
Abstract
CCN1 (CYR61) is a dynamically expressed, multifunctional matricellular protein that plays essential roles in cardiovascular development during embryogenesis, and regulates inflammation, wound healing and fibrogenesis in the adult. Aberrant CCN1 expression is associated with myriad pathologies, including various cancers and diseases associated with chronic inflammation. CCN1 promotes diverse and sometimes opposing cellular responses, which can be ascribed, as least in part, to disparate activities mediated through its direct binding to distinct integrins in different cell types and contexts. Accordingly, CCN1 promotes cell proliferation, survival and angiogenesis by binding to integrin αvβ3, and induces apoptosis and senescence through integrin α6β1 and heparan sulfate proteoglycans. The ability of CCN1 to trigger the accumulation of a robust and sustained level of reactive oxygen species underlies some of its unique activities as a matrix cell-adhesion molecule. Emerging studies suggest that CCN1 might be useful as a biomarker or therapeutic target in certain diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
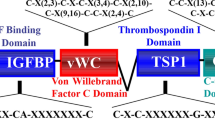
The term “matricellular protein” was first introduced by Bornstein in 1995 to describe a group of extracellular matrix (ECM) proteins that plays minimal roles in matrix structural integrity, but regulates a multitude of cellular responses with protean and sometimes opposing functions [1]. CCN1 (also named CYR61 or cysteine-rich 61) exemplifies many aspects of matricellular proteins by exhibiting diverse and at times seemingly conflicting functions in various cell types. Matricellular proteins are involved in wound healing without exception, and CCN1 is emerging as an important regulator of inflammation and wound repair [2–4]. First identified as a serum-inducible immediate-early gene product in mouse fibroblasts [5–8], CCN1 and other highly conserved homologs comprise the CCN protein family, which contains six members in mammals and nine members in zebrafish [9–11]. The CCN acronym is derived from the first three members of the family identified, namely CYR61, CTGF (connective tissue growth factor), and NOV (nephroblastoma overexpressed), and mammalian members of the family have been renamed CCN1-6 in order of their discovery [12]. CCN proteins share a modular structure, with an N-terminal secretory peptide followed by four conserved domains with sequence homologies to insulin-like growth factor-binding protein (IGFBP), von Willebrand factor type C repeat (vWC), thrombospondin type I repeat (TSR), and a carboxyl-terminal (CT) domain that contains a cysteine knot motif (Fig. 1). Each conserved structural domain is encoded by a separate exon, suggesting that CCN genes arose through exon shuffling [9, 10]. A non-conserved central hinge region bisects the protein into two halves that bind distinct receptors and induce disparate cellular responses (Fig. 1). CCNs were initially thought to function primarily in regulating cell proliferation, as they were identified based on their induction by mitogenic growth factors, oncogenes, or transformation. However, early studies on CCN1 showed that it is tightly associated with the ECM upon secretion [13] and supports cell adhesion through direct binding to integrin receptors [14, 15]. These findings, and the observation that CCN1 does not induce cell proliferation on its own but only enhances DNA synthesis induced by other mitogens [14], led to the appreciation that CCNs are matricellular proteins rather than classical growth factors [10].

Schematic diagram of CCN1, its receptor-binding sites, and biological activities. The modular domain structure of CCN1 and the locations of several identified integrin-binding sites are illustrated. The interaction of CCN1 with αvβ3 in endothelial cells is critical for its angiogenic activities, which underlie biological functions in embryonic development, cell proliferation, and tumor growth. The interaction of CCN1 with α6β1-heparan sulfate proteoglycans (HSPGs) in fibroblasts induces apoptosis or cellular senescence, and may function to regulate the inflammatory response, control fibrosis during wound healing, and suppress tumorigenesis. TBD to be determined
CCN1 is remarkably versatile and has many functions, some seemingly at odds with each other. It promotes cell survival and yet triggers apoptosis, enhances cell proliferation but also induces cell-cycle arrest, and promotes tumor growth and yet suppresses tumorigenesis in various contexts. These disparate activities of CCN1 can now be attributed, in large part, to its interaction with distinct integrins and heparan sulfate proteoglycans (HSPGs) in a cell-type and context-dependent manner (Fig. 1). Some of these activities are unique among cell-adhesion proteins of the ECM, and thus CCN1 serves as a model molecule for understanding novel aspects of matrix signaling. Current data have established CCN1 as being essential for cardiovascular development during embryogenesis, and critical for modulating inflammation, wound healing, and tissue repair in many tissues of the adult. Numerous gene-profiling studies have shown altered CCN1 expression in diverse pathological conditions, including various cancers, inflammatory diseases, and dysfunctional wound healing, suggesting that targeting CCN1 expression and signaling may hold promise in the development of diagnostic markers and therapeutics. Here, I seek to provide an update on the most recent findings on CCN1, focusing on its biological functions and mechanism of actions. Several earlier reviews afford a more comprehensive summary of research on the CCN family [9, 10, 16–18].
Transcriptional and post-transcriptional regulation
CCN1 is an immediate-early gene expressed at a very low level in quiescent fibroblasts, but is transcriptionally activated within minutes of stimulation by serum growth factors without requiring de novo protein synthesis [5, 6]. Exquisitely sensitive to a wide range of extracellular stimuli, CCN1 is transcriptionally activated by platelet-derived growth factor and fibroblast growth factor 2 [8], transforming growth factor β1 (TGF-β1) [19], growth hormone [20], the phorbol ester 12-O-tetradecanoylphorbol-13-acetate, cAMP [8], vitamin D3 [21], estrogen and tamoxifen [22], angiotensin II [23, 24], hypoxia [25], UV light [26], and mechanical stretch [27, 28]. Consistent with a role in inflammation, CCN1 is also induced by bacterial and viral infections [29–32], and by inflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor α (TNF-α) [33]. CCN1 is activated by agonists of G protein-coupled receptors (GPCRs) such as thrombin [34, 35], prostaglandins E2 and F2α [33, 36], and sphinogosine-1-phosphate [37]. Thrombin acts through the GPCR and RhoA signaling pathways to induce CCN1, which in turn interacts with integrins to promote cell proliferation [35]. Thus, CCN1 serves to connect the GPCR and integrin signaling pathways to enhance thrombin-induced cell proliferation [38].
Analysis of the mouse Ccn1 promoter uncovered a serum response element (SRE) (–1,912/–1,933) to which the 67-kDa serum response factor (SRF) binds and mediates transcriptional activation by serum or platelet-derived growth factor in fibroblasts [39]. A 2-kb Ccn1 promoter fragment that includes the SRE is sufficient to confer accurate developmental and tissue-specific expression in transgenic mice when linked to the β-galactosidase gene as a reporter, thus defining the functional Ccn1 promoter for transcriptional regulation in vivo [40]. Mechanical stretch induction of CCN1 in human smooth muscle cells involves activation of the RhoA GTPase, which promotes actin remodeling and consequently recruitment of the transcription factor myocardin-related transcriptional activator (MRTF-A), a G-actin-binding SRF co-activator, to the SRE of the CCN1 promoter [41]. Activation of p38 mitogen-activated protein kinase (MAPK), which enhances the histone acetyltransferase activity of CREB-binding protein (CBP) and its recruitment to the SRF–MRTF-A complex, enhances CCN1 expression. The transcription factor Egr-1, although not sensitive to cytoskeletal remodeling, is also involved in stretch-induced activation of CCN1 in vascular smooth muscle cells [42]. Consistent with its angiogenic functions, CCN1 is highly induced under conditions of hypoxia, mediated through both hypoxia-inducible factor-1α (HIF-1α)-dependent and -independent mechanisms [25, 43, 44]. Recent genomic studies have also shown that CCN1 is a target of the TAZ and YAP transcriptional co-activators, which act in concert with various transcription factors such as TEAD to regulate expression of genes related to proliferation and apoptosis [45, 46].
Specific alternative splicing in CCN1 was first reported in fibroblasts, causing an in-frame deletion within exon 4 (TSR domain) [47]. Another alternative splicing event retains intron 3, resulting in two stop codons that would terminate the CCN1 polypeptide within the central hinge region. However, this alternative splicing is blocked under hypoxic conditions in breast cancer cells, leading to the synthesis of full-length CCN1 [48]. Further post-transcriptional regulation involves microRNA-155, which appears to downregulate CCN1 expression in pre-eclampsia [49].
The CCN1 mRNA contains internal ribosome entry sites (IRES) that allow its preferential translation under conditions of stress, even when the concentration of the cap-binding complex eIF4F is low, for example upon infection by certain viruses [50]. Both the 5′-non coding region of the Ccn1 mRNA and a highly GC-rich region encoding part of the IGFBP domain appear to contain IRES [50, 51]. To date, information on post-translational regulation of CCN1 is scant. Although CCN1 is glycosylated, neither the chemical nature of the glycosylation nor its biological function is well understood. There is no direct evidence indicating whether CCN1 might function as a dimer or oligomer. Anecdotal observations of proteolytic processing have been made, although the biological significance of these modifications in vivo is currently unknown [14, 52].
Multiple receptors and diverse functions
Cell-surface signaling receptors: integrins and HSPGs
The functions of CCN proteins are mediated primarily through their binding to distinct integrins [53], which are bidirectional cell-surface signaling receptors capable of regulating diverse cellular functions [54]. That CCN proteins are ligand of integrins was first demonstrated by the direct binding of CCN1 to integrin αvβ3 to mediate endothelial cell adhesion [15]. Subsequent studies have shown that CCN1 supports cell adhesion by binding integrins α6β1 and HSPGs in fibroblasts and smooth muscle cells [55, 56], αIIbβ3 in activated platelets [57], αMβ2 in monocytes and macrophages [58, 59], and αDβ2 in macrophage foam cells [60]. The functions of CCN1 mediated through integrins are listed in Table 1. CCN1 binds HSPGs with high affinity, which serve as co-receptors with integrins in some contexts [13, 55]. Where examined, syndecan-4 has been identified as the HSPG critical for CCN1 functions [61, 62]. The interactions between CCN1 and integrins-HSPGs have been reviewed in greater detail previously [10, 18, 53].
CCN1 does not contain the canonical RGD sequence that can bind a number of integrins [54]. Instead, specific non-canonical binding sites of CCN1 for several integrins have been identified (Fig. 1) [63–66]. Mutational analysis showed that distinct integrin-binding sites of CCN1 can function independently of one another. CCN1 mutants that disrupt its α6β1-binding sites abolish α6β1-dependent activities without affecting αvβ3-mediated functions [66]. Likewise, disruption of the CCN1 αvβ3-binding site abolishes αvβ3- but not α6β1-mediated activities [65]. However, the synergism of CCN1 with TNFα (see below) requires its interaction with both αvβ5 and α6β1. Interestingly, whereas CCN1 mutants defective for either binding αvβ5 or α6β1 are each incapable of functional synergism with TNFα, a combination of these two mutants can fully reconstitute wild-type activity [62]. This observation suggests that different CCN1 molecules can separately bind distinct integrins on the same cell and generate signaling events that converge inside the cell to produce specific biological outcomes. Aside from integrins and HSPGs, CCN1 may potentially also interact with other cell-surface receptors. The homologous CCN2 has been shown to bind LRP-1 [67] and TrkA [68], whereas CCN3 can bind Notch [69]. Assessing whether CCN1 can interact with these and other receptors will require further investigation.
Cell adhesion, migration, DNA synthesis, and cell survival
Upon secretion, CCN1 is tightly but non-covalently associated with the ECM and the cell surface, an interaction that can be displaced by soluble heparin [13]. This observation led to the finding that CCN1 can support cell adhesion and induce adhesive signaling in many adherent cell types [18, 53]. In human skin fibroblasts, CCN1 supports cell adhesion through α6β1-HSPGs and results in the formation of focal adhesion complexes, activation of focal adhesion kinase, paxillin and Rac, actin cytoskeleton reorganization, and formation of structures critical for cell motility such as filopodia and lamellipodia [70]. A short CCN1 peptide containing the α6β1-HSPG binding sites is sufficient to support cell adhesion and recapitulate the sustained activation of extracellular signal-regulated kinase (ERK) by immobilized CCN1 [66]. Furthermore, CCN1 stimulates cell migration in fibroblasts, smooth muscle cells and endothelial cells [56, 71, 72], and certain cancer cells [73–75]. Although CCN1 does not induce DNA synthesis on its own, it can synergize with other mitogenic growth factors to augment growth factor-induced DNA synthesis in both fibroblasts and endothelial cells through integrin αvβ3 [14, 71]. This activity may contribute to its ability to promote growth factor-induced poliferation in PC3 prostate cancer cells [76, 77] and ovarian carcinoma cells [78], and IL-17-stimulated proliferation in synoviocytes [79]. Consistent with cell-survival functions associated with cell adhesion, CCN1 promotes the survival of endothelial cells through integrin αvβ3 [72], and inhibits apoptosis by activation of Akt in pulmonary epithelial cells [80] or upregulation of XIAP in MCF7 breast cancer cells [81].
Angiogenesis
The strong expression of Ccn1 in endothelial cells during embryonic development in mice suggested a role in vessel growth [82]. Indeed, CCN1 has potent angiogenic activity, first demonstrated in a corneal micropocket implant assay [83] and subsequently confirmed in a rabbit ischemic hindlimb model [84]. The angiogenic activity of CCN1 can be attributed to its binding to integrin αvβ3, a major integrin expressed in endothelial cells responsible for mediating CCN1 function in promoting endothelial cell adhesion, migration, proliferation, survival, and tubule formation [72]. A mutant CCN1 protein specifically mutated in the αvβ3-binding site is devoid of angiogenic activity [65]. Consistent with an important role for CCN1 in angiogenesis in vivo, Ccn1-null mice are defective in placental vessel bifurcation and suffer severe cardiovascular defects [85, 86]. In addition to its direct effects on endothelial cells, CCN1 can also regulate the expression of angiogenic factors such as VEGF-A and VEGF-C [85, 87]. Furthermore, CCN1 stimulates the integrin-dependent recruitment of CD34+ progenitor cells, thereby enhancing endothelial proliferation and neovascularization [88].
Chondrogenesis and osteogenesis
Ccn1 is expressed in pre-chondrocytic mesenchymal condensations of both mesodermal and neuroectodermal origins [82]. Consistent with a role in chondrogenesis, purified CCN1 protein accelerated the differentiation of mouse limb bud mesenchymal cells isolated from E10 embryos, enhanced the extent of differentiation, and lowered the threshold cell density for differentiation [89]. At later stages of skeletal development, CCN1 is highly expressed in the hypertrophic cartilage at the growth plate [40]. Thus, CCN1 may be involved in neovascularization of the hypertrophic cartilage, where vessel invasion provides conduits for osteoblasts. Moreover, CCN1 stimulates osteoblast differentiation through an αvβ3/ILK-dependent pathway but inhibits osteoclastogenesis, suggesting its role as a bifunctional regulator of bone remodeling that promotes bone formation but inhibits bone resorption [90–92].
Potential nuclear functions
In contrast to its prominent localization to the ECM, CCN1 has also been detected in the nucleus of cells, despite the absence of a classical nuclear localization signal [93]. The specific roles of nuclear CCN1 are not well understood. Interestingly, CCN1 was identified in an expression screening of a λgt11 library for proteins that bind the human immunodeficiency virus type 1 (HIV-1) long terminal repeat [47]. Furthermore, recombinant CCN1 was shown to bind DNA with sequence specificity in vitro, and co-transfection of CCN1 expression plasmids inhibited transcription from HIV-1 long terminal repeat reporter constructs [47]. Although the functional implications of these observations have not been investigated further, they suggest the intriguing possibility that CCN1 may bind DNA and regulate transcription under certain conditions. A recent study showed that CCN5, a member of the family that uniquely lacks the CT domain, is able to act as a transcriptional repressor upon a GAL4-dependent reporter when linked to the GAL4 DNA-binding domain, presumably through association with the histone deacetylase HDAC1 [94]. The possibility that CCN1 might regulate nuclear gene transcription certainly warrants further investigation.
CCN1 functions in embryonic development and pregnancy
Ccn1 expression during mouse embryogenesis is tightly associated with development of the skeletal, cardiovascular, and neuronal systems [82], and targeted disruption of Ccn1 in mice results in embryonic lethality. Approximately 30% of Ccn1-null embryos fail to form chorioallantoic fusion at E8.5 and die by E9.5, whereas the remaining embryos perish at mid-gestation from placental vascular insufficiency, loss of embryonic vascular integrity, hemorrhage, and severe cardiac atrioventricular septal defect (AVSD) [85, 86]. At E8.5, Ccn1 is highly expressed in the ectoplacental cone and in trophoblastic giant cells, suggesting a role in implantation and interaction with the decidua [82]. Despite normal vasculogenesis in Ccn1-null mice, specific angiogenic defects are observed. Vessel bifurcation is impaired in the chorionic plate of Ccn1-null embryos, resulting in a dearth of sprouting vessels that penetrate into the labyrinth, and thus an undervascularized placenta [85]. However, trophoblast differentiation and syncytiotrophoblast formation at the chorionic plate occur normally in the Ccn1-null placenta, forming the trilaminar trophoblast barrier between fetal vessels and maternal blood sinuses where fetal vessels are found. Therefore, the labyrinthine vascular deficiency in Ccn1-null mice is not due to trophoblast defects. The endothelium in large vessels of Ccn1-null embryos lack an intact basement membrane and the vascular cells are disorganized and apoptotic, leading to vessel rupture and hemorrhage [85].
In the human placenta, CCN1 is most highly expressed in the second and third trimesters of pregnancy, especially in non-proliferating interstitial extravillous trophoblastic giant cells, vascular endothelial cells, and mesenchymal and stromal cells of the placental villi [95]. CCN1 expression is greatly decreased in placentae associated with pre-eclampsia, a condition thought to result from a shallow invasion of the extravillous trophoblast into the decidua, followed by incomplete remodeling of the maternal vascular structures [95]. This finding is consistent with a role for CCN1 in placental angiogenesis and decidual tissue remodeling, and evidence suggests that serum levels of CCN1 may serve as a marker for early diagnosis of pre-eclampsia [96]. Additionally, CCN1 is upregulated in eutopic and ectopic endometria of women with endometriosis [33], a condition in which endometrial tissue proliferates outside of the uterine cavity, thought to occur most commonly on the ovaries as a result of retrograde menstruation. It is believed that the angiogenic activities of CCN1 may contribute to its role in the pathogenesis of endometriosis [33].
Ccn1-null embryos also exhibit severe defects in atrioventricular valvuloseptal morphogenesis, resulting in a common atrioventricular valve orifice that is the hallmark of complete AVSD [86]. The mesenchymal cells in the endocardial cushion tissue suffer premature apoptosis, preventing them from fusing properly with the atrial and ventricular septa to undergo apposite valvuloseptal morphogenesis. Although Ccn1 +/− mice are largely viable, they exhibit persistent ostium primum atrial septal defects with 20% penetrance [86]. Human AVSD is a common group of congenital disorders often associated with Down’s syndrome, whereas non-syndromic AVSDs are typically inherited with autosomal dominance [97]. The atrial septal defects due to Ccn1 haploinsufficiency resemble those observed in some human patients with mutations in AVSD1, a susceptibility locus for non-syndromic AVSD identified by linkage analysis. The human CCN1 gene and AVSD1 both map within the same chromosomal location at 1p21–31 [97, 98], suggesting that CCN1 may be a candidate gene for human AVSD.
Although Ccn1 is prominently expressed in the skeletal system during embryogenesis [82], Ccn1-null mice succumb too early in development for its role in skeletal development to be assessed fully [85]. Ccn1 is also highly expressed in the developing neuronal system; however, its role in this context has not been thoroughly investigated.
In Xenopus laevis, gain- and loss-of-function experiments have shown that precise control of CCN1 expression is required for normal gastrulation movements and modulation of Wnt signaling [99]. Either overexpression of CCN1 by injection of Ccn1 RNA or CCN1 knockdown by antisense morpholino oligonucleotides caused defects in gastrulation, resulting in severe delay of blastopore closure. Ventral injection of Ccn1 mRNA induces secondary axes formation in Xenopus and activates Wnt/β-catenin signaling, but can also inhibit Wnt8-induced secondary axis formation, indicating that CCN1 can stimulate or inhibit Wnt signaling in a context-dependent manner [99].
Unusual cell adhesive functions: role of reactive oxygen species
Apoptotic synergism with members of the TNF family
Cell adhesion to the ECM is thought to provide a cell-survival function, whereas detachment from the ECM induces rapid cell death by anoikis in many cell types. Although CCN1 promotes cell survival in endothelial cells and some cancer cells [72, 81], it represents the first example of an ECM molecule that can induce apoptosis through the process cell adhesion [61]. In fibroblasts, CCN1 triggers apoptosis by binding α6β1 and syndecan-4, leading to the p53-dependent activation of Bax and cytochrome c release, resulting in apoptosis [61]. This effect is relatively modest in normal fibroblasts, but is greatly enhanced in cells that are defective for p21 and are thus more susceptible to apoptotic signals. More recent studies have found that other ECM proteins can also induce apoptosis, e.g., EMILIN 2 can trigger cell death through a death receptor-mediated mechanism [100].
Physiologically, the apoptotic activities of CCN1 may be most relevant when acting in combination with other apoptotic factors, notably members of the TNF family including TNF-α, FasL, and TRAIL [101]. TNF-α is a pro-inflammatory cytokine and a potent activator of the transcription factor NF-κB, which induces the expression of a multitude of pro-mitogenic, pro-inflammatory, and anti-apoptotic proteins, thus promoting cell proliferation and survival [102]. However, TNF-α is also a powerful apoptotic inducer in vitro when NF-κB signaling is blocked, typically by the addition of cycloheximide. Whereas TNF-α contributes to apoptotic tissue damage in inflammatory diseases, how it induces apoptosis in vivo is not clearly understood [102, 103]. Remarkably, cell adhesion to CCN1 enables TNF-α to induce apoptosis without inhibiting NF-κB signaling or de novo protein synthesis, thus converting TNF-α from a survival-enhancing factor into a potent apoptotic agent [62]. Although CCN1 is a cell-adhesion molecule, these activities are observed when CCN1 is presented to cells either as an adhesion substrate or as a soluble factor. Mechanistically, CCN1 acts by binding integrins α6β1, αvβ5 and syndecan-4, leading to the generation of reactive oxygen species (ROS) via 5-lipoxygenase and mitochondria (Fig. 2) [62]. The high level of ROS induced by CCN1/TNF-α counters the inhibition of JNK activation through the anti-oxidant effect of NF-κB and results in the biphasic activation of JNK necessary for apoptosis, most likely by oxidative inactivation of JNK phosphatases [104]. JNK targets c-FLIP, an inhibitor for the activation of caspase-8/caspase-10, for proteasomal degradation, thus allowing apoptosis to proceed [105]. In addition, CCN1 also strongly enhances the apoptotic activity of other apoptotic members of the TNF family, including FasL and TRAIL [77, 106].

Signaling pathways mediated through CCN1-induced ROS accumulation. The interaction of CCN1 with α6β1 and HSPGs (syndecan-4) is required to trigger the RAC-1-dependent accumulation of ROS through several pathways, including 5-lipoxygenase (5-LOX), NADPH oxidase, and the mitochondria. Neutral sphingomyelinase (nSMase) also contributes to CCN1-induced ROS. CCN1-induced ROS lead to the biphasic activation of JNK, allowing TNF-α to induce apoptosis by targeting c-FLIP for degradation. The high and sustained level of ROS induced by CCN1 also triggers a DNA damage response, leading to p53 activation. ROS also lead to the hyperactivation of ERK and p38 MAPK, which in turn induce p16INK4a and activate pRb. Both p53 and p16INK4a/pRb contribute to senescence
The importance of CCN1 in TNF-α- and FasL-induced apoptosis in vivo has been demonstrated using knockin mice (Ccn1 dm/dm) in which the genomic Ccn1 locus has been replaced by an apoptosis-defective Ccn1 allele with mutations at the α6β1-HSPG-binding sites [62, 101, 106]. In contrast to the embryonic lethality of Ccn1-null mice, these knockin mice are viable, fertile, and do not exhibit any obvious developmental defects. Ccn1 dm/dm mice are substantially resistant to concanavalin A-induced, TNF-α-dependent hepatocyte apoptosis [107, 108]. Dermal apoptosis induced upon direct subcutaneous injection of TNF-α is also substantially reduced in Ccn1 dm/dm mice compared with wild-type mice. Fas-mediated apoptosis is also reduced, as judged by using the monoclonal antibody Jo2 that recognizes and activates the Fas receptor and induces hepatic apoptosis [109], or by using ethanol gavage in mice, a system that mimics binge drinking in humans and triggers FasL-induced hepatocyte apoptosis [110]. In each of these experimental models for TNF-α- or Fas-mediated apoptosis, Ccn1 dm/dm mice consistently show a >60% reduction in apoptosis compared with wild-type mice [62, 106]. These results show that CCN1 is a physiological regulator of TNF-α- and Fas-mediated apoptosis in vivo [111].
CCN1 induces cellular senescence
Another unusual activity of CCN1 as a matrix cell-adhesion molecule is the ability to induce cellular senescence [2]. Through binding to integrin α6β1 and cell-surface HSPGs, CCN1 induces fibroblast senescence by activating RAC1 and the RAC1-dependent NADPH oxidase 1 (NOX1) to trigger a robust level of ROS that is sustained for many hours [2]. Consequently, CCN1 induces a DNA damage response, resulting in the activation of ATM, CHK1, CHK2, and p53 [2]. CCN1 also triggers the ROS-dependent hyperactivation of p38 MAPK and ERK, which in turn activate the p16INK4a/pRb pathway to induce senescence (Fig. 2). Both p53 and p16INK4a/pRb pathways, which are activated by CCN1, contribute to the induction of cellular senescence [112, 113]. Cell adhesion to CCN1 induces a much higher and more sustained level of ROS than cell adhesion to other ECM proteins such as collagen, fibronectin, and laminin, which do not induce senescence. Although CCN1 induces ROS through several pathways, NOX1, but not 5-lipoxygenase, is specifically required for the induction of senescence [2]. It is likely that CCN1 interacts with another receptor(s) in addition to α6β1-HSPGs to elicit these responses, as other matrix proteins such as laminin can also bind α6β1-HSPGs but do not induce sustained ROS accumulation or senescence.
An important feature of senescent cells is the expression of the senescence-associated secretory phenotype (SASP), characterized by the increased expression of ECM-degrading enzymes such as matrix metalloproteinases (MMPs), inflammatory cytokines/chemokines, and down-regulation of ECM components such as collagen [112, 114–116]. Consistently, CCN1-induced senescent fibroblasts express elevated levels of MMPs and downregulate collagen and TGF-β1, thus imposing a matrix-degrading phenotype [2]. These characteristics of senescent cells help to control fibrosis associated with cutaneous wound healing and possibly other pathologies (see below).
CCN1 in pathologies
Wound healing and fibrosis
In the adult, CCN1 expression is associated with many tissues undergoing inflammation and injury repair. Wound healing in virtually all mammalian organ systems is coordinated in three overlapping but distinct phases, initiating with an inflammatory response, followed by ECM deposition and granulation tissue formation, and finally tissue remodeling and maturation to complete the healing process [117–119]. Each of these steps must be tightly regulated for optimal wound healing. In cutaneous wound healing, Ccn1 is highly expressed in myofibroblasts of the granulation tissue [40, 87]. The myofibroblasts, derived from differentiation of resident activated fibroblasts or recruited fibrocytes, proliferate and rapidly synthesize ECM to maintain tissue integrity and help provide a barrier against microbes while the damaged tissue is being repaired, and they also express α-smooth muscle actin and promote wound contraction to accelerate wound closure [120]. However, if ECM synthesis in myofibroblasts is not kept in check, then excessive matrix deposition can lead to fibrosis, scarring, and loss of tissue function [120–122]. Fibrosis is a common problem associated with wound healing, particularly when inflammation and injury become chronic, leading to serious and potentially life-threatening conditions when it occurs in vital organs such as the liver, lung, kidney, and heart [118].
Recent studies show that, as myofibroblasts proliferate in the granulation tissue, CCN1 eventually accumulates to a sufficiently high level to drive the myofibroblasts themselves into senescence [2]. This CCN1-induced senescence switch in later stages of wound healing exerts a self-limiting effect on the synthesis and deposition of ECM by myofibroblasts in several ways (Fig. 3). First, senescent myofibroblasts no longer proliferate, thereby curbing the number of ECM-producing cells. Second, senescent cells express the anti-fibrotic SASP as described above, upregulating MMPs and downregulating collagen expression, leading to matrix degradation [2, 70]. Third, senescent cells can be cleared by natural killer cells, thus accelerating wound resolution [123]. Hence, CCN1 turns the ECM-synthesizing myofibroblasts themselves into ECM-degrading senescent cells, thereby imposing a self-limiting effect on fibrogenesis and promoting wound resolution (Fig. 3) [124]. In knockin mice that express a CCN1 mutant unable to bind α6β1-HSPGs and are thus defective for the induction of senescence, senescent cells do not accumulate in the granulation tissue during wound healing, resulting in exacerbated fibrosis [2]. Topical application of purified CCN1 protein to cutaneous wounds reverses these defects, further establishing the critical role of CCN1 in controlling myofibroblast senescence to limit fibrosis. In a mouse model of carbon tetrachloride-induced liver fibrosis, the accumulation of senescent cells also functions to limit fibrosis [125]. Thus, the induction of cellular senescence during wound healing may be a general mechanism that controls fibrogenesis during wound repair [124, 126].

CCN1 controls fibrosis in wound healing by inducing cellular senescence. In cutaneous wound healing, myofibroblasts are recruited to form the granulation tissue, where they proliferate and rapidly synthesize ECM to provide tissue integrity during repair. At later stages of wound healing, these myofibroblasts are driven into senescence by CCN1, whereupon they express an ECM-degrading phenotype and limit fibrosis. Thus, CCN1 functions as an anti-fibrotic molecular switch that converts ECM-producing myofibroblasts into ECM-degrading senescent cells, thereby imposing a self-limiting control on fibrogenesis during wound healing
In addition to cutaneous wound healing, CCN1 expression is elevated in remodeling cardiomyocytes after myocardial infarctions [24], in vascular injury [56], and in the long bones during fracture repair, notably in proliferating chondrocytes and osteoblasts [127, 128]. Blockade of CCN1 by antibodies inhibits bone fracture healing in mice [129]. In the kidney, CCN1 is expressed in podocytes in normal adult and embryonic glomeruli, but expression is decreased in IgA nephropathy, diabetic nephropathy, and membranous nephropathy, particularly in diseased kidneys with severe mesangial expansion, suggesting that impaired CCN1 function may contribute to the progression of glomerular disease with mesangial expansion [130].
Inflammation
Although the specific roles of CCN proteins in inflammation in vivo are currently not well understood, evidence for their participation in the inflammatory response is beginning to mount [31]. CCN1 expression is induced by inflammatory cytokines such as IL-1 and TNF-α [33, 131], as well as by bacterial and viral infections [29–32]. As described above, CCN1 profoundly alters the activities of inflammatory cytokines such as TNF-α and FasL in vitro and in vivo, and significantly enhances the cytotoxicity of TRAIL [77, 111]. Furthermore, CCN1 supports the adhesion of activated monocytes through integrin αMβ2, and reprograms macrophages towards M1 polarization through αMβ2-mediated activation of NF-κB [58, 59]. These findings suggest that CCN1 can play important roles in inflammation, in part by acting on inflammatory cells such as macrophages, inducing angiogenesis, and collaborating with cytokines such as TNF-α.
CCN1 is upregulated in patients with Crohn’s disease and ulcerative colitis, and in colons of mice treated with dextran sodium sulfate to induce inflammatory colitis [132]. Genomic studies have found that CCN1 is among the most highly differentially expressed genes in rheumatoid arthritis in disease-discordant monozygotic twins [133]. CCN1 is highly expressed in collagen-induced arthritis in rodents, and inhibition of CCN1 expression correlates with suppression of inflammatory arthritis [134]. These findings suggest that CCN1 may play an important role in inflammatory diseases including colitis and rheumatoid arthritis. It is interesting to note that TNF-α is known to be critical in both inflammatory bowel disease and rheumatoid arthritis, and anti-TNF-α therapy is in clinical use for these diseases. The functional interaction between CCN1 and TNF-α may be important in these pathologies.
Vascular diseases
CCN1 is overexpressed in vascular smooth muscle cells of atherosclerotic lesions and in the neointima of restenosis after balloon angioplasty, both in rodent models and in humans [23, 56, 58, 135]. Suppression of CCN1 expression by either small interfering RNA or FOXO3a-mediated transcriptional repression results in reduced neointimal hyperplasia after balloon angioplasty, an effect that is reversed by delivery of CCN1 via gene transfer [136, 137]. Overexpression of CCN1 from an adenoviral vector accelerates re-endothelialization after balloon injury, possibly because CCN1 can recruit CD34+ endothelial progenitor cells to the endothelial monolayer and promote their differentiation [88, 138]. In a mouse model of oxygen-induced retinopathy, overexpressing CCN1 by lentiviral vector delivery, or injecting CCN1-overexpressing hematopoietic stem cells into the vitreous humor, produced significant beneficial effects in repairing damaged vasculature [139]. These findings underscore a critical role for CCN1 in vascular injury repair.
CCN1 in cancer: a double-edged sword?
CCN1 possesses two sets of divergent activities that can contribute to opposing effects on tumorigenesis (Fig. 1). First, CCN1 is a powerful angiogenic inducer in vivo [72, 83], and angiogenesis is essential for the supply of oxygen and nutrients to nourish the growing tumor [140]. With the discovery of its angiogenic activity, CCN1 was shown to promote tumor growth and increase tumor vascularization when overexpressed in human gastric adenocarcinoma cells in SCID mice [83]. These findings demonstrated the ability of CCN1 to promote the growth of tumors generated by transformed cells, typically observed in xenografts transplanted into immunodeficient mice. In addition to angiogenesis, CCN1 can also promote cancer cell proliferation, invasion, survival, and metastasis [74, 75, 141]. Accordingly, subsequent studies have shown that forced expression of CCN1 enhanced tumor growth in xenografts of breast cancer cells [142], prostate cancer cells [74], ovarian carcinoma cells [78], and squamous carcinoma cells [143]. Consistently, silencing of CCN1 expression decreased tumor growth in xenografts of prostate cancer cells [74] and pancreatic cancer cells [144].
The clinical relevance of these observations is supported by the finding of a significant correlation between CCN1 expression and the stage, tumor size, lymph node positivity, and poor prognosis in several cancers, including breast cancer [142, 145–148], prostate cancer [149], glioma [150], gastric adenocarcinoma [151], and squamous cell carcinoma [143]. In MCF7 breast cancer cells, which are dependent on estrogen for growth, overexpression of CCN1 renders these cells estrogen-independent and significantly resistant to apoptosis, in part through upregulation of the anti-apoptotic protein XIAP [81, 152, 153]. The anti-apoptotic functions of CCN1 in breast cancer cells are mediated through its receptor integrin αvβ3 [81, 153], whose expression is also induced by CCN1 [154].
However, CCN1 can also induce apoptosis and cellular senescence [2, 61, 101], two well-established mechanisms of tumor suppression [155]. By inducing apoptosis or senescence, CCN1 may be able to suppress the initial phases of tumorigenesis by preventing the proliferation of damaged cells at risk of transformation. For example, whereas CCN1 can promote the proliferation of prostate cancer cells, CCN1 can also exacerbate apoptosis of these cells in the presence of the immune surveillance molecule TRAIL [74, 76, 77]. Interestingly, high levels of CCN1 expression are found in prostate cancer cell lines with a mutant or null p53 gene, but lower levels are found in cells expressing wild-type p53 [156]. This observation is consistent with the hypothesis that CCN1 and p53 together can promote tumor cell apoptosis or senescence, and thus high CCN1 levels are tolerated only in cells with mutant p53.
Several lines of evidence support the notion that CCN1 can suppress tumor growth. CCN1 expression is decreased in clinical specimens of non-small-cell lung cancer (NSCLC) compared with normal matched lung samples. NSCLC cells overexpressing CCN1 developed smaller tumors than parental cells in nude mice [157]. CCN1 inhibits NSCLC cell proliferation by upregulating p53, p21, and p130/pRb [158], suggesting that CCN1 may induce senescence through a p53- and pRb-dependent pathway [2]. Likewise, overexpression of CCN1 inhibits the proliferation of hepatocellular carcinoma cells, in part through p53 [159]. Furthermore, overexpression of CCN1 in endometrial adenocarcinoma cells decreased cell growth and increased apoptosis [160], and CCN1 expression in melanoma cells reduced tumor growth and metastasis and concomitantly increased apoptosis in tumors [161]. Thus, the role of CCN1 in tumorigenesis may be cell type- and context-dependent, and may hinge on whether the cell types are susceptible to CCN1-induced apoptosis or senescence, or whether angiogenic factors are limiting.
Concluding remarks and future prospects
CCN1 is a multifunctional matricellular protein that is essential for life due to its embryonic developmental functions, including placental angiogenesis, vascular integrity, and cardiac valvuloseptal morphogenesis. In addition to these developmental functions, in the adult it plays critical roles in inflammation, wound healing, and tissue repair, and is aberrantly expressed in myriad diseases associated with deregulation of these processes. How might CCN1 play such diverse roles in seemingly unrelated biological contexts? Based on current information discussed above, we speculate that the remarkable functional diversity of CCN1 may depend on several factors. First, through interaction with distinct integrins preferentially expressed in the target cell types, CCN1 can elicit diverse and at times opposing functions (Fig. 1). For example, interaction through αvβ3 and α6β1 can promote either cell survival or cell death, respectively. Second, CCN1 interacts with multiple receptors, some of which may as yet be unidentified, and the combination of these receptors may be required to generate the signaling events necessary for the biological responses. The modular domains of CCN1 contain binding sites for distinct receptors, and the amalgamation of various receptor-binding sites in a single polypeptide allows the activation of signaling pathways by multiple receptors that converge in the cell to trigger unique functions. Third, the biological responses to CCN1 may be significantly influenced by the presence of other growth factors, cytokines, and morphogens in the cellular microenvironment, as evidenced by the ability of CCN1 to alter the activities of TNF-α and modulate Wnt signaling. The possibility that CCN1 may interact with and thus modify the activities of other growth factors such as BMP, TGF-β, and VEGF, as has been demonstrated for the homologous protein CCN2, remains to be explored [16, 18]. Thus, the biological outcome of CCN1 signaling may be highly context-dependent. Given that CCN1 interacts with inflammatory cells and modulates the activities of cytokines, the absence of an inflammatory response during early embryogenesis may underlie some of the differences in CCN1 activities in the embryo versus the adult.
Despite significant advances in recent years, many questions concerning the functions and mechanisms of action of CCN1 remain. Biochemically, the large number of cysteine residues portends a specific pattern of disulfide bonding and tertiary structure, and suggests potential roles for some of the cysteines in metal chelation or sensing oxidative stress. Much remains to be learned about the structure–function relationships of CCN1, including the significance of any post-translational modification. On the organismal level, CCN1 is likely to participate in the development of the neuronal and skeletal systems and in various other organs, and careful analysis of tissue-specific knockout of Ccn1 will be necessary to assess its roles in the development and functions of various organs and tissues. The specific roles of CCN1 in inflammation, wound healing, and tissue repair are just beginning to emerge, and understanding its functions in these contexts is key to unlocking how CCN1 contributes to the myriad pathologies in which it is aberrantly expressed. Future investigations may validate the potential utility of CCN1 as a biomarker or therapeutic target for diseases associated with its deregulation.
Abbreviations
- AVSD:
-
Atrioventricular septal defects
- CTGF:
-
Connective tissue growth factor
- CYR61:
-
Cysteine-rich 61
- ECM:
-
Extracellular matrix
- ERK:
-
Extracellular signal-regulated kinase
- GPCR:
-
G protein-coupled receptor
- HIF-1α:
-
Hypoxia-inducible factor-1α
- HIV-1:
-
Human immunodeficiency virus type 1
- HSPG:
-
Heparan sulfate proteoglycan
- IL:
-
Interleukin
- IRES:
-
Internal ribosome entry sites
- MMP:
-
Matrix metalloproteinase
- MRTF-A:
-
Myocardin-related transcriptional activator
- NSCLC:
-
Non-small-cell lung cancer
- NOV:
-
Nephroblastoma overexpressed
- ROS:
-
Reactive oxygen species
- SASP:
-
Senescence-associated secretory phenotype
- SRE:
-
Serum response element
- SRF:
-
Serum response factor
- TGF-β:
-
Transforming growth factor β
- TNF-α:
-
Tumor necrosis factor α
- TSR:
-
Thrombospondin type I repeat
References
Bornstein P (1995) Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol 130:503–506
Jun J-I, Lau LF (2010) The matricellular protein CCN1/CYR61 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12:676–685
Kyriakides TR, Bornstein P (2003) Matricellular proteins as modulators of wound healing and the foreign body response. Thromb Haemost 90:986–992
Chiodoni C, Colombo MP, Sangaletti S (2010) Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev 29:295–307
Lau LF, Nathans D (1985) Identification of a set of genes expressed during the G0/G1 transition of cultured mouse cells. EMBO J 4:3145–3151
Lau LF, Nathans D (1987) Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc Natl Acad Sci USA 84:1182–1186
Simmons DL, Levy DB, Yannoni Y, Erikson RL (1989) Identification of a phorbol ester-repressible v-src-inducible gene. Proc Natl Acad Sci USA 86:1178–1182
O’Brien TP, Yang GP, Sanders L, Lau LF (1990) Expression of cyr61, a growth factor-inducible immediate-early gene. Mol Cell Biol 10:3569–3577
Brigstock DR (1999) The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr Rev 20:189–206
Lau LF, Lam SC (1999) The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res 248:44–57
Fernando CA, Conrad PA, Bartels CF, Marques T, To M, Balow SA, Nakamura Y, Warman ML (2010) Temporal and spatial expression of CCN genes in zebrafish. Dev Dyn 239:1755–1767
Brigstock DR, Goldschmeding R, Katsube KI, Lam SC, Lau LF, Lyons K, Naus C, Perbal B, Riser B, Takigawa M et al (2003) Proposal for a unified CCN nomenclature. Mol Pathol 56:127–128
Yang GP, Lau LF (1991) Cyr61, product of a growth factor-inducible immediate early gene, is associated with the extracellular matrix and the cell surface. Cell Growth Differ 2:351–357
Kireeva ML, Mo F-E, Yang GP, Lau LF (1996) Cyr61, product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol Cell Biol 16:1326–1334
Kireeva ML, Lam SCT, Lau LF (1998) Adhesion of human umbilical vein endothelial cells to the immediate-early gene product Cyr61 is mediated through integrin αvβ3. J Biol Chem 273:3090–3096
Leask A, Abraham DJ (2006) All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci 119:4803–4810
Holbourn KP, Acharya KR, Perbal B (2008) The CCN family of proteins: structure-function relationships. Trends Biochem Sci 33:461–473
Chen C–C, Lau LF (2009) Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol 41:771–783
Brunner A, Chinn J, Neubauer M, Purchio AF (1991) Identification of a gene family regulated by transforming growth factor-β. DNA Cell Biol 10:293–300
Cui TX, Lin G, Lapensee CR, Calinescu AA, Rathore M, Streeter C, Piwien-Pilipuk G, Lanning N, Jin H, Carter-Su C et al. (2011) C/EBPβ mediates growth hormone-regulated expression of multiple target genes. Mol Endocrinol 25:681–693
Schutze N, Lechner A, Groll C, Siggelkow H, Hufner M, Kohrle J, Jakob F (1998) The human analog of murine cysteine-rich protein 61 is a 1α, 25-dihydroxyvitamin D3 responsive immediate early gene in human fetal osteoblasts: regulation by cytokines, growth factors, and serum. Endocrinology 139:1761–1770
Rivera-Gonzalez R, Petersen DN, Tkalcevic G, Thompson DD, Brown TA (1998) Estrogen-induced genes in the uterus of ovariectomized rats and their regulation by droloxifene and tamoxifen. J Steroid Biochem Molec Biol 64:13–24
Hilfiker A, Hilfiker-Kleiner D, Fuchs M, Kaminski K, Lichtenberg A, Rothkotter HJ, Schieffer B, Drexler H (2002) Expression of CYR61, an angiogenic immediate early gene, in arteriosclerosis and its regulation by angiotensin II. Circulation 106:254–260
Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, Grote K, Kiian I, Wollert KC, Hilfiker A et al (2004) Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation 109:2227–2233
Kunz M, Moeller S, Koczan D, Lorenz P, Wenger RH, Glocker MO, Thiesen HJ, Gross G, Ibrahim SM (2003) Mechanisms of hypoxic gene regulation of angiogenesis factor Cyr61 in melanoma cells. J Biol Chem 278:45651–45660
Quan T, He T, Shao Y, Lin L, Kang S, Voorhees JJ, Fisher GJ (2006) Elevated cysteine-rich 61 mediates aberrant collagen homeostasis in chronologically aged and photoaged human skin. Am J Pathol 169:482–490
Chaqour B, Goppelt-Struebe M (2006) Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J 273:3639–3649
Kivela R, Kyrolainen H, Selanne H, Komi PV, Kainulainen H, Vihko V (2007) A single bout of exercise with high mechanical loading induces the expression of Cyr61/CCN1 and CTGF/CCN2 in human skeletal muscle. J Appl Physiol 103:1395–1401
Kim SM, Park JH, Chung SK, Kim JY, Hwang HY, Chung KC, Jo I, Park SI, Nam JH (2004) Coxsackievirus B3 infection induces cyr61 activation via JNK to mediate cell death. J Virol 78:13479–13488
Wiedmaier N, Muller S, Koberle M, Manncke B, Krejci J, Autenrieth IB, Bohn E (2008) Bacteria induce CTGF and CYR61 expression in epithelial cells in a lysophosphatidic acid receptor-dependent manner. Int J Med Microbiol 298:231–243
Kular L, Pakradouni J, Kitabgi P, Laurent M, Martinerie C (2010) The CCN family: a new class of inflammation modulators? Biochimie 93:377–388
Kurozumi K, Hardcastle J, Thakur R, Shroll J, Nowicki M, Otsuki A, Chiocca EA, Kaur B (2008) Oncolytic HSV-1 infection of tumors induces angiogenesis and upregulates CYR61. Mol Ther 16:1382–1391
Gashaw I, Stiller S, Boing C, Kimmig R, Winterhager E (2008) Premenstrual regulation of the pro-angiogenic factor CYR61 in human endometrium. Endocrinology 149:2261–2269
Pendurthi UR, Allen KE, Ezban M, Rao LV (2000) Factor VIIa and thrombin induce the expression of Cyr61 and connective tissue growth factor, extracellular matrix signaling proteins that could act as possible downstream mediators in factor VIIa x tissue factor-induced signal transduction. J Biol Chem 275:14632–14641
Walsh CT, Radeff-Huang J, Matteo R, Hsiao A, Subramaniam S, Stupack D, Brown JH (2008) Thrombin receptor and RhoA mediate cell proliferation through integrins and cysteine-rich protein 61. FASEB J 22:4011–4021
Liang Y, Li C, Guzman VM, Evinger AJ III, Protzman CE, Krauss AH, Woodward DF (2003) Comparison of PGF2α, Bimatoprost (prostamide) and butaprost (EP2 agonist) on Cyr61 and CTGF gene expression. J Biol Chem 278:27267–27277
Kim YM, Lim SC, Han CY, Kay HY, Cho IJ, Ki SH, Lee MY, Kwon HM, Lee CH, Kim SG (2011) Gα12/13 induction of CYR61 in association with arteriosclerotic intimal hyperplasia: effect of sphingosine-1-phosphate. Arterioscler Thromb Vasc Biol 31:861–869
Walsh CT, Stupack D, Brown JH (2008) G protein-coupled receptors go extracellular: RhoA integrates the integrins. Mol Interv 8:165–173
Latinkic BV, O’Brien TP, Lau LF (1991) Promoter function and structure of the growth factor-inducible immediate early gene cyr61. Nucleic Acids Res 19:3261–3267
Latinkic BV, Mo F-E, Greenspan JA, Copeland NG, Gilbert DJ, Jenkins NA, Lau LF (2001) Promoter function of the angiogenic inducer Cyr61 gene in transgenic mice: tissue specificity, inducibility during wound healing, and role of the serum response element. Endocrinology 142:2549–2557
Hanna M, Liu H, Amir J, Sun Y, Morris SW, Siddiqui MA, Lau LF, Chaqour B (2009) Mechanical regulation of the proangiogenic factor CCN1/CYR61 gene requires the combined activities of MRTF-A and CREB-binding protein histone acetyltransferase. J Biol Chem 284:23125–23136
Grote K, Bavendiek U, Grothusen C, Flach I, Hilfiker-Kleiner D, Drexler H, Schieffer B (2004) Stretch-inducible expression of the angiogenic factor CCN1 in vascular smooth muscle cells is mediated by Egr-1. J Biol Chem 279:55675–55681
Meyuhas R, Pikarsky E, Tavor E, Klar A, Abramovitch R, Hochman J, Lago TG, Honigman A (2008) A key role for cyclic AMP-responsive element binding protein in hypoxia-mediated activation of the angiogenesis factor CCN1 (CYR61) in tumor cells. Mol Cancer Res 6:1397–1409
Wolf N, Yang W, Dunk CE, Gashaw I, Lye SJ, Ring T, Schmidt M, Winterhager E, Gellhaus A (2010) Regulation of the matricellular proteins CYR61 (CCN1) and NOV (CCN3) by hypoxia-inducible factor-1α and transforming-growth factor-β3 in the human trophoblast. Endocrinology 151:2835–2845
Chan SW, Lim CJ, Chong YF, Venkatesan PA, Huang C, Hong W (2011) Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J Biol Chem 286:7018–7026
Zhang H, Pasolli HA, Fuchs E (2011) Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA 108:2270–2275
Leng E, Malcolm T, Tai G, Estable M, Sadowski I (2002) Organization and expression of the cyr61 gene in normal human fibroblasts. J Biomed Sci 9:59–67
Hirschfeld M, zur HA, Bettendorf H, Jager M, Stickeler E (2009) Alternative splicing of Cyr61 is regulated by hypoxia and significantly changed in breast cancer. Cancer Res 69:2082–2090
Zhang Y, Diao Z, Su L, Sun H, Li R, Cui H, Hu Y (2010) MicroRNA-155 contributes to preeclampsia by down-regulating CYR61. Am J Obstet Gynecol 202:466–467
Johannes G, Carter MS, Eisen MB, Brown PO, Sarnow P (1999) Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc Natl Acad Sci USA 96:13118–13123
Mukudai Y, Kubota S, Eguchi T, Sumiyoshi K, Janune D, Kondo S, Shintani S, Takigawa M (2010) A coding RNA segment that enhances the ribosomal recruitment of chicken ccn1 mRNA. J Cell Biochem 111:1607–1618
Pendurthi UR, Tran TT, Post M, Rao LV (2005) Proteolysis of CCN1 by plasmin: functional implications. Cancer Res 65:9705–9711
Lau LF, Lam SCT (2005) Integrin-mediated CCN functions. In: Perbal B, Takigawa M (eds) CCN proteins: a new family of cell growth and differentiation regulators. Imperial College Press, London, pp 61–79
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687
Chen N, Chen CC, Lau LF (2000) Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin α6β1 and cell surface heparan sulfate proteoglycans. J Biol Chem 275:24953–24961
Grzeszkiewicz TM, Lindner V, Chen N, Lam SCT, Lau LF (2002) The angiogenic factor CYR61 supports vascular smooth muscle cell adhesion and stimulates chemotaxis through integrin α6β1 and cell surface heparan sulfate proteoglycans. Endocrinology 143:1441–1450
Jedsadayanmata A, Chen CC, Kireeva ML, Lau LF, Lam SC (1999) Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/Mouse connective tissue growth factor is mediated through integrin αIIbβ3. J Biol Chem 274:24321–24327
Schober JM, Chen N, Grzeszkiewicz TM, Emeson EE, Ugarova TP, Ye RD, Lau LF, Lam SCT (2002) Identification of integrin αMβ2 as an adhesion receptor on peripheral blood moncytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2), immediate-early gene products expressed in atherosclerotic lesions. Blood 99:4457–4465
Bai T, Chen C–C, Lau LF (2010) The matricellular protein CCN1 activates a pro-inflammatory genetic program in murine macrophages. J Immunol 184:3223–3232
Yakubenko VP, Yadav SP, Ugarova TP (2006) Integrin αDβ2, an adhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood 107:1643–1650
Todorovic V, Chen C–C, Hay N, Lau LF (2005) The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol 171:559–568
Chen C–C, Young JL, Monzon RI, Chen N, Todorovic V, Lau LF (2007) Cytotoxicity of TNFα is regulated by integrin-mediated matrix signaling. EMBO J 26:1257–1267
Schober JM, Lau LF, Ugarova TP, Lam SC (2003) Identification of a novel integrin αMβ2 binding site in CCN1 (CYR61), a matricellular protein expressed in healing wounds and atherosclerotic lesions. J Biol Chem 278:25808–25815
Leu S-J, Liu Y, Chen N, Chen CC, Lam SC, Lau LF (2003) Identification of a novel integrin α6β1 binding site in the angiogenic Inducer CCN1 (CYR61). J Biol Chem 278:33801–33808
Chen N, Leu S-J, Todorovic V, Lam SCT, Lau LF (2004) Identification of a novel integrin αvβ3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem 279:44166–44176
Leu S-J, Chen N, Chen C–C, Todorovic V, Bai T, Juric V, Liu Y, Yan G, Lam SCT, Lau LF (2004) Targeted mutagenesis of the matricellular protein CCN1 (CYR61): selective inactivation of integrin α6β1-heparan sulfate proteoglycan coreceptor-mediated cellular activities. J Biol Chem 279:44177–44187
Segarini PR, Nesbitt JE, Li D, Hayes LG, Yates JR III, Carmichael DF (2001) The low density lipoprotein receptor-related protein/α2-Macroglobulin receptor is a receptor for connective tissue growth factor (CTGF). J Biol Chem 276:40659–40667
Wahab NA, Weston BS, Mason RM (2005) Connective tissue growth factor CCN2 interacts with and activates the tyrosine kinase receptor TrkA. J Am Soc Nephrol 16:340–351
Sakamoto K, Yamaguchi S, Ando R, Miyawaki A, Kabasawa Y, Takagi M, Li CL, Perbal B, Katsube K (2002) The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J Biol Chem 277:29399–29405
Chen C–C, Chen N, Lau LF (2001) The angiogenic factors Cyr61 and CTGF induce adhesive signaling in primary human skin fibroblasts. J Biol Chem 276:10443–10452
Grzeszkiewicz TM, Kirschling DJ, Chen N, Lau LF (2001) CYR61 stimulates human skin fibroblasts migration through integrin αvβ5 and enhances mitogenesis through integrin αvβ3, independent of its carboxyl-terminal domain. J Biol Chem 276:21943–21950
Leu S-J, Lam SCT, Lau LF (2002) Proangiogenic activities of CYR61 (CCN1) mediated through integrins αvβ3 and α6β1 in human umbilical vein endothelial cells. J Biol Chem 277:46248–46255
Lin BR, Chang CC, Chen LR, Wu MH, Wang MY, Kuo IH, Chu CY, Chang KJ, Lee PH, Chen WJ et al (2007) Cysteine-rich 61 (CCN1) enhances chemotactic migration, transendothelial cell migration, and intravasation by concomitantly up-regulating chemokine receptor 1 and 2. Mol Cancer Res 5:1111–1123
Sun ZJ, Wang Y, Cai Z, Chen PP, Tong XJ, Xie D (2008) Involvement of Cyr61 in growth, migration, and metastasis of prostate cancer cells. Br J Cancer 99:1656–1667
Kassis JN, Virador VM, Guancial EA, Kimm D, Ho AS, Mishra M, Chuang EY, Cook J, Gius D, Kohn EC (2009) Genomic and phenotypic analysis reveals a key role for CCN1 (CYR61) in BAG3-modulated adhesion and invasion. J Pathol 218:495–504
Sakamoto S, Yokoyama M, Aoki M, Suzuki K, Kakehi Y, Saito Y (2004) Induction and function of CYR61 (CCN1) in prostatic stromal and epithelial cells: CYR61 is required for prostatic cell proliferation. Prostate 61:305–317
Franzen CA, Chen CC, Todorovic V, Juric V, Monzon RI, Lau LF (2009) The matrix protein CCN1 is critical for prostate carcinoma cell proliferation and TRAIL-induced apoptosis. Mol Cancer Res 7:1045–1055
Gery S, Xie D, Yin D, Gabra H, Miller C, Wang H, Scott D, Yi WS, Popoviciu ML, Said JW et al (2005) Ovarian carcinomas: CCN genes are aberrantly expressed and CCN1 promotes proliferation of these cells. Clin Cancer Res 11:7243–7254
Zhang Q, Wu J, Cao Q, Xiao L, Wang L, He D, Ouyang G, Lin J, Shen B, Shi Y et al (2009) A critical role of Cyr61 in interleukin-17-dependent proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Rheum 60:3602–3612
Jin Y, Kim HP, Ifedigbo E, Lau LF, Choi AM (2005) Cyr61 protects against hyperoxia induced cell death via Akt pathway in pulmonary epithelial cells. Am J Respir Cell Mol Biol 33:297–302
Lin MT, Chang CC, Chen ST, Chang HL, Su JL, Chau YP, Kuo ML (2004) Cyr61 expression confers resistance to apoptosis in breast cancer MCF-7 cells by a mechanism of NF-κB-dependent XIAP up-regulation. J Biol Chem 279:24015–24023
O’Brien TP, Lau LF (1992) Expression of the growth factor-inducible immediate early gene cyr61 correlates with chondrogenesis during mouse embryonic development. Cell Growth Differ 3:645–654
Babic AM, Kireeva ML, Kolesnikova TV, Lau LF (1998) CYR61, product of a growth factor-inducible immediate-early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci USA 95:6355–6360
Fataccioli V, Abergel V, Wingertsmann L, Neuville P, Spitz E, Adnot S, Calenda V, Teiger E (2002) Stimulation of angiogenesis by cyr61 gene: a new therapeutic candidate. Hum Gene Ther 13:1461–1470
Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF (2002) CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol 22:8709–8720
Mo F-E, Lau LF (2006) The matricellular protein CCN1 is essential for cardiac development. Circ Res 99:961–969
Chen C–C, Mo F-E, Lau LF (2001) The angiogenic inducer Cyr61 induces a genetic program for wound healing in human skin fibroblasts. J Biol Chem 276:47329–47337
Grote K, Salguero G, Ballmaier M, Dangers M, Drexler H, Schieffer B (2007) The angiogenic factor CCN1 promotes adhesion and migration of circulating CD34 + progenitor cells: potential role in angiogenesis and endothelial regeneration. Blood 110:877–885
Wong M, Kireeva ML, Kolesnikova TV, Lau LF (1997) Cyr61, product of a growth factor-inducible immediate-early gene, regulates chondrogenesis in mouse limb bud mesenchymal cells. Dev Biol 192:492–508
Si W, Kang Q, Luu HH, Park JK, Luo Q, Song WX, Jiang W, Luo X, Li X, Yin H et al (2006) CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol Cell Biol 26:2955–2964
Crockett JC, Schutze N, Tosh D, Jatzke S, Duthie A, Jakob F, Rogers MJ (2007) The matricellular protein CYR61 inhibits osteoclastogenesis by a mechanism independent of αvβ3 and αvβ5. Endocrinology 148:5761–5768
Su JL, Chiou J, Tang CH, Zhao M, Tsai CH, Chen PS, Chang YW, Chien MH, Peng CY, Hsiao M et al (2010) CYR61 regulates BMP-2-dependent osteoblast differentiation through the αvβ3 integrin/integrin-linked kinase/ERK pathway. J Biol Chem 285:31325–31336
Tamura I, Rosenbloom J, Macarak E, Chaqour B (2001) Regulation of Cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am J Physiol Cell Physiol 281:C1524–C1532
Sabbah M, Prunier C, Ferrand N, Megalophonos V, Lambein K, De WO, Nazaret N, Lachuer J, Dumont S, Redeuilh G (2011) CCN5, a novel transcriptional repressor of transforming growth factor-β signaling pathway. Mol Cell Biol 31:1459–1469
Gellhaus A, Schmidt M, Dunk C, Lye SJ, Kimmig R, Winterhager E (2006) Decreased expression of the angiogenic regulators CYR61 (CCN1) and NOV (CCN3) in human placenta is associated with pre-eclampsia. Mol Hum Reprod 12:389–399
Gellhaus A, Schmidt M, Dunk C, Lye SJ, Winterhager E (2007) The circulating proangiogenic factors CYR61 (CCN1) and NOV (CCN3) are significantly decreased in placentae and sera of preeclamptic patients. Reprod Sci 14:46–52
Sheffield VC, Pierpont ME, Nishimura D, Beck JS, Burns TL, Berg MA, Stone EM, Patil SR, Lauer RM (1997) Identification of a complex congenital heart defect susceptibility locus by using DNA pooling and shared segment analysis. Hum Mol Genet 6:117–121
Jay P, Berge-Lefranc JL, Marsollier C, Mejean C, Taviaux S, Berta P (1997) The human growth factor-inducible immediate early gene, CYR61, maps to chromosome 1p. Oncogene 14:1753–1757
Latinkic BV, Mercurio S, Bennett B, Hirst EM, Xu Q, Lau LF, Mohun TJ, Smith JC (2003) Xenopus Cyr61 regulates gastrulation movements and modulates Wnt signalling. Development 130:2429–2441
Mongiat M, Ligresti G, Marastoni S, Lorenzon E, Doliana R, Colombatti A (2007) Regulation of the extrinsic apoptotic pathway by the extracellular matrix glycoprotein EMILIN2. Mol Cell Biol 27:7176–7187
Chen CC, Lau LF (2010) Deadly liaisons: fatal attraction between CCN matricellular proteins and the tumor necrosis factor family of cytokines. J Cell Commun Signal 4:63–69
Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3:745–756
Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104:487–501
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005) Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120:649–661
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M (2006) The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIP(L) turnover. Cell 124:601–613
Juric V, Chen CC, Lau LF (2009) Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol Cell Biol 29:3266–3279
Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP, Tiegs G (1998) Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology 114:1035–1045
Wolf D, Hallmann R, Sass G, Sixt M, Kusters S, Fregien B, Trautwein C, Tiegs G (2001) TNF-α-induced expression of adhesion molecules in the liver is under the control of TNFR1—relevance for concanavalin A-induced hepatitis. J Immunol 166:1300–1307
Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S (1993) Lethal effect of the anti-Fas antibody in mice. Nature 364:806–809
Zhou Z, Sun X, Kang YJ (2001) Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol 159:329–338
Chen CC, Juric V, Lau LF (2011) The extracellular matrix protein CCN1 dictates TNFα and FasL cytotoxicity in vivo. Adv Exp Med Biol 691:595–603
Campisi J, d’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8:729–740
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130:223–233
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6:2853–2868
Kuilman T, Peeper DS (2009) Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 9:81–94
Young AR, Narita M (2009) SASP reflects senescence. EMBO Rep 10:228–230
Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J Med 341:738–746
Gurtner GC, Werner S, Barrandon Y, Longaker MT (2008) Wound repair and regeneration. Nature 453:314–321
Shaw TJ, Martin P (2009) Wound repair at a glance. J Cell Sci 122:3209–3213
Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214:199–210
Stramer BM, Mori R, Martin P (2007) The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol 127:1009–1017
Eming SA, Krieg T, Davidson JM (2007) Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 127:514–525
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445:656–660
Jun JI, Lau LF (2010) Cellular senescence controls fibrosis in wound healing. Aging (Albany, NY) 2:627–631
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134:657–667
Rodier F, Campisi J (2011) Four faces of cellular senescence. J Cell Biol 192:547–556
Hadjiargyrou M, Ahrens W, Rubin CT (2000) Temporal expression of the chondrogenic and angiogenic growth factor CYR61 during fracture repair. J Bone Miner Res 15:1014–1023
Nakata E, Nakanishi T, Kawai A, Asaumi K, Yamaai T, Asano M, Nishida T, Mitani S, Inoue H, Takigawa M (2002) Expression of connective tissue growth factor/hypertrophic chondrocyte-specific gene product 24 (CTGF/Hcs24) during fracture healing. Bone 31:441–447
Athanasopoulos AN, Schneider D, Keiper T, Alt V, Pendurthi UR, Liegibel UM, Sommer U, Nawroth PP, Kasperk C, Chavakis T (2007) Vascular endothelial growth factor (VEGF)-induced up-regulation of CCN1 in osteoblasts mediates proangiogenic activities in endothelial cells and promotes fracture healing. J Biol Chem 282:26746–26753
Sawai K, Mukoyama M, Mori K, Kasahara M, Koshikawa M, Yokoi H, Yoshioka T, Ogawa Y, Sugawara A, Nishiyama H et al (2007) Expression of CCN1 (CYR61) in developing, normal, and diseased human kidney. Am J Physiol Renal Physiol 293:F1363–F1372
Cooker LA, Peterson D, Rambow J, Riser ML, Riser RE, Najmabadi F, Brigstock D, Riser BL (2007) TNF-alpha, but not IFN-gamma, regulates CCN2 (CTGF), collagen type I, and proliferation in mesangial cells: possible roles in the progression of renal fibrosis. Am J Physiol Renal Physiol 293:F157–F165
Koon HW, Zhao D, Xu H, Bowe C, Moss A, Moyer MP, Pothoulakis C (2008) Substance P-mediated expression of the pro-angiogenic factor CCN1 modulates the course of colitis. Am J Pathol 173:400–410
Haas CS, Creighton CJ, Pi X, Maine I, Koch AE, Haines GK, Ling S, Chinnaiyan AM, Holoshitz J (2006) Identification of genes modulated in rheumatoid arthritis using complementary DNA microarray analysis of lymphoblastoid B cell lines from disease-discordant monozygotic twins. Arthr Rheum 54:2047–2060
Kok SH, Hou KL, Hong CY, Wang JS, Liang PC, Chang CC, Hsiao M, Yang H, Lai EH, Lin SK (2011) Simvastatin inhibits cytokine-stimulated Cyr61 expression in osteoblastic cells: a therapeutic benefit for arthritis. Arthr Rheum 63:1010–1020
Sigala F, Georgopoulos S, Papalambros E, Chasiotis D, Vourliotakis G, Niforou A, Kotsinas A, Kavantzas N, Patsouris E, Gorgoulis VG et al (2006) Heregulin, cysteine rich-61 and matrix metalloproteinase 9 expression in human carotid atherosclerotic plaques: relationship with clinical data. Eur J Vasc Endovasc Surg 32:238–245
Lee HY, Chung JW, Youn SW, Kim JY, Park KW, Koo BK, Oh BH, Park YB, Chaqour B, Walsh K et al (2007) Forkhead transcription factor FOXO3a is a negative regulator of angiogenic immediate early gene CYR61, leading to inhibition of vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res 100:372–380
Matsumae H, Yoshida Y, Ono K, Togi K, Inoue K, Furukawa Y, Nakashima Y, Kojima Y, Nobuyoshi M, Kita T et al (2008) CCN1 knockdown suppresses neointimal hyperplasia in a rat artery balloon injury model. Arterioscler Thromb Vasc Biol 28:1077–1083
Yu Y, Gao Y, Qin J, Kuang CY, Song MB, Yu SY, Cui B, Chen JF, Huang L (2010) CCN1 promotes the differentiation of endothelial progenitor cells and reendothelialization in the early phase after vascular injury. Basic Res Cardiol 105:713–724
Hasan A, Pokeza N, Shaw L, Lee HS, Lazzaro D, Chintala H, Rosenbaum D, Grant MB, Chaqour B (2011) The matricellular protein cysteine-rich protein 61 (CCN1/Cyr61) enhances physiological adaptation of retinal vessels and reduces pathological neovascularization associated with Ischemic retinopathy. J Biol Chem 286:9542–9554
Folkman J (2006) Angiogenesis. Annu Rev Med 57:1–18
Monnier Y, Farmer P, Bieler G, Imaizumi N, Sengstag T, Alghisi GC, Stehle JC, Ciarloni L, ndrejevic-Blant S, Moeckli R et al (2008) CYR61 and αvβ5 integrin cooperate to promote invasion and metastasis of tumors growing in preirradiated stroma. Cancer Res 68:7323–7331
Xie D, Miller CW, O’Kelly J, Nakachi K, Sakashita A, Said JW, Gornbein J, Koeffler HP (2001) Breast cancer. Cyr61 is overexpressed, estrogen-inducible, and associated with more advanced disease. J Biol Chem 276:14187–14194
Kok SH, Chang HH, Tsai JY, Hung HC, Lin CY, Chiang CP, Liu CM, Kuo MY (2010) Expression of Cyr61 (CCN1) in human oral squamous cell carcinoma: an independent marker for poor prognosis. Head Neck 32:1665–1673
Haque I, Mehta S, Majumder M, Dhar K, De A, McGregor D, Vanveldhuizen PJ, Banerjee SK, Banerjee S (2011) Cyr61/CCN1 signaling is critical for epithelial-mesenchymal transition and stemness and promotes pancreatic carcinogenesis. Mol Cancer 10:8
Tsai MS, Hornby AE, Lakins J, Lupu R (2000) Expression and function of CYR61, an angiogenic factor, in breast cancer cell lines and tumor biopsies. Cancer Res 60:5603–5607
Xie D, Nakachi K, Wang H, Elashoff R, Koeffler HP (2001) Elevated levels of connective tissue growth factor, WISP-1, and CYR61 in primary breast cancers associated with more advanced features. Cancer Res 61:8917–8923
Jiang WG, Watkins G, Fodstad O, Douglas-Jones A, Mokbel K, Mansel RE (2004) Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr Relat Cancer 11:781–791
O’Kelly J, Chung A, Lemp N, Chumakova K, Yin D, Wang HJ, Said J, Gui D, Miller CW, Karlan BY et al (2008) Functional domains of CCN1 (Cyr61) regulate breast cancer progression. Int J Oncol 33:59–67
D’Antonio KB, Toubaji A, Albadine R, Mondul AM, Platz EA, Netto GJ, Getzenberg RH (2010) Extracellular matrix associated protein CYR61 is linked to prostate cancer development. J Urol 183:1604–1610
Goodwin CR, Lal B, Zhou X, Ho S, Xia S, Taeger A, Murray J, Laterra J (2010) Cyr61 mediates hepatocyte growth factor-dependent tumor cell growth, migration, and Akt activation. Cancer Res 70:2932–2941
Lin MT, Zuon CY, Chang CC, Chen ST, Chen CP, Lin BR, Wang MY, Jeng YM, Chang KJ, Lee PH et al (2005) Cyr61 induces gastric cancer cell motility/invasion via activation of the integrin/nuclear factor-κB/cyclooxygenase-2 signaling pathway. Clin Cancer Res 11:5809–5820
Tsai MS, Bogart DF, Castaneda JM, Li P, Lupu R (2002) Cyr61 promotes breast tumorigenesis and cancer progression. Oncogene 21:8178–8185
Menendez JA, Vellon L, Mehmi I, Teng PK, Griggs DW, Lupu R (2005) A novel CYR61-triggered ‘CYR61-αvβ3 integrin loop’ regulates breast cancer cell survival and chemosensitivity through activation of ERK1/ERK2 MAPK signaling pathway. Oncogene 24:761–779
Vellon L, Menendez JA, Lupu R (2005) αvβ3 integrin regulates heregulin (HRG)-induced cell proliferation and survival in breast cancer. Oncogene 24:3759–3773
Schmitt CA (2003) Senescence, apoptosis and therapy—cutting the lifelines of cancer. Nat Rev Cancer 3:286–295
Lv H, Fan E, Sun S, Ma X, Zhang X, Han DM, Cong YS (2009) Cyr61 is up-regulated in prostate cancer and associated with the p53 gene status. J Cell Biochem 106:738–744
Tong X, Xie D, O’Kelly J, Miller CW, Muller-Tidow C, Koeffler HP (2001) Cyr61, a member of CCN family, is a tumor suppressor in non-small cell lung cancer. J Biol Chem 276:47709–47714
Tong X, O’Kelly J, Xie D, Mori A, Lemp N, McKenna R, Miller CW, Koeffler HP (2004) Cyr61 suppresses the growth of non-small-cell lung cancer cells via the β-catenin-c-myc-p53 pathway. Oncogene 23:4847–4855
Feng P, Wang B, Ren EC (2008) Cyr61/CCN1 is a tumor suppressor in human hepatocellular carcinoma and involved in DNA damage response. Int J Biochem Cell Biol 40:98–109
Chien W, Kumagai T, Miller CW, Desmond JC, Frank JM, Said JW, Koeffler HP (2004) Cyr61 suppresses growth of human endometrial cancer cells. J Biol Chem 279:53087–53096
Dobroff AS, Wang H, Melnikova VO, Villares GJ, Zigler M, Huang L, Bar-Eli M (2009) Silencing cAMP-response element binding protein (CREB) identifies cysteine-rich protein 61 (CYR61) as a tumor suppressor gene in melanoma. J Biol Chem 284:26194–26206
Yoshida Y, Togi K, Matsumae H, Nakashima Y, Kojima Y, Yamamoto H, Ono K, Nakamura T, Kita T, Tanaka M (2007) CCN1 protects cardiac myocytes from oxidative stress via β1 integrin-Akt pathway. Biochem Biophys Res Commun 355:611–618
Tan TW, Yang WH, Lin YT, Hsu SF, Li TM, Kao ST, Chen WC, Fong YC, Tang CH (2009) Cyr61 increases migration and MMP-13 expression via αvβ3 integrin, FAK, ERK and AP-1-dependent pathway in human chondrosarcoma cells. Carcinogenesis 30:258–268
Acknowledgments
I thank Chih-Chiun Chen and Joon-Il Jun for helpful comments on the manuscript. This work was supported by grants (CA46565, GM78492 and HL81390) from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lau, L.F. CCN1/CYR61: the very model of a modern matricellular protein. Cell. Mol. Life Sci. 68, 3149–3163 (2011). https://doi.org/10.1007/s00018-011-0778-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0778-3