
Abstract
Transforming growth factor (TGF)-β treatment of human vascular smooth-muscle cells increases the expression of biglycan and causes marked elongation of its glycosaminoglycan (GAG) chains. We investigated the role of MAP kinases and Smad transcription factors in this response. TGF-β-stimulated phosphorylation of p38, ERK, and JNK as well as Smad2 at both its carboxy terminal (phospho-Smad2C) and in the linker region (phospho-Smad2L). Pharmacological inhibition of ERK and p38 blocked TGF-β-mediated GAG elongation and expression of biglycan whereas inhibition of JNK had no effect. Inhibition of ERK and p38 but not JNK attenuated the effect of TGF-β to increase phospho-Smad2L. High levels of phospho-Smad2L were detected in a nuclear fraction of TGF-β treated cells. Thus, MAP kinase signaling through ERK and p38 and via phosphorylation of the linker region of Smad2 mediates the effects of TGF-β on biglycan synthesis in vascular smooth-muscle cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proteoglycans are ubiquitous extracellular matrix molecules that play a major role in vascular biology and pathology [1, 2]. Proteoglycans are composed of a core protein and covalently attached glycosaminoglycan (GAG) chains and they have a range rather than a discrete molecular weight due to the range of sizes of the GAG chains [2]. The major proteoglycans produced by vascular smooth-muscle cells (VSMC) are the chondroitin sulfate/dermatan sulfate (CS/DS) proteoglycans versican, biglycan, and decorin, and the heparan sulfate (HS) proteoglycan, perlecan [3]. Vasoactive factors stimulate the expression of proteoglycan core proteins and independently modify the biochemistry of the GAG chains using cell-surface tyrosine and serine/threonine kinase [4, 5] and G protein-coupled receptors [6, 7]. These intracellular signaling pathways control the transcription and translation of unidentified factors which regulate proteoglycan and specifically GAG synthesis and structure [8].
Alterations in the synthesis and structure of proteoglycans lead to enhanced binding to apolipoproteins on lipids in vitro [9–11] and enhanced trapping of atherogenic low-density lipoproteins (LDL) in the vessel wall. The early trapping of LDL in the vessel wall by modified biglycan is the initiating factor in animal models [12, 13] and in human coronary artery atherosclerosis [14, 15]. Preventing the changes in proteoglycan synthesis and structure has been proposed as a therapeutic target for the prevention of atherosclerosis [11, 16].
One of the major growth factors associated with vascular disease is transforming growth factor (TGF)-β [17], which acts via ubiquitous serine/threonine kinase receptors [18, 19]. TGF-β stimulates proteoglycan synthesis, specifically the expression of biglycan and an elongation of its DS GAG chains and this results in an increased binding of the proteoglycan to LDL [4, 5, 10]. TGF-β signals changes in biglycan synthesis through the canonical TGF-β receptor I (TβRI/Alk 5)-mediated phosphorylation of Smad transcription factors, specifically Smad2 [5, 18]. TGF-β signaling is also known to involve (positively and negatively) interactions with the mitogen-activated protein (MAP) kinase pathways [18, 20, 21]. TGF-β-stimulated expression of biglycan in pancreatic carcinoma cells involves p38 MAP kinase signaling downstream of Smad activation [22, 23], however, the relationship between TβRI/Alk 5, Smad phosphorylation and MAP kinase(s) activation in VSMCs remains to be determined.
As well as stimulating phosphorylation of Smad transcription factors on Ser residues in their carboxy terminal, TGF-β can also stimulate the phosphorylation of ERK 1/2 [20]. Furthermore, activated ERK 1/2 can phosphorylate Smad transcription factors on Thr [20] and possibly Ser residues in the linker region of the transcription factor, a response which can hinder the translocation of the Smad to the nucleus [20]. We have made preliminary observations that inhibitors of the MAP kinase, p38, can inhibit TGF-β-stimulated proteoglycan synthesis in VSMCs but the role of TGF-β in the phosphorylation of p38 and the extended role of p38, ERK, and JNK MAP kinases has not been reported.
In this study, we investigate the ability of TGF-β to stimulate the phosphorylation of MAP kinases and determine the activity of a range of inhibitors to block the action of TGF-β on proteoglycan synthesis as well as further explore the downstream interactions between MAP kinases and Smad transcription factors. We report that all three prototypical MAP kinases are phosphorylated by treatment of VSMCs with TGF-β and that ERK and p38 MAP kinase but not JNK are involved in proteoglycan synthesis. We also report that there are interactions between the canonical Smad pathway and the MAP kinase pathway in mediating the effects on proteoglycan synthesis in VSMCs.
Materials and methods
Materials
Whatman 3MM chromatography paper, dimethylsulfoxide (DMSO), DEAE Sephacel, SB431542, and SP600125 were obtained from Sigma-Aldrich (St. Louis, MO). FR180204, PD98059, and SB202190 were from Calbiochem (La Jolla, CA, USA). UO126 was from Promega (Madison, WI, USA) Human recombinant TGF-β1 was obtained from R&D systems (Minneapolis, USA). Foetal bovine serum (FBS) was obtained from CSL (Parkville, Australia). Scintillation fluid Instagel was from Packard (Groningen, The Netherlands). Carrier-free [35S]-SO4 was from ICN Biomedicals (Irvine, CA, USA). [35S]-methionine/cysteine was obtained from MP Biomedicals (California, USA). Cetylpyridinium chloride (CPC) was from Uni-lab Chemicals and Pharmaceuticals, India. Cell culture materials were from GIBCO BRL (Grand Island, USA). Anti-phospho-p44/p42 (ERK1/2) antibody, anti-p44/p42 (ERK1/2) antibody, anti-phospho-p38 antibody, anti-phospho-Smad2 Ser245/255/250 antibody, anti-phospho-Smad2 Ser465/467 antibody and anti-GAPDH antibody were from Cell Signalling Technology (USA). Anti-phospho-JNK 1/2/3 antibody was from Santa Cruz Biotechnology, Inc. (California, USA). Anti-smooth muscle α-actin antibody was from Dako Corporation (California, USA). Peroxidase-labeled anti-rabbit IgG was from GE Healthcare, Buckinghamshire, UK.
Cell culture
Human VSMCs were isolated using the explant technique from discarded segments of the saphenous veins from a variety of patient donors undergoing surgery at the Alfred Hospital (Melbourne, Australia) [24]. The acquisition of the vessels was approved by the Alfred Hospital Ethics Committee. Cells were seeded into 24-well plates at 50,000 cells/well in low-glucose (5 mM) DMEM with 10% FBS and antibiotics and maintained until confluent. Cells were serum deprived by culturing in low glucose (5 mM) DMEM with 0.1% FBS for 48 h prior to experimentation. Experiments were conducted on cells passage 5–22. Human VSMC were treated with various drugs under basal conditions and in the presence of TGF-β for 24 h with [35S]-SO4 (50 μCi/ml) or 35S-methionine/cysteine (50 μCi/ml). Parallel plates without radiolabel were established to assess cell numbers, which were then used to normalize the data. The drugs used at the concentrations shown did not affect cell numbers in these experiments.
Measurement of [35S]-SO4 incorporation into proteoglycans
Proteoglycans were extracted from the medium as previously described [25]. Incorporation of [35S]-SO4 into proteoglycans was measured by CPC precipitation as previously described [26]. Briefly, aliquots (50 μl) of the medium were spotted on filter paper and washed five times for 40 min in 1% CPC with 0.05 M NaCl. The amount of precipitate on the dried filter paper was determined by liquid scintillation counting. The CPC precipitation method elutes highly sulfated macromolecules, of which proteoglycans make up the large majority of these, and we have shown that biglycan is the dominant form.
Ion-exchange chromatography
Extracted proteoglycans were purified on DEAE-Trisacryl minicolumns, which were washed extensively with 8 M urea, 2 mM disodium EDTA, 0.5% Triton X-100, 0.25 M NaCl, 50 mM Tris/HCl, pH 7.5 to remove glycoproteins and free [35S]-SO4 and the proteoglycans were eluted with the same buffer containing a higher salt concentration (3 M NaCl). The isolated material was further concentrated by ethanol/potassium acetate precipitation.
Gel electrophoresis
SDS-PAGE was performed according to the procedure of Laemmli [27] on 4–13% acrylamide gradient gels with a 3% stacking gel. The labeled proteoglycans were visualized by exposing dried gels to a phosphoimaging screen (Fuji Photo Film Co., Japan) for approximately 3 days, and then scanned on a Bio-imaging analyzer BAS-1000 MacBas (Fuji Photo Film Co., Japan). Samples are run in parallel with a 14C radioactive protein ladder, which reveals a band at 220 kDa, corresponding to biglycan.
Western blotting
Total cell lysates (50 μg of protein) were resolved on 10% SDS-PAGE and transferred onto a PVDF membrane. Membranes were blocked with 5% skim milk and incubated with primary antibody followed by horseradish peroxidase-anti IgG then ECL detection. Each experiment was conducted at least three times.
Analysis of TGF-β-mediated gene expression: real-time, reverse transcription polymerase chain reaction (real-time RT-PCR)
Quiescent cells were treated with TGF-β (2 ng/ml) for 0–24 h. Total RNA was extracted using Qiagen RNeasy mini kit. cDNA was synthesized from DNase-treated RNA (1.8 μg/μl), using 10 ng/μl random hexamers (Takawa Bio Inc., Japan). For the analysis of TGF-β-mediated gene expression of biglycan, TaqMan Fast Universal PCR Master Mix (Applied Biosystems, California USA) was used with primers and a FAM-labeled probe, with the following sequences:
Forward primer 5′-CTCAACTACCTGCGCATCTCAG-3′; Reverse primer 5′-GATGGCCTGGATTTTGTTGTG-3′; Probe 5′-FAMCCAAAGACCTCCCTGAGACCCTGAATGA-3′. Data was normalized to the 18S housekeeping gene. Experiments were performed at least two times and analyzed in duplicate. Amplification was performed on 96-well plates using the 7500 Fast Real-Time PCR System (Applied Biosystems, California, USA) and analyzed with 7500 Fast sequence detection software v1.3.1 (Applied Biosystems, California USA).
Statistics
Proteoglycan data are presented as mean ± S.E.M. from at least three independent experiments conducted in triplicate. Data from three independent experiments were statistically analyzed using a one-way ANOVA for the determination of the least significant difference upon which statistical significance is reported.
Results
Inhibition of ERK by MEK1/2 inhibitors blocks TGF-β-mediated GAG elongation and core protein synthesis in VSMCs
To determine whether ERK is a mediator of TGF-β signaling of GAG elongation in human VSMC, we used two inhibitors, UO126 (MEK 1/2 inhibitor) and PD98059 (MEK 1 inhibitor) that specifically block activity of MEK1/2 and consequently block ERK. Cells treated with TGF-β showed an increase of almost two fold in [35S]-SO4 incorporation into secreted proteoglycans (Figs. 1a, 2a) compared to control cells. Treatment of cells with UO126 (0.1–10 μM) or PD98059 (1–100 μM) showed a concentration-dependent decrease of TGF-β-mediated [35S]-SO4 incorporation into secreted proteoglycans (Figs. 1a, 2a) with a maximum decrease of 93% of TGF-β-stimulated component at 10 μM UO126 (p < 0.01) and slightly more than 100% of TGF-β-stimulated component at 100 μM PD98059 (p < 0.01) (Figs. 1a, 2a). The TβRI/Alk 5 inhibitor, SB431542 [28] was used as a positive control. The TGF-β-stimulated [35S]-SO4 incorporation was totally abolished by SB431542 (Figs. 1a, 2a). Separation of proteoglycans on an SDS–PAGE showed a decrease in electrophoretic mobility of biglycan in the presence of TGF-β compared to control cells (Figs. 1b, 2b, lane2 vs. lane1). The positive control, SB431542 completely blocked this response (Figs. 1b, 2b, lane 8 vs. lane 2). SDS–PAGE showed a concentration-dependent increase in electrophoretic mobility in the presence of UO126 or PD98059 relative to TGF-β treatment alone (Figs. 1b, 2b, lane 3–7), indicating a concentration-dependent decrease in proteoglycan size upon treatment of VSMCs with UO126 or PD98059.

Determination of the role of ERK activity in TGF-β-mediated proteoglycan synthesis in human VSMCs using the MEK1/2 inhibitor UO126. VSMCs were treated with UO126 (0.1–10 μM) in the presence of TGF-β (2 ng/ml) and [35S]-SO4 (50 μCi/ml) for 24 h. a Medium containing secreted proteoglycans was harvested and spotted onto 3MM paper and CPC precipitation method applied to assess radiolabel incorporation. Results are the mean ± S.E. of data normalized to control from three separate experiments in triplicate. **p < 0.01 versus TGF-β alone using one-way ANOVA. b Secreted proteoglycans were isolated using ion exchange chromatography (DEAE-Sephacel) and concentrated by ethanol/potassium acetate precipitation. Electrophoretic mobility of complete proteoglycans was assessed by SDS-PAGE on a 4–13% acrylamide gradient gel. The gel shows the analysis of biglycan as the proteoglycan of interest. The gel is representative of three identical experiments. c VSMCs were treated with TGF-β (2 ng/ml) and UO126 (0.1–10 μM) and radiolabeled with [35S]-Met/Cys (50 μCi/ml) for 24 h to assess total proteoglycan core protein synthesis. Medium containing secreted proteoglycans was spotted onto 3MM paper and assayed as above. Results shown are the mean ± S.E. of normalized data from three separate experiments in triplicate. **p < 0.01 versus TGF-β alone using one-way ANOVA. d VSMCs were treated with TGF-β (2 ng/ml) and UO126 (10 μM) for 24 h. Total RNA was harvested and biglycan mRNA expression was analyzed using RT-PCR. Results shown are the mean ± S.E. of normalized data from two separate experiments in duplicate. # p < 0.05 versus control and **p < 0.01 versus TGF-β alone using one-way ANOVA

Determination of the role of ERK activity in TGF-β-mediated proteoglycan synthesis in human VSMCs using the MEK1 inhibitor PD98059. MEK1/2 inhibitor blocks TGF-β-mediated GAG elongation in human VSMCs. VSMCs were treated with PD98059 (1–100 μM) in the presence of TGF-β (2 ng/ml) and [35S]-SO4 (50 μCi/ml) for 24 h. a [35S]-SO4 incorporation. b SDS-PAGE analysis. c [35S]-Met/Cys incorporation into proteoglycan core protein synthesis. d Biglycan mRNA expression was analyzed exactly as described in the legend of Fig. 1. **p < 0.01 versus treated control; # p < 0.05 versus untreated control
Changes in total proteoglycan core protein synthesis were assessed by [35S]-Met/Cys incorporation and CPC precipitation for the specific quantitation of proteoglycans. TGF-β-stimulated VSMCs showed an increase of [35S]-Met/Cys incorporation into secreted proteoglycans of approximately 20% (Figs. 1c, 2c) compared to control cells. Treatment of cells with UO126 (0.1–10 μM) or PD98059 (1–100 μM) showed a concentration-dependent decrease of TGF-β-mediated [35S]-Met/Cys incorporation into secreted proteoglycans (Figs. 1c, 2c) with a maximum decrease of 100% of TGF-β-stimulated component at 10 μM UO126 (p < 0.01) and 90% TGF-β-stimulated component at 100 μM PD98059 (p < 0.01). The TGF-β-stimulated [35S]-Met/Cys incorporation was inhibited by SB431542 by more than 81% (p < 0.01) (Figs. 1c, 2c).
To more specifically explore the effect of ERK inhibition on total core protein, we examined the expression of biglycan. TGF-β has been reported to increase expression of biglycan in VSMCs [5]. Hence we assessed the effect of ERK inhibition on TGF-β-stimulated VSMC biglycan mRNA levels. There are no significant changes of biglycan mRNA expression at early time points (data not shown). TGF-β-stimulated VSMCs showed increased biglycan mRNA expression by 30% compared to control cells (Figs. 1d, 2d) at 24 h. The TGF-β-stimulated biglycan mRNA expression was reduced to less than the basal level by UO126 and PD98059 (10 μM) (Figs. 1d, 2d) at 24 h, indicating inhibition of ERK blocks TGF-β-stimulated biglycan expression, which contributes to the effect on total proteoglycan core protein shown above.
Direct inhibition of ERK by FR180204 blocks TGF-β-mediated GAG elongation and core protein synthesis in VSMC
To further confirm whether ERK is a mediator of TGF-β signaling in proteoglycan synthesis in human VSMC, we used the direct ERK inhibitor, FR180204, which specifically blocks downstream activity of ERK. Cells treated with TGF-β showed a 64% increase of [35S]-SO4 incorporation into secreted proteoglycans (Fig. 3a) compared to control cells. Treatment of cells with FR180204 (0.1–10 μM) showed a concentration-dependent decrease of TGF-β-mediated [35S]-SO4 incorporation into secreted proteoglycans (Fig. 3a) with a maximum decrease of 100% of TGF-β-stimulated component at 10 μM FR180204 (p < 0.01) (Fig. 3a). The TGF-β-stimulated [35S]-SO4 incorporation was totally abolished by SB431542 (Fig. 3a). Separation of proteoglycans by SDS-PAGE showed a large decrease in electrophoretic mobility of biglycan in the presence of TGF-β compared to control cells (Fig. 3b, lane 2 vs. lane 1). Electrophoretic mobility of biglycan in TGF-β-treated cells was inhibited to control the level in the presence of SB431542 (Fig. 3b, lane 8 vs. lane 2). Separation of proteoglycans on SDS-PAGE showed a concentration-dependent increase in electrophoretic mobility in the presence of FR180204 relative to TGF-β treatment alone (Fig. 3b, lane 3–7), indicating a concentration-dependent decrease in proteoglycan size upon treatment with FR180204.

Determination of the role of ERK activity in TGF-β-mediated proteoglycan synthesis in human VSMCs using the direct ERK inhibitor FR180204. The ERK inhibitor blocks TGF-β-mediated GAG elongation in human VSMCs. VSMCs were treated with FR180204 (0.1–10 μM) in the presence of TGF-β (2 ng/ml) and [35S]-SO4 (50 μCi/ml) for 24 h. a [35S]-SO4 incorporation. b SDS-PAGE analysis. c [35S]-Met/Cys incorporation into proteoglycan core protein synthesis. d Biglycan mRNA expression was analyzed exactly as described in the legend of Fig. 1. e TGF-β/Alk 5-mediated ERK1/2 phosphorylation in human VSMCs. VSMCs were treated with TGF-β (2 ng/ml) and UO126 (3 μM), PD98059 (30 μM), FR180204 (10 μM) and SB431542 (3 μM) for 4 h. PDGF (50 ng/ml)-treated VSMCs for 4 h were used as positive control. Cell lysates were collected, and proteins were resolved by SDS-PAGE on a 10% acrylamide gel and transferred onto a polyvinylidene difluoride membrane. The membrane was incubated with anti-phospho-ERK1/2 polyclonal antibody (1:1,000 dilution) overnight at 4°C followed by incubation with peroxidase-labeled anti-rabbit IgG (1:5,000 dilution) for 1 h at room temperature. The membrane was then probed with anti-ERK1/2 rabbit polyclonal antibody (1:1,000 dilution) for 1 h at room temperature followed by incubation with peroxidase-labeled anti-rabbit IgG (1:4,000 dilution) for 1 h. The Western-blot analysis is representative of three independent experiments. *p < 0.05 versus treated control; **p < 0.01 versus treated control; # p < 0.05 versus untreated control
TGF-β-stimulated VSMCs showed an increase of [35S]-Met/Cys incorporation into secreted proteoglycan core proteins of approximately 17% (Fig. 3c) compared to control cells. Treatment of cells with FR180204 (0.1–10 μM) showed a concentration-dependent decrease of TGF-β-mediated [35S]-Met/Cys incorporation into secreted proteoglycans (Fig. 3c) with a maximum decrease of 100% of the TGF-β-stimulated component at 10 μM FR180204 (p < 0.01). The TGF-β-stimulated [35S]-Met/Cys incorporation was reduced to less than basal level by SB431542 (Fig. 3c).
There are no significant changes of biglycan mRNA expression at early time points (data not shown). TGF-β-stimulated VSMCs showed an increase in biglycan mRNA expression by 30% compared to control cells (Fig. 3d) at 24 h. The TGF-β-stimulated biglycan mRNA expression was decreased to 95% compared to TGF-β-stimulated component by FR180204 at 10 μM 24 h (Fig. 3d), which is consistent with our results that inhibition of ERK blocks TGF-β-stimulated biglycan expression, which contributes to the effect on total proteoglycan core protein shown above.
To determine whether ERK phosphorylation is a mediator of TGF-β/Alk 5 signaling in VSMCs, we used the three inhibitors above. A time course study indicates that TGF-β/Alk 5 stimulates maximum phosphorylation of ERK1 and ERK2 at 4 h, which is the time point chosen for subsequent studies. Bands of 42 and 44 kDa corresponding to phosphorylated ERK1 and ERK2 were observed in Western blot of whole-cell lysate from VSMCs (Fig. 3e, lane 1), indicating the presence of phospho-ERK1/2 under basal conditions. The phospho-ERK1/2 band intensities in TGF-β (2 ng/ml)-treated cells were increased compared to control cells (Fig. 3e, lane 1 vs. lane 2), indicating the stimulation of ERK1/2 phosphorylation. Treatment with UO126 (3 μM) and PD98059 (30 μM) decreased the ERK1/2 phosphorylation to below the basal level in TGF-β-treated cells (Fig. 3e, lane 3, 4 vs. lane 2), indicating that ERK1/2 phosphorylation is down-regulated by inhibition of MEK1/2. Consistent with its mechanism of action, no change in ERK1/2 phosphorylation in FR180204/TGF-β-treated cells was observed (Fig. 3e, lane 5 vs. lane 2). Phosphorylation of ERK1/2 is decreased to basal levels by the TGF-β/Alk 5 inhibitor SB431542 (3 μM) (Fig. 3e, lane 6 vs. 2). PDGF (50 ng/ml)-treated VSMCs were used as positive control and ERK phosphorylation was greatly enhanced in these cells (Fig. 3e, lane 7). Taken together with our earlier findings, this implies that TGF-β/Alk 5 signals via MEK1/2 and ERK to induce biglycan expression and GAG elongation in human VSMCs.
Direct inhibition of p38 MAP kinase by SB202190 blocks TGF-β-mediated GAG elongation and core protein synthesis in VSMC
p38 MAP kinase was demonstrated to act as a mediator of TGF-β signaling in GAG elongation in human VSMC [5]. To further investigate TGF-β-mediated p38 MAP kinase signaling of GAG elongation in human VSMC, we used the direct p38 MAP kinase inhibitor, SB202190. In these experiments, cells treated with TGF-β showed a 148% increase of [35S]-SO4 incorporation into secreted proteoglycans (Fig. 4a) compared to control cells. Treatment of cells with SB202190 (0.1–10 μM) showed a concentration-dependent decrease of TGF-β-mediated [35S]-SO4 incorporation into secreted proteoglycans (Fig. 4a) with a maximum decrease of 95% of TGF-β-stimulated component at 10 μM SB202190 (p < 0.01) (Fig. 4a). The TGF-β-stimulated [35S]-SO4 incorporation was totally abolished by SB431542 (Fig. 4a). Separation of proteoglycans on an SDS-PAGE showed a decrease in electrophoretic mobility of biglycan in the presence of TGF-β compared to control cells (Fig. 4b, lane 2 vs. lane 1). Electrophoretic mobility of biglycan in TGF-β-treated cells was inhibited to control levels in the presence of SB431542 (Fig. 4b, lane 8 vs. lane 2). Separation of proteoglycans with SDS-PAGE showed a concentration-dependent increase in electrophoretic mobility in the presence of SB202190 relative to TGF-β treatment alone (Fig. 4b, lanes 2–7), indicating a concentration-dependent decrease in proteoglycan size upon treatment with SB202190.

Determination of the role of p38 MAP kinase activity in TGF-β-mediated proteoglycan synthesis in human VSMCs using the direct p38 inhibitor SB202190. VSMCs were treated with SB202190 (0.1–10 μM) in the presence of TGF-β (2 ng/ml) and [35S]-SO4 (50 μCi/ml) for 24 h. a [35S]-SO4 incorporation. b SDS-PAGE analysis. c [35S]-Met/Cys incorporation into proteoglycan core protein synthesis. d Biglycan mRNA expression were analyzed exactly as described in the legend of Fig. 1. SB SB431542. e Temporal effects of TGF-β/Alk 5-mediated p38 phosphorylation in human VSMCs. VSMCs were treated with TGF-β (2 ng/ml) for up to 4 h. H2O2 (500 μM)-treated VSMCs for 30 min were used as positive control. Cell lysates were collected, and proteins were resolved by SDS-PAGE on a 10% acrylamide gel and transferred onto a polyvinylidene difluoride membrane. The membrane was incubated with anti-phospho-p38 MAP kinase (Thr180/Tyr 182) polyclonal antibody (1:500 dilution) overnight at 4°C followed by incubation with peroxidase-labeled anti-rabbit IgG (1:5,000 dilution) for 1 h at room temperature. The membrane was then probed with anti-GAPDH rabbit monoclonal antibody (1:1,000 dilution) for 1 h at room temperature followed by incubation with peroxidase-labeled anti-mouse IgG (1:5,000 dilution) for 1 h. The Western blot is representative of three independent experiments. *p < 0.05 versus treated control; **p < 0.01 versus treated control; # p < 0.05 versus untreated control
TGF-β-stimulated VSMCs showed an increase of [35S]-Met/Cys incorporation into secreted proteoglycans of approximately 30% (Fig. 4c) compared to control cells. Treatment of cells with SB202190 (0.1–10 μM) showed a concentration-dependent decrease of TGF-β-mediated [35S]-Met/Cys incorporation into secreted proteoglycans (Fig. 4c) with a maximum decrease of 95% of TGF-β-stimulated component at 10 μM SB202190 (p < 0.01). The TGF-β-stimulated [35S]-Met/Cys incorporation was reduced to basal levels by SB431542 (Fig. 4c).
The effect of p38 inhibition by SB202190 on TGF-β-stimulated VSMC biglycan mRNA levels was investigated. TGF-β-stimulated VSMCs showed an increase in biglycan mRNA expression of 30% compared to control cells (Fig. 4d). The TGF-β-stimulated biglycan mRNA expression was decreased to 100% compared to TGF-β-stimulated component by SB202190 (10 μM) (Fig. 4d), which is consistent with our results that inhibition of p38 blocks TGF-β-stimulated biglycan expression which contributes to the effect on total proteoglycan core protein shown above.
TGF-β/Alk 5-mediated p38 MAP kinase phosphorylation in human VSMCs
We undertook a time course study of TGF-β/Alk 5-mediated p38 MAP kinase phosphorylation in human VSMCs using Western blot. The phospho-p38 MAP kinase band intensity in TGF-β (2 ng/ml) treated cells at 30 min, 1 h, and 2 h were increased significantly compared to control cells (Fig. 4e, lanes 2–4 vs. lane 1) followed by a decrease to basal level at 4 h (Fig. 4e, lane 5). p38 MAP kinase is known to be activated by cell stress [29], therefore we used H2O2-induced oxidative stress as a positive control. Treatment with H2O2 (500 μM) increased p38 MAP kinase phosphorylation compared to control cells (Fig. 4e, lane 6 vs. lane 1). These results demonstrate that in human VSMCs, TGF-β/Alk 5 stimulates the phosphorylation of p38 MAP kinase. We could not examine the action of the inhibitor SB202190 on phospho-p38 as its mode of action is to block the enzymatic activity of p38 but not its phosphorylation, this is similar to the ERK1/2 inhibitor FR180204 (Fig. 3e).
Inhibition of JNK MAP kinase has no effect on TGF-β-induced proteoglycan synthesis
To examine whether JNK MAP kinase is a mediator of TGF-β signaling of GAG elongation in human VSMC, we used the direct JNK MAP kinase inhibitor, SP600125 that specifically blocks the activity of JNK MAP kinase. Cells treated with TGF-β showed 115% increase of [35S]-SO4 incorporation into secreted proteoglycans (Fig. 5a) compared to control cells. Treatment of cells with SP600125 (0.01–1 μM) showed no effect of TGF-β-mediated [35S]-SO4 incorporation into secreted proteoglycans (Fig. 5a). The TGF-β-stimulated [35S]-SO4 incorporation was totally abolished by SB431542 (Fig. 5a). Separation of proteoglycans on an SDS-PAGE showed a decrease in electrophoretic mobility of biglycan in the presence of TGF-β compared to control cells (Fig. 5b, lane 2 vs. lane 1). Electrophoretic mobility of biglycan in TGF-β-treated cells was inhibited in the presence of SB431542 (Fig. 5b, lane 8 vs. lane 2). There was no effect on electrophoretic mobility in the presence of SP600125 relative to TGF-β treatment alone (Fig. 5b, lanes 3–7 vs. lane 2). The radio sulphate data indicated that JNK MAP kinase has no effect on proteoglycan synthesis therefore further analysis of core protein synthesis is not necessary.

Determination of the role of JNK activity in TGF-β-mediated proteoglycan synthesis in human VSMCs using the JNK inhibitor SP600125. VSMCs were treated with SP600125 (0.01–1 μM) in the presence of TGF-β (2 ng/ml) and [35S]-SO4 (50 μCi/ml) for 24 h. a [35S]-SO4 incorporation. b SDS-PAGE analysis were analyzed exactly as described in the legend of Fig. 1. c No effect of JNK inhibitor SP600125 on TGF-β/ALK5-mediated JNK phosphorylation in human VSMCs. VSMCs were treated with TGF-β (2 ng/ml) and SP600125 (1 μM) for 4 h. Cell lysates were collected, and proteins were resolved by SDS-PAGE on a 10% acrylamide gel and transferred onto a polyvinylidene difluoride membrane. The membrane was incubated with anti-phospho-JNK monoclonal antibody (1:200 dilution) overnight at 4°C followed by incubation with peroxidase-labeled anti-mouse IgG (1:4,000 dilution) for 1 h at room temperature. The membrane was then reprobed with anti-smooth-muscle α-actin mouse monoclonal antibody (1:1,000 dilution) for 1 h at room temperature followed by incubation with peroxidase-labeled anti-mouse IgG (1:5,000 dilution) for 1 h. The Western blot is representative of three identical experiments
To determine whether JNK MAP kinase is a potential mediator of TGF-β/Alk 5 signaling in VSMCs, we used direct JNK MAP kinase inhibitor SP600125. A time-course study indicates that TGF-β/Alk 5 stimulates maximum phosphorylation of JNK1/2/3 MAP kinase at 4 h, which is the time point chosen for subsequent studies. A band of 46 kDa, corresponding to phosphorylated JNK1/2/3 MAP kinase, was observed (Fig. 5c, lane 1), indicating the presence of phospho-JNK1/2/3 MAP kinase under basal conditions. The phospho-JNK1/2/3 MAP kinase band intensity in TGF-β (2 ng/ml) treated cell was increased compared to control cells (Fig. 5c, lane 1 vs. lane 2). Treatment with SP600125 (1 μM) decreased the JNK phosphorylation to near basal levels in TGF-β-treated cells (Fig. 5c, lane 3 vs. lane 2). JNK MAP kinase is known to be activated by cell stress [30], therefore we used H2O2-induced oxidative stress as a positive control. Treatment with H2O2 (200 μM) increased JNK1/2/3 MAP kinase phosphorylation significantly compared to control cells (Fig. 5c, lane 4). These data indicated that TGF-β/Alk 5 signals via JNK MAP kinase, but this signaling pathway is not involved in proteoglycan synthesis in human VSMCs.
Involvement of Smad2 carboxyl terminal and linker region phosphorylation in TGF-β signaling in human VSMCs
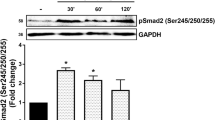
We undertook a time-course study of TGF-β/Alk 5-mediated Smad2 C-terminus phosphorylation in human VSMCs using Western blot. The phospho-Smad2C band intensities in TGF-β (2 ng/ml) treated cells at 30 min, 1 h, 2 h, 4 h, and 6 h were increased compared to control cells (Fig. 6a, lane 1 vs. lane 2–6) followed by a decrease at 24 h to a low level as we have previously reported [5] (Fig. 6a, lane 7). These results demonstrate that in human VSMCs, TGF-β/Alk 5 stimulates rapid phosphorylation of Smad2 C-terminus.

Temporal aspects and effects of MAP kinase inhibitors on TGF-β-stimulated phosphorylation of C-terminal and linker region of Smad2 in human VSMCs. VSMCs were treated with TGF-β (2 ng/ml) for up to 24 h. Cell lysates were collected, and proteins were resolved by SDS-PAGE on a 10% acrylamide gel and transferred onto a polyvinylidene difluoride membrane. The membrane was incubated with anti-phospho-Smad2 Ser 465/467 polyclonal antibody (1:1,000 dilution) (a) or anti-phospho-Smad2 Ser 245/250/255 polyclonal antibody (1:1,000 dilution) (b) overnight at 4°C followed by incubation with peroxidase-labeled anti-rabbit IgG for 1 h at room temperature. The membrane was then reprobed with anti-GAPDH rabbit monoclonal antibody (1:1,000 dilution) for 1 h at room temperature followed by incubation with peroxidase-labeled anti-mouse IgG for 1 h. The Western blot is representative of three independent experiments. c Effects of MAP kinase inhibitors on TGF-β/Alk 5-mediated C-terminal (top row) and linker region (middle row) Smad2 phosphorylation in human VSMCs. VSMCs were treated with TGF-β (2 ng/ml) and UO126 (3 μM), SB202190 (3 μM, 10 μM), SP600125 (1 μM) and SB431542 (3 μM) for 1 h. Cell lysates were collected, and proteins were resolved by SDS-PAGE on a 10% acrylamide gel and transferred onto a polyvinylidene difluoride membrane. The membrane was incubated with anti-phospho-Smad2 Ser 465/467 polyclonal antibody (1:1,000 dilution) (top row) or anti-phospho-Smad2 Ser 245/250/255 polyclonal antibody (1:1,000 dilution) (middle row) overnight at 4°C followed by incubation with peroxidase-labeled anti-rabbit IgG (1:1,000 dilution) for 1 h at room temperature. The membrane was then reprobed with anti-GAPDH rabbit monoclonal antibody (1:1,000 dilution) for 1 h at room temperature followed by incubation with peroxidase-labeled anti-mouse IgG (1:5,000 dilution) for 1 h. The Western blot is representative of three independent experiments. d Effect of TGF-β stimulation on nucleus accumulation of phospho-Smad2L in VSMCs. VSMCs were treated with TGF-β (2 ng/ml) for 1 h. Cell lysates were collected, nucleus proteins were extracted and resolved by SDS-PAGE followed by transfer onto a polyvinylidene difluoride membrane. The membrane was incubated with anti-phospho-Smad2 Ser 245/250/255 polyclonal antibody (1:1,000 dilution) overnight at 4°C followed by incubation with peroxidase-labeled anti-rabbit IgG (1:1,000 dilution) for 1 h at room temperature. The membrane was then reprobed with anti-GAPDH rabbit monoclonal antibody (1:1,000 dilution) for 1 h at room temperature followed by incubated with peroxidase-labeled anti-mouse IgG (1:5,000 dilution) for 1 h. The Western blot is representative of three independent experiments
A time course of TGF-β/Alk 5-mediated Smad2L phosphorylation was determined in human VSMCs using Western blot. The Smad2 linker region phosphorylation (phospho-Smad2L) band intensities in TGF-β (2 ng/ml)-treated cells at 1 h, 2 h, 4 h, and 6 h were increased significantly compared to control cells (Fig. 6b, lanes 2–5 vs. lane 1) followed by a decrease at 24 h to a low level (Fig. 6b, lane 6). The increased level of cytosolic phospho-Smad2L in TGF-β-treated cells at 1 h was completely prevented in cells treated with SB431542 (Fig. 6b, lane 7 vs. lane 2). These results demonstrate that in human VSMCs, TGF-β/Alk 5 stimulates maximum phosphorylation of Smad2L at 1 h, which is sustained for at least 6 h.
Experiments then focused on whether Smad2 phosphorylation is involved in the TGF-β-mediated MAP kinase signaling pathway. MEK1/2 inhibitor UO126, p38 MAP kinase inhibitor SB202190 and JNK MAP kinase inhibitor SP600125 were used. A band of 58 kDa corresponding to phosphorylated Smad2 at the C-terminal region was observed (Fig. 6c, lane 1), indicating the presence of phospho-Smad2C under basal conditions. The phospho-Smad2C band intensity in TGF-β (2 ng/ml; 1 h)-treated cells was greatly increased compared to control cells (Fig. 6c, lane 1 vs. lane 2), indicating the stimulation of Smad2 C-terminal phosphorylation. Treatment with UO126 (3 μM) or SB202190 (3 μM) or SP600125 (1 μM) had no effect on the Smad2 C-terminus phosphorylation in TGF-β-treated cells (Fig. 6c, lane 3–5 vs. lane 2). Interestingly, our preliminary results indicated that SB202190 does not block activity of C-terminal Smad2 phosphorylation via TGF-β/Alk 5 at 10 μM in VSMCs (data not shown), a concentration which was found to block the activity of TGF-β/Alk 5 in A498 renal epithelial carcinoma cells [31]. Treatment with SB431542 (3 μM) reduced Smad2C phosphorylation to the basal level (Fig. 6c, lane 6 vs. lane 1). These results indicate that Smad2C phosphorylation does not involve TGF-β-mediated p38 MAP kinase, MEK1/2 and JNK MAP kinase signaling.
Experiments then focused on whether Smad2 linker region phosphorylation is involved in TGF-β-mediated MAP kinase signaling pathway. In TGF-β-treated cells, UO126 (3 μM) and SB202190 (3 μM) inhibited the Smad2 linker region phosphorylation, approximately 30 and 40%, respectively, (Fig. 6c, lane 3, 4 vs. lane 2) whereas there was no effect of SP600125 (Fig. 6c, lane 5 vs. lane 2). The inhibitory response to the MAP kinase inhibitors was relatively small but fairly consistent. In five separate experiments we observed inhibition of phospho-Smad2L levels by ERK inhibition on five occasions, inhibition due to p38 inhibition on three occasions and we never observed inhibition using a JNK inhibitor. In one experiment, combined inhibition of ERK and p38 had an additive effect compared to each inhibitor alone. As expected, treatment with SB431542 (3 μM) reduced Smad2 linker region phosphorylation to near basal levels in all experiments (Fig. 6c, lane 6 vs. lane 1). We then further investigated if TGF-β stimulation of VSMCs leads to accumulation of phospho-Smad2L in the nucleus. Cells were treated with TGF-β and a nuclear fraction isolated and assessed for phospho-Smad2L by Western blotting. TGF-β stimulation of VSMCs leads to marked accumulation of phospho-Smad2L in the nucleus compared to control cells (Fig. 6d, lane 2 vs. lane 1). These results suggest that phosphorylation of the linker region of Smad2 by p38 and ERK1/2 is involved in TGF-β-mediated biglycan synthesis in human VSMCs.
Discussion
Proteoglycan synthesis in VSMCs is of interest because the binding of lipids to modified proteoglycans is an initiating step in the development of atherosclerosis [14, 15]. Multiple vasoactive growth factors regulate the synthesis and structure of proteoglycans and their cellular signaling pathways are of interest as potential therapeutic targets [11, 16]. TGF-β is involved in multiple mechanisms of the development of atherosclerosis including actions on proteoglycan synthesis [5, 17]. TGF-β primarily elicits its effects through the canonical Smad/phospho-Smad transcription factor pathway [18]. Recent studies have shown a potentially important role for MAP kinases in determining the response to TGF-β [18, 20, 21] and in this report we have investigated the role of MAP kinases in TGF-β-mediated proteoglycan synthesis in human VSMCs. TGF-β treatment stimulates the phosphorylation of the three major MAP kinases, p38, ERK, and JNK, all within a relatively short time frame, but not as rapid as the phosphorylation of Smad2 in its carboxyl terminal. TGF-β stimulates the elongation of GAG chains on DS proteoglycans, specifically biglycan and this response requires p38 and ERK but not JNK MAP kinase. TGF-β also stimulates the expression of proteoglycan core proteins including at least the critical lipid binding proteoglycan, biglycan and the TGF-β-stimulated expression of biglycan is mediated via p38 and ERK but also not JNK. Inhibition of both ERK and p38 alone completely abrogates the effect of TGF-β on proteoglycan synthesis, suggesting that they may be working synergistically and/or may be able to replace each other's influence. TGF-β mediates rapid phosphorylation of Smad2 on its carboxy terminal and this is not antagonized by inhibitors of ERK, p38 or JNK indicating as expected that the MAP kinase is not involved in this response. TGF-β also increases phosphorylation of Smad2 in the linker region which is, as expected, blocked by SB431542 whose target is Alk 5. The TGF-β-mediated phosphorylation of Smad2 in the linker region is weakly blocked by inhibitors of ERK, p38 but not by a JNK MAP kinase inhibitor. Linker region phosphorylation has previously been described to involve ERK and JNK [32, 33]. The data indicates that p38 and ERK are involved in the TGF-β-mediated modification of proteoglycan synthesis in VSMCs and although JNK is phosphorylated it is not involved in proteoglycan synthesis. Furthermore, it appears that the mechanisms differentiating the activity of the three MAP kinases to mediate TGF-β-stimulated proteoglycan synthesis might relate to the activity (or lack thereof) of the MAP kinases to phosphorylate Smad2 in its linker region a point that requires further investigation. It is specifically unclear as to how some but not all MAP kinases are able to phosphorylate Smad2 in its linker region.
The role of Smad transcription factors in TGF-β-mediated cellular responses has been researched and reviewed [18, 34], but the role of MAP kinases is a nascent area of interest [18, 20, 21]. The earliest observations were the rapid activation of p21Ras by TGF-β [35] and the identification of the MAP kinase kinase kinase (MAPKKK) TAK1 (TGF-β-activated kinase) as an early response to TGF-β [36]. Subsequently the activation (phosphorylation) of the three major MAP kinases has been described [18, 20, 21]. The direct nature of the activation, meaning that it was independent of Smad phosphorylation, arose in cells in which Smad function was compromised and TGF-β could still mediate JNK activation and phosphorylation. Nevertheless, Smad and MAP kinase pathways downstream of TβRI/Alk 5 are known to integrate. The canonical TGF-β activation of Smads involves Ser phosphorylation in the carboxy terminal [18] essential for allowing the interaction with Smad4 and thus translocation to the nucleus and regulation of transcription [18]. MAP kinases can phosphorylate Smads outside the carboxy terminal region and specifically in what is termed the linker region [32, 37]. The convergence of Smad and MAP kinase signaling can result in both cooperativity and antagonism and as such is context dependent [38].
The carboxy terminal phosphorylation and activation of Smads is strong and rapid, occurring in minutes after receptor stimulation, and we made similar findings in human VSMCs (see Fig. 6a). The temporal aspects and the strength of the phosphorylation of MAP kinases by TGF-β are much more variable. Some responses are rapid, but others are delayed and prolonged, possibly suggesting the involvement of pathways downstream of Smad phosphorylation and transcription and indeed the potential exists for both responses to co-exist. In vivo, the presence of multiple growth factors in diseased vessels raises the possibility that the much stronger phosphorylation of MAP kinases via tyrosine kinase receptors (see Fig. 3e) may mask the TGF-β-mediated activation, notwithstanding that both pathways may play a role in cellular responses. Further mechanistic studies in vitro and in vivo are required to define the contribution of TGF-β-mediated MAP kinase signaling in vascular disease.
Phosphorylation of the carboxyl terminal of Smad2/3 is associated with the translocation of the Smad to the nucleus, but the role of linker region phosphorylation remains unresolved and is an interesting question in the cell biology of TGF-β actions. Linker region phosphorylation can prevent the translocation of the Smad to the nucleus [37] and thus inhibit TGF-β signaling. Conversely, linker region phosphorylation can modify and enhance the transcription activity of the Smad hetero-complex and hence regulate and intensify TGF-β signaling [33]. We have demonstrated that TGF-β stimulation of VSMCs leads to marked accumulation of phospho-Smad2 in the nucleus and this Smad is phosphorylated in the linker region. This strongly suggests that the MAP kinase-mediated phosphorylation of the linker region of Smad2 in VSMCs leads to the stimulation of growth factor-mediated GAG hyperelongation. Further work will be aimed at identifying the genes and their targets that upregulate the mechanism of GAG synthesis leading to the hyperelongation of GAG chains associated with increased lipid binding and atherosclerosis.
Li et al. [32] have recently reported that ERK2 is involved in the TGF-β-mediated phosphorylation of the linker region but not the carboxy terminal region of Smad2, and by correlation TGF-β stimulation of collagen expression also involves the ERK2 and linker region phosphorylation [32]. The NIH/3T3 fibroblasts showed much higher expression of ERK2 compared to ERK1 whereas in our cells expression and phosphorylation of ERK1 and ERK2 and consequently, after stimulation, of phospho-ERK1 and phospho-ERK2 were approximately equal. Our studies demonstrate a role of ERK and phospho-ERK in proteoglycan synthesis by VSMCs. As the work of Li et al. [32] demonstrated, specific roles for individual ERK isoforms then application of their methodologies to study the specific roles of ERK1 and ERK2 (and their phospho derivatives) should be a worthwhile extension of the current studies. Also, the over expression of a Smad2 mutant that lacks the ability to be phosphorylated in the linker region would provide an interesting means to evaluate the mechanistic implication of the MAP kinase/Smad2 interaction.
ERK can be negatively or positively involved in TGF-β induced cellular responses [38]. In our VSMCs, inhibition of ERK and p38 blocks the effect of TGF-β to mediate elongation of GAG chains (see Figs. 3, 4). It is therefore likely that ERK is positively implicated in the response to TGF-β, implying that there is a downstream ERK target that is involved in proteoglycan synthesis. However, the biochemical mechanism through which growth factors cause hyperelongation of GAG chains is unknown, at least in part, because the mechanism of polymerization of CS/DS GAG chains is not resolved [7, 39–41]. We have recently demonstrated that the GAG elongation to a multitude of growth factors, including TGF-β, is dependent upon both transcription and translation [8]. Our studies on growth factors have pointed to some of the likely transcription factors involved, such as phospho-Smad2 and its binding partners but the targets, presumably the transcripts for the enzymes involved in polymerization remain unknown [41]. Linking the transcription factors of the growth factor-signaling pathways with the activation of the biochemical mechanisms mediating GAG hyperelongation is an ongoing aim of our laboratory.
In conclusion, continuing evidence emerges to support the role of proteoglycans in the initiation and progression of atherosclerosis, particularly human atherosclerosis and cardiovascular disease [15, 42]. The role might not be so prominent in animal models because as recently pointed out, the mechanisms of atherosclerosis are quite different in rodent models compared to humans [43]. Whereas the animal model favors the so-called inflammatory response of atherosclerosis, human atherosclerosis is dependent upon events occurring in a proteoglycan-rich layer of the neo-intima [43]. Nevertheless, we have recently been able to utilize a standard rodent model, the high-fat-fed ApoE−/− model to provide proof of principle that an agent which inhibits proteoglycan synthesis, specifically inhibits GAG hyperelongation, reduces LDL binding in vitro, and ex vivo can reduce the deposition of lipid in the wall of the blood vessel [13]. Notwithstanding the limitations of the animal model, this suggests that targeting proteoglycan synthesis may be a pathway for the prevention of atherosclerosis in humans. The major limitations would be hepatic effects; however, these are unlikely to be relevant due to the different metabolism in that tissue [42]. Growth factor-signaling pathways provide valid targets for the prevention of atherosclerosis, and the data provided in this study further elucidates the important signaling pathways through which TGF-β mediates its effects on proteoglycan synthesis in human VSMCs.
References
Wight TN (1989) Cell biology of arterial proteoglycans. Arteriosclerosis 9:1–20
Hascall VC, Heinegard DK, Wight TN (1991) Proteoglycans: metabolism and pathology. In: Hay ED (ed) Cell biology of extracellular matrix. Plenum Press, New York, pp 149–175
Wight T (1996) The vascular extracellular matrix. In: Fuster V, Ross R, Topol EJ (eds) Atherosclerosis and coronary artery disease. Lippincott-Raven, Philadelphia, pp 421–440
Schonherr E, Jarvelainen HT, Kinsella MG, Sandell LJ, Wight TN (1993) Platelet-derived growth factor and transforming growth factor-beta 1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler Thromb 13:1026–1036
Dadlani H, Ballinger ML, Osman N, Getachew R, Little PJ (2008) Smad and p38 MAP kinase-mediated signaling of proteoglycan synthesis in vascular smooth muscle. J Biol Chem 283:7844–7852
Ivey ME, Little PJ (2008) Thrombin regulates vascular smooth muscle cell proteoglycan synthesis via PAR-1 and multiple downstream signalling pathways. Thromb Res 123:288–297
Ballinger ML, Ivey ME, Osman N, Thomas WG, Little PJ (2009) Endothelin-1 activates ETA receptors on human vascular smooth muscle cells to yield proteoglycans with increased binding to LDL. Atherosclerosis 205:451–457
Yang SN, Burch ML, Getachew R, Ballinger ML, Osman N, Little PJ (2009) Growth factor-mediated hyper-elongation of glycosaminoglycan chains on biglycan requires transcription and translation. Arch Physiol Biochem 115:147–154
Camejo G (1982) The interaction of lipids and lipoproteins with the intercellular matrix of arterial tissue: its possible role in atherogenesis. Adv Lipid Res 19:1–53
Little PJ, Tannock L, Olin KL, Chait A, Wight TN (2002) Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler Thromb Vasc Biol 22:55–60
Ballinger ML, Nigro J, Frontanilla KV, Dart AM, Little PJ (2004) Regulation of glycosaminoglycan structure and atherogenesis. Cell Mol Life Sci 61:1296–1306
Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J (2002) Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417:750–754
Ballinger ML, Osman N, Hashimura K, de Hann J, Jandeleit-Dahm K, Allen TJ, Tannock LR, Rutledge JC, Little PJ (2009) Imatinib inhibits vascular smooth muscle proteoglycan synthesis and reduces LDL binding in vitro and aortic lipid deposition in vivo. J Cell Mol Med. doi:10.1111/j.1582-4934.2009.00902.x)
Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K (2007) Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol 27:1159–1165
Little PJ, Osman N, O’Brien KD (2008) Hyperelongated biglycan: the surreptitious initiator of atherosclerosis. Curr Opin Lipidol 19:448–454
Little PJ, Ballinger ML, Osman N (2007) Vascular wall proteoglycan synthesis and structure as a target for the prevention of atherosclerosis. Vasc Health Risk Manag 3:1–8
Bobik A, Agrotis A, Kanellakis P, Dilley R, Krushinsky A, Smirnov V, Tararak E, Condron M, Kostolias G (1999) Distinct patterns of transforming growth factor-beta isoform and receptor expression in human atherosclerotic lesions. Colocalization implicates TGF-beta in fibrofatty lesion development. Circulation 99:2883–2891
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577–584
Massague J (1998) TGF-beta signal transduction. Annu Rev Biochem 67:753–791
Kretzschmar M, Doody J, Timokhina I, Massague J (1999) A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev 13:804–816
Moustakas A, Heldin CH (2005) Non-Smad TGF-beta signals. J Cell Sci 118:3573–3584
Ungefroren H, Lenschow W, Chen WB, Faendrich F, Kalthoff H (2003) Regulation of biglycan gene expression by transforming growth factor-beta requires MKK6-p38 mitogen-activated protein Kinase signaling downstream of Smad signaling. J Biol Chem 278:11041–11049
Ungefroren H, Groth S, Ruhnke M, Kalthoff H, Fandrich F (2005) Transforming growth factor-beta (TGF-beta) type I receptor/ALK5-dependent activation of the GADD45beta gene mediates the induction of biglycan expression by TGF-beta. J Biol Chem 280:2644–2652
Neylon CB, Little PJ, Cragoe EJ Jr, Bobik A (1990) Intracellular pH in human arterial smooth muscle. Regulation by Na+/H+ exchange and a novel 5-(N-ethyl-N-isopropyl)amiloride-sensitive Na(+)- and HCO3(−)-dependent mechanism. Circ Res 67:814–825
Chang Y, Yanagishita M, Hascall VC, Wight TN (1983) Proteoglycans synthesized by smooth muscle cells derived from monkey (Macaca nemestrina) aorta. J Biol Chem 258:5679–5688
Nigro J, Dilley RJ, Little PJ (2002) Differential effects of gemfibrozil on migration, proliferation and proteoglycan production in human vascular smooth muscle cells. Atherosclerosis 162:119–129
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS (2002) SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 62:65–74
Jiang J, Song CP (2008) MEK1/2 and p38-like MAP kinase successively mediate H(2)O(2) signaling in Vicia guard cell. Plant Signal Behav 3:996–998
Zhang JY, Jiang H, Gao W, Wu J, Peng K, Shi YF, Zhang XJ (2008) The JNK/AP1/ATF2 pathway is involved in H2O2-induced acetylcholinesterase expression during apoptosis. Cell Mol Life Sci 65:1435–1445
Laping NJ, Grygielko E, Mathur A, Butter S, Bomberger J, Tweed C, Martin W, Fornwald J, Lehr R, Harling J, Gaster L, Callahan JF, Olson BA (2002) Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol 62:58–64
Li F, Zeng B, Chai Y, Cai P, Fan C, Cheng T (2009) The linker region of Smad2 mediates TGF-beta-dependent ERK2-induced collagen synthesis. Biochem Biophys Res Commun 386:289–293
Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, Fujisawa J, Okazaki K (2004) TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 23:7416–7429
Agrotis A, Kalinina N, Bobik A (2005) Transforming growth factor-beta, cell signaling and cardiovascular disorders. Curr Vasc Pharmacol 3:55–61
Mulder KM, Morris SL (1992) Activation of p21ras by transforming growth factor beta in epithelial cells. J Biol Chem 267:5029–5031
Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K (1995) Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270:2008–2011
Massague J (2003) Integration of Smad and MAPK pathways: a link and a linker revisited. Genes Dev 17:2993–2997
Verrecchia F, Tacheau C, Wagner EF, Mauviel A (2003) A central role for the JNK pathway in mediating the antagonistic activity of pro-inflammatory cytokines against transforming growth factor-beta-driven SMAD3/4-specific gene expression. J Biol Chem 278:1585–1593
Victor XV, Nguyen TK, Ethirajan M, Tran VM, Nguyen KV, Kuberan B (2009) Investigating the elusive mechanism of glycosaminoglycan biosynthesis. J Biol Chem 284:25842–25853
Gotoh M, Yada T, Sato T, Akashima T, Iwasaki H, Mochizuki H, Inaba N, Togayachi A, Kudo T, Watanabe H, Kimata K, Narimatsu H (2002) Molecular cloning and characterization of a novel chondroitin sulfate glucuronyltransferase that transfers glucuronic acid to N-acetylgalactosamine. J Biol Chem 277:38179–38188
Little PJ, Ballinger ML, Burch ML, Osman N (2008) Biosynthesis of natural and hyperelongated chondroitin sulfate glycosaminoglycans: new insights into an elusive process. Open Biochem J 2:135–142
Tabas I, Williams KJ, Boren J (2007) Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116:1832–1844
Finn AV, Kramer MC, Vorpahl M, Kolodgie FD, Virmani R (2009) Pharmacotherapy of coronary atherosclerosis. Expert Opin Pharmacother 10:1587–1603
Acknowledgments
This work was supported by research grants from the National Health and Medical Research Council of Australia Project Grants (#268928 and #472611) and Development Grant #418934) (PJL), project grant from the Diabetes Australia Research Trust (NO) and a Grant-in-Aid from the National Heart Foundation of Australia (PJL). The PhD Program of MLB generously received support through a National Heart Foundation of Australia post-graduate scholarship and a post-graduate supervisor award from GlaxoSmithKline Australia.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. L. Burch and S. N. Y. Yang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Burch, M.L., Yang, S.N.Y., Ballinger, M.L. et al. TGF-β stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cell. Mol. Life Sci. 67, 2077–2090 (2010). https://doi.org/10.1007/s00018-010-0315-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0315-9