
Abstract
Introduction
Mitochondrial dysfunction is a common denominator of neuroinflammation recognized by neuronal oxidative stress-mediated apoptosis that is well recognized by common intracellular molecular pathway-interlinked neuroinflammation and mitochondrial oxidative stress, a feature of epileptogenesis. In addition, the neuronal damage in the epileptic brain corroborated the concept of brain injury-mediated neuroinflammation, further providing an interlink between inflammation, mitochondrial dysfunction, and oxidative stress in epilepsy.
Materials and methods
A systematic literature review of Bentham, Scopus, PubMed, Medline, and EMBASE (Elsevier) databases was carried out to provide evidence of preclinical and clinically used drugs targeting such nuclear, cytosolic, and mitochondrial proteins suggesting that the correlation of mechanisms linked to neuroinflammation has been elucidated in the current review. Despite that, the evidence of elevated levels of inflammatory mediators and pro-apoptotic protein levels can provide the correlation of inflammatory responses often concerned with hyperexcitability attributing to the fact that mitochondrial redox mechanisms and higher susceptibilities to neuroinflammation result from repetitive recurring epileptic seizures. Therefore, providing an understanding of seizure-induced pathological changes read by activating neuroinflammatory cascades like NF-kB, RIPK, MAPK, ERK, JNK, and JAK-STAT signaling further related to mitochondrial damage promoting hyperexcitability.
Conclusion
The current review highlights the further opportunity for establishing therapeutic interventions underlying the apparent correlation of neuroinflammation mediated mitochondrial oxidative stress might contribute to common intracellular mechanisms underlying a future prospective of drug treatment targeting mitochondrial dysfunction linked to the neuroinflammation in epilepsy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The epileptic brain is susceptible to oxidative stress due to high oxygen-metabolic activity and its high iron content, which is needed for neurological processes. The changes in the oxygen concentration are observed during an excessive prolonged seizure-like insult in the brain [1]. Excessive glutamatergic neurotransmission-mediated neuronal injury in the epileptic brain leads to the activation of glial cells as a neuroimmune response activating inflammatory cascades involved in neuroinflammation [1, 2]. The significant increase of neuroinflammation is one of the causes of secondary seizures, further leading to neuronal apoptotic death. Under the adverse and traumatic conditions associated with prolonged repetitive epileptic seizures, glial cells migrate as a neuroimmune response mechanism and release pro-inflammatory mediators like cytokines (IL-1β, IL-18, and HMGB1), the excessive release of such neurotoxic substances results in neuronal degeneration [3, 4]. The high expression of HMGB1 and IL-1β is seen among patient’s brain tissue damage in status epilepticus and temporal epilepsy, indicating the excessive neuroinflammatory processes mediating neuronal death [3, 4]. Therefore, the immunoreactivity such as migration of microglial cells in response to the seizure-induced brain tissue damage represents the oxidative stress in epilepsy. The numerous mechanisms are associated with prolonged epileptic seizures causing neuronal injury resulting in inflammatory responses and production of reactive oxygen species involved in neuronal cell death [5, 6]. These include the elevation of intracellular calcium ion concentration disrupting mitochondrial membrane-mediated neuronal death, a prominent feature of seizures-induced neuronal damage [7, 8]. The increased intracellular calcium influx is a key for initiating the mitochondrial apoptotic pathway. The mitochondrial calcium uniporter (MCU) takes up the calcium rapidly to maintain cytosolic calcium homeostasis. MCU mutation causes mitochondrial calcium overload inducing mitochondrial oxidative stress by increasing ROS production representing mDNA alterations with increased ROS and depletion ATP production-mediated neuronal death [9,10,11]. The increased mitochondrial reactive oxygen species (ROS) initiates inflammatory responses, aggravating mitochondrial dysfunction and ultimately contributing to neuronal death through an increase in Bim protein (pro-apoptotic protein) and mitochondrial permeability transfer pore (MPTP).
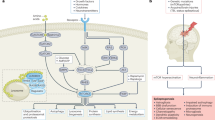
Additionally, this results in the release of cytochrome C (Cyt C) into the cytosol forming Apaf-1/cytochrome c complex with subsequent strong activation of caspase-9 to caspase-3 following neuronal DNA fragmentation exhibiting neurodegeneration in epilepsy [10, 12] (Fig. 1). Thus, in response to mitochondrial damage, the strong activation of system glial cells releases neurotoxin substances IL-1β, IL-6, IL-18, and TNF-alpha, HMGB1 inducing neuronal death [13,14,15]. The increased cytokines participate in carrying downstream signaling of inflammation via Toll-like receptor (TL-1R) activating transcriptional factor NF-kB further responsible for elevating pro-inflammatory mediators provoking neurodegeneration in epilepsy [16,17,18]. Thus, the diverse stimuli of such neurotoxic cytokines further initiating the downstream intracellular inflammatory signaling by binding to the Toll-like receptor recruiting TRAF2/3 (TNF receptor-associated factor) as well activating nuclear factor-kappa beta (NF-kB) increasing neurotoxic cytokines levels induced oxidative stress [4, 13,14,15]. Furthermore, the excessive cellular ROS-dependent TRAF protein stimulates Src kinase protein to implicate downstream PI3K/Akt/mTOR signaling pathways of neuronal death. The other stress-activated proteins like c-Jun N-terminal kinase (JNK), mitogen-activated protein kinase (MAPK) P38 get stimulated by ROS-dependent TRAF protein increasing pro-inflammatory mediators levels as well as trigger the mitochondrial apoptosis pathway inducing neuronal death through apoptotic-inducing factors and increased expression of pro-apoptotic proteins (BAX, BCL2) [16,17,18,19].

Representation of correlation between neuroinflammation and mitochondrial dysfunction in epilepsy
However, the mitochondria are a major factor in the production of the reactive oxygen species with elevated cytokine levels and contribute to apoptotic neuronal death [17, 20]. Therefore, chronic epileptic seizures result in alterations of intracellular calcium levels meditated impairment of mitochondrial bioenergetics, contributing to declining ATP-mediated brain damage in epilepsy [19, 21]. The prolonged electrical discharge in the epileptic brain promotes the homeostatic alterations of intracellular calcium accumulation and increased reactive oxygen species production with chronic activation of microglial cells representing neuroinflammation in epilepsy. Therefore, the neuroimmune system causes early inflammatory changes in the brain and the increased release of glutamate, causing persistent hyperactivation of metabotropic receptors (NMDA, AMPA) that result in increased intracellular calcium concentration mediated excitotoxicity. Together, the excessive migration of glial cells, metabotropic receptors, and voltage-gated calcium channels prolong the intracellular calcium concentration which triggers the mitochondrial calcium release inducing excitotoxicity and eventually initiating mitochondrial apoptotic pathway-mediated neurodegeneration in epilepsy [22, 23]. The increased intracellular calcium levels potentiate mPTP opening leading to calcium trafficking across the mitochondrial membrane causing mitochondrial swelling and initiating the pro-apoptotic signaling with increased levels (Bcl-2, Bax) [23, 24]. The mitochondrial membrane damage causes the release of the cytochrome C (cyt C) into the cytosol, further causing neuronal death by different mechanisms, i.e., activating the caspase-3 than caspase-9-mediating activation of nucleases promoting DNA damage and apoptosis [25,26,27]. The release of cytochrome C results in a deficiency of ATP (bioenergetic deficits), with a corresponding rise in ROS and activation of the neuroimmune system, which further initiates the neuronal dysfunction in epilepsy. The rise in ROS increases the neuroimmune response to activation of glial cells releasing neurotoxic substances and contributing to neuronal death in epilepsy through apoptotic-inducing factors. [28, 29]. Thus, targeting the neuroinflammation causing mitochondrial oxidative stress or mitochondrial oxidative stress causing neuroinflammation might act as a novel treatment neuroprotective approach in attenuating neuronal death by prolonged excessive seizures.
Pathways correlating neuroinflammation and mitochondrial dysfunction and neuronal death in epilepsy
The persistent activation of neuroglial cells in response to the concurrent seizures further causes mitochondrial dysfunction-mediated neuronal death in epilepsy [30,31,32]. Therefore, the various neuroinflammatory cascades get initiated in response to the prolonged seizure-induced neuronal injury. The elevated levels of pro-inflammatory mediators’ levels represent the aberrant migration of astrocytes. Microglial cells release cytotoxic substances like cytokines (IL-1 beta, TNF-alpha, IL-6, etc.) factors exacerbate the mitochondrial oxidative stress in epilepsy [31, 32]. Seizure-induced physiological changes in the epileptic brain include dysregulation of glia immune-inflammatory activity that is characterized by activation of various neuroinflammatory cascades like NF-kB, RIPK, MAPK, ERK, JNK, and JAK–STAT signaling involved in causing brain inflammation further related to mitochondrial damage promoting hyperexcitability with elevated levels of cytokines, chemokine-mediated neuronal dysfunction in epilepsy, or vice versa [32, 33]. Elevated levels of TNF-alpha stimulate the neuronal hyperexcitability focusing mitochondrial oxidative stress under prolonged seizures (status epilepticus) which have seemed to be regulating activator protein 1 (AP-1)-mediated apoptotic neuronal death through activation of NF-kB and MAPK/ERK and p38/ JNK signaling as inflammatory response provides a clear correlation of neuroinflammation-mediated mitochondrial apoptotic signaling in epilepsy [32, 33].
Additionally, the other inflammatory mediator, Interleukin-1 bet, initiates PI3K/mTOR/Akt signaling which seems to be inducing aberrant apoptotic hippocampal neuronal death by interacting with mitochondria-derived activators caspase-3, BCL2-associated X, BH3 proteins causing cognitive dysfunction in temporal lobe epilepsy or status epilepticus [34]. Whereas the cytokine HMGB1 also seems to be involved in intracellular calcium ironic concentration by impacting GABA-ergic inhibitory transmission and increasing NMDA transmission linked to increased calcium influx further overloads mitochondria-mediated excitotoxicity in epilepsy [33, 35, 36]. The current review provided the correlation of neuroinflammatory pathways, and evidence of elevated levels of inflammatory mediators mediated mitochondrial oxidative stress underlying excitotoxicity in epilepsy.
Correlation of neuroinflammatory PI3k/Akt and Bad/Bcl (XL) mediated neuronal mitochondrial apoptotic death in epilepsy
Under the seizure-induced neuronal cell stress response, there is an activation of adenosine monophosphate-activated protein kinase that has a role in the up-regulation of pro-apoptotic BH3-only protein Bcl-2-modifying factor (Bmf) [37, 38]. AMPK enzyme is activated in response to the cell stress-like depletion of ATP in the affected brain region caused by rapid prolonged epileptic seizures [37, 39]. In epilepsy, the increased intracellular calcium concentration induces excitotoxicity injury by disrupting mitochondrial membrane-mediated neuronal apoptotic death [39]. Therefore, the AMPK sensitizes mitochondrial dysfunction induces apoptosis by up-regulation of Bax. The activated Bax and Bak further promote mitochondrial dysfunction by forming MPTP and allowing the release of cytochrome c, further activating caspase cascade-mediated apoptotic neuronal death [38, 39]. Along with apoptosis regulation, the AMPK also tends to attenuate neuroinflammation by inhibiting the NF-kB and activating neuroprotective signaling through SIRT1, PPARG coactivator 1-alpha, AMPK/p53/NF-κB, and AMPK/FoxO/NF-κB pathways [40]. Therefore, the studies provided the correlation of AMPK with inflammation, i.e., the activation of AMPK suppressing NF-kB and decreasing pro-inflammatory mediators IL-6, TNF-α, and iNOS that are further involved in mitochondrial oxidative stress. In turn, AMPK inhibits the NF-κB signaling pathway and increases cellular NAD+ levels by activating sirtuin 1 (SIRT1), FO XO, and PGC1α [41]. Thus, it should consider the dual role of AMPK in mitochondrial dysfunction and neuroinflammation either as a neuroprotective effect or neuronal death depending upon stress stimuli [40].
Role of inflammasome and mitochondrial dysfunctioning in epilepsy
The recurrence of epileptic seizures and aberrant activation of neuroimmune system of excessive migration of glial cells releasing the pro-inflammatory mediators like cytokines (IL-1β, IL-18, and HMGB1) give rise to the neuronal excitability and neurodegeneration seen among status epilepticus and temporal lobe epileptic patients [15, 17, 42]. The NLRP3 inflammasome protein is highly expressed in microglial cells. It gets activated by increased intercellular calcium or reactive oxygen species, mitochondrial and autophagolysosomal system dysfunctioning results in neuronal death [15, 43, 44]. The inflammasome NLRP3 gets activated in response to the increased intracellular calcium-induced cellular stress and further initiates the caspase-dependent secretion of pro-inflammatory mediators. In epilepsy, the hyperactivation of metabotropic receptors focuses on excitotoxicity by increased intracellular calcium influx induces the mitochondrial Ca2+ overload leading to mitochondrial oxidative stress associating increased ROS-mediated neuronal apoptotic death [23, 24]. The MPT further releases the cytochrome C by altering cardiolipin molecules abundantly found on the mitochondrial membrane binds cytochrome C to the mitochondrial membrane [45]. Under the mitochondrial oxidative stress, cardiolipin binding gets alters that encourage cytochrome C to release by detaching the cytochrome C from the mitochondrial membrane and subsequently initiates neuronal apoptotic death [46, 47]. In this process of neuronal death, the cardiolipin also initiates the activation of inflammasome NLRP3, triggers caspase-1 maturation, and increases the secretion of pro-inflammatory cytokines (IL-1β and IL-18) [45, 46]. Thus, the increased pro-inflammatory mediator indicates the mitochondrial oxidative stress with increased expression of NF-κB or mitochondrion-derived inflammation-induced neuronal death under the stress condition of prolonged epileptic seizures.
Alteration of sirtuin regulating neuroinflammation and mitochondrial dysfunction in epilepsy
Increased oxidative stress and activation of the cytokines are the hallmarks of neurodegeneration in epilepsy. Growing evidence shows that decreased levels of sirtuin (SIRT1 and SIRT3) protein in the epileptic brain indicate mitochondrial dysfunction, causing neurodegeneration under the stressful condition of prolonged seizures. Sirtuin proteins are highly expressed in neuronal and non-neuronal cells (glial cells) that tend to possess neuroprotective mechanisms against seizure-induced neuronal death followed by prolonged repetitive seizures in the brain [48,49,50]. Evidenced data concluded the involvement of sirtuin in inhibiting the mitochondrial permeability transition pore (mPTP) formation. Downregulation of sirtuin protein during recurrent spontaneous seizures resulted in mitochondrial permeability transition pore (mPTP) formation with subsequent ATP depletion caused by increased intracellular calcium trafficking triggering apoptotic neuronal death [51, 52]. Therefore, Sirtuin protein regulates the mitochondrial bioenergetic by activating peroxisome proliferator-activated receptor coactivator 1-α (PGC-1α), modulating mitochondrial function with increased activation nuclear respiratory factor (NRF). Alteration in Sirtuin protein is linked to the alteration in the redox reaction indicating the mitochondrial oxidative stress under the prolonged repetitive seizures [48]. Sirtuin protein also regulated oxidative stress and neuroinflammation by influencing the deacetylation of (PGC-1α) and suppresses the oxidative stress through overexpression of MnSOD anti-oxidant that has a role in scavenging-free radicals [53,54,55,56]. Perhaps, the most prominent function of sirtuin in the regulation of mitochondrial function also regulates the neuroinflammation by deacetylation of NF-κB p65 subunit elucidating anti-inflammatory activity [53, 57]. Therefore, SIRT3 prevents the glial cell secretion of pro-inflammatory mediators contributing to neuronal damage in epilepsy [58]. Increased inflammatory mediators are the components of epileptogenesis that aggravate secondary recurrent seizures [59]. Therefore, sirtuin acts as a potential neuroprotective strategy in epilepsy.
Neuroinflammatory JAK–STAT signaling promoting mitochondrial dysfunction
Atypical activation of JAK–STAT signaling is mainly related to the neuroinflammatory processes aggravating neuronal impairment in Epilepsy. Diverse stimuli of cytotoxic substances released by activated glial cells in response to prolonged seizure-induced brain damage like axon sprouting represent neuroinflammation in epilepsy [60]. The elevated levels of cytokines (TNF-α, IL-6, IL-1β) cause secondary development of seizures that are adversely associated with neuronal apoptosis by activating JAK–STAT in glial cells favoring neuroinflammation in epilepsy [32, 61]. Activation of JAK/STAT pathway illustrates an overview of the mechanism of increased neuronal stress with progressive ultrastructural alteration of mitochondrial dysfunction promoting apoptosis with increased Bcl-2 apoptotic protein enduring apoptotic neuronal death under the prolonged seizures-induced CNS insult [62,63,64]. The pre-clinical findings uncover an activation of the JAK/STAT pathway under excessively prolonged seizures induced neuronal injury and likely to be involved in causing neurodegeneration in epilepsy. The correlation of the JAK/STAT pathway with mitochondrial dysfunction was screened using WP1066, an inhibitor of the JAK/STAT pathway that resulted in the prevention of the development of epileptic seizures in rodents [64]. Therefore, the present target STAT3 tends to be involved in mitochondrial oxidative stress in epilepsy, as concluded by the decreased levels of pro-survival proteins Mcl-1 mRNA, Bcl-xl, c-myc mRNA that regulates mitochondrial function, which gets decreased under chronic epileptic seizures [62]. Therefore, targeting STAT3 tends to be an effective strategy in preventing epileptic seizures and providing the mechanism of mitochondrial dysfunction under prolonged repetitive epileptic seizures mediating apoptotic death in Epilepsy.
NF-kB/caspase-1, NF-kB/RIPK, and P38MAPK/ERK/JNK signaling correlated mitochondrial dysfunction in epilepsy
Neuroinflammation is an innate neuroimmune response to chronic seizure-induced brain damage with adequate activation of glial cells contributing to secondary seizures [65]. Invasive activation of microglia cells secretes pro-inflammatory mediators like interleukin-6, TNF-alpha; cyclooxygenase-2 gets secreted in response to the seizures-induced neuronal damage. Nuclear factor-kappa B (NF-kB) is a modulator of transcribing pro-inflammatory mediator genes involved in neuroinflammation and its progression. The correlation of NF-kB with mitochondrial dysfunction in epilepsy has not been yet explored. Still, the other disease study evidence suggested the translocation of NF-kB into mitochondrial DNA, releasing cytochrome C into the cytosol, and triggering caspases mediated apoptotic neuronal death [66]. Altered NF-kB, which is the primary regulator of neuroinflammation that gets activated by diverse stimuli (like TNF-alpha, Interleukin-1 beta, and growth factors) is responsible for increasing the pro-inflammatory cytokines (TNF-alpha IL-1, IL-6), ROS (MnSOD, CuZn SOD) [67, 68]. Thus, the increased cytokine levels further cause the mitochondrial dysfunction-mediated neuronal apoptotic death by activating the downstream JAK–STAT pathway that gets activated during excessive repetitive epileptic seizures [69]. Therefore, the activation of the neuroinflammatory pathway JAK–STAT and elevated pro-inflammatory mediators' levels indicate increased neuroinflammation undergoes pyroptotic neuronal death through caspase-1-dependent mitochondrial damage [70, 72, 73]. The diverse stimuli of TNF-alpha-activated Toll-like receptor also further recruits RIP1 signaling phosphorylating NF-KB, causing necroptosis by the formation of mitochondrial permeability transition (MPT) pore formation resulted in mitochondrial neuronal apoptotic death pathway with increased apoptotic proteins (caspase 3 and BAX) [72, 73]. There is also a subsequent stimulation of the P38MAPK/ERK/JNK pathway carrying the downstream signaling of apoptotic neuronal death as a secondary response of neuroinflammation with elevated levels of NF-kB transcribing inflammatory mediators (TNF-alpha, IL-1, IL-6) contributing to mitochondrial oxidative stress by forming a mitochondrial permeability transition pore (MPTP) leading to neuronal impairment in epilepsy [53, 72].
Correlation of AMPK pathway in neuroinflammation-mediated mitochondrial dysfunction
AMP-activated protein kinase (AMPK) is neuronal stress sensor-activated under chronic seizure-induced stress exacerbating neuronal death resulting from ATP depletion and dysregulated intracellular calcium homeostasis [74]. Apart from the pathological involvement of AMPK, it also modulates mitochondrial functioning having a role in neuronal survival depending upon the stress level with immense energy demands. AMPK has a dual role in neuronal survival as well as neuronal death [75, 76]. The sustained epileptic seizures induce excitotoxic stress by accumulating cytotoxic reactive oxygen species that are the consequences of structural damages in the brain associated with mitochondrial DNA with a significant decline of ATP. Thus, such impairment in the epileptic brain is the hallmark of neuronal death observed after repetitive, continuous seizures [77, 78]. Under the pathological condition, AMPK integrates energy balance by increasing neuronal biogenesis via upregulating the AMPK/PGC-1α pathway [79].
Furthermore, the activation of the AMPK/PGC-1α pathway correlates with NRF-1, and TFAM levels are increased to maintain the normal mitochondrial membrane potential and mitochondrial biogenesis under prolonged excessive epileptic seizures [79]. Activation of AMPK increases the ability of mitochondrial biogenesis to produce ATP and further regulating the activity of SIRT1 activating PCG1-alpha, decreasing mitochondrial oxidative stress by increasing mitochondrial anti-oxidant enzymes (UCP2/SOD2) [54]. AMPK activation also regulates neuroinflammation by inhibiting the NF-kB and decreasing the expression of CCL2, TNF-α, and inducible nitric oxide synthase (iNOS) levels that are implicated during epileptogenesis responsible for causing neurodegeneration by activation of STAT1 signaling [79]. This further provides a correlation of the relation between bioenergetics and inflammation-mediated neuronal death. Therefore, the activation of the AMPK/PGC-1α/ SIRT1 pathway is a treatment strategy for neurodegenerative diseases that showed a neuroprotective effect against status epilepticus-induced seizure damage [48, 54].
Peroxisome proliferator-activated receptors γ/mitochondrial uncoupling protein 2 signaling correlating neuroinflammation and mitochondrial dysfunction in epilepsy
Peroxisome proliferator-activated receptor γ is a ligand-activated transcriptional factor that acts a therapeutic potential in epilepsy, regulating the expression of pro-inflammatory levels [TNF-α, IL-1β, IL-6, iNOS, inducible cyclooxygenase (COX) 2] by direct inhibition of NFκB pathway [80,81,82,83]. Apart from the regulation of neuroinflammation, the PPAR γ molecule also enhances the mitochondrial respiratory chain activity, further preventing neuronal death by increasing the expression of mitochondrial UCP2 [84]. Mitochondrial UCP2 regulates mitochondrial ROS production, further preventing the mitochondrial oxidative stress and reducing the neuronal damage by controlling the expression of pro-apoptotic protein Bax and releasing cytochrome C-mediated apoptotic neuronal death [84]. The activation of PPAR γ tends to be neuroprotective in reducing the status epilepticus-mediated neurodegeneration by preventing the mitochondrial oxidative stress releasing cytochrome C initiating apoptotic signaling [85]. Therefore, the PPAR-γ has been demonstrated as a therapeutic strategy in epilepsy and reducing the cellular injury caused by repetitive prolonged chronic seizures and reducing oxidative stress and neuroinflammation [84, 89, 90].
Involvement of purinergic receptors in inflammation-mediated mitochondrial dysfunction
Purinergic receptors are ATP-gated non-selective cationic channels highly expressed in neurons and non-neuronal cells (astrocytes and oligodendrocytes) [88]. In response to prolonged seizure-induced brain damage, the elevated levels of P2X7 receptor represent the migration of microglial cells releasing cytotoxic pro-inflammatory mediators (IL-1β, IL-6, IL-18, and TNF-α) mediated neuronal death [89]. Thus, the P2X7 is a significant driver of immunoreactivity contributing to mitochondrial oxidative stress. Second, the hyperactivation of the P2X7 receptor allows the increased permeability of Ca2+ ions, leading to increased intracellular calcium ion concentration inducing mitochondrial Ca2+ overload [90]. In addition, the mitochondrial Ca2+ overload promotes mitochondrial-derived ROS and inducing the opening of the permeability transition pore, releasing cytochrome C into the cytosol with increased expression of pro-apoptotic proteins suggesting mitochondrial dysfunction-mediated neuronal death [32, 94, 95]. Targeting the P2X7 receptor provides a future perspective in decreasing the immunoreactivity and preventing mitochondrial oxidative stress-mediated neuronal death in epilepsy.
An illustration of glial cell migration releasing neurotoxic inflammatory mediators (TNF-alpha, IL-1, IL-6, and IL-18) activates purinergic receptor-mediated increased intracellular calcium influx causing mitochondrial oxidative stress. Further activating neuroimmune response with increased expression of pro-inflammatory mediators by activated nuclear factor kappa beta (NF-kB) expressing increased neurotoxic inflammatory mediators initiating multiple neuroinflammatory cascades (NF-kB/MAPK/JNK/P38/ERK) correlating the altered levels of intracellular molecules (SIRT, PGC-1-alpha, PPAR gamma, AMPK, and PGC-1) mediated mitochondrial oxidative stress or vice versa, i.e., increased influx of calcium through hyperactivation of voltage-gated ion channels causing mitochondrial calcium overload mediated mitochondrial oxidative stress increasing the ROS as well elevated levels of inflammatory mediators suggesting the activation of neuroinflammatory pathways initiating neuronal apoptosis by activation of caspase-9, 3 mediated excitotoxicity in epilepsy.
Preclinical evidence and potential novel therapeutic interventions targeting neuroinflammation and mitochondrial dysfunction in epilepsy (Fig. 2)

Pharmacological drugs targeting interlinked pathways between mitochondrial dysfunction and neuroinflammation in epilepsy
The various pre-clinical studies provided evidence of elevated levels of inflammatory mediators [TNF-alpha, (IL)-1β, and IL-6] under the chronic epileptic seizures along with increased levels of mitochondrial-derived activators caspase-3, BCL2-associated X, BH3 proteins resulted in neurodegeneration [32, 33]. Elevated levels of inflammatory mediators in epileptic brain tissue represent the activation of the neuroimmune system in response to the prolonged seizures produced excitotoxicity and increased levels of pro-apoptotic proteins are the key indicators of the relation of mitochondrial dysfunction and neuroinflammation-mediated neurodegeneration in epilepsy [33]. The altered neuronal network with synchronous discharging of neurons in epilepsy induces neuroinflammation and oxidative stress-like excitotoxicity events underlying neurodegeneration with structural changes like hippocampal cell degeneration and mossy fiber sprouting in TLE and status epilepticus [78]. Therefore, the review summarizes the pre-clinical pharmacotherapy targeting neuroinflammatory signaling cascades linked to mitochondrial dysfunction involved in epilepsy (Table 1; Fig. 2).
Future prospective and conclusion
Excessive glutamatergic neurotransmission-mediated neuronal injury in the epileptic brain leads to the activation of glial cells as a neuroimmune response activating inflammatory cascades involved in neuroinflammation. The significant increase of neuroinflammation is one of the causes of initiation of secondary seizures associated with the production of reactive oxygen species (ROS/RNS) further increasing the risk of mitochondrial oxidative stress leading to neuronal apoptotic death. The elevated levels of inflammatory mediators such as chemokines, cytokines, and prostaglandin are the biomarkers in chronic epileptic seizure-induced neuronal dysfunctioning, indicating the activation of astrocytes and microglia and altering the blood–brain barrier that seems to be altered in anti-epileptic drug resistance. Various pre-clinical studies have explored the mechanistic correlation of inflammatory mediators released by glial cells releasing NADPH oxidase in epilepsy which is the consequence of increased intracellular calcium ion concentration altering mitochondrial membrane permeability causing mitochondrial permeability transition pore (MPTP) formation-mediated neurodegeneration in Epilepsy. This indicates that targeting neuroinflammation can prevent mitochondrial apoptotic neuronal death in epilepsy or vice versa. Dysregulation of the miRNA system has emerged as a mechanism that underlies epileptogenesis. Alterations of mRNAs are associated with mitochondrial dysfunction and targeting mRNAs studies preventing mitochondrial oxidative stress in epilepsy. Pharmacological modulation or manipulation of miRNAs offers a novel, multi-targeting approach to regulate the gene expression patterns in epileptogenesis. Selective targeting of miRNAs offers the therapeutic possibility for the management of epilepsy (Fig. 3). Therefore, the current review has summarized the evidenced-based targets and correlation with multiple neuroinflammatory pathway activation in response to the seizure-induced mitochondrial apoptotic neuronal death signaling that might act as a future target for the treatment of epilepsy. Nevertheless, further investigations are essential to clarify the correlation of neuroinflammatory pathways and mitochondrial dysfunction-mediated excitotoxicity in epilepsy.

Pictorial presentation of alterations of mRNAs associated with mitochondrial dysfunction and targeting mRNAs preventing mitochondrial oxidative stress in epilepsy
Availability of data and materials
Not applicable.
Abbreviations
- ERK:
-
Extracellular regulated kinase
- NF-κB:
-
Nuclear factor-kappa light chain enhancer of activated B cells
- JAK–STAT:
-
Janus kinases signal transducer and activator of transcription proteins
- MAPK:
-
Mitogen-activated protein kinases
- P38:
-
P38 MAP KINASE
- PPARs:
-
Peroxisome proliferator-activated receptors
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- AMPK:
-
AMP-activated protein kinase
- SIRT1:
-
Silent mating type information regulation 2 homolog
- PGC-1-alpha:
-
Peroxisome proliferator-activated receptor-γ
- COX-2:
-
Cyclooxygenase2
- ASK-1:
-
Apoptosis signal-regulating kinase-1
- TNF α:
-
Tumor necrosis factor alpha
- IL-1:
-
Interleukin-1
- INOS:
-
Inducible nitric oxide synthase
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- UCP2:
-
Uncoupling protein 2
- CCL2:
-
Chemokine (C–C motif) ligand 2
- MnSOD:
-
Manganese-dependent superoxide dismutase
- DNA:
-
Deoxyribonucleic acid
- NAD:
-
Nicotinamide adenine dinucleotide
- FOXO:
-
Forkhead box transcription factors
References
Brown GC, Murphy MP, Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. https://doi.org/10.1042/bse0470069.
Onyango IG, Lu J, Rodova M, Lezi E, Crafter AB, Swerdlow RH. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta Mol Basis Dis. 2010;1802:228–34. https://doi.org/10.1042/bse0470069.
Udhayabanu T, Manole A, Rajeshwari M, Varalakshmi P, Houlden H, Ashokkumar B. Riboflavin responsive mitochondrial dysfunction in neurodegenerative diseases. J Clin Med. 2017. https://doi.org/10.3390/jcm6050052.
Zsurka G, Kunz WS. Mitochondrial dysfunction and seizures: the neuronal energy crisis. Lancet Neurol. 2015;14:956–66. https://doi.org/10.1016/S1474-4422(15)00148-9.
Kovac S, Dinkova Kostova AT, Herrmann AM, Melzer N, Meuth SG, Gorji A. Metabolic and homeostatic changes in seizures and acquired epilepsy—mitochondria, calcium dynamics and reactive oxygen species. J Mol Sci. 2017;18:1935. https://doi.org/10.3390/ijms18091935.
Singh S, Singh TG, Rehni AK. An insight into molecular mechanisms and novel therapeutic approaches in epileptogenesis. CNS Neurol Disord Drug Targets. 2020;19(10):750–79. https://doi.org/10.2174/1871527319666200910153827.
Yang H, Wu J, Guo R, Peng Y, Zheng W, Liu D, Song Z. Glycolysis in energy metabolism during seizures. Neural Regen Res. 2013;8:1316. https://doi.org/10.3969/j.issn.1673-5374.2013.14.008.
Kovács R, Gerevich Z, Friedman A, Otáhal J, Prager O, Gabriel S, Berndt N. Bioenergetic mechanisms of seizure control. Front Cell Neurosci. 2018;12:335. https://doi.org/10.3389/fncel.2018.00335.
Sharma VK, Singh TG, Mehta V. Stressed mitochondria: A target to intrude Alzheimer’s disease. Mitochondrion. 2021;59:48–57. https://doi.org/10.1016/j.mito.2021.04.004.
McDonald T, Puchowicz M, Borges K. Impairments in oxidative glucose metabolism in epilepsy and metabolic treatments thereof. Front Cell Neurosci. 2018;12:274. https://doi.org/10.3389/fncel.2018.00274.
Gleichmann M, Mattson MP. Neuronal calcium homeostasis and dysregulation. Antioxid Redox Signal. 2011;14:1261–73. https://doi.org/10.1089/ars.2010.3386.
Roganovic M, Pantovic S, Dizdarevic S. Role of the oxidative stress in the pathogenesis of epilepsy. Brain. 2019;1:3. https://doi.org/10.5152/NSN.2019.11632.
López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13:106–18. https://doi.org/10.1016/j.mito.2013.01.003.
Rose J, Brian C, Woods J, Pappa A, Panayiotidis MI, Powers R, Franco R. Mitochondrial dysfunction in glial cells: implications for neuronal homeostasis and survival. Toxicology. 2017;391:109–15. https://doi.org/10.1016/j.tox.2017.06.011.
Meng XF, Tan L, Tan MS, Jiang T, Tan CC, Li MM, Wang HF, Yu JT. Inhibition of the NLRP3 inflammasome provides neuroprotection in rats following amygdala kindling-induced status epilepticus. J Neuroinflamm. 2014;11:1–2. https://doi.org/10.1186/s12974-014-0212-5.
Rehni AK, Singh TG, Chand P. Amisulpride-induced seizurogenic effect: a potential role of opioid receptor-linked transduction systems. Basic Clin Pharmacol Toxicol. 2011;108(5):310–7. https://doi.org/10.1111/j.1742-7843.2010.00655.x.
Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun. 2011;25:1281–9. https://doi.org/10.1016/j.bbi.2011.03.018.
Brodie MJ. Antiepileptic drug therapy the story so far. Seizure. 2010;19:650–5. https://doi.org/10.1016/j.seizure.2010.10.027.
Guimarães J, Ribeiro JA. Pharmacology of antiepileptic drugs in clinical practice. Neurologist. 2010;16:353–7. https://doi.org/10.1097/NRL.0b013e3181dba5d3.
Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. https://doi.org/10.1038/sj.embor.7400779.
Sugiura Y, Ugawa Y. Epilepsy and ion channels. Clin Neurol. 2016;16(57):1–8. https://doi.org/10.5692/clinicalneurol.cn-000963.
Nigar S, Pottoo FH, Tabassum N, Verma SK, Javed MN. Molecular insights into the role of inflammation and oxidative stress in epilepsy. J Adv Med Pharm Sci. 2016;12:1–9. https://doi.org/10.9734/JAMPS/2016/24441.
Waldbaum S, Patel M. Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy? J Bioenerg Biomembr. 2010;1(42):449–55. https://doi.org/10.1007/s10863-010-9320-9.
Pu X, Storr SJ, Zhang Y, Rakha EA, Green AR, Ellis IO, Martin SG. Caspase-3 and caspase-8 expression in breast cancer: caspase-3 is associated with survival. Apoptosis. 2017;22:357–68. https://doi.org/10.1007/s10495-016-1296-4.
Maroso M, Balosso S, Ravizza T, Liu J, Bianchi ME, Vezzani A. Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J Intern Med. 2011;270:319–26. https://doi.org/10.1111/j.1365-2796.2011.02431.x.
Rana A, Musto AE. The role of inflammation in the development of epilepsy. J Neuroinflamm. 2018;15:1–2. https://doi.org/10.1186/s12974-018-1192-7.
Lalani AI, Zhu S, Gokhale S, Jin J, Xie P. TRAF molecules in inflammation and inflammatory diseases. Curr Pharmacol Rep. 2018. https://doi.org/10.1007/s40495-017-0117-y.
Zhang XM, Zhu J. Kainic acid-induced neurotoxicity: targeting glial responses and glia-derived cytokines. Curr Neuropharmacol. 2011;9:388–98. https://doi.org/10.2174/157015911795596540.
Rehni AK, Singh TG, Kalra R, Singh N. Pharmacological inhibition of inducible nitric oxide synthase attenuates the development of seizures in mice. Nitric Oxide. 2009;21(2):120–5. https://doi.org/10.1016/j.niox.2009.06.001.
Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–35. https://doi.org/10.1016/j.tins.2007.07.007.
Scott RC. What are the effects of prolonged seizures in the brain? Epileptic Disord. 2014;16:S6-11. https://doi.org/10.1684/epd.2014.0689.
Sanz P, Garcia-Gimeno MA. Reactive glia inflammatory signaling pathways and epilepsy. Int J Mol Sci. 2020;21:4096. https://doi.org/10.1016/j.mito.2021.03.009.
Singh S, Singh TG, Rehni AK, Sharma V, Singh M, Kaur R. Reviving mitochondrial bioenergetics: a relevant approach in epilepsy. Mitochondrion. 2021. https://doi.org/10.1016/j.mito.2021.03.009.
Han T, Qin Y, Mou C, Wang M, Jiang M, Liu B. Seizure induced synaptic plasticity alteration in hippocampus is mediated by IL-1β receptor through PI3K/Akt pathway. Am J Transl Res. 2016;8:4499.
Balosso S, Liu J, Bianchi ME, Vezzani A. Disulfide-containing high mobility group box-1 promotes N-methyl-d-aspartate receptor function and excitotoxicity by activating Toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal. 2014;21:1726–40. https://doi.org/10.1089/ars.2013.5349.
Paudel YN, Shaikh M, Chakraborti A, Kumari Y, Aledo-Serrano Á, Aleksovska K, Alvim MK, Othman I. HMGB1: a common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front Behav Neurosci. 2018;11(12):628. https://doi.org/10.3389/fnins.2018.00628.
Moran C, Sanz-Rodriguez A, Jimenez-Pacheco A, Martinez-Villareal J, McKiernan RC, Jimenez-Mateos EM, Mooney C, Woods I, Prehn JH, Henshall DC, Engel T. Bmf upregulation through the AMP-activated protein kinase pathway may protect the brain from seizure-induced cell death. Cell Death Differ. 2013;4:e606. https://doi.org/10.1038/cddis.2013.136.
Wu SB, Wu YT, Wu TP, Wei YH. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochimica Biochim Biophys Acta Gen Subj. 2014;1840:1331–44. https://doi.org/10.1016/j.bbagen.2013.10.034.
Kim SH, Yu HS, Park S, Park HG, Ahn YM, Kang UG, Kim YS. Electroconvulsive seizures induce autophagy by activating the AMPK signaling pathway in the rat frontal cortex. Int J Neuropsychopharmacol. 2020;23:42–52. https://doi.org/10.1093/ijnp/pyz055.
Peixoto CA, de Oliveira WH, da Racho Araújo SM, Nunes AK. AMPK activation: role in the signaling pathways of neuroinflammation and neurodegeneration. Exp Neurol. 2017;298:31–41. https://doi.org/10.1016/j.expneurol.2017.08.013.
Velagapudi R, El-Bakoush A, Lepiarz I, Ogunrinade F, Olajide OA. AMPK and SIRT1 activation contribute to inhibition of neuroinflammation by thymoquinone in BV2 microglia. Mol Cell Biochem. 2017;435:149–62. https://doi.org/10.1007/s11010-017-3064-3.
Zhang XM, Duan RS, Chen Z, Quezada HC, Mix E, Winblad B, Zhu J. IL-18 deficiency aggravates kainic acid-induced hippocampal neurodegeneration in C57BL/6 mice due to an overcompensation by IL-12. Exp Neurol. 2007;205:64–73. https://doi.org/10.1016/j.expneurol.2007.01.019.
Thawkar BS, Kaur G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J Neuroimmunol. 2019;326:62–74. https://doi.org/10.1016/j.jneuroim.2018.11.010.
Wang BH, Hou Q, Lu YQ, Jia MM, Qiu T, Wang XH, Zhang ZX, Jiang Y. Ketogenic diet attenuates neuronal injury via autophagy and mitochondrial pathways in pentylenetetrazol-kindled seizures. Brain Res. 2018;1678:106–15. https://doi.org/10.1016/j.brainres.2017.10.009.
Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. 2018;103:115–24. https://doi.org/10.1016/j.molimm.2018.09.010.
Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Ca2+-induced ROS production promotes cytochrome c release from rat liver mitochondria via MPT-dependent and MPT-independent mechanisms. Role of cardiolipin . J Biol Chem. 2004. https://doi.org/10.1074/jbc.M407500200.
Perkins G, Bossy-Wetzel E, Ellisman MH. New insights into mitochondrial structure during cell death. Exp Neurol. 2009;218:183–92. https://doi.org/10.1016/j.expneurol.2009.05.021.
Chuang YC, Chen SD, Jou SB, Lin TK, Chen SF, Chen NC, Hsu CY. Sirtuin 1 regulates mitochondrial biogenesis and provides an endogenous neuroprotective mechanism against seizure-induced neuronal cell death in the hippocampus following status epilepticus. Int J Mol Sci. 2019;20:3588. https://doi.org/10.3390/ijms20143588.
Pallàs M, Casadesús G, Smith MA, Coto-Montes A, Pelegri C, Vilaplana J, Camins A. Resveratrol and neurodegenerative diseases: activation of SIRT1 as the potential pathway towards neuroprotection. Curr Neurovasc Res. 2009;6:70–81. https://doi.org/10.2174/156720209787466019.
Javadov S, Kuznetsov A. Mitochondrial permeability transition and cell death: the role of cyclophilin d. Front physiol. 2013;4:76. https://doi.org/10.3389/fphys.2013.00076.
Grewal AK, Singh N, Singh TG. Neuroprotective effect of pharmacological postconditioning on cerebral ischaemia-reperfusion-induced injury in mice. J Pharm Pharmacol. 2019;71(6):956–70. https://doi.org/10.1111/jphp.13073.
Sun J, Gao X, Meng D, Xu Y, Wang X, Gu X, Guo M, Shao X, Yan H, Jiang C, Zheng Y. Antagomirs targeting mirorna-134 attenuates epilepsy in rats through regulation of oxidative stress, mitochondrial functions and autophagy. Front Pharmacol. 2017;8:524. https://doi.org/10.3389/fphar.2017.00524.
Chuang YC, Chen SD, Hsu CY, Chen SF, Chen NC, Jou SB. Resveratrol promotes mitochondrial biogenesis and protects against seizure-induced neuronal cell damage in the hippocampus following status epilepticus by activation of the PGC-1α signaling pathway Int J Mol Sci. 2019;20:998. https://doi.org/10.3390/ijms20040998.
Wang SJ, Zhao XH, Chen W, Bo N, Wang XJ, Chi ZF, Wu W. Sirtuin 1 activation enhances the PGC-1α/mitochondrial anti-oxidant system pathway in status epilepticus. Mol Med. 2015;11:521–6. https://doi.org/10.3892/mmr.2014.2724.
Sidorova-Darmos E, Sommer R, Eubanks JH. The role of SIRT3 in the brain under physiological and pathological conditions. Front Cell Neurosci. 2018;12:196. https://doi.org/10.3389/fncel.2018.00196.
Borowicz-Reutt KK, Czuczwar SJ. Role of oxidative stress in epileptogenesis and potential implications for therapy. Pharmacol Rep. 2020. https://doi.org/10.1007/s43440-020-00143-w.
Khan H, Tiwari P, Kaur A, Singh TG. Sirtuin acetylation and deacetylation: a complex paradigm in neurodegenerative disease. Mol Neurobiol. 2021;58(8):3903–17. https://doi.org/10.1007/s12035-021-02387-w.
Cho I, Jeong KH, Zhu J, Choi YH, Cho KH, Heo K, Kim WJ. Sirtuin3 protected against neuronal damage and cycled into nucleus in status epilepticus model. Mol Neurobiol. 2019;56:4894–903. https://doi.org/10.1007/s12035-018-1399-8.
Shimada T, Takemiya T, Sugiura H, Yamagata K. Role of inflammatory mediators in the pathogenesis of epilepsy. Mediat Inflamm. 2014. https://doi.org/10.1155/2014/901902.
Hixson KM, Cogswell M, Brooks-Kayal AR, Russek SJ. Evidence for a non-canonical JAK/STAT signaling pathway in the synthesis of the brain’s major ion channels and neurotransmitter receptors. BMC Genom. 2019;20:1–6. https://doi.org/10.1186/s12864-019-6033-2.
Nicolas CS, Amici M, Bortolotto ZA, Doherty A, Csaba Z, Fafouri A, Dournaud P, Gressens P, Collingridge GL, Peineau S. The role of JAK–STAT signaling within the CNS. JAK–STAT. 2013;2: e22925. https://doi.org/10.4161/jkst.22925.
Grabenstatter HL, Del Angel YC, Carlsen J, Wempe MF, White AM, Cogswell M, Russek SJ, Brooks-Kayal AR. The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol Dis. 2014;62:73–85. https://doi.org/10.1016/j.nbd.2013.09.003.
Hokenson KE. BDNF signaling in epilepsy: TRKB-induced JAK/STAT pathway and phosphorylation of LSF in neurons (doctoral dissertation). Diss. https://hdl.handle.net/2144/16740. Accessed 2016.
Han CL, Liu YP, Guo CJ, Du TT, Jiang Y, Wang KL, Shao XQ, Meng FG, Zhang JG. The lncRNA H19 binding to let-7b promotes hippocampal glial cell activation and epileptic seizures by targeting Stat3 in a rat model of temporal lobe epilepsy. Cell Prolif. 2020;53: e12856. https://doi.org/10.1111/cpr.12856.
Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. 2019;15:459–72. https://doi.org/10.1038/s41582-019-0217-x.
Singh S, Singh TG. Role of nuclear factor kappa B (NF-κB) signalling in neurodegenerative diseases: an mechanistic approach. Curr Neuropharmacol. 2020;18(10):918–35. https://doi.org/10.2174/1570159X18666200207120949.
Kothur K, Bandodkar S, Wienholt L, Chu S, Pope A, Gill D, Dale RC. Etiology is the key determinant of neuroinflammation in epilepsy: elevation of cerebrospinal fluid cytokines and chemokines in febrile infection-related epilepsy syndrome and febrile status epilepticus. Epilepsia. 2019. https://doi.org/10.1111/epi.16275.
Granat L, Hunt RJ, Bateman JM. Mitochondrial retrograde signalling in neurological disease. Philos Trans R Soc B. 1801;2020(375):20190415. https://doi.org/10.1098/rstb.2019.0415.
Heuser K, Szokol K, Taubøll E. The role of glial cells in epilepsy. Tidsskr Nor Legeforen. 2014. https://doi.org/10.4045/tidsskr.12.1344.
Rehni AK, Singh TG. Modulation of leukotriene D4 attenuates the development of seizures in mice. Prostaglandins Leukot Essent Fatty Acids. 2011;85(2):97–106. https://doi.org/10.1016/j.plefa.2011.04.003.
Rehni AK, Singh TG, Singh N, Arora S. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(1):11–9. https://doi.org/10.1007/s00210-009-0476-y.
Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22:225–36. https://doi.org/10.1038/cdd.2014.126.
Lin DQ, Cai XY, Wang CH, Yang B, Liang RS. Optimal concentration of necrostatin-1 for protecting against hippocampal neuronal damage in mice with status epilepticus. Neural Regen Res. 2020;15:936. https://doi.org/10.4103/1673-5374.268903.
Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010. https://doi.org/10.1152/ajpendo.00745.2009.
Rousset CI, Leiper FC, Kichev A, Gressens P, Carling D, Hagberg H, Thornton C. A dual role for AMP-activated protein kinase (AMPK) during neonatal hypoxic–ischaemic brain injury in mice. J Neurochem. 2015;133:242–52. https://doi.org/10.1111/jnc.13034.
Jiang S, Li T, Ji T, Yi W, Yang Z, Wang S, Yang Y, Gu C. AMPK: potential therapeutic target for ischemic stroke. Theranostics. 2018;8:4535. https://doi.org/10.7150/thno.25674.
Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109:17–23. https://doi.org/10.1111/j.1471-4159.2009.05916.x.
Waldbaum S, Patel M. Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy? J Bioenerg Biomembr. 2010;42:449–55. https://doi.org/10.1007/s10863-010-9320-9.
Xu W, Yan J, Ocak U, Lenahan C, Shao A, Tang J, Zhang J, Zhang JH. Melanocortin 1 receptor attenuates early brain injury following subarachnoid hemorrhage by controlling mitochondrial metabolism via AMPK/SIRT1/PGC-1α pathway in rats. Theranostics. 2021;11:522. https://doi.org/10.7150/thno.49426.
Bernardo A, Minghetti L. PPAR-γ agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des. 2006;12:93–109. https://doi.org/10.2174/138161206780574579.
Wilson G, Bryan J, Cranston K, Kitzes J, Nederbragt L, Teal TK. Good enough practices in scientific computing. PLoS Comput Biol. 2017;13: e1005510. https://doi.org/10.1371/journal.pone.0144806.
Villapol S. Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol Neurobiol. 2018;38:121–32. https://doi.org/10.1007/s10571-017-0554-5.
Meng QQ, Feng ZC, Zhang XL, Hu LQ, Wang M, Zhang HF, Li SM. PPAR-γ activation exerts an anti-inflammatory effect by suppressing the nlrp3 inflammasome in spinal cord-derived neurons. Mediat Inflamm. 2019. https://doi.org/10.1155/2019/6386729.
Chuang YC, Lin TK, Huang HY, Chang WN, Liou CW, Chen SD, Chang AY, Chan SH. Peroxisome proliferator-activated receptors γ/mitochondrial uncoupling protein 2 signaling protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus. J Neuroinflamm. 2012. https://doi.org/10.1186/1742-2094-9-184.
Abdallah DM. Anticonvulsant potential of the peroxisome proliferator-activated receptor gamma agonist pioglitazone in pentylenetetrazole-induced acute seizures and kindling in mice. Brain Res. 2010;1351:246–53. https://doi.org/10.1016/j.brainres.2010.06.034.
Lucchi C, Costa AM, Giordano C, Curia G, Piat M, Leo G, Vinet J, Brunel L, Fehrentz JA, Martinez J, Torsello A. Involvement of PPARγ in the anticonvulsant activity of EP-80317, a ghrelin receptor antagonist. Front Pharmacol. 2017;8:676. https://doi.org/10.3389/fphar.2017.00676.
Hung TY, Chu FL, Wu DC, Wu SN, Huang CW. The protective role of peroxisome proliferator-activated receptor-gamma in seizure and neuronal excitotoxicity. Mol Neurobiol. 2019;56:5497–506. https://doi.org/10.1007/s12035-018-1457-2.
Saha L, Bhandari S, Bhatia A, Banerjee D, Chakrabarti A. Anti-kindling effect of bezafibrate, a peroxisome proliferator-activated receptors alpha agonist, in pentylenetetrazole induced kindling seizure model. J Epilepsy Res. 2014;4:45. https://doi.org/10.14581/jer.14011.
Huang C, Chi XS, Li R, Hu X, Xu HX, Li JM, Zhou D. Inhibition of P2X7 receptor ameliorates nuclear factor-kappa B mediated neuroinflammation induced by status epilepticus in rat hippocampus. J Mol Neurosci. 2017;63:173–84. https://doi.org/10.1007/s12031-017-0968-z.
Liang X, Samways DS, Wolf K, Bowles EA, Richards JP, Bruno J, Dutertre S, DiPaolo RJ, Egan TM. Quantifying Ca2+ current and permeability in ATP-gated P2X7 receptors. J Biol Chem. 2015;290:7930–42. https://doi.org/10.1074/jbc.M114.627810.
Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Poletti F, Rimessi A, Suski JM. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion. 2012;12:77–85. https://doi.org/10.1016/j.mito.2011.07.004.
Kent AC, El Baradie KB, Hamrick MW. Targeting the mitochondrial permeability transition pore to prevent age-associated cell damage and neurodegeneration. Oxid Med Cell Longev. 2021. https://doi.org/10.1155/2021/6626484.
Zhang Z, Sun T, Niu JG, He ZQ, Liu Y, Wang F. Amentoflavone protects hippocampal neurons: anti-inflammatory, antioxidative, and antiapoptotic effects. Neural Regen Res. 2015;10:1125. https://doi.org/10.4103/1673-5374.160109.
Panigrahy SR, Pradhan S, Maharana CS. Amelioration of oxidative stress and neuroinflammation by Saroglitazar, a dual PPARα/γ agonist in MES induced epileptic rats. Biomed Pharmacol J. 2019;12:1985–91. https://doi.org/10.13005/bpj/1830.
Tambe R, Jain P, Patil S, Ghumatkar P, Sathaye S. Antiepileptogenic effects of borneol in pentylenetetrazole-induced kindling in mice. Naunyn-Schmiedeberg’s Arch Pharmacol. 2016;389:467–75. https://doi.org/10.1007/s00210-016-1220-z.
Shu Y, Zhu C, Zeng M, Zhan Q, Hu Z, Wu X. The protective effect of carbenoxolone on gap junction damage in the hippocampal CA1 area of a temporal lobe epilepsy rat model. Ann Transl Med. 2019. https://doi.org/10.21037/atm.2019.11.04.
Hino H, Takahashi H, Suzuki Y, Tanaka J, Ishii E, Fukuda M. Anticonvulsive effect of paeoniflorin on experimental febrile seizures in immature rats: possible application for febrile seizures in children. PLoS ONE. 2012;7: e42920. https://doi.org/10.1371/journal.pone.0042920.
Chen YF, Lee MM, Fang HL, Yang JG, Chen YC, Tsai HY. Paeoniflorin inhibits excitatory amino acid agonist-and high-dose morphine-induced nociceptive behavior in mice via modulation of N-methyl-d-aspartate receptors. BMC Complement Altern Med. 2016;16:1–4. https://doi.org/10.1186/s12906-016-1230-x.
Hu PF, Chen WP, Bao JP, Wu LD. Paeoniflorin inhibits IL-1β-induced chondrocyte apoptosis by regulating the Bax/Bcl-2/caspase-3 signaling pathway. Mol Med. 2018;17:6194–200. https://doi.org/10.3892/mmr.2018.8631.
Cong C, Kluwe L, Li S, Liu X, Liu Y, Liu H, Gui W, Liu T, Xu L. Paeoniflorin inhibits tributyltin chloride-induced apoptosis in hypothalamic neurons via inhibition of MKK4-JNK signaling pathway. J Ethnopharmacol. 2019;237:1–8. https://doi.org/10.1016/j.jep.2019.03.030.
Singh N, Saha L, Kumari P, Singh J, Bhatia A, Banerjee D, Chakrabarti A. Effect of dimethyl fumarate on neuroinflammation and apoptosis in pentylenetetrazol kindling model in rats. Brain Res Bull. 2019;144:233–45. https://doi.org/10.1016/j.brainresbull.2018.11.013.
Kahn-Kirby AH, Amagata A, Maeder CI, Mei JJ, Sideris S, Kosaka Y, Hinman A, Malone SA, Bruegger JJ, Wang L, Kim V. Targeting ferroptosis: a novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS ONE. 2019;14: e0214250. https://doi.org/10.1371/journal.pone.0214250.
Elsayed AA, Menze ET, Tadros MG, Ibrahim BM, Sabri NA, Khalifa AE. Effects of genistein on pentylenetetrazole-induced behavioral and neurochemical deficits in ovariectomized rats. Naunyn-Schmiedeberg’s Arch Pharmacol. 2018;391:27–36. https://doi.org/10.1007/s00210-017-1435-7.
Jiang G, Wu H, Hu Y, Li J, Li Q. Gastrodin inhibits glutamate-induced apoptosis of PC12 cells via inhibition of CaMKII/ASK-1/p38 MAPK/p53 signaling cascade. Cell Mol Neurobiol. 2014;34:591–602. https://doi.org/10.1007/s10571-014-0043-z.
Chen L, Liu X, Wang H, Qu M. Gastrodin attenuates pentylenetetrazole-induced seizures by modulating the mitogen-activated protein kinase-associated inflammatory responses in mice. Neurosci Bull. 2017;33:264–72. https://doi.org/10.1007/s12264-016-0084-z.
Yamamoto HA, Mohanan PV. Ganglioside GT1B and melatonin inhibit brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Brain Res. 2003;964:100–6. https://doi.org/10.1016/S0006-8993(02)04083-0.
Yang N, Guan QW, Chen FH, Xia QX, Yin XX, Zhou HH, Mao XY. Antioxidants targeting mitochondrial oxidative stress: promising neuroprotectants for epilepsy. Oxid Med Cell Longev. 2020. https://doi.org/10.1155/2020/6687185.
Hellmich HL, Rojo DR, Micci MA, Sell SL, Boone DR, Crookshanks JM, DeWitt DS, Masel BE, Prough DS. Pathway analysis reveals common pro-survival mechanisms of metyrapone and carbenoxolone after traumatic brain injury. PLoS ONE. 2013;8: e53230. https://doi.org/10.1371/journal.pone.0053230.
García-García L, Shiha AA, de la Rosa RF, Delgado M, Silván Á, Bascuñana P, Bankstahl JP, Gomez F, Pozo MA. Metyrapone prevents brain damage induced by status epilepticus in the rat lithium-pilocarpine model. Neuropharmacology. 2017;123:261–73. https://doi.org/10.1016/j.neuropharm.2017.05.007.
Reddy SD, Younus I, Sridhar V, Reddy DS. Neuroimaging biomarkers of experimental epileptogenesis and refractory epilepsy. Int J Mol Sci. 2019;20:220. https://doi.org/10.3390/ijms20010220.
Castro OW, Upadhya D, Kodali M, Shetty AK. Resveratrol for easing status epilepticus induced brain injury, inflammation, epileptogenesis, and cognitive and memory dysfunction—are we there yet? Front Neurol. 2017;8:603. https://doi.org/10.3389/fneur.2017.00603.
Gao X, Zhao XL, Zhu YH, Li XM, Xu Q, Lin HD, Wang MW. Tetramethylpyrazine protects palmitate-induced oxidative damage and mitochondrial dysfunction in C2C12 myotubes. Life Sci. 2011;88:803–9. https://doi.org/10.1016/j.lfs.2011.02.025.
Jin Y, Cai S, Jiang Y, Zhong K, Wen C, Ruan Y, Chew LA, Khanna R, Xu Z, Yu J. Tetramethylpyrazine reduces epileptogenesis progression in electrical kindling models by modulating hippocampal excitatory neurotransmission. ACS Chem Neurosci. 2019;10:4854–63. https://doi.org/10.1021/acschemneuro.9b00575.
Grewal AK, Singh TG, Sharma D, Sharma V, Singh M, Rahman MH, Najda A, Walasek-Janusz M, Kamel M, Albadrani GM, Akhtar MF, Saleem A, Abdel-Daim MM. Mechanistic insights and perspectives involved in neuroprotective action of quercetin. Biomed Pharmacother. 2021;140: 111729. https://doi.org/10.1016/j.biopha.2021.111729.
Acknowledgements
The authors are grateful to the Chitkara College of Pharmacy, Chitkara University, Rajpura, Patiala, Punjab, India for providing the necessary facilities to carry out the research work.
Funding
Nil.
Author information
Authors and Affiliations
Contributions
Conceptualization: conceived and designed the experiments: TGS. Analyzed the data: SS and TGS. Wrote the manuscript: SS. Editing of the manuscript: TGS; critically reviewed the article: TGS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Singh, S., Singh, T.G. Emerging perspectives on mitochondrial dysfunctioning and inflammation in epileptogenesis. Inflamm. Res. 70, 1027–1042 (2021). https://doi.org/10.1007/s00011-021-01511-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-021-01511-9