
Abstract
Cyclin dependent kinase 5 regulatory subunit-associated protein 2 (CDK5RAP2) has gained attention in the last years following the discovery, in 2005, that recessive mutations cause primary autosomal recessive microcephaly. This disease is seen as an isolated developmental defect of the brain, particularly of the cerebral cortex, and was thus historically also referred to as microcephalia vera. Unraveling the pathomechanisms leading to this human disease is fascinating scientists because it can convey insight into basic mechanisms of physiologic brain development (particularly of cortex formation). It also finds itself in the spotlight because of its implication in trends in mammalian evolution with a massive increase in the size of the cerebral cortex in primates. Here, we provide a timely overview of the current knowledge on the function of CDK5RAP2 and mechanisms that might lead to disease in humans when the function of this protein is disturbed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyclin-dependent kinase 5, regulatory subunit-associated protein 2 (CDK5RAP2), also referred to as centrosome-associated protein 215 (CEP215), CDK5 activator binding protein C48 (C48; a fragment), KIAA1633 and DKFZp686D1070, has moved into the spotlight because of its central function in human brain development and evolution. Mutations in the CDK5RAP2 gene cause the rare human disease primary autosomal recessive microcephaly (MCPH, for MicroCephaly Primary Hereditary) type 3 (Figs. 1, 2; Table 1). Here, we provide a timely overview of current knowledge on the function of CDK5RAP2 and mechanisms that might lead to disease in humans when the function of this protein is disturbed.

Main features of patients with MCPH. a Illustration depicting typical coronal brain sections from a patient with MCPH and a control. Note the significant reduction in cerebral cortex volume (as shown by arrows) and the simplification of the gyral pattern in the MCPH patient in comparison to a control patient. b Main features seen in MCPH patients are listed

Position of mutations within the CDK5RAP2 gene and protein domains. The coding and non-coding regions of the CDK5RAP2 gene are drawn to scale, and the exons are depicted as boxes (reference sequences according to NCBI genome viewer build 35.1 and Ensembl release 54): the localization and type of published mutations are given as well as the number of times a specific mutation has been reported. Known and predicted MCPH protein domains are presented, and the full-length protein is depicted even though four isoforms have been described (see Fig. 3). The exact positions of the two reported structural maintenance-of-chromosome (SMC) domains [7] within the protein (SMC AA137-499, SMC_N AA793-1040; accession no. CAI16963) as well as that of the interaction site with CDK5R1 (AA1726–1768) are not clear and can only be predicted. The C-terminal Cnn Motif 2 (CM2) domain is known for Golgi complex interaction and binding to calmodulin. γTuRC gamma tubulin ring complex, EB1 plus-end binding protein 1, CDK5R1 cyclin dependent kinase 5 regulatory kinase 1
CDK5RAP2 gene and protein structure
Human CDK5RAP2 was first cloned and designated as KIAA1633 by Nagase et al. [1] through sequencing clones obtained from a size-fractionated fetal brain cDNA library. In the same year, Ching et al. workers obtained a partial, 24 kDa CDK5RAP2 clone, designated as C48 through a yeast two-hybrid screen of two rat brain cDNA libraries using CDK5 regulatory kinase 1 CDK5R1 as bait (CDK5R1; syn. Cdk5 activator binding protein or p23; two CDK5R1 isoforms: p35 and truncated form p25). They isolated the full sequence using the novel cDNA sequence as a probe and demonstrated its interaction with CDK5R1 in an affinity pulldown assay [2]. CDK5RAP2 interacts with the hydrophilic face of CDK5R1 [3], while the hydrophobic face of CDK5R1 is involved in CDK5 activation [4].
The human full length, 1893 aa CDK5RAP2 protein (NP_060719.4, Q96SN8) contains a predicted N-terminal interaction site with the gamma tubulin ring complex (γTuRC, 59-133 aa; syn. CM1 [5]), two predicted structural maintenance-of-chromosomes (SMC) sites (137–499 aa, 1395–1653 aa) known to be important for chromatid cohesion and DNA recombination during meiosis and mitosis [6–8], a Ser-rich motif for interaction with plus-end binding protein EB1 (926-1208 aa; [9]), an interaction site with pericentrin (within 1726-1893 aa; [10]), a C-terminal Cnn Motif 2 (CM2) domain for Golgi complex interaction and binding to calmodulin (within 1861-1870 aa; [10]) and a C-terminal interaction site with CDK5R1, an activator or modulator of CDK5 ([2]; Fig. 2).
Structure prediction using the method of Lupas et al. [11] revealed that CDK5RAP2 contains predominantly coiled-coil domains and may form a filamentous structure, which is a characteristic of pericentriolar matrix scaffold proteins [5]. Indeed, alignment of CDK5RAP2 to the NCBI GenBank conserved domains database reveals homologies to SMC proteins (COG1196, TIGR02168) involved in cell division and chromosome partitioning. These weak but significant homologies span approximately sequences aa 150–600 and aa 1300–1650 of CDK5RAP2. A direct alignment of full-length human CDK5RAP2 to human SMC1 (NP_006297) identifies stretches of homology that lie on both sides of the hinge region of SMC1 in the two long coiled-coil motifs connecting the highly conserved nucleotide binding N-terminal Walker A and C-terminal Walker B domains (for review [12]). Both CDK5RAP2 and SMC1 contain long coiled-coil motifs interrupted by a rather unstructured domain (Fig. 3). The latter has been discussed, in the case of SMC, to be a flexible hinge that allows the protein to gain its V-shaped structure necessary for the C- and the N-terminal domains to come in close contact. For this purpose, a hetero- or homodimer is generated using the coiled-coil domains to interact in an anti-parallel fashion. These structural similarities between CDK5RAP2 and SMC1 indicate potentially similar mechanism (dimer formation) for CDK5RAP2.

Predicted coils in human SMC1 and CDK5RAP2. Coiled coil regions were predicted in (a) human SMC1 and (b) human CDK5RAP2 proteins using the corresponding EMBnet program (http://www.ch.embnet.org/software/COILS_form.html) according to Lupas et al. [11]. COILS is a program that compares a sequence to a database of known parallel two-stranded coiled-coils and derives a similarity score. By comparing this score to the distribution of scores in globular and coiled-coil proteins, the program then calculates the probability that the sequence (amino acid position represented in x-axis) will adopt a coiled-coil conformation (probability 0–1 represented in y-axis). A comparison of the predicted conformation of human SMC1 (structural maintenance of chromosomes protein 1A, NP_006297.2) and human CDK5RAP2 (NP_060719.4) reveals structural similarities between the two proteins. Both are composed of two long coiled-coil motifs connected by a rather unstructured domain that in the case of SMC proteins has been predicted to act as a flexible hinge that allows the proteins to gain a V-shaped structure necessary for the C-and N-terminal domains to come in close contact. The settings of the coils program were set to default, using a window width of 14, 21 and 28 amino acids
Four isoforms produced by alternative splicing have been reported so far: (1) isoform 1, Q96SN8-1, the full-length form; (2) isoform 2, Q96SN8-2 missing 702–733 aa (exon 19); (3) isoform 3, Q96SN8-3 missing 1009–1049 (exon 23); (4) isoform 4, Q96SN8-4 missing 1576–1654 (exon 32) (Fig. 4). The in vivo existence of these isoforms in humans is confirmed by Genbank EST data. None of the functional motifs identified so far are encoded by the exons alternatively spliced to generate the CDK5RAP2 isoforms. Thus, the specific functions of the four isoforms remain to be elucidated. Similarly to the situation in humans, comparable isoforms also exist in other organisms such as mice and rats, and have been confirmed by GenBank in silico analysis. For mice and rats, the full-length isoform containing all three alternatively spliced exons is not represented in the “non-redundant nucleotide” or “non-redundant protein database” GenBank; however, a search of GenBank sequences shows the presence of the respective homologous exons on the genomic level. Further isoforms generated through co- and/or posttranslational modifications or degradation may exist as authors have described bands of 70, 90, 105 and 230 kDa in mice depending on the age and organ investigated as well as the anti-Cdk5rap2 antibody applied (see below and [13, 14]).

Alignment of the four human CDK5RAP2 isoforms. The human CDK5RAP2 gene comprises 38 exons, and isoform Q96SN8-1 contains all of these 38 exons. Three further isoforms produced by alternative splicing have been reported so far: (1) isoform Q96SN8-2 missing exon 19 (aa 702–733); (2) isoform Q96SN8-3 missing exon 23 (aa 1009–1049); and (3) isoform Q96SN8-4 lacking exon 32 (aa 1576–1654). The in vivo existence of these isoforms in humans is confirmed by GenBank EST evidence. None of the functional motifs identified so far (see Fig. 2) are encoded by the alternatively spliced exons of the CDK5RAP2 isoforms. Similarly to the situation in humans, comparable isoforms also exist in other organisms such as mice and rats, and have been confirmed by GenBank in silico analysis. In mice and rats, the isoform 1 is not represented in the GenBank; however, a search of GenBank sequences shows the presence of the respective homologous exon on the genomic level
CDK5RAP2 in other organisms
CDK5RAP2 is a highly conserved protein, and ortholog genes with similar domain structures are also found in other organisms such as in apes, cows, dogs, rats, mice and chicken (Fig. 5; Supplementary Figure 1).


CDK5RAP2 sequence in various organisms. Sequences of ortholog gene products are compared to that of human CDK5RAP2: orangutan (Pongo abelii), rhesus monkey (Macaca mulatta), cow (Bos taurus), dog (Canis lupus familiaris), rat (Rattus norvegicus), mouse (Mus musculus) and chicken (Gallus gallus). Detailed information on sequences used for alignments can be found in the supplementary information box 1
Other CDK5RAP proteins
In addition to CDK5RAP2, two further CDK5RAPs exist and are referred to as CDK5RAP1 and CDK5RAP3 or C42 and C53, respectively. While CDK5RAP1–3 compete for a common N-terminal region of (free or CDK5-bound) CDK5R1 (p35), no common CDK5RAP structural motif specific for CDK5R1 (p35) binding has been identified so far [3]. Various studies underline the importance of these CDK5RAPs in the regulation of the cell cycle, DNA damage checkpoints, and thus proliferation and apoptosis (e.g., [15, 16]), and they might thus have an interesting connection to developmental defects of the brain. Similar to CDK5RAP2, the other two CDK5RAPs are expressed in various organs.
Expression and localization of CDK5RAP2
CDK5RAP2 mRNA is widely expressed in human and in embryonal mouse tissue [2, 14, 17]. In murine embryos at E15.5, the highest levels were detected in the central nervous system (CNS; [17]). In humans, moderate to high expression was detected in all adult tissues and specific brain regions examined by quantitative RT-PCR ELISA with the highest levels in skeletal muscle, fetal liver, brain, kidney and ovary [1]. Within the brain, expression was particularly high within the thalamus, corpus callosum, substantia nigra, hippocampus and caudate nucleus [1]. In murine whole-brain protein extracts, Cdk5rap2 was detected at high levels between E10.5 and E13.5, remained robust at E15.5 and subsequently decreased with development [14]. At the same time, the authors detected a smaller band of about 90 kDa through Western blotting that first appeared at E13.5 and then increased in intensity, paralleling the decrease of the 200 kDa band. Similarly, Lizarraga et al. [13] detected a large, 230-kDa isoform in most tissues in addition to a smaller, approximately 105-kDa isoform in adult testis, thymus and brain as well as a 70-kDa testis-specific isoform through Western blots using adult tissues. In these organs, a 2.3-kb transcript was found to be expressed in most tissues, a 5.8-kb transcript in several tissues (particularly in testis and thymus) and a 1.4-kb transcript only in the testis by Northern blots. This indicates that there may be a change of gene expression and/or protein isoform synthesis throughout murine brain development and when comparing various organs. At E14.5, punctuate, centrosomal Cdk5rap2 signal was detected in young, Tuj1-positve neurons (Tuj1 = neuron-specific class III beta-tubulin) within the cortex. The most intense centrosomal signal was present at the luminal surface of the ventricular zone in neural stem cell marker nestin-positive progenitors.
CDK5RAP2 was localized to the centrosomes in the interphase and also to the spindle poles during mitosis in the human epithelial cervical cancer cell line HeLa [5, 17, 18]. Recently, the subcellular localization of Cdk5rap2 during the cell cycle was described in detail via immuncytology in murine fibroblasts (antibody directed against the N-terminus, [19]). The authors found that Cdk5rap2 levels were relatively low at interphase centrosomes in a microtubule-independent way. The centrosomal Cdk5rap2 levels increased in the prophase and remained high throughout mitosis until the telophase, when the signal dropped again to interphase levels. These results underline that Cdk5rap2 is a centrosomal protein, and they illustrate that centrosomal Cdk5rap2 is regulated in a cell cycle-dependent manner. In addition to its centrosomal localization, CDK5RAP2 has been described to localize to the distal, growing tips of microtubules in human tumor cells (embryonic kidney cell line HEK239; HeLa and human osteosarcoma cell line U2OS) but not in rodent cells [9]. In these human tumor cells, CDK5RAP2 has been shown to regulate the plus-end dynamics via interaction with plus-end binding protein EB1, a prototypic member of the microtubule plus-end tracking proteins [9]. Only recently, the localization of CDK5RAP2 to the Golgi complex lasting throughout the interphase until mitosis has been demonstrated through colocalization with trans-Golgi network marker TGN46 and cis/medial-Golgi marker mannosidase II in HeLa, MCF7 and RPE1 cells (MCF7 = breast cancer cell line; RPE1 = human telomerase-immortalized retinal epithelial cell line [10]; Fig. 6). It has been further shown that the Golgi-localization of CDK5RAP2 is independent of microtubules, but dependent on centrosome function and energy supply [10]. It will be intriguing to identify further interaction partners of CDK5RAP2.

Localization of CDK5RAP2 protein during the interphase and in mitosis. CDK5RAP2 protein is found at the centrosome and the Golgi complex during the interphase, and is localized at the centrosome in the mitotic cell (shown in tumor cells). Also, this protein has been localized to the plus end of microtubules. During the interphase cells are growing and duplicate their DNA (blue) in order to later divide during mitosis through the mitotic spindle apparatus (orange). Growth and division of cells needs a constant supply of new proteins and lipids to the site of synthesis in the endoplasmatic reticulum via the Golgi apparatus (green). During mitosis, the Golgi apparatus is fragmented into many small vesicles that are subsequently divided upon the later daughter cells
CDK5RAP2 as a regulator of CDK5R1 and CDK5
CDK5RAP2 has been suggested to inhibit or regulate CDK5 indirectly through in vitro studies. Ching et al. [2] demonstrated that high concentrations of C48 (resembling the C-terminal fragment of Cdk5rap2) were able to inhibit Cdk5 kinase activity up to about 20%, and their data also indicate that Cdk5rap2 can be phosphorylated by Cdk5. Apart from controlling Cdk5 kinase activity, a regulation of CDK5R1 isoform activity may also be important for the intracellular localization and substrate specificity of Cdk5 [20–26]. Still, the mechanism of the CDK5RAP2 inhibitory effect has not been completely elucidated, and convincing data illustrating especially an in vivo interaction with CDK5/p35 in the context of human brain development are lacking. While CDK5RAPs show a widespread tissue distribution, their target CDK5R1 is expressed mainly in the CNS. This indicates that CDK5R1-binding may not represent the primary function of these proteins in non-CNS tissues and might thus not explain the phenotype seen in patients with CDK5RAP2 gene mutations.
Human disease caused by CDK5RAP2 gene mutations
Homozygous mutations of the CDK5RAP2 gene have been identified as a cause for MCPH3 in three families (MIM*608201): (1) a nonsense mutation in exon 4 (243T>A, S81X) and (2) an A to G transition in intron 26 (IVS26-15A>G, E385fsX4) introducing a new splice acceptor site, a frame shift and a premature stop codon [17, 27]. Both mutations are assumed to lead to non-functional, truncated protein products [5], but so far no study has clearly shown this. Other MCPH subtypes can be caused by homozygous mutations in the genes encoding microcephalin (MCPH1 [28, 29]), WD-repeat containing protein 62 WDR62 (MCPH2 [30–32]), centrosomal protein of 152 kDa CEP152 (MCPH4 [33, 34]), abnormal spindle-like, microcephaly associated ASPM (MCPH5 [35, 36]), centromeric protein J CENPJ (MCPH6 [17, 37]) and SCL/TAL1-interrupting locus STIL (MCPH7 [38]). Moreover, further genetic heterogeneity likely exists as in our experience about 20% of patients/families with a MCPH phenotype do not show linkage to any of the known loci (Table 1).
Patients with MCPH present typically at birth with microcephaly, a small cranium with significantly reduced occipito-frontal head circumference (OFC), because of reduced brain volume (Fig. 1). Individual patients can have an OFC in the lower normal range at birth followed by the development of a ‘secondary microcephaly’ within the first year of life [39]. Many patients display mental retardation, and some have further neurological findings including speech delay, hyperactivity and attention deficit, aggressiveness, delay of developmental milestones, focal or generalized seizures, and pyramidal signs on clinical exam [39]. Moreover, abnormal height and weight [39, 40], and congenital hearing loss have been detected in some patients. We refer to Kaindl et al. [41] for further details.
Imaging studies reveal typically brains of ‘normal architecture’ but of reduced cerebral cortex (and white matter) volume [40]. The cerebral cortex structure has been described as simplified, and individual patients with MCPH provide evidence of periventricular neuronal heterotopias, infra-tentorial anomalies such as brainstem or cerebellar hypoplasia, dysmorphic and/or enlarged lateral ventricles, corpus callosum agenesis, and focal micropolygyria and/or dysplasia [39, 40]. Histological analysis, performed at a time when a genetic diagnosis was not possible, revealed a significantly reduced brain volume with an almost preserved convolution pattern [42, 43]. The manifestation of MCPH already at fetal age, its assumed non-progressiveness and the specific effect on the cerebral cortex have supported the hypothesis that the disease is caused by a deficiency in proliferation of cortical neural progenitor cells.
Mouse phenotype caused by mutation in CDK5RAP2 homologous gene Cdk5rap2 or shRNAi in vivo
The Hertwig’s anemia (an) mouse was identified recently as a model of MCPH3 by positional cloning [13] (Table 2). These mice have an inversion of exon 4 of the Cdk5rap2 gene resulting in exon 4 skipping and have been well known for years for their hematopoietic phenotype of macrocytic, hypoproliferative anemia and leucopenia with an increase of severity in more mature hematopoietic progenitors as well as for their predisposition to hematopoietic tumors in homozygous mutants. Inbred Cdk5rap2 an/an B6.Cg mice rarely survive beyond 1 week of age, have reduced brain and body sizes already at E12.5 and defects in multiple organs including the brain, thymus and testis. While male Cdk5rap2 an/an B6.Cg mice are infertile because of a severe germ cell deficiency; females have less severe defects in their oocytes, but cannot deliver pups. In these mice, a high level of aneuploidy (abnormal number of chromosomes) in primary cell cultures of several organs derived from mutant animals has been reported [13].
In these mutant mice, Lizzaraga et al. detected small brains that display a thinning of the cerebral cortex, especially of the superficial, later-born Brn1/Cux1-positive neuronal layers (Brn1 = brain-specific homeobox/POU domain protein 1; Cux1 = homeobox protein cut-like 1), and a reduced size of the ganglionic eminence, hippocampus and olfactory bulbs. The cortex layering was preserved, but there was a significant reduction of neurogenesis, a reduction in the number of Tbr2-immunopositive basal progenitors (Tbr2 = T-box brain protein 2), an increase of M-phase cells coupled with a delay in mitotic progression and defective mitotic spindle orientation and abnormal mitotic figures (aneupolar spindles) in cortical progenitors. At the ventricular surface, dividing cells in Cdk5rap2 an/an B6.Cg mice were found to have an increase of horizontal and a reduction of perpendicular cell division planes (thought to produce side-by-side daughter cells) compared to controls at E14.5 [13]. This finding was assumed to indicate a relative increase of asymmetric cell division [13].
Similarly, knockdown of Cdk5rap2 through shRNAi plamids introduced via in utero electroporation of mouse embryos led to (1) an altered cell distribution in the developing neocortex with significantly fewer cells remaining in the ventricular/subventricular zone and more entering the intermediate zone or cortical plate; (2) significantly more postmitotic electroporated cells when compared to control cells; (3) a higher number of electroporated cells expressing layer VI marker Foxp2 and (4) a trend towards fewer electroporated GFP-positive cells in the upper layers (layers II/III and IV; no layer-specific markers applied); (5) increased neuronal differentiation; (6) a significant decrease in cell proliferation; (7) an increase of neurogenesis; (8) an altered makeup of the progenitor pool with a decrease of Pax6-positive apical and an increase of Tbr2-positive basal progenitors (this is in contrast to the finding in KO mice, and the authors discuss a possible transient effect that might not be associated with an enduring phenotype); (9) an alteration of cell cycle parameters in the mouse neuroblastoma cell line Neuro-2A with a shift from S to G0/1 phase, a reduction of proliferation, an increase of cell-cycle length and an increase of cell-cycle exit, together leading to a depletion of the progenitor pool [14].
In comparison to the knockout mice mentioned above, Barrera et al. [19] recently published the phenotype of Cdk5rap2 mutant mice that were viable, had no obvious behavioral deficits, and had normal brain size and morphology as well as normal body size and weight. These mice were derived from two Bay Genomics embryonic stem cell clones RRU0301 and RRF465, harboring transposon splice-trap insertions within introns 3 and 12, respectively, and resulting in translation of the first 64 and 435 amino acids of Cdk5rap2, respectively [19]. While males showed variable fertility (1/3 infertility), female mice were fertile. The difference in phenotype to the Cdk5rap2 an/an B6.Cg mice remains to be elucidated, but might be caused by a leaky mutation and/or a difference in genetic backgrounds of the mice.
Drosophila phenotype caused by loss of CDK5RAP2 homologous gene Centrosomin Cnn
In Drosophila, a loss of the CDK5RAP2 homologous gene centrosomin cnn causes centrosome malfunction in centrosomin mutant Drosophila embryos (cnn −/−, [44, 45]; Table 2). In cnn −/− flies, Lucas and Raff reported a failure of centrioles not to establish, but to maintain a stable connection to the pericentriolar matrix PCM (i.e., an attachment to the nuclear envelope in interphase and to the spindle poles in mitosis [44]). They further showed that this leads to microtubule-dependent rocketing of centrioles within the cells [44]. It has been suggested that the primary function of cnn may be to mechanically strengthen the PCM and that a weakened PCM in the absence of cnn initiates such centriole rocketing. Moreover, in cnn −/− flies, centrosomes were able to organize astral microtubule arrays, but were unable to maintain a connection with them. Thus, when the embryos entered mitosis, many nuclei were not associated with centrioles, and astral spindles assembled around the mitotic chromatin. Further, it has been demonstrated that cnn is required for centrosome cohesion (centrosome duplicates during the cell cycle but functions as a single microtubule-organizing center until shortly before mitosis [46]) and for docking of the γTuRC to the centrosome via its N-terminal domain (but not for γTuRC assembly) [44, 47]. While loss of cnn function results in ‘catastrophic’ failures in mitosis in syncytial embryos, somatic cells that lack cnn have few mitotic defects but have errors in centriole segregation [48]. Amazingly, only subtle defects of asymmetric divisions occurred in cnn mutant neuroblasts, there was no reduction of brain size, and cnn mutant flies were viable [44]. Flies with mutant cnn genes are able to compensate for the defect in a way that perhaps humans with homozygous CDK5RAP2 mutations cannot. Recent studies have illustrated that mitotic spindle can form even in the absence of centrosomes and that individual cells can divide without this organelle [49]. However, it has been hypothesized that the ability of centrioles to recruit PCM has evolved to ensure equal partitioning of the centrioles during cell division rather than the efficient assembly of the mitotic spindle [50].
Further experimental data regarding the functions of CDK5RAP2
From the human phenotype and animal models it has become clear that CDK5RAP2 has an influence on brain size regulation during fetal development. It is involved in cell cycle control and plays a role in regulating the shift in symmetric to asymetric proliferation of progenitors and the proliferative potential of neuroblasts during neurogenesis. Moreover, CDK5RAP2 may also be involved in brain enlargement during evolution. Several studies have focused on the functions of CDK5RAP2 on a molecular level, and in the following we describe these functions in greater detail.
Centrosomal attachment of the γ-tubulin ring complex
CDK5RAP2 is a pericentriolar structural component that functions in gamma tubulin ring complex (γTuRC) attachment and therefore in the microtubule organizing function of the centrosome [5]. Centromsomes consist of a pair of centrioles surrounded by amorphous pericentriolar material (PCM) including proteins such as gamma tubulin, the γTuRC and pericentrin, and provide a docking site for the minus end of microtubules [51, 52]. CDK5RAP2 is localized throughout the PCM in all stages of the cell cycle in somatic cells, as demonstrated by Fong et al. [5]. As mentioned above, Cdk5rap2 levels at the centrosome have been found to be regulated in a cell cycle-dependent manner in murine fibroblasts (low levels in interphase, high levels in prophase until telophase; [19]). CDK5RAP2 is associated with the γTuRC by a short conserved sequence present in several related proteins found in a broad range of organisms (Fig. 7). The binding of CDK5RAP2 is required for γTuRC attachment to the centrosome but not for γTuRC assembly [5]. The centrosome is the primary microtubule organizing center (MTOC), which plays a key role in the control of the temporal and spatial distribution of microtubule networks [46, 53, 54]. Hence, through its interaction with the γTuRC, CDK5RAP2 plays a role in the microtubule organizing function of the centrosome. A disrupted CDK5RAP2 function inhibits centrosomal microtubule nucleation, which leads to disorganization of interphase microtubule arrays and formation of anastral mitotic spindles. It has been shown for other gamma tubulin binding proteins that a failure of γ tubulin to localize to the centrosome is associated with (1) a G2–M arrest of some cells followed by apoptosis or (2) highly defecitve mitoses with increased monopolar spindles for those cells bypassing the G2-M arrest [55]. It has been recently demonstrated that the γ tubulin depletion (along with the depletion of multiple other centrosomal constituents) results in intra-G1 arrest and cellular senescence via a p53-dependent mechanism [56].

Alignment of the gamma tubulin ring complex (γTuRC) site sequence in various organisms. Sequences of the γTuRC in ortholog gene products of various organisms are compared to that of human CDK5RAP2: chimpanzee (Pan troglodytes), horse (Equus caballus), dog (Canis lupus familiaris), rhesus monkey (Macaca mulatta), mouse (Mus musculus), rat (Rattus norvegicus), opossum (Monodelphis domestica), chicken (Gallus gallus), cow (Bos taurus), fruit fly (Drosophila melanogaster). Detailed information on sequences used for alignments can be found in the supplementary info box 1
Dynein-dependent accumulation of further pericentriolar matrix proteins for spindle pole formation
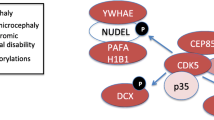
Cdk5rap2 has been implicated in dynein-dependent transport regulation (Fig. 8; [51]). Eukaryotic cells necessitate active, directional transport for the efficient and accurate localization of cellular components and accomplish this with the help of motor proteins such as dyneins, kinesins and myosins that ‘walk’ along cytoskeletal tracks. Dyneins move along microtubules towards the microtubule minus ends, which in most cells are collected into the microtubule-organizing center (MTOC) near the nucleus. Dynactin is an essential dynein adaptor that targets dynein to specific cellular locations and activates dynein. A knockdown of Cdk5rap2 by siRNA has been shown to lead to an increase of cell proliferation of HeLa cells (increase of phospho H3 levels) accompanied by abnormal spindle formations such as unfocused spindle poles and centrosomes that were detached from the spindle poles [51]. Such a depletion of Cdk5rap2 further led to a reduction of dynein at the centrosome, but did not affect its total protein levels or its levels at microtubule [51]. On the other hand, the centrosomal localization of Cdk5rap2 was also shown to depend on the function of the dynein-dynactin complex. In view of these findings, Lee and Rhee thus hypothesized that Cdk5rap2 regulates a dynein-dependent transport of the pericentriolar matrix proteins during centrosome maturation.

Cdk5rap2 and dynein-dependent transport regulation: a hypothesis. Dynein and its activating adaptor dynactin move along microtubules towards the microtubule minus ends. It has been suggested that (1) Cdk5rap2 may be transported to the centrosome by the dynein-dynactin complex and that, on the other hand, Cdk5rap2 may regulate the dynein-dependent transport of other pericentriolar matrix (PCM) proteins during centrosome maturation. (2) In addition, Cdk5rap2 can attract PCM proteins such as gamma tubulin in a dynein-dynactin-independent fashion
Spindle checkpoint function
CDK5RAP2 is essential for spindle formation [17] and proper chromosome segregation during mitosis, and has been suggested to be required for the spindle checkpoint-signaling pathway [57]. The mitotic spindle checkpoint prevents chromosome missegregation by delaying sister chromatid separation and thus entry of a mitotic cell into anaphase until all chromosomes are bipolarly attached via their kinetochores to spindle microtubules [58, 59]. This pattern of attachment ensures that each daughter cell receives one copy of each chromosome. Inhibition of Cdk5rap2 via siRNA led to spindle checkpoint failure with chromosome missegregation, anastral mitotic spindles and reduced expression of the spindle checkpoint proteins BUBR1 (budding uninhibited by benzimidazoles 1 homolog beta) and MAD2 (mitotic arrest-deficient 2) via interaction with their promoters in HeLa cells (Fig. 9; [57]). CDK5RAP2 has been reported to reside on the BUBR1 and MAD2 promoters and to function as a positive transcriptional regulator [57]. Moreover, loss of CDK5RAP2 function was associated with an increase in chromatin-associated CDC20 [57]. Chromosome segregation is initiated by activation of the spindle checkpoint target APC (anaphase-promoting complex), which functions as an E3 ligase when bound to its activator CDC20 and drives cells from metaphase into anaphase by inducing degradation of securin and mitotic cyclins [57, 59, 60]. The spindle checkpoint ensures that the CDC20-dependent activation of APC does not occur until all chromosomes have achieved bipolar kinetochore-microtubule attachment [57]. The spindle checkpoint proteins BUBR1 and MAD2 are part of an inhibitory complex for APC by sequestering CDC20 through directly binding and thus inhibiting APC activity [57]. Zhang et al. suggested a model in which downregulation of CDK5RAP2 reduces its binding to CDC20 and inhibits transcription of BUBR1 and MAD2, consequently decreases BUBR1 and MAD2 protein levels, and releases BUBR1- and MAD2-sequestered CDC20. Thereby, more free CDC20 would be available to bind chromatin, to activate the APC and thus to allow the transition from metaphase to anaphase in the presence of unattached kinetochores and/or kinetochores that lack tension in CDK5RAP2-inhibited cells. Additionally, cytosolic CDK5RAP2 sequesters CDC20 from chromatin. These intriguing results regarding the role of Cdk5rap2 remain to be repeated independently and to be complemented by further detailed studies.

Cdk5rap2 and spindle checkpoint function: a hypothesis. The mitotic spindle checkpoint prevents chromosome missegregation by delaying sister chromatid separation and thus entry of a mitotic cell into anaphase until all chromosomes are bipolarly attached via their kinetochores to spindle microtubules. Chromosome segregation is initiated by spindle checkpoint target APC, which is activated by binding to its activator CDC20 and drives cells from metaphase into anaphase by inducing degradation of securin and mitotic cyclins. The spindle checkpoint proteins BUBR1 and MAD2 are part of an inhibitory complex for APC by sequestering CDC20 through directly binding and thus inhibiting APC activity. It has been suggested that Cdk5rap2 downregulation reduces its binding to CDC20 and inhibits transcription of BUBR1 and MAD2, consequently decreases BUBR1 and MAD2 protein levels, and releases BUBR1-and MAD2-sequestered CDC20. Thereby, more free CDC20 would be available to bind chromatin, to activate the APC and thus to allow transition from metaphase to anaphase in the presence of unattached kinetochores and/or kinetochores that lack tension in CDK5RAP2-inhibited cells
Microtubule dynamics
CDK5RAP2 localizes to microtubules and concentrates at their distal tips in addition to its centrosomal localization in human tumor cell lines HeLa, HEK293T and U2OS, but not in rodent cells [9]. CDK5RAP2 interacts with EB1, a prototypic member of microtubule plus-end tracking proteins (+TIPs), and contains the basic and Ser-rich motif responsible for EB1 binding [61, 62]. The CDK5RAP2-EB1 complex regulates microtubule dynamics and stability. Fong et al. [9] also showed that the CDK5RAP2–EB1 complex induces microtubule bundling and acetylation when expressed in cell cultures, and stimulates microtubule polymerization, assembly and bundle formation, but does not induce nucleation in vitro. Therefore, CDK5RAP2 may participate in microtubule reorganization through the selective stabilization of microtubules and thus play a role in microtubule-dependent processes such as cell polarization and directional movement.
The EB-1 binding motif, spanning aa 926–956 in the human sequence, is well conserved in higher mammals such as apes, cattle and dogs. However, it shows increasing divergence in lower mammals such as mice and rats as well as in non-mammals such as chickens (Fig. 10). Fong et al. [9] recently suggested a gain of function of the EB1 site during mammalian evolution. This hypothesis was based on their finding that mouse Cdk5rap2 does not show EB1-binding and plus-end tracking properties in a respective assay and the fact that two residues including the proline of a critical Ile/Leu-Pro motif (position 14 in Fig. 10) are substituted in the rat and mouse sequences. Moreover, they showed that a mutation of these two residues to Ala in the human sequence resulted in a loss of activity in an EB1-binding assay. However, as shown in Fig. 10, the chicken sequence has a conserved Ile/Leu-Pro motif. This indicates that other residues may also need to be conserved for EB1 binding if one assumes that the chicken motif is not capable of binding EB1. This will need to be analyzed through further experiments. The basic and Ser-rich motif was not found in the sequences of Cnn, Mto1p and Pcp1p, implying that the tip-tracking function may not be conserved in Drosophila and S. pombe.

Alignment of the EB1 binding site sequence in various organisms. Sequences of the EB1 binding site in ortholog gene products of various organisms are compared to that of human CDK5RAP2: chimpanzee (Pan troglodytes), orangutan (Pongo abelii), horse (Equus caballus), dog (Canis lupus familiaris), cow (Bos taurus), opossum (Monodelphis domestica), rat (Rattus norvegicus), mouse (Mus musculus) and chicken (Gallus gallus). Detailed information on sequences used for alignments can be found in the supplementary info box 1
Centriole engagement and cohesion
Only recently it has been demonstrated by Barrera et al. [19] that Cdk5rap2 is required for the maintenance of centriole engagement and cohesion in mice. Centrosomes contain two structurally distinct centrioles, a mature ‘mother’ centriole and a ‘daughter’ centriole, that are amplified in a tightly regulated centriole duplication cycle to ensure that it occurs only once per cell cycle (brief overview in [19]): (1) G1 phase: mother centriole initiates formation of the primary cilium; (2) S phase: each centriole templates the assembly of a single daughter centriole, which grows from its proximal base and remains tightly bound, or "engaged," until disengagement occurs at mitosis; (3) G2 phase: engaged centriole pairs remain tethered by cohesion. Centriole cohesion refers to the tethering of centriole pairs by cohesion fibers during interphase, and it is physiologically lost at mitotic onset to allow centrosome separation in preparation for mitotic spindle assembly. After cell division, a cell inherits a pair of disengaged but cohered centrioles. Barrera et al. reported that loss of Cdk5rap2 function in murine embryonic fibroblasts (MEFs) is concomitant with centriole amplification; however, centrioles are frequently single, unpaired and showed an increase in daughter-daughter centriole pairs. Centrioles were disengaged in mutant MEFs, and early in mitosis, amplified centrosomes assembled multipolar spindles in Cdk5rap2 mutant cells. With their results, the authors indicate that Cdk5rap2 is required to maintain centriole engagement and cohesion, thereby restricting centriole replication.
Switch from symmetric to asymmetric cell division and neuronal differentiation
During neurogenesis, the length of the cell cycle and the proportion of symmetric and cell fate-determining asymmetric divisions of neural progenitors crucially affect the brain size. In the MCPH3 Cdk5rap2 an/an B6.Cg mouse model, Lizarraga et al. [13] reported a change in cleavage plane orientation with an increase of horizontal cleavage plane positioning in knockout cells. The authors argue that this is likely associated with an increase of asymmetric and a decrease of symmetric cell proliferation [13]. In other mouse models, such a shift from symmetric to asymmetric proliferation has been shown to lead to a depletion of the progenitor pool, a reduction of brain size, and an increase of precursor and neuronal cell death [13, 63–65].
Some of the CDK5RAP2 effects on neuronal differentiation may be elicited via its interaction with CDK5R1 and indirectly CDK5, though it needs to be pointed out that binding of CDK5RAP2 to human CDK5R1 and regulation of CDK5 has not been seriously studied in human tissues. For CDK5, a pivotal role in neuronal differentiation, cell polarity and synaptic function has been reported [22, 66–68]. Moreover, the direct Cdk5rap2 interaction partner Cdk5r1 (p35) not only interacts with Cdk5, but also with Notch signaling pathway regulators [69]. It would be intriguing to analyze the role of Cdk5rap2 in these pathways. Cdk5 is otherwise an unusual member of the Cdk protein family since, unlike other Cdks, it is not believed to regulate a ‘typical cell cycle.’
Does CDK5RAP2 play a role in migration?
While migration defects are not mainstream features of patients with MCPH3 and also not a striking feature in the Cdk5rap2 an/an B6.Cg mice or cnn −/− flies, brain malformations that indicate migration defects, such as heterotopias, have been described in individual patients with other MCPH subtypes (only three families with MCPH3 have been described so far). It thus remains to be elucidated whether Cdk5rap2 plays a subtle role in migration in humans. Cdk5rap2 is believed to inhibit (or regulate) Cdk5/Cdk5r1(p35), a complex required for the proper migration of neurons to the cortical plate during corticogenesis [22]. Cdk5 and its regulatory subunits Cdk5r1 (p35) and Cdk5r2 (p39) are essential for one of the two pathways for radial migration identified in mice (reviewed in [22, 70]). This Cdk5 pathway is of considerable importance in radial migration as Cdk5, p35 and p39 knockout mice reveal cortical inversion to varying degrees (but apparently no change of brain size; [71–74]). It remains to be elucidated whether Cdk5rap2 interacts with other regulators of p35 such as Brain-1 (Brn1) and Brain-2 (Brn2) [75], and Heat Shock Factor 2 (Hsf2) [76].
Does CDK5RAP2 play a role in cerebellar development?
Developmentally controlled proliferation, differentiation, migration and apoptosis of cells through processes such as centrosome function, cell cycle control, spindle assembly and division plane orientation are not unique for the cerebral cortex. They also occur throughout the development of the cerebellar cortex. Subtle cerebellar defects have been described in individual patients with MCPH such as cerebellar hypoplasia. However, why a loss of CDK5RAP2 protein function does not lead to a gross reduction of the cortex in the cerebellum as it does in the cerebrum is unknown and raises questions regarding the difference in cortex development within the brain. Moreover, the role of CDK5RAP2 has not been studied in cerebellar development, and it is thus not known whether the CDK5RAP2 gene is expressed in this context.
It thus remains to be elucidated what role Cdk5rap2 plays in cerebellar development, especially in light of the crystallizing respective role of Cdk5. In the perinatally lethal Cdk5 knockout mice, cerebellar development was disrupted with regard to cell migration, especially of the Purkinje cells (the Purkinje layer could not be distinguished at E18.5) [77]. Cdk5 chimeric knockout mice, containing both Cdk5 wild type and Cdk5 knockout cells, have hypoplastic cerebella with aberrantly localized, Cdk5 mutant Purkinje cells in the white matter. Moreover, some of the Cdk5 mutant granule cells in chimeric mice displayed a migration defect, as they did not migrate to the internal granule layer (IGL). On the other hand, Cdk5 mutant cells of the deep cerebellar nuclei (DCN) had normal configuration, indicating that the cells of the nuclei migrate by a Cdk5 independent pathway, even though they are generated at a similar time and in the same location as the Purkinje cells [77]. The Cdk5 mutation divides neuronal migration into different types: an early Cdk5-independent and a late Cdk5-dependent form. Since the granule cells can migrate from the rhombic lip to the cerebellum even in the absence of Cdk5, it seems the same cell can make use of different mechanisms of migration at different times [77].
CDK5RAP2 and evolution
The rapid increase in brain size over the past 3–4 million years together with the associated increase in cognitive skills is one of the most striking features of hominid evolution. MCPH genes are obvious candidates for this strong evolutionary movement and several, sometimes controversial, studies have attended to this point [7, 78–96]. From an evolutionary perspective, primary microcephaly can be viewed as an example of ‘evolutionary regression’ whereby the brain dimensions of the affected individuals revert back to the level resembling that of very early hominids [85, 97], a probably simplistic interpretation as hominids had small brains.
Evans et al. (2006) compared the evolutionary rates of CDK5RAP2 among primates, rodents and carnivores to verify the hypothesis that genes regulating brain size during development might also play a role in brain evolution in primates and especially humans [98]. They showed that the protein evolutionary rate of CDK5RAP2 is significantly higher in primates than in rodents or carnivores, especially in functional domains that are implicated in chromosome cohesion and condensation, such as the SMC domains [7]. Moreover, the plus-end binding protein EB1 motif detected in CDK5RAP2 that is important for microtubule dynamics and stability [9] is conserved in the gene sequences of chimpanzees, bovines and dogs, but not in those of rats and mice (see discussion above). Fong et al. [9] argue that CDK5RAP2 is possibly engaged in evolutionary brain development in part by association with microtubule distal tips. Rimol et al. [99] described sex-specific associations between SNPs and cortical surface area; such SNPs may have effects upon specific groups of neuronal progenitor populations. Still, the functional role of the adaptive evolution of MCPH genes remains speculative, and no clear answer has emerged from genetic epidemiology.
Conclusion
Brain size at birth is largely determined by the relative rates of proliferation, differentiation and cell death. CDK5RAP2 holds a role in cell cycle regulation, cell cycle checkpoint control and DNA repair, chromosome condensation, centrosome function, spindle formation and dynamics, kinetochore attachment to spindles, cellular abscission and apoptosis. It thereby contributes to the human phenotype. The phenotype of humans with MCPH and of animal models indicates that pathomechanisms underlying MCPH include a decrease or abnormal cell proliferation/differentiation, increased rates of cell death and possibly neuronal migration defects. All MCPH genes encode centrosomal proteins such as Cdk5rap2, implicating a critical role for the centrosome in brain development/growth. Further centrosomal proteins such as pericentrin have been associated with diseases that involve microcephaly, in the latter case Seckel syndrome (MIM*210600) and microcephalic osteodysplastic primordial dwarfism type II (MIM*210720). Future work will need to address the molecular function of Cdk5rap2 and its interaction with other proteins in greater detail.
References
Nagase T, Kikuno R, Nakayama M, Hirosawa M, Ohara O (2000) Prediction of the coding sequences of unidentified human genes. XVIII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 7(4):273–281
Ching YP, Qi Z, Wang JH (2000) Cloning of three novel neuronal Cdk5 activator binding proteins. Gene 242(1–2):285–294
Wang X, Ching YP, Lam WH, Qi Z, Zhang M, Wang JH (2000) Identification of a common protein association region in the neuronal Cdk5 activator. J Biol Chem 275(41):31763–31769. doi:10.1074/jbc.M004358200
Chin KT, Ohki SY, Tang D, Cheng HC, Wang JH, Zhang M (1999) Identification and structure characterization of a Cdk inhibitory peptide derived from neuronal-specific Cdk5 activator. J Biol Chem 274(11):7120–7127
Fong KW, Choi YK, Rattner JB, Qi RZ (2008) CDK5RAP2 is a pericentriolar protein that functions in centrosomal attachment of the gamma-tubulin ring complex. Mol Biol Cell 19(1):115–125
Revenkova E, Eijpe M, Heyting C, Gross B, Jessberger R (2001) Novel meiosis-specific isoform of mammalian SMC1. Mol Cell Biol 21(20):6984–6998
Evans PD, Vallender EJ, Lahn BT (2006) Molecular evolution of the brain size regulator genes CDK5RAP2 and CENPJ. Gene 375:75–79
Hirano T (2005) SMC proteins and chromosome mechanics: from bacteria to humans. Philos Trans R Soc Lond B Biol Sci 360(1455):507–514
Fong KW, Hau SY, Kho YS, Jia Y, He L, Qi RZ (2009) Interaction of CDK5RAP2 with EB1 to track growing microtubule tips and to regulate microtubule dynamics. Mol Biol Cell. doi:10.1091/mbc.E09-01-0009
Wang Z, Wu T, Shi L, Zhang L, Zheng W, Qu JY, Niu R, Qi RZ (2010) A conserved motif of CDK5RAP2 mediates its localization to centrosomes and the Golgi complex. J Biol Chem. doi:10.1074/jbc.M110.105965
Lupas A, Van Dyke M, Stock J (1991) Predicting coiled coils from protein sequences. Science 252(5010):1162–1164
Hirano T (2002) The ABCs of SMC proteins: two-armed ATPases for chromosome condensation, cohesion, and repair. Genes Dev 16(4):399–414. doi:10.1101/gad.955102
Lizarraga SB, Margossian SP, Harris MH, Campagna DR, Han AP, Blevins S, Mudbhary R, Barker JE, Walsh CA, Fleming MD (2010) Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137(11):1907–1917. doi:10.1242/dev.040410
Buchman JJ, Tseng HC, Zhou Y, Frank CL, Xie Z, Tsai LH (2010) Cdk5rap2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron 66(3):386–402. doi:10.1016/j.neuron.2010.03.036
Jiang H, Luo S, Li H (2005) Cdk5 activator-binding protein C53 regulates apoptosis induced by genotoxic stress via modulating the G2/M DNA damage checkpoint. J Biol Chem 280(21):20651–20659. doi:10.1074/jbc.M413431200
Jiang H, Wu J, He C, Yang W, Li H (2009) Tumor suppressor protein C53 antagonizes checkpoint kinases to promote cyclin-dependent kinase 1 activation. Cell Res 19(4):458–468. doi:10.1038/cr.2009.14
Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SM, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennett C, Corry P, Walsh CA, Woods CG (2005) A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37(4):353–355
Graser S, Stierhof YD, Nigg EA (2007) Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J Cell Sci 120(Pt 24):4321–4331
Barrera JA, Kao LR, Hammer RE, Seemann J, Fuchs JL, Megraw TL (2010) CDK5RAP2 regulates centriole engagement and cohesion in mice. Dev Cell 18(6):913–926. doi:10.1016/j.devcel.2010.05.017
Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402(6762):615–622. doi:10.1038/45159
Cicero S, Herrup K (2005) Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci 25(42):9658–9668. doi:10.1523/JNEUROSCI.1773-05.2005
Dhariwala FA, Rajadhyaksha MS (2008) An unusual member of the Cdk family: Cdk5. Cell Mol Neurobiol 28(3):351–369. doi:10.1007/s10571-007-9242-1
Hahn CM, Kleinholz H, Koester MP, Grieser S, Thelen K, Pollerberg GE (2005) Role of cyclin-dependent kinase 5 and its activator P35 in local axon and growth cone stabilization. Neuroscience 134(2):449–465. doi:10.1016/j.neuroscience.2005.04.020
Pareek TK, Keller J, Kesavapany S, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB (2006) Cyclin-dependent kinase 5 activity regulates pain signaling. Proc Natl Acad Sci USA 103(3):791–796. doi:10.1073/pnas.0510405103
Rakic S, Yanagawa Y, Obata K, Faux C, Parnavelas JG, Nikolic M (2009) Cortical interneurons require p35/Cdk5 for their migration and laminar organization. Cereb Cortex 19(8):1857–1869. doi:10.1093/cercor/bhn213
Zhang J, Cicero SA, Wang L, Romito-Digiacomo RR, Yang Y, Herrup K (2008) Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc Natl Acad Sci USA 105(25):8772–8777
Moynihan L, Jackson AP, Roberts E, Karbani G, Lewis I, Corry P, Turner G, Mueller RF, Lench NJ, Woods CG (2000) A third novel locus for primary autosomal recessive microcephaly maps to chromosome 9q34. Am J Hum Genet 66(2):724–727
Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, Karbani G, Jafri H, Rashid Y, Mueller RF, Markham AF, Woods CG (2002) Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet 71(1):136–142. doi:10.1086/341283
Jackson AP, McHale DP, Campbell DA, Jafri H, Rashid Y, Mannan J, Karbani G, Corry P, Levene MI, Mueller RF, Markham AF, Lench NJ, Woods CG (1998) Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am J Hum Genet 63(2):541–546. doi:10.1086/301966
Roberts E, Jackson AP, Carradice AC, Deeble VJ, Mannan J, Rashid Y, Jafri H, McHale DP, Markham AF, Lench NJ, Woods CG (1999) The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1–13.2. Eur J Hum Genet 7(7):815–820. doi:10.1038/sj.ejhg.5200385
Nicholas AK, Khurshid M, Desir J, Carvalho OP, Cox JJ, Thornton G, Kausar R, Ansar M, Ahmad W, Verloes A, Passemard S, Misson JP, Lindsay S, Gergely F, Dobyns WB, Roberts E, Abramowicz M, Woods CG (2010) WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet 42(11):1010–1014. doi:10.1038/ng.682
Yu TW, Mochida GH, Tischfield DJ, Sgaier SK, Flores-Sarnat L, Sergi CM, Topcu M, McDonald MT, Barry BJ, Felie JM, Sunu C, Dobyns WB, Folkerth RD, Barkovich AJ, Walsh CA (2010) Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet 42(11):1015–1020. doi:10.1038/ng.683
Jamieson CR, Govaerts C, Abramowicz MJ (1999) Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15. Am J Hum Genet 65(5):1465–1469. doi:10.1086/302640
Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau-Sturk T, Beis J, Dumas N, Evans SC, Ferguson M, Matsuoka M, Macgillivray C, Nightingale M, Patry L, Rideout AL, Thomas A, Orr A, Hoffmann I, Michaud JL, Awadalla P, Meek DC, Ludman M, Samuels ME (2010) Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet 87(1):40–51. doi:10.1016/j.ajhg.2010.06.003
Pattison L, Crow YJ, Deeble VJ, Jackson AP, Jafri H, Rashid Y, Roberts E, Woods CG (2000) A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31. Am J Hum Genet 67(6):1578–1580. doi:10.1086/316910
Shen J, Eyaid W, Mochida GH, Al-Moayyad F, Bodell A, Woods CG, Walsh CA (2005) ASPM mutations identified in patients with primary microcephaly and seizures. J Med Genet 42(9):725–729. doi:10.1136/jmg.2004.027706
Leal GF, Roberts E, Silva EO, Costa SM, Hampshire DJ, Woods CG (2003) A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2. J Med Genet 40(7):540–542
Kumar A, Girimaji SC, Duvvari MR, Blanton SH (2009) Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet 84(2):286–290. doi:10.1016/j.ajhg.2009.01.017
Passemard S, Titomanlio L, Elmaleh M, Afenjar A, Alessandri JL, Andria G, de Villemeur TB, Boespflug-Tanguy O, Burglen L, Del Giudice E, Guimiot F, Hyon C, Isidor B, Megarbane A, Moog U, Odent S, Hernandez K, Pouvreau N, Scala I, Schaer M, Gressens P, Gerard B, Verloes A (2009) Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology 73(12):962–969. doi:10.1212/WNL.0b013e3181b8799a
Woods CG, Bond J, Enard W (2005) Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet 76(5):717–728
Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, Gerard B, Verloes A, Mani S, Gressens P (2009) Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol. doi:10.1016/j.pneurobio.2009.11.002
Bamatter F, Rabinowicz T (1969) Study of a familial case of microcephaly and micrencephaly. Clinical and anatomo-pathologic considerations on a preliminary basis. J Genet Hum 17(3):247–274
Robain O, Lyon G (1972) Familial microcephalies due to cerebral malformation. Anatomical and clinical study. Acta Neuropathol 20(2):96–109
Lucas EP, Raff JW (2007) Maintaining the proper connection between the centrioles and the pericentriolar matrix requires Drosophila centrosomin. J Cell Biol 178(5):725–732. doi:10.1083/jcb.200704081
Megraw TL, Li K, Kao LR, Kaufman TC (1999) The centrosomin protein is required for centrosome assembly and function during cleavage in Drosophila. Development 126(13):2829–2839
Doxsey S, Zimmerman W, Mikule K (2005) Centrosome control of the cell cycle. Trends Cell Biol 15(6):303–311. doi:10.1016/j.tcb.2005.04.008
Zhang J, Megraw TL (2007) Proper recruitment of gamma-tubulin and D-TACC/Msps to embryonic Drosophila centrosomes requires Centrosomin Motif 1. Mol Biol Cell 18(10):4037–4049. doi:10.1091/mbc.E07-05-0474
Megraw TL, Kao LR, Kaufman TC (2001) Zygotic development without functional mitotic centrosomes. Curr Biol 11(2):116–120. doi:S0960-9822(01)00017-3
Mahoney NM, Goshima G, Douglass AD, Vale RD (2006) Making microtubules and mitotic spindles in cells without functional centrosomes. Curr Biol 16(6):564–569. doi:10.1016/j.cub.2006.01.053
Rieder CL, Faruki S, Khodjakov A (2001) The centrosome in vertebrates: more than a microtubule-organizing center. Trends Cell Biol 11(10):413–419. doi:S0962-8924(01)02085-2
Lee S, Rhee K (2010) CEP215 is involved in the dynein-dependent accumulation of pericentriolar matrix proteins for spindle pole formation. Cell Cycle 9(4):774–783. doi:10667
Bornens M (2002) Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol 14(1):25–34. doi:S0955067401002903
Luders J, Stearns T (2007) Microtubule-organizing centres: a re-evaluation. Nat Rev Mol Cell Biol 8(2):161–167
Ou Y, Rattner JB (2004) The centrosome in higher organisms: structure, composition, and duplication. Int Rev Cytol 238:119–182
Zimmerman WC, Sillibourne J, Rosa J, Doxsey SJ (2004) Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol Biol Cell 15(8):3642–3657. doi:10.1091/mbc.E03-11-0796
Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S (2007) Loss of centrosome integrity induces p38–p53-p21-dependent G1–S arrest. Nat Cell Biol 9(2):160–170
Zhang X, Liu D, Lv S, Wang H, Zhong X, Liu B, Wang B, Liao J, Li J, Pfeifer GP, Xu X (2009) CDK5RAP2 is required for spindle checkpoint function. Cell Cycle 8(8):1206–1216
Musacchio A, Salmon ED (2007) The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8(5):379–393
Nakayama KI, Nakayama K (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6(5):369–381
McGrogan BT, Gilmartin B, Carney DN, McCann A (2008) Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta 1785(2):96–132
Galjart N (2005) CLIPs and CLASPs and cellular dynamics. Nat Rev Mol Cell Biol 6(6):487–498
Akhmanova A, Steinmetz MO (2008) Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol 9(4):309–322
Feng Y, Walsh CA (2004) Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44(2):279–293. doi:10.1016/j.neuron.2004.09.023
Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB (2006) Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA 103(27):10438–10443. doi:10.1073/pnas.0604066103
Sanada K, Tsai LH (2005) G protein betagamma subunits and AGS3 control spindle orientation and asymmetric cell fate of cerebral cortical progenitors. Cell 122(1):119–131. doi:10.1016/j.cell.2005.05.009
Jessberger S, Aigner S, Clemenson GD Jr, Toni N, Lie DC, Karalay O, Overall R, Kempermann G, Gage FH (2008) Cdk5 regulates accurate maturation of newborn granule cells in the adult hippocampus. PLoS Biol 6(11):e272
Causeret F, Jacobs T, Terao M, Heath O, Hoshino M, Nikolic M (2007) Neurabin-I is phosphorylated by Cdk5: implications for neuronal morphogenesis and cortical migration. Mol Biol Cell 18(11):4327–4342
Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2(10):749–759
Choe EA, Liao L, Zhou JY, Cheng D, Duong DM, Jin P, Tsai LH, Peng J (2007) Neuronal morphogenesis is regulated by the interplay between cyclin-dependent kinase 5 and the ubiquitin ligase mind bomb 1. J Neurosci 27(35):9503–9512. doi:10.1523/JNEUROSCI.1408-07.2007
Gupta A, Tsai LH (2003) Cyclin-dependent kinase 5 and neuronal migration in the neocortex. Neurosignals 12(4–5):173–179. doi:10.1159/000074618
Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna PantHC, Brady RO, Martin LJ, Kulkarni AB (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 93(20):11173–11178
Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18(1):29–42. doi:S0896-6273(01)80044-1
Gupta A, Sanada K, Miyamoto DT, Rovelstad S, Nadarajah B, Pearlman AL, Brunstrom J, Tsai LH (2003) Layering defect in p35 deficiency is linked to improper neuronal–glial interaction in radial migration. Nat Neurosci 6(12):1284–1291. doi:10.1038/nn1151
Kwon YT, Tsai LH (1998) A novel disruption of cortical development in p35(−/−) mice distinct from reeler. J Comp Neurol 395(4):510–522. doi:10.1002/(SICI)1096-9861(19980615)395:4<510:AID-CNE7>3.0.CO;2-4
McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG (2002) Transcriptional regulation of cortical neuron migration by POU domain factors. Science 295(5559):1528–1532
Chang Y, Ostling P, Akerfelt M, Trouillet D, Rallu M, Gitton Y, El Fatimy R, Fardeau V, Le Crom S, Morange M, Sistonen L, Mezger V (2006) Role of heat-shock factor 2 in cerebral cortex formation and as a regulator of p35 expression. Genes Dev 20(7):836–847
Ohshima T, Gilmore EC, Longenecker G, Jacobowitz DM, Brady RO, Herrup K, Kulkarni AB (1999) Migration defects of cdk5(−/−) neurons in the developing cerebellum is cell autonomous. J Neurosci 19(14):6017–6026
Ali F, Meier R (2008) Positive selection in ASPM is correlated with cerebral cortex evolution across primates but not with whole-brain size. Mol Biol Evol 25(11):2247–2250. doi:10.1093/molbev/msn184
Balter M (2005) Evolution. Are human brains still evolving? Brain genes show signs of selection. Science 309(5741):1662–1663. doi:10.1126/science.309.5741.1662
Balter M (2006) Bruce Lahn profile. Links between brain genes, evolution, and cognition challenged. Science 314(5807):1872. doi:10.1126/science.314.5807.1872
Balter M (2006) Bruce Lahn profile. Brain man makes waves with claims of recent human evolution. Science 314(5807):1871–1873. doi:10.1126/science.314.5807.1871
Bond J, Woods CG (2006) Cytoskeletal genes regulating brain size. Curr Opin Cell Biol 18(1):95–101. doi:10.1016/j.ceb.2005.11.004
Cotoi S, Incze A, Georgescu C, Carasca E (1980) Pacing below the ventricular rate in terminating ventricular tachycardia and flutter. Am Heart J 100(5):763–764
Currat M, Excoffier L, Maddison W, Otto SP, Ray N, Whitlock MC, Yeaman S (2006) Comment on “Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens” and “Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans”. Science 313(5784):172. doi:10.1126/science.1122712 author reply 172
Evans PD, Anderson JR, Vallender EJ, Choi SS, Lahn BT (2004) Reconstructing the evolutionary history of microcephalin, a gene controlling human brain size. Hum Mol Genet 13(11):1139–1145. doi:10.1093/hmg/ddh126
Evans PD, Anderson JR, Vallender EJ, Gilbert SL, Malcom CM, Dorus S, Lahn BT (2004) Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum Mol Genet 13(5):489–494. doi:10.1093/hmg/ddh055
Evans PD, Gilbert SL, Mekel-Bobrov N, Vallender EJ, Anderson JR, Vaez-Azizi LM, Tishkoff SA, Hudson RR, Lahn BT (2005) Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science 309(5741):1717–1720. doi:10.1126/science.1113722
Frost P (2008) The spread of alphabetical writing may have favored the latest variant of the ASPM gene. Med Hypotheses 70(1):17–20. doi:10.1016/j.mehy.2007.04.039
Kouprina N, Pavlicek A, Mochida GH, Solomon G, Gersch W, Yoon YH, Collura R, Ruvolo M, Barrett JC, Woods CG, Walsh CA, Jurka J, Larionov V (2004) Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLoS Biol 2(5):E126. doi:10.1371/journal.pbio.0020126
Mekel-Bobrov N, Gilbert SL, Evans PD, Vallender EJ, Anderson JR, Hudson RR, Tishkoff SA, Lahn BT (2005) Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 309(5741):1720–1722. doi:10.1126/science.1116815
Ponting C, Jackson AP (2005) Evolution of primary microcephaly genes and the enlargement of primate brains. Curr Opin Genet Dev 15(3):241–248. doi:10.1016/j.gde.2005.04.009
Ponting CP (2006) A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics 22(9):1031–1035. doi:10.1093/bioinformatics/btl022
Richards GD (2006) Genetic, physiologic and ecogeographic factors contributing to variation in Homo sapiens: Homo floresiensis reconsidered. J Evol Biol 19(6):1744–1767. doi:10.1111/j.1420-9101.2006.01179.x
Stern R, Woods CG (2006) Evolutionary genetics: is brain evolution still continuing in modern humans? Eur J Hum Genet 14(7):799–800. doi:10.1038/sj.ejhg.5201624
Wang JK, Li Y, Su B (2008) A common SNP of MCPH1 is associated with cranial volume variation in Chinese population. Hum Mol Genet 17(9):1329–1335. doi:10.1093/hmg/ddn021
Zhang J (2003) Evolution of the human ASPM gene, a major determinant of brain size. Genetics 165(4):2063–2070
Wang YQ, Su B (2004) Molecular evolution of microcephalin, a gene determining human brain size. Hum Mol Genet 13(11):1131–1137. doi:10.1093/hmg/ddh127
Gilbert SL, Dobyns WB, Lahn BT (2005) Genetic links between brain development and brain evolution. Nat Rev Genet 6(7):581–590
Rimol LM, Agartz I, Djurovic S, Brown AA, Roddey JC, Kahler AK, Mattingsdal M, Athanasiu L, Joyner AH, Schork NJ, Halgren E, Sundet K, Melle I, Dale AM, Andreassen OA (2010) Sex-dependent association of common variants of microcephaly genes with brain structure. Proc Natl Acad Sci USA 107(1):384–388. doi:10.1073/pnas.0908454107
Acknowledgments
Our research is supported by the German Research Foundation (DFG; SFB665 and IB of the BMBF), the Sonnenfeld Stiftung and the Berliner Krebsgesellschaft e.V.
Author information
Authors and Affiliations
Corresponding author
Additional information
N. Kraemer and L. Issa contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kraemer, N., Issa, L., Hauck, S.C.R. et al. What’s the hype about CDK5RAP2?. Cell. Mol. Life Sci. 68, 1719–1736 (2011). https://doi.org/10.1007/s00018-011-0635-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0635-4