
Abstract
Chromosome 22q11 deletion is the most common chromosomal deletion syndrome and is found in the majority of patients with DiGeorge syndrome and velo-cardio-facial syndrome. Patients with CHARGE syndrome may share similar features. Cardiac malformations, speech delay, and immunodeficiency are the most common manifestations. The immunological phenotype may vary widely between patients. Severe T lymphocyte immunodeficiency is rare—thymic transplantation offers a new approach to treatment, as well as insights into thymic physiology and central tolerance. Combined partial immunodeficiency is more common, leading to recurrent sinopulmonary infection in early childhood. Autoimmunity is an increasingly recognized complication. New insights into pathophysiology are reviewed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Deletion of the q11.2 region of chromosome 22 is found in a heterogeneous group of disorders sharing a common genetic basis. It is the most common chromosomal deletion syndrome with an estimated incidence of 1:4,000 live births [1]. The majority of patients with DiGeorge syndrome and velo-cardio-facial syndrome have monosomic deletions of chromosome 22q11.2 [1]. Other syndromes have occasionally been associated with this deletion including conotruncal anomaly face syndrome, Opitz/GBB, and CHARGE (coloboma, heart disease, choanal atresia, retarded growth and central nervous system development, genital hypoplasia and ear abnormalities and/or deafness) syndrome [2]. Cardiac malformations, speech delay, and immunodeficiency are the most common manifestations [2] but there is a wide phenotypic spectrum including neuropsychiatric disorders and otolaryngological disorders. No clinical features are diagnostic and size of the deletion does not predict disease severity [3]. The clinical phenotype may vary widely between patients, including monozygotic twins with identical mutations, suggesting non-genetic factors contribute to disease phenotype [4].
The pharyngeal arches and pouches are a common embryonic precursor for the thymus, parathyroid, and conotruncal regions of the heart. Defects in these organs in 22q11.2 deletion syndrome are believed to be caused by impaired migration of neural crest cells into pouch ectoderm [5]. Disruption in the pathways of neural crest cell development in mice results in malformations similar to the 22q11.2 deletion phenotype [4]. This review will focus on the immunological features associated with 22q11.2 deletion.
Genetics of 22q11.2 deletion syndrome
The clinical association of thymic aplasia and hypoparathyroidism was first recognized as a syndrome when Dr. Angelo DiGeorge described the postmortem findings of a group of infants with a congenital absence of the thymus and parathyroid glands [6]. Subsequently, conotruncal cardiac abnormalities and facial dysmorphism were added as characteristic features, but it was not until the early 1980s that deletions within chromosome 22 were identified as the cause of DiGeorge syndrome [7, 8]. Subsequently, the same deletion has been discovered in other disorders including velo-cardio-facial syndrome [9], conotruncal anomaly face syndrome [10], and sporadic cases of Opitz/GBB syndrome [11] and CHARGE syndrome [12]. The classic features of DiGeorge syndrome have been described in patients with point mutations in the gene TBX1, found in the 22q11.2 region [13], and with other chromosomal re-arrangements including chromosome 10p deletion [14–16]. The nomenclature is somewhat confusing and evolving. Patients without the classic 22q11 deletion, but phenotypic features of DiGeorge syndrome are often described as having 22q11.2 deletion syndrome, and the immunological phenotype bears no relationship to the size of the deletion, or its absence [17]. Many patients with CHARGE syndrome have a deletion in the gene CHD7 [18], and some have a similar immunological phenotype [19]. This review will focus on immunological defects found in patients with genetic or clinical features found above, using the term 22q11.2 deletion syndrome to encompass all of the phenotypes and diagnoses, particularly for DiGeorge and CHARGE syndrome.
Approximately 90% of individuals with the classical 22q11 deletion have a heterozygous deletion of identical three million base pairs (3 Mb) of 22q11.2, the ‘common deletion’ [20]. This region is prone to rearrangements, likely due to homologous recombination events occurring within two complex highly homologous low-copy repeat areas that are 3 Mb apart [21]. The 3 Mb sequence of genomic DNA contains approximately 30–50 genes [22]. A further 8% of patients have smaller deletions of 1.5 Mb encompassing 24 genes [21]. However, there are no consistent phenotype differences associated with smaller deletions [23]. Numerous candidate genes have been proposed based on their location within the region or expression pattern consistent with a DiGeorge syndrome phenotype. So far it has not been possible to identify which, if any, features of the phenotype can be specifically linked to one gene or combinations of genes affected by this deletion [24].
Murine models identified Tbx1 as a candidate gene for the 22q11.2 deletion syndrome. TBX1 belongs to a family of transcription factors that contain a DNA-binding domain called “T-box”. A homozygous deletion of the murine Tbx1 gene is embryonic lethal. However, phenotypic features of 22q11.2 deletion were detectable in the embryos including abnormal facial features, thymic and parathyroid hypoplasia, and cardiac abnormalities. Heterozygous mutants exhibited a less penetrant phenotype, with varying degrees of absence or reduction in fourth pharyngeal arches [25]. Other implicated genes include the Crkl gene, which encodes an adaptor protein implicated in growth factor and adhesion molecule signaling, which is highly expressed in neural crest derived tissue during development. Homozygous Crkl gene deletion results in gestational death in mice with multiple defects in neural crest derivatives including aortic arch arteries, thymus, and craniofacial structures [26], but heterozygous mutants do not display this phenotype. Compound heterozygosity of murine homologs of CRKL and TBX1, results in a more penetrant phenotype compared to heterozygosity at either locus, suggesting that dose-sensitive interaction of these genes is important to establish the phenotype [27]. It is clear that at least some of the genes in the deleted region are critical for development of pharyngeal ectoderm, endoderm, and mesenchyme, and disruption of these genes leads to disruption of structures of the pharyngeal arch, including the cardiac outflow tract, parathyroid glands, and thymus, as well as craniofacial morphology. Some of these effects may be mediated through downstream signaling [28], and it is not yet clear why there is such a variable morphological and clinical phenotype.
CHD7, mutated in CHARGE syndrome, is a member of the chromodomain helicase DNA-binding domain family of adenosine-5′-triphosphate-dependent chromatin remodeling enzymes. The related CHD1, CHD3, and CHD4 proteins contribute to nucleosome remodeling and histone deacetylation, which regulates dynamic changes in chromatin structure during transcription, recombination, repair, and replication [29, 30], and is important in regulating early embryonic development and cell-cycle control. These proteins lead to transcriptional activation or repression of a gene or region. It is likely that CHD7 has a similar function [31]. CHD7 is expressed throughout the neural crest containing mesenchyme of the pharyngeal arches [31, 32]. Seven of ten CHARGE fetuses were found to have thymic hypoplasia or agenesis [32]. The clinical overlap of CHARGE and 22q11.2 deletion syndrome reported previously [33, 34] suggests a common neural crest defect. In 22q11.2 deletion syndrome, defects in thymus, parathyroid and conotruncal regions of the heart are caused by impaired migration of neural crest cells into pouch exoderm. TBX1 contains a DNA-binding domain, and may be a functional target for CHD7, perhaps explaining some of the overlapping features in CHARGE syndrome and 22q11.2 deletion syndrome.
Thymic development
Segmentation of the posterior pharynx initiates thymic development. There follows a series of incompletely understood developmental events culminating in endodermally derived epithelial cells of the third pharyngeal pouch forming the thymus anlage [35]. Neural crest-derived mesenchymal cells of the pharyngeal arches give rise to the thymic connective tissue as well as smooth muscles of the great cephalic arteries and aorto-pulmonary septum, and the parathyroid gland connective tissue [36]. Thymic mesenchyme promotes thymic epithelium development and signaling between the two cell types controls initial thymic morphogenesis and mesenchymal cells regulate proliferation and differentiation of immature thymic epithelial cells. Once thymic development is able to support immature thymocytes, further thymic epithelial differentiation is largely independent of mesenchymal cells.
Lymphoid stem cells begin to populate the thymus by the eighth week of embryogenesis [37]. T lymphocyte progenitors colonize the thymus before it is vascularized, transmigrating through the mesenchyme to the epithelial cells. Molecular interaction between the developing lymphoid and the epithelial cell is critical for further thymic development [38]. Defects in genes that promote T lymphocyte development lead to defective thymic development. Complete deficiency of thymocytes (e.g., null mutations in AK2, IL2RG, ADA) results in a severely atrophic thymus with lack of corticomedullary demarcation. Thymocyte development arrested at a later developmental stage (e.g., null mutations in RAG) leads to normal cortical development but only rudimentary medullary regions [39].
T lymphocyte development
The thymus plays a crucial role in T lymphocyte development. Immature T lymphocytes develop from the common lymphoid progenitor in the bone marrow and traffic to the thymus. Developing CD4–CD8- (double negative) T lymphocytes (thymocytes) enter the thymus at the cortico-medullary junction. During cortical migration, double-positive thymocytes express both CD4 and CD8. Following successful T lymphocyte receptor gene rearrangement, positive selection occurs in the thymic cortex upon successful engagement of the T lymphocyte receptor in the context of MHC class I or class II molecules to differentiate into single positive CD8+ or CD4+ T lymphocytes, respectively. Only cells that recognize MHC expressed by thymic cortical epithelial cells are selected. Single positive cells are predominantly found in the medulla where they closely interact with thymic epithelial cells. Bi-directional cross-talk between thymocytes positively selected in the cortex is essential to preserve thymic architecture, and hence function. Positively selected thymocytes promote maturation of thymic epithelial cells and formation of thymic medullary regions through molecular signaling [40]. Some mature medullary thymic epithelial cells and thymic dendritic cells, directed by the transcriptional regulator, AIRE, express organ-specific self-antigen. Autoreactive thymocytes that recognize these self-antigens are deleted (negative selection) [41], although some auto-reactive thymocytes may be converted into FOXP3+ regulatory T lymphocytes through interaction with thymic dendritic cells [42]. Surviving thymocytes leave the thymic medulla and enter the peripheral circulation.
The importance of all of these elements of immune development is emphasized by examining human disease severely affecting thymic and T lymphocyte development. Null mutations leading to absence of T lymphocyte development lead to thymic atrophy, reversed when developing thymocytes re-populate the thymus, which then develops normally [43]. Hypomorphic mutations, which lead to restricted T lymphocyte development, give rise to abnormal thymic development, resulting in partial immuno-deficiency in which T lymphocyte and B lymphocyte deficiency are manifest, as well as autoimmune features [38]. Defects in AIRE expression give rise to normal T and B cell immunity, but a predisposition to T lymphocyte-mediated organ-specific autoimmune polyendocrinopathy [44], caused by the escape of normally deleted auto-reactivce thymocytes into the peripheral circulation. Lack of FOXP3+ regulatory T lymphocytes, as seen in IPEX syndrome, gives rise to auto-immunity manifest particularly by cytopenias, severe autoimmune enteritis, and insulin-dependent diabetes mellitus caused by pancreatic islet cell destruction by auto-reactive T lymphocytes [45].
Given the critical nature of molecular interaction between developing lymphoid and thymic epithelial cells for thymic development, and the disordered immune function that can arise from primary partially permissive T lymphocyte development defects leading to secondary disordered thymic architecture [46], it can be conjectured that primary disordered thymic architecture may give rise to secondary disordered immunity. It is likely that the following descriptions of immunodeficiency found in patients with 22q11.2 deletion syndrome and CHARGE syndrome relate to this.
Immunological features of 22q11.2 deletion syndrome
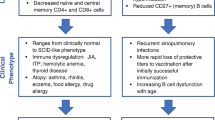
Immunodeficiency is one of the key features of 22q11.2 deletion syndrome, and was one of the initial triad described by Angelo DiGeorge [6]. Defective immunity is also increasingly recognized in CHARGE syndrome [19, 47]. The observed immunodeficiency is secondary to thymic aplasia or hypoplasia with subsequent impaired thymocyte development. The spectrum of severity of the immune deficit varies widely and the frequency and severity of the immunodeficiency does not appear to be related to other phenotypic features [17]. Patients with complete thymic aplasia present with severe or complete T lymphopenia, manifesting with a severe combined immunodeficiency phenotype. However, the degree of immunodeficiency seen in patients with 22q11.2 deletion is highly variable and may include defects of T lymphocyte number and function as well as humoral defects (Table 1). Careful evaluation of patients is necessary to ensure prompt and appropriate treatment (Table 2).
Severe T lymphocyte defects
Presenting features
Complete thymic aplasia with severe or complete T lymphopenia, presenting with a severe combined immunodeficiency phenotype (T-B + NK+) is the most widely appreciated immunodeficiency associated with 22q11.2 deletion. It is however, extremely rare, with less than 1.5% of patients having this phenotype [48]. Patients present within the first few months of life with similar clinical features to infants with other forms of severe combined immunodeficiency, namely, failure to clear infection and presentation with persistent viral respiratory tract or gut infection, and failure to thrive. Occasionally, infants present with disseminated BCG. Other features of 22q11.2 deletion syndrome may be apparent, including facial dysmorphism, hypocalcemia due to hypoparathyroidism, occasionally presenting with seizures or neonatal tetany, and congenital cardiac defects. The diagnosis may be delayed, as normal cytogenetic analysis may fail [49], due to the fact that leucocyte cultures for chromosome analysis rely on phytohemagglutinin to stimulate T lymphocytes into a transient blastic transformation so that metaphase chromosomes can be obtained after 3 days in culture. Similarly, cytogenetic analysis specifying fluorescent in situ hybridization for the 22q11.2 region will fail to give a result. Investigation usually shows severe lymphopenia from birth, demonstrated on a complete blood count and differential leucocyte count. Lymphocyte phenotyping shows severely depleted T-lymphocyte numbers, typically completely absent or present in extremely low numbers (<50 CD3+ cells/mm3); B lymphocytes and NK cells are normal. There is an absent or extremely reduced proliferative response to mitogens such as phytohemagglutinin. Mitogen responses are usually absent. Immunoglobulin estimations show low levels of IgG, IgA, and IgM although residual trans-placental maternal IgG in the first few weeks of life may give a falsely reassuring result. Chest radiographs show an absent thymus and, if infection is present, hyperinflation and/or interstitial pneumonia (Table 1).
Occasional patients show unusual patterns of mature T lymphocyte markers; in such cases, maternal engraftment should be excluded [50]. Congenital GvHD occurs because infants with severe combined immunodeficiency are unable to reject foreign lymphocytes acquired either from the mother in utero [51], or from a non-irradiated blood transfusion, particularly likely if severe congenital heart disease, requiring urgent open heart surgery, is present. When clinically apparent, there is typically a mild reticular skin rash, sometimes with abnormal liver function tests. GvHD following transfusion with non-irradiated blood or white cell or platelet concentrates is generally more severe and can be fatal. In these cases, the skin rash is severe and lymphadenopathy and hepatosplenomegaly may be present.
In atypical forms, characterized by oligoclonal peripheral T lymphocyte expansion, often associated with erythroderma and lymphadenopathy giving an Omenn syndrome-like picture, the diagnosis is based on a lack of naive T lymphocytes (<50 CD3+ CD45RA+ CD62L+ cells/mm3), indicating absence of recent thymic emigrants [52].
Investigation and treatment
General
Children with absent T lymphocytes should be referred urgently to a designated specialist immunology center for further evaluation and treatment. If erythrocyte transfusions (for cardiac surgery) are required before the immunological results are available, they should be from cytomegalovirus seronegative donors, and irradiated to prevent potential transfusion-associated graft versus host disease. Anti-pneumocystis, antiviral, and anti-fungal prophylaxis, and immunoglobulin replacement therapy should be commenced.
Adoptive transfer of mature T lymphocytes
Athymic patients with completely absent T lymphocytes, or with an atypical oligoclonal T lymphocyte expansion, present a therapeutic challenge. Unlike patients with other forms of severe combined immunodeficiency, the absence of T lymphocytes is due to an absence of thymic environment rather than an intrinsic hematopoietic defect. Patients receiving hematopoietic stem cells can achieve peripheral engraftment of post-thymic donor T lymphocytes, but do not demonstrate ongoing T lymphopoiesis [53]. Indeed, T lymphocyte-depleted grafts unsurprisingly fail to demonstrate donor T lymphopoiesis [19]. Adoptive transfer of cells of hematopoietic origin, achieving long-term survival of the patient, has been published in a few cases only [53, 54]. Overall survival is poor (41–48%) as compared to other forms of severe combined immunodeficiency (>80%) (Table 3) [55]. Severe pre-existing medico-surgical conditions are a major cause of death, as is graft versus host disease (GvHD), which often seems more severe than expected given that most patients receive cells from well-HLA-matched donors, and not all receive chemotherapy conditioning [53]. Indeed, it could be argued that there is no rational for giving chemotherapy prior to transplantation, except perhaps in patients with oligoclonal T lymphocytes and an Omenn-like presentation, as there is no requirement for donor myeloid cells or B lymphocytes, and lymphocyte progenitors will not lead to T lymphopoiesis given the lack of thymic machinery.
Most patients who survive long-term remain well without frequent infections. Humoral immunity in survivors shows good antibody responses to protein or conjugate vaccines when tested. Few survivors require ongoing immunoglobulin replacement. Information on T lymphocyte-mediated immunity is incomplete, but where tested, the majority had CD3+ and CD4+ lymphocytopenia and an absence of naive T lymphocytes with a skewed T lymphocyte receptor repertoire. Data from other studies indicates that these patients may remain at risk of late complications, particularly if lymphocyte numbers decline with time [56].
There are a number of possible explanations for the severity of GvHD. Firstly, the underlying condition may predispose to chronically inflamed tissue acting as a substrate for GvHD, for instance lung inflammation secondary to gastro-esophageal reflux and pulmonary aspiration. Secondly, an absence of thymus-derived FOXP3 + regulatory T lymphocytes may predispose to prolonged and severe GvHD. Thirdly, GvHD could be related to homeostatic expansion of donor T lymphocytes as neo-T lymphopoiesis does not take place in completely athymic patients.
The failure of neo-lymphopoiesis may also explain the poor outcome of patients presenting with pre-existing viral infection, with only 2/6 surviving [53], compared with an overall survival of 59% for patients with other forms of severe combined immunodeficiency, even after haploidentical transplantation, in the European series [55].
Thymic transplantation
In athymic individuals with a typical or atypical presentation, an alternative treatment to hematopoietic stem cell transplantation is allogeneic thymic transplantation. Initially, fetal thymic tissue was used [57, 58]. While some success was achieved, with evidence of T lymphocyte reconstitution, hematopoietic stem cell transplantation superseded as the treatment of choice. More recently, trials of postnatal allogeneic thymus transplantation have been pioneered at Duke University Medical Center, North Carolina [59], and Great Ormand Street Hospital, London, UK (G Davies, pers. comm.). Thymic tissue, taken from infants less than 9 months old undergoing open cardiac surgery, is cultured for 12–21 days to ensure removal of donor thymocytes, which might otherwise cause GvHD, before being transplanted into the quadriceps muscle of the recipient [60]. Immunosuppression is not required, unless the recipient has atypical, Omenn-like features, in which case the patient is pre-treated with anti-thymocyte globulin and receives a calcineurin inhibitor until there is evidence of thymopoiesis.
While direct comparison with the results following hematopoietic stem cell transplantation is not possible, as some patients are excluded from the thymic transplantation on the basis of pre-existing medical complications, the procedure seems successful. The results from 60 patients have been reported [34, 60]. Forty-three of 60 subjects are alive (72%). The main cause of death was infection, with four others dying from causes unrelated to the transplant. Immune reconstitution was very good, with evidence of thymopoiesis in the donor thymus (Fig. 1). T lymphocyte numbers are lower than normal for age, but naive T lymphocytes are present, indicating thymopoiesis, the cells proliferate when stimulated with PHA and there is a diverse T lymphocyte repertoire, which persists [34, 60–62]. Tolerance of recipient T lymphocytes to donor thymic tissue, has been demonstrated [63]. Subsequent to transplantation, tolerance to donor lymphocytes is also seen.

Biopsy from the thigh of a patient with thymic aplasia, following thymic transplantation. Skeletal muscle is shown with thymic tissue at the top right. Cytokeratin (CK) staining identifies thymus tissue (inset a), CD1a and Ki-67 identify cortical thymocytes (inset b, c), and CD3 identifies T lymphocytes (inset d). (Courtesy of Dr. Graham Davies, Great Ormand Street Hospital, London—copyright retained)
The major long-term adverse events in this patient group relate to autoimmunity. Thirteen patients had autoimmune thyroid disease, and nine patients experienced 1-, 2-, or 3-lineage hematological cytopenia. Other events included nephritic syndrome and autoimmune enteritis.
The most interesting concept surrounds the issue of HLA matching. Thymus transplantation is performed without matching the HLA antigens of the thymus donor to the recipient, yet outcome is not dependent on the degree of HLA matching between the donor thymus, and the recipient, and, perhaps surprisingly, there seem to be no adverse events associated with transplanting across HLA barriers [64]. Despite the thymus being the site at which developing T lymphocytes undergo both negative and positive selection, a completely HLA-mismatched allogeneic thymus permits development of recipient T lymphocytes that protect the recipient from infection. The process by which this occurs is incompletely understood. Negative selection is effected by CD83+ dendritic cells, derived from hematopoietic stem cells and which migrate from the bone marrow to the thymus, and are located throughout the medulla and at the corticomedullary junction [65]. Recipient dendritic cells are likely to populate a transplanted thymus, and so mediate negative selection, allowing deletion of T lymphocytes with receptors demonstrating high affinity to self MHC. As these dendritic cells also present organ-specific self-antigens regulated by AIRE, auto-reactive T lymphocytes will be deleted [66]. The process of positive selection presents more of a dilemma in the HLA-mismatched thymic transplant. It has been believed that only T lymphocytes that recognize MHC expressed by thymic cortical epithelial cells are positively selected. Thus, when the donor thymus is HLA-mismatched with respect to the recipient, it can be predicted that the positively selected T lymphocytes will be restricted to thymus donor MHC. Subsequently, T lymphocytes emigrating from the thymus should not be able to interact with antigen-presenting cells carrying recipient MHC in the periphery and these T lymphocytes, because of MHC mismatching, will not be functional. The successful reconstitution of recipient T lymphocyte function in the thymus recipients suggests that alternative cells may provide positive selection in the allogeneic donor thymus. Recipient cells may contribute to positive selection in the transplanted thymus. Studies in mice have demonstrated thymocytes that present self MHC to each other [67, 68], which enables positive selection to recipient MHC. At present, however, the mechanisms of positive selection in allogeneic thymus transplantation are not defined and further investigation is required. The opportunity to further unravel the mechanism of central tolerance is anticipated.
Partial combined immunodeficiency
Presenting and immunological features
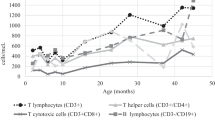
While the majority of pediatricians associate DiGeorge syndrome with severe immunodeficiency, it is clear that the severe phenotype as described above is rare. Much more common is partial combined immunodeficiency, manifest predominantly as recurrent upper respiratory tract infection (URTI) and more rarely, lower respiratory tract infection. This presents after the first 6 months of life with frequent coughs, colds, and sino-pulmonary infections, often due to encapsulated bacteria. Concomitant velo-pharyngeal dysfunction contributes to the increased frequency of URTI, which are common in these patients even in the absence of immunological abnormalities. There is often mild to moderate antibody impairment often with a T lymphocytopenia. This is most marked in infancy [69, 70] and there is a normal age-related decline in T lymphocyte counts, although blunted in patients [71]. There is a reduction in naive cells exiting the thymus [72–74] and in older patients, an accelerated post-thymic conversion of naive to memory T lymphocytes due to homeostatic proliferation [75].
Consequently, abnormalities in the TCRVB repertoire have been described including expanded or contracted TCRVB families in CD4+ and CD8+ sub-populations, and oligoclonal TCRVB populations demonstrated on flow cytometry and by VDJ region sequencing. Restricted TCR diversity has been demonstrated by spectratyping, with altered CDR3 profiles in most TCRVB families investigated, with oligoclonality demonstrated by VDJ region sequencing [75, 76, McClean-Tooke, pers. comm.]. There are conflicting accounts regarding the gdTCR population with reductions and expansions reported [71, 77]. A few patients with T lymphocytopenia appear to be at greater risk of significant viral or candidal infection or early infectious death, particularly those with a combined CD4 and CD8 lymphocytopenia and diminished thymic output, or associated hypoparathyroidism [78, 79].
A wide spectrum of antibody deficiencies has been described. Isolated low IgM may be associated with recurrent infection [80–83], and there may be poor specific IgM responses, with low isohemagglutinins. Low IgG with low IgG subclasses are also described [69, 83]. Poor or absent specific antibody responses to polysaccharide antigen, particularly pneumococcal antigen, are relatively common [82–84], which may be associated with otherwise normal IgG levels. Absent, low, or elevated IgA levels are described [69, 71, 81, 83, 85, 86] and may be associated with recurrent infection or autoimmunity. In many patients, initial low immunoglobulin levels normalize with increasing age [71]. In some patients, B lymphocytopenia is a feature, particularly in infancy, but with numbers normalizing over time [69]. Others have described normal B lymphocyte numbers in patients but low CD27+ IgM+ IgD+ (memory) and CD27+ IgM− IgD− (class-switched memory) B lymphocyte numbers. It is not clear if this is a direct effect of 22q11.2 deletion on B lymphocytes or impaired T lymphocyte/B lymphocyte interaction [73, 77, 81]. There is also evidence that in patients with humoral deficiency, there is a normal IgVH repertoire, but somatic hypermutation is reduced [87]. Interestingly, in a patient with severe T lymphocyte immunodeficiency, there was also a normal IgVH repertoire, but no somatic hypermutation.
Investigation and treatment
In pre-school children, lymphocyte phenotype should be evaluated, immunoglobulin levels measured, and the IgG antibody response to tetanus, Hib, and pneumococcal antigens evaluated. Inadequate responses should be re-evaluated after further vaccination. Patients with recurrent or persistent lower respiratory tract infection should be considered for thoracic high-resolution computerized tomographic imaging of the chest and lung function assessment.
Treatment for this partial form of 22q11 deletion is largely symptomatic. Patients generally improve as they become older. Bacterial sinopulmonary infection should be treated promptly. Prophylactic antibiotics may be given, particularly over the winter months, but may be required throughout the year in occasional patients. For a few with symptomatic hypogammaglobulinemia, or with recurrent bacterial infection despite adequate antibiotic prophylaxis, immunoglobulin replacement may be required [83]. Live vaccines are generally safe to give, even in those with CD4 counts <600cells/μl and are effective at preventing disease [88–90]. However, vaccine-related infection has occasionally been described in patients with low T lymphocyte counts [91], and poor T lymphocyte responses to specific antigens can be found in patients with T lymphocyte counts <10th centile of normal values [92]. It seems prudent, therefore, that live vaccines are withheld in patients with total CD4 counts less than 400 cells/μl [93].
Autoimmunity
Presenting and immunological features
As more patients are identified with 22q11 deletion, it is becoming clear that autoimmunity is found more commonly than in the normal population. Currently, there are no biomarkers to identify or predict which patients will develop autoimmune features [71]. A range of autoimmune features have been described including cytopenias, systemic autoimmunity, particularly rheumatoid arthritis, and organ-specific autoimmunity, most notably autoimmune thyroid disease [71, 83, 84, 86, 94–97]. Other immunological abnormalities, including IgA deficiency may be associated [85].
The mechanism by which autoimmunity occurs is unclear, and is likely to be multifactorial. Infections may lead to autoimmune disease through a variety of mechanisms including the release of sequestered antigens through tissue damage; ‘bystander’ activation of autoreactive T lymphocytes by inflammatory cytokines and microbial products; and ‘molecular mimicry’ due to structural similarity between microbial and endogenous peptides [98]. Patients with immunodeficiency are less able to clear infectious agents. This results in chronic immune responses and tissue damage, a situation which favors the breaking of peripheral tolerance [99]. However, the development of autoimmunity does not appear to be related to the number of infections in 22q11-deleted patients. There is some evidence to implicate skewing of T lymphocyte subsets to a TH2 phenotype through homeostatic peripheral expansion, leading to a dysregulated B lymphocyte compartment, which may contribute to autoimmunity [100].
As previously noted, abnormal thymic development, resulting in partial immuno-deficiency, can lead to autoimmune features [38]. Detailed histological examination of thymii from patients with 22q11 deletion and partial T and B lymphocyte deficiency has not been reported, but it is possible that abnormal expression of AIRE may contribute to some autoimmune features, at least in some patients. There is little work detailing FOXP3 + regulatory T lymphocytes in these patients, but in one study, patients had proportionately fewer cells than normal controls [73], although there were no differences in patients with or without autoimmune features. Interestingly, comparison of TCRVB repertoire abnormalities between the CD4+ and CD4+ CD25Bright population showed no significant differences for individual patients, but significant differences were seen between the CD8+ and CD4+ CD25Bright T lymphocytes and expansions/contractions were seen (McLean-Tooke submitted).
Investigation and treatment
Beyond early childhood, it is a good practice to review patients annually for evidence of autoimmune disease. History and examination should be directed towards symptoms of autoimmunity. Investigations, directed by the clinical picture, should include appropriate auto-antibodies, thyroid function, full blood count, and film and direct anti-globulin test. Treatment should be directed toward specific symptoms.
Conclusions
Immunological disorders are common in patients with 22q11 deletion, and are not restricted to severe or recurrent infection. However, most studies are cross-sectional, and there is still little data on the longitudinal immunological history. While it is clear that autoimmunity is more common in these patients, it is not possible to predict who is at risk of developing disease. Further work on defining mechanisms of autoimmunity is needed. An awareness of these problems by physicians caring for children and for adults is important, so that patients are managed appropriately. For patients with a severe phenotype, thymic transplantation offers an exciting alternative to hematopoietic stem cell transplantation, although it may be that the procedure transforms a patient with complete T lymphocyte deficiency into one with partial combined immunodeficiency, with the associated problems of autoimmunity and immune dysregulation.
References
Kobrynski LJ, Sullivan KE (2007) Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet 370:1443–1452
Perez E, Sullivan KE (2002) Chromosome 22q11.2 deletion syndrome (DiGeorge and velocardiofacial syndromes). Curr Opin Pediatr 14:678–683
Shprintzen RJ, Higgins AM, Antshel K, Fremont W, Roizen N, Kates W (2005) Velo-cardio-facial syndrome. Curr Opin Pediatr 17:725–730
Yamagashi H, Srivastava D (2003) Unravelling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med 9:383–389
Jerome LA, Papaioannou VE (2001) DiGeorge syndrome phenotype in mice mutant for the T box gene, Tbx1. Nat Genet 27:286–291
Cooper MD, Peterson RDA, Good RA (1965) A new concept of the cellular basis of immunity. J Pediatr 67:907–908
de la Chapelle A, Herva R, Koivisto M, Aula P (1981) A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet 57:253–256
Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH (1982) The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatr 101:197–200
Driscoll DA, Spinner NB, Budarf ML, McDonald-McGinn DM, Zackai EH, Goldberg RB, Shprintzen RJ, Saal HM, Zonana J, Jones MC (1992) Deletions and microdeletions of 22q11.2 in velo-cardio-fa-cial syndrome. Am J Med Genet 44:261–268
Matsuoka R, Takao A, Kimura M, Imamura S, Kondo C, Joh-o K, Ikeda K, Nishibatake M, Ando M, Momma K (1994) Confirmation that the conotruncal anomaly face syndrome is associated with a deletion within 22q11.2. Am J Med Genet 53:285–289
McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J (1995) Autosomal dominant “Opitz” GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet 59:103–113
Devriendt K, Swillen A, Fryns JP (1998) Deletion in chromosome region 22q11 in a child with CHARGE association. Clin Genet 53:408–410
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R (2003) Role of TBX1 in human del22q11.2 syndrome. Lancet 362:1366–1373
Greenberg F, Valdes C, Rosenblatt HM, Kirkland JL, Ledbetter DH (1986) Hypoparathyroidism and T cell immune defect in a patient with 10p deletion syndrome. J Pediatr 109:489–492
Monaco G, Ciccimarra F, Pignata C, Garofalo S (1989) T cell immunodeficiency in a patient with 10p deletion syndrome. J Pediatr 115:330
Monaco G, Pignata C, Rossi E, Mascellaro O, Cocozza S, Ciccimarra F (1991) DiGeorge anomaly associated with 10p deletion. Am J Med Genet 39:215–216
Sullivan KE, Jawad AF, Randall P, Driscoll DA, Emanuel BS, McDonald-McGinn DM, Zackai EH (1998) Lack of correlation between impaired T cell production, immunodeficiency, and other phenotypic features in chromosome 22q11.2 deletion syndromes. Clin Immunol Immunopathol 86:141–146
Johnson D, Morrison N, Grant L, Turner T, Fantes J, Connor JM, Murday V (2006) Confirmation of CHD7 as a cause of CHARGE association identified by mapping a balanced chromosome translocation in affected monozygotic twins. J Med Genet 43:280–284
Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, McLean-Tooke A, Montgomery T, Goodship JA, Burt AD, Flood TJ, Abinun M, Cant AJ, Johnson D (2008) Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell+ severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol 153:75–80
Carlson C, Sirotkin H, Pandita R, Goldberg R, McKie J, Wadey R, Patanjali SR, Weissman SM, Anyane-Yeboa K, Warburton D, Scrambler P, Shprintzen R, Kucherlapati R, Morrow BE (1997) Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet 61:620–629
Edelmann L, Pandita RK, Morrow BE (1999) Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet 64:1076–1086
Meechan DW, Maynard TM, Gopalakrishna D, Wu Y, LaMantia AS (2007) When half is not enough: gene expression and dosage in the 22q11 deletion syndrome. Gene Expr 13:299–310
Taddei I, Morishima M, Huynh T, Lindsay EA (2001) Genetic factors are major determinants of phenotypic variability in a mouse model of the DiGeorge/del22q11 syndromes. Proc Natl Acad Sci USA 98:11428–11431
Schinke M, Izumo S (2001) Deconstructing DiGeorge syndrome. Nat Genet 27:238–240
Jerome LA, Papaioannou VE (2001) DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet 27:286–291
Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A (2001) Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet 27:293–298
Guris DL, Duester G, Papaioannou VE, Imamoto A (2006) Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell 10:81–92
van Bueren KL, Papangeli I, Rochais F, Pearce K, Roberts C, Calmont A, Szumska D, Kelly RG, Bhattacharya S, Scambler PJ (2010) Hes1 expression is reduced in Tbx1 null cells and is required for the development of structures affected in 22q11 deletion syndrome. Dev Biol 340:369–380
Marfella CG, Imbalzano AN (2007) The Chd family of chromatin remodelers. Mutat Res 618:30–40
Sillibourne JE, Delaval B, Redick S, Sinah M, Doxsey SJ (2007) Chromatin remodeling proteins interact with pericentrin to regulate centrosome integrity. Mol Biol Cell 18:3667–3680
Aramaki M, Kimura T, Udaka T, Kosaki R, Mitsuhashi T, Okada Y, Takahashi T, Kosaki K (2007) Embryonic expression profile of chicken CHD7, the ortholog of the causative gene for CHARGE syndrome. Birth Defects Res A Clin Mol Teratol 79:50–57
Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, Audollent S, Esculpavit C, Goudefroye G, Ozilou C, Fredouille C, Joye N, Morichon-Delvallez N, Dumez Y, Weissenbach J, Munnich A, Amiel J, Encha-Razavi F, Lyonnet S, Vekemans M, Attié-Bitach T (2006) Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet 43:211–217
de Lonlay-Debeney P, Cormier-Daire V, Amiel J, Abadie V, Odent S, Paupe A, Couderc S, Tellier AL, Bonnet D, Prieur M, Vekemans M, Munnich A, Lyonnet S (1997) Features of DiGeorge syndrome and CHARGE association in five patients. J Med Genet 34:986–989
Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, Chinn IK, Hale LP, Kepler TB, He M, Sarzotti M, Skinner MA, Rice HE, Hoehner JC (2007) Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood 109:4539–4547
Holländer G, Gill J, Zuklys S, Iwanami N, Liu C, Takahama Y (2006) Cellular and molecular events during early thymus development. Immunol Rev 209:28–46
Le Lievre CS, Le Douarin NM (1975) Mesenchymal derivatives of the neural crest: analysis of chimeric quail and chick embryos. J Embryol Exp Morphol 34:125–154
Haynes BF, Heinly CS (1995) Early human T cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J Exp Med 181:1445–1458
Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM, Notarangelo LD (2009) Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood 114:105–108
Gill J, Malin M, Sutherland J, Gray D, Hollander G, Boyd R (2003) Thymic generation and regeneration. Immunol Rev 195:28–50
Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, Asaumi Y, Kitazawa J, Takayanagi H, Penninger JM, Matsumoto M, Nitta T, Takahama Y, Inoue J (2008) The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity 29:423–437
Zuklys S, Balciunaite G, Agarwal A, Fasler-Kan E, Palmer E, Holländer GA (2000) Normal thymic architecture and negative selection are associated with Aire expression, the gene defective in the autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). J Immunol 165:1976–1983
Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, Liu YJ (2005) Hassall’s corpuscles instruct dendritic cells to induce CD4+ CD25+ regulatory T cells in human thymus. Nature 436:1181–1185
Müller SM, Kohn T, Schulz AS, Debatin KM, Friedrich W (2000) Similar pattern of thymic-dependent T-cell reconstitution in infants with severe combined immunodeficiency after human leukocyte antigen (HLA)-identical and HLA-nonidentical stem cell transplantation. Blood 96:4344–4349
Consortium ***Finnish-German APECED (1997) An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 17:399–403
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27:20–21
Poliani PL, Vermi W, Facchetti F (2009) Thymus microenvironment in human primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol 9:489–495
Writzl K, Cale CM, Pierce CM, Wilson LC, Hennekam RC (2007) Immunological abnormalities in CHARGE syndrome. Eur J Med Genet 50:338–345
Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brøndum-Nielsen K, Scambler PJ (1997) Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 34:798–804
Barber JCK, Walker JM, Barker MR, McNinch AW, Hallett RJ (1996) Repeated cytogenetic culture failure as an indicator of immunodeficiency. Lancet 348:1518
Ocejo-Vinyals JG, Lozano MJ, Sánchez-Velasco P, Escribano de Diego J, Paz-Miguel JE, Leyva-Cobián F (2000) An unusual concurrence of graft versus host disease caused by engraftment of maternal lymphocytes with DiGeorge anomaly. Arch Dis Child 83:165–169
Müller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W (2001) Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood 98:1847–1851
Markert ML, Alexieff MJ, Li J, Sarzotti M, Ozaki DA, Devlin BH, Sempowski GD, Rhein ME, Szabolcs P, Hale LP, Buckley RH, Coyne KE, Rice HE, Mahaffey SM, Skinner MA (2004) Complete DiGeorge syndrome: development of rash, lymphadenopathy, and oligoclonal T cells in 5 cases. J Allergy Clin Immunol 113:734–741
Janda A, Sedlacek P, Hönig M, Friedrich W, Champagne M, Matsumoto T, Fischer A, Neven B, Contet A, Bensoussan D, Bordigoni P, Loeb D, Savage W, Jabado N, Bonilla FA, Slatter MA, Davies EG, Gennery AR (2010) Multicenter survey on the outcome of transplantation of hematopoietic cells in patients with the complete form of DiGeorge anomaly. Blood 116:2229–2236
McGhee SA, Lloret MG, Stiehm ER (2009) Immunologic reconstitution in 22q deletion (DiGeorge) syndrome. Immunol Res 45:37–45
Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, Amrolia PJ, Gaspar HB, Davies EG, Friedrich W, Hoenig M, Notarangelo LD, Mazzolari E, Porta F, Bredius RG, Lankester AC, Wulffraat NM, Seger R, Güngör T, Fasth A, Sedlacek P, Neven B, Blanche S, Fischer A, Cavazzana-Calvo M, Landais P, Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation, European Society for Immunodeficiency (2010) Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol 126, 602-10.e1-11
Neven B, Leroy S, Decaluwe H, Le Deist F, Picard C, Moshous D, Mahlaoui N, Debré M, Casanova JL, Dal Cortivo L, Madec Y, Hacein-Bey-Abina S, de Saint Basile G, de Villartay JP, Blanche S, Cavazzana-Calvo M, Fischer A (2009) Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood 113:4114–4124
Cleveland WW, Fogel BJ, Brown WT, Kay HE (1968) Foetal thymic transplant in a case of DiGeorge’s syndrome. Lancet (7580), 1211–1214
August CS, Berkel AI, Levey RH, Rosen FS, Kay HE (1970). Establishment of immunological competence in a child with congenital thymic aplasia by a graft of fetal thymus. Lancet (7656):1080–1083
Markert ML, Boeck A, Hale LP, Kloster AL, McLaughlin TM, Batchvarova MN, Douek DC, Koup RA, Kostyu DD, Ward FE, Rice HE, Mahaffey SM, Schiff SE, Buckley RH, Haynes BF (1999) Transplantation of thymus tissue in complete DiGeorge syndrome. N Engl J Med 341:1180–1189
Markert ML, Devlin BH, McCarthy EA (2010) Thymus transplantation. Clin Immunol 135:236–246
Markert ML, Sarzotti M, Ozaki DA, Sempowski GD, Rhein ME, Hale LP, Le Deist F, Alexieff MJ, Li J, Hauser ER, Haynes BF, Rice HE, Skinner MA, Mahaffey SM, Jaggers J, Stein LD, Mill MR (2003) Thymus transplantation in complete DiGeorge syndrome: immunologic and safety evaluations in 12 patients. Blood 102:1121–1130
Markert ML, Alexieff MJ, Li J, Sarzotti M, Ozaki DA, Devlin BH, Sedlak DA, Sempowski GD, Hale LP, Rice HE, Mahaffey SM, Skinner MA (2004) Postnatal thymus transplantation with immunosuppression as treatment for DiGeorge syndrome. Blood 104:2574–2581
Chinn IK, Devlin BH, Li YJ, Markert ML (2008) Long-term tolerance to allogeneic thymus transplants in complete DiGeorge anomaly. Clin Immunol 126:277–281
Markert ML, Devlin BH, Chinn IK, McCarthy EA, Li YJ (2008) Factors affecting success of thymus transplantation for complete DiGeorge anomaly. Am J Transplant 8:1729–1736
Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC (2003) Aire regulates negative selection of organ-specific T cells. Nat Immunol 4:350–354
Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D (2005) The cellular mechanism of Aire control of T cell tolerance. Immunity 23:227–239
Li W, Kim MG, Gourley TS, McCarthy BP, Sant’Angelo DB, Chang CH (2005) An alternate pathway for CD4 T cell development: thymocyte-expressed MHC class II selects a distinct T cell population. Immunity 23:375–386
Choi EY, Jung KC, Park HJ, Chung DH, Song JS, Yang SD, Simpson E, Park SH (2005) Thymocyte–thymocyte interaction for efficient positive selection and maturation of CD4 T cells. Immunity 23:387–396
Junker AK, Driscoll DA (1995) Humoral immunity in DiGeorge syndrome. J Pediatr 127:231–237
Sullivan KE, McDonald-McGinn D, Driscoll DA, Emanuel BS, Zackai EH, Jawad AF (1999) Longitudinal analysis of lymphocyte function and numbers in the first year of life in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol 6:906–911
Jawad AF, McDonald-Mcginn DM, Zackai E, Sullivan KE (2001) Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr 139:715–723
Cancrini C, Romiti ML, Finocchi A, Di Cesare S, Ciaffi P, Capponi C, Pahwa S, Rossi P (2005) Post-natal ontogenesis of the T-cell receptor CD4 and CD8 Vbeta repertoire and immune function in children with DiGeorge syndrome. J Clin Immunol 25:265–274
McLean-Tooke A, Barge D, Spickett GP, Gennery AR (2008) Immunological defects in 22q11.2 deletion syndrome. J Allergy Clin Immunol 122:362–367
Lima K, Abrahamsen TG, Foelling I, Natvig S, Ryder LP, Olaussen RW (2010) Low thymic output in the 22q11.2 deletion syndrome measured by CCR9+ CD45RA+ T cell counts and T cell receptor rearrangement excision circles. Clin Exp Immunol 161:98–107
Piliero LM, Sanford AN, McDonald-McGinn DM, Zackai EH, Sullivan KE (2004) T-cell homeostasis in humans with thymic hypoplasia due to chromosome 22q11.2 deletion syndrome. Blood 103:1020–1025
Pierdominic IM, Mazzetta F, Caprini E, Marziali M, Digilio MC, Marino B, Aiuti A, Amati F, Russo G, Novelli G, Pandolfi F, Luzi G, Giovannetti A (2003) Biased T-cell receptor repertoires in patients with chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Exp Immunol 132:323–331
Sedivá A, Bartůnková J, Zachová R, Poloucková A, Hrusák O, Janda A, Kocárek E, Novotná D, Novotná K, Klein T (2005) Early development of immunity in DiGeorge syndrome. Med Sci Monit 11: CR182–187
Eberle P, Berger C, Junge S, Dougoud S, Büchel EV, Riegel M, Schinzel A, Seger R, Güngör T (2009) Persistent low thymic activity and non-cardiac mortality in children with chromosome 22q11.2 microdeletion and partial DiGeorge syndrome. Clin Exp Immunol 155:189–198
Herwadkar A, Gennery AR, Moran AS, Haeney MR, Arkwright PD (2010) Association between hypoparathyroidism and defective T cell immunity in 22q11.2 deletion syndrome. J Clin Pathol 63:151–155
Kung SJ, Gripp KW, Stephan MJ, Fairchok MP, McGeady SJ (2007) Selective IgM deficiency and 22q11.2 deletion syndrome. Ann Allergy Asthma Immunol 99:87–92
Finocchi A, Di Cesare S, Romiti ML, Capponi C, Rossi P, Carsetti R, Cancrini C (2006) Humoral immune responses and CD27+ B cells in children with DiGeorge syndrome (22q11.2 deletion syndrome). Pediatr Allergy Immunol 17:382–388
Schubert MS, Moss RB (1992) Selective polysaccharide antibody deficiency in familial DiGeorge syndrome. Ann Allergy 69:231–238
Gennery AR, Barge D, O’Sullivan JJ, Flood TJ, Abinun M, Cant AJ (2002) Antibody deficiency and autoimmunity in 22q11.2 deletion syndrome. Arch. Dis Child 86:422–425
Brown JJ, Datta V, Browning MJ, Swift PJF (2004) Graves’ disease in Di George Syndrome: Patient report with a review of endocrine autoimmunity associated with 22q11.2 deletion. J Pediatr Endocrinol Metab 17: 1575–1579
Smith CA, Driscoll DA, Emmanuel BS, McDonald-McGinn DM, Zackai EH, Sullivan KE (1998) Increased prevalence of immunoglobulin A deficiency in patients with a chromosome 22q11.2 deletion syndrome (DiGeorge Syndrome/ Velo-Cardio-Facial Syndrome). Clin Diagn Lab Immunol 5:415–417
Davies K, Stiehm RE, Woo P, Murray KJ (2001) Juvenile idiopathic polyarticular arthritis and IGA deficiency in the 22q11 deletion syndrome. J Rheumatol 28:2326–2334
Haire RN, Buell RD, Litman RT, Ohta Y, Fu SM, Honjo T, Matsuda F, de la Morena M, Carro J, Good RA, Litman GW (1993) Diversification, not use, of the immunoglobulin VH gene repertoire is restricted in DiGeorge syndrome. J Exp Med 178:825–834
Perez EE, Bokszczanin A, McDonald-McGinn D, Zackai EH, Sullivan KE (2003) Safety of live viral vaccines in patients with chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Pediatrics 112:e325
Moylett EH, Wasan AN, Noroski LM, Shearer WT (2004) Live viral vaccines in patients with partial DiGeorge syndrome: clinical experience and cellular immunity. Clin Immunol 112:106–112
Azzari C, Gambineri E, Resti M, Moriondo M, Betti L, Saldias LRG, Gelli AM, Vierucci A (2005) Safety and immunogenicity of measles-mumps-rubella vaccine in children with congenital immunodeficiency (DiGeorge syndrome). Vaccine 23:1668–1671
Waters V, Peterson KS, LaRussa P (2007) Live viral vaccines in a DiGeorge syndrome patient. Arch Dis Child 92:519–520
Davis CM, Kancherla VS, Reddy A, Chan W, Yeh HW, Noroski LM, Rosenblatt H, Shearer WT, Chinen J (2008) Development of specific T-cell responses to Candida and tetanus antigens in partial DiGeorge syndrome. J Allergy Clin Immunol 122:1194–1199
Al-Sukaiti N, Reid B, Lavi S, Al-Zaharani D, Atkinson A, Roifman CM, Grunebaum E (2010) Safety and efficacy of measles, mumps, and rubella vaccine in patients with DiGeorge syndrome. J Allergy Clin Immunol 126:868–869
Kratz CP, Niehues T, Lyding S, Heusch A, Janssen G, Gobel U (2003) Evans Syndrome in a patient with Chromosome 22q11.2 deletion syndrome: a case report. Pediatr Hematol Oncol 20:167–172
Rasmussen SA, Williams CA, Ayoube M, Sleasman JW, Gray BA, Bent-Williams A, Stalker HJ, Zorir T (1996) Juvenile rheumatoid arthritis in velo-cardio-facial syndrome: coincidence or unusual complication. Am J Med Genet 64:546–550
DePiero AD, Louire EM, Berman BW, Robin NH, Zinn EB, Hostoffer RW (1997) Recurrent immune cytopenias in 2 patients with Di George/velo cardio facial syndrome. J Paediatr 13:484–486
Verloes A, Curry C, Jamar M, Herens C, O’Lague P, Marks J, Sarda P, Blanchet P (1998) Juvenile rheumatoid arthritis and del (22q11) syndrome: a non-random association. J Med Genet 35:943–947
Bach JF (2005) Infections and autoimmune diseases. J Autoimmun 25(Suppl):74–80
Schurman SH, Candotti F (2003) Autoimmunity in Wiskott-Aldrich syndrome. Curr Opin Rheumatol 15:446–453
Zemble R, Luning Prak E, McDonald K, McDonald-McGinn D, Zackai E, Sullivan K (2010) Secondary immunologic consequences in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Immunol 136:409–418
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gennery, A.R. Immunological aspects of 22q11.2 deletion syndrome. Cell. Mol. Life Sci. 69, 17–27 (2012). https://doi.org/10.1007/s00018-011-0842-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0842-z