitivity, the anti-inflammatory adiponectin improves insulin sensitivity. In treating two models of obesity-induced insulin resistance with adiponectin, Holland et al. [44] found that the usual accumulation of tissue ceramides evident in Lep ob/ob and HFD-fed mice was conspicuously absent, and the reduction in ceramides was associated with a significant improvement in whole-body insulin sensitivity in both models. Moreover, tissue ceramide levels in disparate adiponectin models (transgenic overexpression or adiponectin-null mice) reveal that adiponectin function is inversely correlated with ceramide levels, and reduced ceramides convey protection against insulin resistance [44].
Given the widely acknowledged benefits of AMPK activation, pharmacological activators of AMPK are actively studied. In particular, metformin, the most widely prescribed anti-diabetic drug, activates AMPK, which has been shown to potently inhibit inflammatory mediators [30, 112–114]. Moreover, AMPK inhibits de novo ceramide synthesis in astrocytes [115] and myotubes (Bikman BT, unpublished observation) via SPT inhibition. However, as is the case with adiponectin, it is yet unknown whether the beneficial effects of metformin-induced AMPK activation require the subsequent actions of S1P [44].
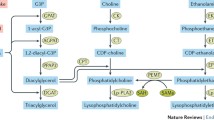
Non-ceramide sphingolipids
While most research centered on sphingolipids and insulin resistance has focused on ceramides as the dominant sphingolipid responsible for insulin resistance, atherosclerosis, etc., caution is required. Because the majority of interventions used to explore ceramides as a mediator of insulin resistance have used inhibitors of de novo sphingolipid synthesis, it is likely that all sphingolipids downstream of ceramide are also reduced and, hence, may be important mediators of effects assigned exclusively to ceramide. Indeed, evidence exists to support a role for both glucosylceramides [116] and sphingomyelin [117] as necessary for a host of health complications, including insulin resistance and NAFLD. However, while other sphingolipids are clearly capable of inducing deleterious consequences, and indeed may be the prime sphingolipids mediating these effects, there is somewhat conflicted evidence concerning these lipids and their responsiveness to inflammatory signals (Fig. 5). The sphingomyelin-degrading actions of TNFα and other inflammatory cytokines via sphingomyelinase are well established [39–41] and covered above (See “Cytokines and sphingolipids”). The data surrounding glucosylceramides is less clear. While we and others have shown that TLR4 activation in macrophages elicits an increase in glucosylceramides [23, 105], unlike ceramides, this effect is not observed in other cell types or whole tissues (e.g., myocytes or whole muscle)[23]. However, given that macrophages reside in tissues that play a large role in metabolic function (e.g., muscle and liver), this may prove to be an important factor. Nevertheless, until the effects of macrophage-derived sphingolipids are more clearly understood, it is unknown whether inflammation-induced macrophage-derived glucosylceramides mediate deleterious metabolic outcomes.

Several sphingolipids elicit metabolically deleterious results, similar to ceramide. However, only ceramide formation (red pathways) has been shown to mediate inflammation-induced disturbances. Inhibition of the indicated enzymes invites the indicated results. Serine palmitoyltransferase (SPT); dihydroceramide desaturase 1 (Des1); sphingomyelinase synthase (SMS); sphingomyelinase (SMase); glucosylceramide synthase (GCS); sphingosine kinase (SK)
Ceramides and inflammation
Although the majority of research, and the paradigm of this review, indicates that inflammatory signals activate ceramide biosynthesis, a conflicting perspective exists. After observing that the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (Nlrp3) inflammasome, a Nod-like receptor, correlated with degree of obesity-induced insulin resistance, Vandanmagsar et al. [118] found that ablation of the Nlrp3 inflammasome prevents HFD-induced insulin resistance. Interestingly, they report that ceramide activates the Nlrp3 inflammasome, though it is noteworthy that ceramide is not used alone in treatments, but only in combination with LPS [118]. This combination of lipid and LPS requires the reported results to be viewed with an added measure of caution given the synergistic amplification of the inflammatory response in cells exposed to SFA and LPS together compared to either insult alone [119], which may be a result of a lipid-induced TLR4 dimerization [120].
Conclusions
Since its initial discovery, obesity-induced inflammation has lead to an eruption in research implicating inflammatory pathways in the etiology of several prominent obesity-related diseases. The purpose of this review has been to highlight the role of inflammatory pathways in sphingolipid biosynthesis and, to a degree, establish a role for sphingolipids as critical mediators of many of the deleterious effects of obesity-induced inflammation. While other mechanisms may exist, it is clear that sphingolipids play important roles in the etiology and lethality of obesity-related disease states commonly linked with inflammation, namely cardiovascular complications, NAFLD, and insulin resistance. Future efforts will not only further elucidate the role of sphingolipids in the pathophysiology of obesity-induced inflammation, but also likely find even greater obesity-related disease states mediated by sphingolipids.
References
Finkelstein EA, Trogdon JG, Cohen JW, Dietz W (2009) Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff (Millwood) 28:w822–w831
Fontaine KR, Redden DT, Wang C, Westfall AO, Allison DB (2003) Years of life lost due to obesity. JAMA 289:187–193
Flegal KM, Graubard BI, Williamson DF, Gail MH (2007) Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA 298:2028–2037
Thevissen K, Francois IE, Winderickx J, Pannecouque C, Cammue BP (2006) Ceramide involvement in apoptosis and apoptotic diseases. Mini Rev Med Chem 6:699–709
Johns DG, Charpie JR, Webb RC (1998) Is ceramide signaling a target for vascular therapeutic intervention? Curr Pharm Des 4:481–488
Unger RH (2002) Lipotoxic diseases. Annu Rev Med 53:319–336
Amati F, Dube JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, Switzer GE, Bickel PE, Stefanovic-Racic M, Toledo FG et al (2011) Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60:2588–2597
Das A, Mukhopadhyay S (2011) The evil axis of obesity, inflammation and type-2 diabetes. Endocr Metab Immune Disord Drug Targets 11:23–31
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259:87–91
Ferrante AW Jr (2007) Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J Intern Med 262:408–414
Bikman BT, Zheng D, Pories WJ, Chapman W, Pender JR, Bowden RC, Reed MA, Cortright RN, Tapscott EB, Houmard JA et al (2008) Mechanism for improved insulin sensitivity after gastric bypass surgery. J Clin Endocrinol Metab 93:4656–4663
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95:2409–2415
Fried SK, Bunkin DA, Greenberg AS (1998) Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 83:847–850
Perreault M, Marette A (2001) Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 7:1138–1143
Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB (1999) Elevated C-reactive protein levels in overweight and obese adults. JAMA J Am Med Assoc 282:2131–2135
Rull A, Camps J, Alonso-Villaverde C, Joven J (2010) Insulin resistance, inflammation, and obesity: role of monocyte chemoattractant protein-1 (or CCL2) in the regulation of metabolism. Mediat Inflamm. doi:10.1155/2010/326580
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Investig 112:1821–1830
Fain JN, Cheema PS, Bahouth SW, Lloyd Hiler M (2003) Resistin release by human adipose tissue explants in primary culture. Biochem Biophys Res Commun 300:674–678
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Investig 112:1796–1808
Jung SH, Park HS, Kim KS, Choi WH, Ahn CW, Kim BT, Kim SM, Lee SY, Ahn SM, Kim YK et al (2008) Effect of weight loss on some serum cytokines in human obesity: increase in IL-10 after weight loss. J Nutr Biochem 19:371–375
Yang WS, Lee WJ, Funahashi T, Tanaka S, Matsuzawa Y, Chao CL, Chen CL, Tai TY, Chuang LM (2001) Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J Clin Endocrinol Metab 86:3815–3819
Kolak M, Westerbacka J, Velagapudi VR, Wagsater D, Yetukuri L, Makkonen J, Rissanen A, Hakkinen AM, Lindell M, Bergholm R et al (2007) Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes 56:1960–1968
Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR et al (2011) Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121:1858–1870
Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A et al (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5:167–179
Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK, Sriwijitkamol A et al (2008) Elevated Toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57:2595–2602
Adams JM 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ (2004) Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53:25–31
Huang H, Kasumov T, Gatmaitan P, Heneghan HM, Kashyap SR, Schauer PR, Brethauer SA, Kirwan JP (2011) Gastric bypass surgery reduces plasma ceramide subspecies and improves insulin sensitivity in severely obese patients. Obesity 19:2235–2240
Promrat K, Longato L, Wands JR, de la Monte SM (2011) Weight loss amelioration of non-alcoholic steatohepatitis linked to shifts in hepatic ceramide expression and serum ceramide levels. Hepatol Res Off J Jpn Soc Hepatol 41:754–762
Dube JJ, Amati F, Toledo FG, Stefanovic-Racic M, Rossi A, Coen P, Goodpaster BH (2011) Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 54:1147–1156
Bikman BT, Zheng D, Kane DA, Anderson EJ, Woodlief TL, Price JW, Dohm GL, Neufer PD, Cortright RN (2010) Metformin improves insulin signaling in obese rats via reduced IKKbeta action in a fiber-type specific manner. J Obes. doi:10.1155/2010/970865
Shoelson SE, Lee J, Yuan M (2003) Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord J Int Assoc Study Obes 27(Suppl 3):S49–S52
Lee JY, Sohn KH, Rhee SH, Hwang D (2001) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 276:16683–16689
Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, Hwang DH (2003) Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res 44:479–486
Bikman BT, Summers SA (2011) Ceramides as modulators of cellular and whole-body metabolism. J Clin Investig 121:4222–4230
Erridge C, Samani NJ (2009) Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol 29:1944–1949
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015–3025
Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M (2011) Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147:173–184
Meyer SG, de Groot H (2003) Cycloserine and threo-dihydrosphingosine inhibit TNF-alpha-induced cytotoxicity: evidence for the importance of de novo ceramide synthesis in TNF-alpha signaling. Biochim Biophys Acta 1643:1–4
Chatterjee S (1993) Neutral sphingomyelinase. Adv Lipid Res 26:25–48
Zeidan YH, Hannun YA (2009) The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr Mol Med 10:454–466
Hofmeister R, Wiegmann K, Korherr C, Bernardo K, Kronke M, Falk W (1997) Activation of acid sphingomyelinase by interleukin-1 (IL-1) requires the IL-1 receptor accessory protein. The Journal of biological chemistry 272:27730–27736
Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M et al (2000) Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 102:1296–1301
Kobayashi H, Ouchi N, Kihara S, Walsh K, Kumada M, Abe Y, Funahashi T, Matsuzawa Y (2004) Selective suppression of endothelial cell apoptosis by the high molecular weight form of adiponectin. Circ Res 94:e27–e31
Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM et al (2010) Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17:55–63
Villa NY, Kupchak BR, Garitaonandia I, Smith JL, Alonso E, Alford C, Cowart LA, Hannun YA, Lyons TJ (2009) Sphingolipids function as downstream effectors of a fungal PAQR. Mol Pharmacol 75:866–875
Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA (2005) Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem 280:20148–20153
Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Natl Rev Mol Cell Biol 4:397–407
Morales-Ruiz M, Lee MJ, Zollner S, Gratton JP, Scotland R, Shiojima I, Walsh K, Hla T, Sessa WC (2001) Sphingosine 1-phosphate activates Akt, nitric oxide production, and chemotaxis through a Gi protein/phosphoinositide 3-kinase pathway in endothelial cells. J Biol Chem 276:19672–19677
Zheng DM, Kitamura T, Ikejima K, Enomoto N, Yamashina S, Suzuki S, Takei Y, Sato N (2006) Sphingosine 1-phosphate protects rat liver sinusoidal endothelial cells from ethanol-induced apoptosis: role of intracellular calcium and nitric oxide. Hepatology 44:1278–1287
Igarashi J, Michel T (2001) Sphingosine 1-phosphate and isoform-specific activation of phosphoinositide 3-kinase beta. Evidence for divergence and convergence of receptor-regulated endothelial nitric-oxide synthase signaling pathways. J Biol Chem 276:36281–36288
Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH Jr, Futerman AH (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem 283:5677–5684
Lavieu G, Scarlatti F, Sala G, Levade T, Ghidoni R, Botti J, Codogno P (2007) Is autophagy the key mechanism by which the sphingolipid rheostat controls the cell fate decision? Autophagy 3:45–47
Bolick DT, Srinivasan S, Kim KW, Hatley ME, Clemens JJ, Whetzel A, Ferger N, Macdonald TL, Davis MD, Tsao PS et al (2005) Sphingosine-1-phosphate prevents tumor necrosis factor-{alpha}-mediated monocyte adhesion to aortic endothelium in mice. Arterioscler Thromb Vasc Biol 25:976–981
Lin CI, Chen CN, Lin PW, Lee H (2007) Sphingosine 1-phosphate regulates inflammation-related genes in human endothelial cells through S1P1 and S1P3. Biochem Biophys Res Commun 355:895–901
Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH (1997) Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 336:973–979
Ridker PM, Cook NR, Lee IM, Gordon D, Gaziano JM, Manson JE, Hennekens CH, Buring JE (2005) A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 352:1293–1304
Spijkers LJ, van den Akker RF, Janssen BJ, Debets JJ, De Mey JG, Stroes ES, van den Born BJ, Wijesinghe DS, Chalfant CE, MacAleese L et al (2011) Hypertension is associated with marked alterations in sphingolipid biology: a potential role for ceramide. PLoS One 6:e21817
Fenger M, Linneberg A, Jorgensen T, Madsbad S, Sobye K, Eugen-Olsen J, Jeppesen J (2011) Genetics of the ceramide/sphingosine-1-phosphate rheostat in blood pressure regulation and hypertension. BMC Genet 12:44
Park TS, Panek RL, Mueller SB, Hanselman JC, Rosebury WS, Robertson AW, Kindt EK, Homan R, Karathanasis SK, Rekhter MD (2004) Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 110:3465–3471
Jiang XC, Paultre F, Pearson TA, Reed RG, Francis CK, Lin M, Berglund L, Tall AR (2000) Plasma sphingomyelin level as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol 20:2614–2618
Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I (1996) Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Investig 98:1455–1464
Williams KJ, Tabas I (2005) Lipoprotein retention and clues for atheroma regression. Arterioscler Thromb Vasc Biol 25:1536–1540
Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED et al (2008) Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 49:2101–2112
Park TS, Rosebury W, Kindt EK, Kowala MC, Panek RL (2008) Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol Res 58:45–51
Glaros EN, Kim WS, Garner B (2010) Myriocin-mediated up-regulation of hepatocyte apoA-I synthesis is associated with ERK inhibition. Clin Sci 118:727–736
Klingenberg R, Nofer JR, Rudling M, Bea F, Blessing E, Preusch M, Grone HJ, Katus HA, Hansson GK, Dengler TJ (2007) Sphingosine-1-phosphate analogue FTY720 causes lymphocyte redistribution and hypercholesterolemia in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 27:2392–2399
Liu J, Huan C, Chakraborty M, Zhang H, Lu D, Kuo MS, Cao G, Jiang XC (2009) Macrophage sphingomyelin synthase 2 deficiency decreases atherosclerosis in mice. Circ Res 105:295–303
Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL (1997) Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation 95:1247–1252
Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL (1992) Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257:387–389
Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, Takeshita A, Sunagawa K (2005) Blockade of NF-kappaB improves cardiac function and survival without affecting inflammation in TNF-alpha-induced cardiomyopathy. Cardiovasc Res 66:520–529
Neuschwander-Tetri BA, Caldwell SH (2003) Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology 37:1202–1219
Kim CH, Younossi ZM (2008) Nonalcoholic fatty liver disease: a manifestation of the metabolic syndrome. Cleve Clin J Med 75:721–728
Farrell GC, Larter CZ (2006) Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43:S99–S112
Baranova A, Gowder SJ, Schlauch K, Elariny H, Collantes R, Afendy A, Ong JP, Goodman Z, Chandhoke V, Younossi ZM (2006) Gene expression of leptin, resistin, and adiponectin in the white adipose tissue of obese patients with non-alcoholic fatty liver disease and insulin resistance. Obes Surg 16:1118–1125
Jarrar MH, Baranova A, Collantes R, Ranard B, Stepanova M, Bennett C, Fang Y, Elariny H, Goodman Z, Chandhoke V et al (2008) Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol Ther 27:412–421
Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ (2004) Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 40:185–194
Peng Y, Rideout D, Rakita S, Sajan M, Farese R, You M, Murr MM (2009) Downregulation of adiponectin/AdipoR2 is associated with steatohepatitis in obese mice. J Gastrointest Surg: Off J Soc Surg Alimentary Tract 13:2043–2049
Kaser S, Moschen A, Cayon A, Kaser A, Crespo J, Pons-Romero F, Ebenbichler CF, Patsch JR, Tilg H (2005) Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 54:117–121
Ma H, Gomez V, Lu L, Yang X, Wu X, Xiao SY (2009) Expression of adiponectin and its receptors in livers of morbidly obese patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol 24:233–237
Desai MS, Mariscalco MM, Tawil A, Vallejo JG, Smith CW (2008) Atherogenic diet-induced hepatitis is partially dependent on murine TLR4. J Leukoc Biol 83:1336–1344
Memon RA, Holleran WM, Moser AH, Seki T, Uchida Y, Fuller J, Shigenaga JK, Grunfeld C, Feingold KR (1998) Endotoxin and cytokines increase hepatic sphingolipid biosynthesis and produce lipoproteins enriched in ceramides and sphingomyelin. Arterioscler Thromb Vasc Biol 18:1257–1265
Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F (2009) Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab 297:E211–E224
Deevska GM, Rozenova KA, Giltiay NV, Chambers MA, White J, Boyanovsky BB, Wei J, Daugherty A, Smart EJ, Reid MB et al (2009) Acid sphingomyelinase deficiency prevents diet-induced hepatic triacylglycerol accumulation and hyperglycemia in mice. J Biol Chem 284:8359–8368
Dressler KA, Mathias S, Kolesnick RN (1992) Tumor necrosis factor-alpha activates the sphingomyelin signal transduction pathway in a cell-free system. Science 255:1715–1718
Andrieu-Abadie N, Levade T (2002) Sphingomyelin hydrolysis during apoptosis. Biochim Biophys Acta 1585:126–134
Schutze S, Wiegmann K, Machleidt T, Kronke M (1995) TNF-induced activation of NF-kappa B. Immunobiology 193:193–203
Wiegmann K, Schutze S, Machleidt T, Witte D, Kronke M (1994) Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell 78:1005–1015
Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Kronke M (1992) TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 71:765–776
Mathias S, Dressler KA, Kolesnick RN (1991) Characterization of a ceramide-activated protein kinase: stimulation by tumor necrosis factor alpha. Proc Natl Acad Sci USA 88:10009–10013
Chatterjee S (1994) Neutral sphingomyelinase action stimulates signal transduction of tumor necrosis factor-alpha in the synthesis of cholesteryl esters in human fibroblasts. J Biol Chem 269:879–882
Vandenabeele P, Declercq W, Beyaert R, Fiers W (1995) Two tumour necrosis factor receptors: structure and function. Trends Cell Biol 5:392–399
Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y et al (2006) Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 55:415–424
DeFronzo RA (1997) Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidaemia and atherosclerosis. Neth J Med 50:191–197
Hsing AW, Gao YT, Chua S Jr, Deng J, Stanczyk FZ (2003) Insulin resistance and prostate cancer risk. J Natl Cancer Inst 95:67–71
Bruning PF, Bonfrer JM, van Noord PA, Hart AA, de Jong-Bakker M, Nooijen WJ (1992) Insulin resistance and breast-cancer risk. Int J Cancer 52:511–516
Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271:665–668
Hotamisligil GS (1999) Mechanisms of TNF-alpha-induced insulin resistance. Exp Clin Endocrinol Diabetes Off J Ger Soc Endocrinol Ger Diabetes Assoc 107:119–125
Williamson RT (1901) On the treatment of glycosuria and diabetes mellitus with sodium salicylate. Br Med J 1:760–762
Reid J, Macdougall AI, Andrews MM (1957) Aspirin and diabetes mellitus. Br Med J 2:1071–1074
Yin MJ, Yamamoto Y, Gaynor RB (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396:77–80
Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293:1673–1677
Hoehn KL, Hohnen-Behrens C, Cederberg A, Wu LE, Turner N, Yuasa T, Ebina Y, James DE (2008) IRS1-independent defects define major nodes of insulin resistance. Cell Metabolism 7:421–433
Wellen KE, Hotamisligil GS (2003) Obesity-induced inflammatory changes in adipose tissue. J Clin Investig 112:1785–1788
Barber SA, Perera PY, Vogel SN (1995) Defective ceramide response in C3H/HeJ (Lpsd) macrophages. J Immunol 155:2303–2305
Sims K, Haynes CA, Kelly S, Allegood JC, Wang E, Momin A, Leipelt M, Reichart D, Glass CK, Sullards MC et al (2010) Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J Biol Chem 285:38568–38579
Rovina P, Graf C, Bornancin F (2010) Modulation of ceramide metabolism in mouse primary macrophages. Biochem Biophys Res Commun 399:150–154
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K et al (1999) Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79–83
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N et al (2001) The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7:941–946
Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Investig 116:1784–1792
Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS, Kaestner KH, Foretz M, Viollet B, Birnbaum MJ (2011) Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1–AMPK signaling. J Clin Investig 121:2518–2528
Holland WL, Scherer PE (2009) PAQRs: a counteracting force to ceramides? Mol Pharmacol 75:740–743
Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, Schonbeck U, Libby P (2006) Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol 26:611–617
Cacicedo JM, Yagihashi N, Keaney JF Jr, Ruderman NB, Ido Y (2004) AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun 324:1204–1209
Bikman BT, Zheng D, Reed MA, Hickner RC, Houmard JA, Dohm GL (2010) Lipid-induced insulin resistance is prevented in lean and obese myotubes by AICAR treatment. Am J Physiol Regul Integr Comp Physiol 298:R1692–R1699
Blazquez C, Geelen MJ, Velasco G, Guzman M (2001) The AMP-activated protein kinase prevents ceramide synthesis de novo and apoptosis in astrocytes. FEBS Lett 489:149–153
van Eijk M, Aten J, Bijl N, Ottenhoff R, van Roomen CP, Dubbelhuis PF, Seeman I, Ghauharali-van der Vlugt K, Overkleeft HS, Arbeeny C et al (2009) Reducing glycosphingolipid content in adipose tissue of obese mice restores insulin sensitivity, adipogenesis and reduces inflammation. PLoS One 4:e4723
Li Z, Zhang H, Liu J, Liang CP, Li Y, Teitelman G, Beyer T, Bui HH, Peake DA, Zhang Y et al (2011) Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol Cell Biol 31:4205–4218
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17:179–188
Schwartz EA, Zhang WY, Karnik SK, Borwege S, Anand VR, Laine PS, Su Y, Reaven PD (2010) Nutrient modification of the innate immune response: a novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler Thromb Vasc Biol 30:802–808
Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH (2009) Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem 284:27384–27392
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bikman, B.T. A role for sphingolipids in the pathophysiology of obesity-induced inflammation. Cell. Mol. Life Sci. 69, 2135–2146 (2012). https://doi.org/10.1007/s00018-012-0917-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-0917-5