
Abstract
Influenza A virus (IAV) is a relevant respiratory tract pathogen leading to a great number of deaths and hospitalizations worldwide. Secondary bacterial infections are a very common cause of IAV associated morbidity and mortality. The robust inflammatory response that follows infection is important for the control of virus proliferation but is also associated with lung damage, morbidity and death. The role of the different components of immune response underlying protection or disease during IAV infection is not completely elucidated. Overall, in the context of IAV infection, inflammation is a ‘double edge sword’ necessary to control infection but causing disease. Therefore, a growing number of studies suggest that immunomodulatory strategies may improve disease outcome without affecting the ability of the host to deal with infection. This review summarizes recent aspects of the inflammatory responses triggered by IAV that are preferentially involved in causing severe pulmonary disease and the anti-inflammatory strategies that have been suggested to treat influenza induced immunopathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza virus infection is one of the leading causes of mortality and morbidity worldwide. Seasonal influenza infections causes three to five million cases of severe illness every year, and approximately 250,000–500,000 deaths worldwide [1]. Furthermore, due to major genetic changes in influenza virus, the so-called antigenic shift, when whole segments of different virus strains are interchanged in the same host, the virus can become highly pathogenic and cause global pandemics with great number of deaths [2].
Influenza A is the best studied member of the Orthomyxoviridae family, comprised of negative-sense single-stranded RNA (ssRNA) enveloped virus. It contains a genome composed of eight segments of ssRNA enclosed by nucleoprotein forming the so-called ribonucleoprotein (RNP) complexes that carry also their own polymerases PA, PB1 and PB2 [3]. The subtypes of the virus are defined by the differences in viral glycoproteins hemagglutinin (HA) and neuraminidase (NA) and virtually all the various virus subtypes can be found in migratory birds—the natural reservoir of influenza A virus [4]. Different subtypes can infect and cause disease in a diverse number of species including birds, pigs and humans [5]. In humans, influenza virus infection is in most cases confined to the respiratory tract [6], and most commonly caused by influenza A H1N1, influenza A H3N2, influenza B Victoria and influenza B Yamagata [7]. The virus reaches the human respiratory epithelium and attaches to the N-acetylneuraminic acid (also called sialic acid) of epithelial cells. HA is the protein needed for virus binding to the host cell and differences in structure and conformation of this viral glycoprotein determines the receptor specificity of influenza A virus [8]. After infecting epithelial cells the virus can spread and infect immune or non-immune cells in the respiratory tract.
The lung is in constant contact with the environment and in consequence with different potential pathogens. Therefore, there are several barriers that can limit the growth and establishment of a microorganism in the respiratory surface, including the mucus and the immune system [9]. The inflammatory response is an important defense mechanism against influenza A virus infection (IAV) preventing the replication and spread of the virus. However, an uncontrolled and exacerbated response to the virus may be associated with intense lung injury and death [10–12]. Indeed, there are several studies which have suggested a tight association between inflammation and the most severe cases of IAV infection [13–15]. Therefore, the inflammatory response triggered by IAV infections can be described as a double edge sword—it is necessary to protect against viral infection but may also cause severe pulmonary injury (Fig. 1). It is our hypothesis that there are mediators of the inflammatory response that are preferentially associated with severe disease but not necessary for protection against the virus. Based on this tenet we might be able to develop novel therapeutic targets to treat the severe manifestations of the disease. This review will focus on features of the inflammatory innate responses against influenza A virus that are preferentially involved in causing severe pulmonary disease.

Inflammation as a double edge sword during Influenza A infection. The low response leads to an insufficient control of the virus and can also predisposes to secondary bacterial infections. On the other hand, an excessive uncontrolled inflammatory response leads to increased immunopathology, morbidity and mortality
Influenza A virus recognition
Once inside the cells of the respiratory tract, IAV is recognized by the innate immune system during the steps of virus infection and replication because of the presence of the pattern recognition receptors (PRRs). These receptors can sense the so-called pathogen-associated molecular patterns (PAMPs) that are common molecules to different types of microorganisms, including IAV (Fig. 2). There are three classes of PRRs that can recognize and signal the sensing of IAV infection: toll-like receptors (TLRs), nod-like receptors (NLRs) and Retinoic acid Induced Gene 1 like receptor (RIG-I). A myriad of these receptors is activated during the different steps of IAV infection and replication and act synergistically to activate transcription factors, including NF-κB and IRF3/7 resulting in a pro-inflammatory response and local antiviral state [2, 16, 17].

Schematic representation of IAV initial recognition by pattern recognition receptors (PRRs). The endosome-localized TLR3 and TLR7/8 are activated by virus RNA upon infection. TLR4 is activated by oxidized phospholipids (OxPLs) produced during the oxidative burst triggered by IAV infection. The cytoplasmic NLRP3 can sense and signal modifications of protons concentrations due to the ion channel M2 viral protein and also is activated by ssRNA. RIG-I, also expressed in cytoplasm, recognizes viral RNA due to its 5′-triphosphate. The activation of PRRs initiates an antiviral and pro-inflammatory response with transcription of pro-inflammatory mediators and IFN
Toll-like receptors (TLRs) are the most well-studied and characterized class of PRRs [18]. Based on their cellular localization, TLRs can sense different types of virus-derived or induced molecules. The endosome-localized TLRs (TLR 3, 7, 8 and 9) play an important role in IAV recognition. Due to the intracellular localization of these receptors, they can sense and signal the presence of IAV nucleic acids. On the other hand, the role of surface localized TLRs (TLR 1, 2, 4, 5 and 6) in IAV infection recognition is indirect. This type of TLRs can contribute to the IAV infection response due to the recognition of damage associated molecular patterns (DAMPs) that are produced in response to infection [19].
Respiratory tract epithelial cells constitutively express TLR3 which can be activated during IAV infection by double-strain RNA produced during virus replication [20]. The activation of TLR3 contributes to the production of pro-inflammatory chemokines and cytokines and despite generating an antiviral response it also contributes to the increased immunopathology and mortality in mice [20–22]. TLR7 signaling is also important for the development of inflammatory responses during IAV infection. Plasmocytoid dendritic cells (pDCs) can sense viral ssRNA through TLR7 leading to maturation of these cells that are an important source of type I interferons [23]. ssRNA is also an agonist of TLR8. However, the specific role of this receptor is not well defined during IAV infection.
TLR10 was recently shown to be important for the development of innate immune responses during IAV infection. During influenza infection the expression of TLR10 is increased and the activation of this receptor induces secretion of pro-inflammatory cytokines and interferons. Therefore, these receptors may also contribute to the viral clearance and immunopathology associated with IAV infection [24].
The second group of PRRs that can recognize IAV infection are the NLRs. NLRs, together with the adaptor ASC and pro-caspase-1, compose a multiprotein platform called inflammasome. This complex is responsible for the cleavage of pro-caspase-1 in its active form which cleaves pro-IL1β and pro-IL-18 into IL-1β and IL-18 that contribute to the inflammatory response against IAV infection [25]. NLRP3 is highly expressed during IAV infection in the lungs and immune cells recruited to the airways and can be activated by ssRNA, proton influx through the influenza virus-encoded matrix 2 (M2) ion channel [26] and the virulence protein PB1-F2 [27]. Activation of NLRP3 inflammasome is important for virus clearance, but may also contribute to the intense inflammation associated with severe cases of IAV infection [26, 27].
Lastly, the other group of PRRs that sense IAV infection are the inducible cytoplasm sensors called retinoic acid inducible gene-I receptors (RIG-I) that recognize the 5′-triphosphate viral ssRNA that is produced during viral replication [28]. These receptors are expressed in several cell types such as alveolar macrophages, conventional dendritic cells and lung epithelial cells. Its activation and interaction with adaptor protein mitochondrial antiviral signalling (MAVS/IPS-1) and virus induced signaling adaptor (cardif/VISA) leads to translocation of NFKB and/or IRF3/7 to the nucleus and production of pro-inflammatory cytokines and type I and III interferon. Therefore, RIG-I is very important for the development of an antiviral response by regulation of production of IFNs and mediating the activation of dendritic cells and T cells responses [29, 30].
Inflammatory mediators
The signaling pathways activated by all classes of PRRs lead to the transcription of pro-inflammatory genes and production of large amounts of cytokines and chemokines that orchestrate the inflammation in the lungs during infection. The equilibrium of the inflammatory response in the lungs is determinant for the outcome of IAV infection. Pro-inflammatory cytokines, such as interferons, interleukins, chemokines and tumor necrosis factor are the main molecules controlling the lung environment during the infection [31]. These molecules are responsible for the communication between immune or non-immune cells and can drive several important response steps against IAV, including epithelium activation, leukocyte recruitment, cell proliferation and differentiation and development of adaptive immunity [31]. Thus, cytokines control the immune events during infection and are associated not only with the protective responses and viral clearance but also with the pathology associated with inflammation during IAV infection. Most of the effects of some inflammatory mediators in immunity or disease during IAV infection are summarized in Table 1.
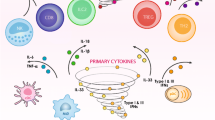
Pro-inflammatory cytokines and chemokines activate and recruit leukocytes into the lungs and airways, and these cells can produce large amounts of these molecules in a positive feedback loop. If this cycle is not controlled, it can lead to the exacerbation of the inflammatory response. The systemic presence and large levels of these signaling molecules lead to an event known as “cytokine storm” which is one of the causes of increased mortality during severe IAV infections [32–34]. “Cytokine storm” is a term largely used to characterize the intense cytokine response against distinct subtypes of influenza, such as the pandemic avian A (H5N1) and A(H1N1)pdm09 [12, 13, 35, 36]. The cytokine storm is correlated with the emergence of severe clinical symptoms, including alveolar hemorrhage, acute pneumonia, extensive pulmonary edema, acute respiratory distress syndrome and death [13]. The higher amounts of chemokines and cytokines produced after IAV infection leads to an excessive recruitment and activation of neutrophils, macrophages and T lymphocytes resulting in an intense production of proteases, oxygen and nitrogen reactive species that can directly contribute to tissue damage [37, 38]. It is observed an increase in cells apoptosis and necrosis and increased tissue permeability which results in lung edema [39, 40]. In addition, the excessive production of cytokines can lead to a rare event called haemophagocytosis which is associated with the more severe cases of H5N1 IAV infection [41]. This disorder characterized by an intense efferocytosis of leukocytes, erythrocytes and platelets and the proliferation of histiocytes and is correlated with the multiple organ failure observed during the severe cases of pandemic avian flu [42].Although the inflammatory responses triggered by seasonal and less severe types of IAV is much less intense than that triggered by the pandemic subtypes, this response is also correlated with the symptoms and morbidity of patients [43, 44].
During infection, there appears to be different cytokine waves as infection progresses. The first cytokines to be produced in the lungs after IAV infection are the chemokines CCL5/RANTES, CCL2/MCP-1 and CXCL8/IL-8 and the main antiviral cytokines IFN-α, IFN-β and IFN-λ [20, 45, 46]. When the virus reaches the lower respiratory tract, it can infect alveolar macrophages that are activated and secrete even greater levels of pro-inflammatory and antiviral cytokines including IL-1α/β, IL-6, TNF-α, IL-18, IFN type I, CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES, CCL7/MCP-3, CCL20/MIP-3α, CXCL10/IP-10 and CXCL8/IL-8 [47].
Much knowledge about IAV induced pathogenesis comes from murine models. In mice, as in humans, levels of pro-inflammatory cytokines are directly associated with the poor prognosis. H5N1 virus infection evokes a more robust and acute inflammatory response than inflammation triggered by seasonal IAV strains. H5N1 infection resulted in higher levels of IL-6, TNF-α, IFN-γ and chemokines in serum of patients, particularly in the fatal cases [12, 48]. The 2009 H1N1 pandemic virus was also associated with the development of a stronger and more sustained inflammation than the disease observed during seasonal IAV infections [49].
Interferons (IFNs) compose a family of cytokines that have a key antiviral role during the innate immune response against viral infections [50]. There are three major types of interferons: type I (IFN-α and IFN-β), type II (IFN-γ) and type III (IFN-λ) [51]. Type I IFN plays an important role in protection during IAV infection [52–54]. Some studies have suggested an essential role of type I IFNs in the development of antiviral responses [55], whereas other studies have also suggested that type I IFNs play an inhibitory role on CXCL1 and CXCL2 chemokine production and thus control neutrophil influx during IAV infection protecting against lung injury [56]. However, a recent study showed that an excess of release of these mediators are correlated with overwhelming inflammation and severity during IAV infection [54]. Indeed, another recent study demonstrated that type I IFN is necessary for neutrophil and Ly6Chigh inflammatory monocyte recruitment to the IAV infected lungs and clearance of virus but is also important to prevent the exaggerated inflammatory response caused by IFN y [57].
IFN-γ is known to play an important role in the response against several viral infections [58, 59]. This cytokine exerts a stimulatory effect on macrophage function, upregulates the major histocompatibility complex molecules (MHC) on antigen-presenting cells and also is important to regulate the cytokine production and CD4+ T cells activation during infection [60, 61]. However, IFN-γ seems to have a minor role in mounting the immune response and mediating viral clearance during IAV infection [52]. Despite the inability to compromise the response against IAV, the absence of this cytokine during IAV infection leads to an unregulated specific CD8+ T cell trafficking to the lungs [62]. Furthermore, during adoptive transfer of CD8+ T cells to mice with alveolar epithelial cells that express an influenza antigen, the resulted immunopathology was associated with IFN-γ production [63]. Accordingly, in vivo blockage of IFN-γ in mice led to a significant reduction in the number of infiltrating leukocytes into the lungs after IAV infection [64]. The study from Stifter and colleagues also demonstrated that type I and III IFN inhibit neutrophil trafficking during IAV infection in a synergic way [57]. Type III interferons have an important role in inducing antiviral state in respiratory epithelial cells [65] and were shown to contribute to the innate immunity against IAV [66]. Type I and III IFN stimulate the transcription of over 300 interferon induced genes (ISGs) that are responsible for the production of several antiviral proteins (including MxA, ISG15, OAS and PKR) that interfere with different steps of viral replication [67, 68].
Interleukins are another important family of mediators of inflammation. They are a group of cytokines with several biological functions and can drive pro-inflammatory or anti-inflammatory responses [31]. IL-1α and IL-1β are pro-inflammatory interleukins produced after the activation of NLRP3 inflammasome and cleavage of the pro-IL-1 form. These cytokines are important for the development of immunity against viral infections by inducing a cascade of inflammatory events (recruitment of leukocytes and induction of production of other cytokines) and also lead to T cell activation [69–71]. Indeed, the absence of IL-1 signaling is associated with increased mortality during IAV infection in mice and genetic variants of the genes for IL-1α and IL-1β are correlated with IAV infection susceptibility in human patients [72, 73]. It has also been shown that reduced levels of IL-1β can contribute to the increased risk of secondary bacterial infections after IAV infection [74, 75]. However, other studies have shown that these molecules also have a role in driving lung injury and their levels in the lung fluid correlate with the severity of symptoms during IAV infection. These studies are consistent with a dual role of IL-1 during infection [72, 76].
Interleukin-6 (IL-6), together with tumor necrosis factor alpha (TNF-α), are pro-inflammatory cytokines that are also involved in the onset of inflammation during IAV infection [31]. These cytokines contribute to endothelium activation leading to expression of P-selectin, E-selectin, and integrins that are essential for leukocyte recruitment to the lungs [77]. The production of these mediators during IAV infection is correlated with symptoms in human patients [42, 43] and the blockade of IL-6 or TNF-α protects against the severity of IAV infection in mice [78, 79]. Indeed, TNF receptor 1-deficient mice show significantly reduced morbidity following IAV H5N1 infection compared with wild-type mice, despite no difference in viral titers [80]. Therefore, pro-inflammatory cytokines, such as TNF-α, contribute to the exacerbation of inflammation during IAV infection with no or a minor contribution to the defense against the virus. In this regard, targeting TNF may have potential therapeutic benefits in patients with IAV infection.
The participation of IL-17 during the immune responses against IAV infection has also been reported. IL-17A was shown to protect mice from lethal challenge with IAV H1N1 and H3N2 and it was related with an increased and early neutrophil influx into the lungs of animals [81, 82]. Another study showed that IL-17 can play a critical role in mediating the recruitment of B cells to the lungs during a H5N1 infection in mice [83]. However, its role in protecting against IAV infection is controversial. Another study indicated that IL-17 rather than acting to improve viral clearance, contributed to the immunopathology associated with IAV infection by augmenting inflammatory responses [84]. In agreement with the latter finding, elevated levels of IL-17 were found in the serum of patients with severe pandemic H1N1 infection possibly contributing to overwhelming inflammation, morbidity and poor prognosis [85].
IL-27 and IL-22, two regulatory cytokines, also have been suggested to play a significant role during IAV infection by controlling the inflammatory responses and the return to lung homeostasis [86]. IL-27 was shown to increase the levels of IFN-γ in T CD8+ cells during infection [87], and an antiviral activity of this cytokine was also reported during an in vitro study [88]. IL-22 was shown to have an important protection role during sublethal IAV infection as the production of this cytokine reduced lung injury and inflammation and decreased mice lethality during secondary bacterial infections [89]. In addition, this cytokine is important for lung repair responses after IAV infection contributing to restoration of lung function [90].
Another family of inflammatory mediators that deserves attention in the context of IAV infection is the family of lipid mediators, including platelet-activation factor (PAF) and leukotriene B4 (LTB4). PAF is an inflammatory phospholipid that acts through a G protein-coupled receptor (PAFR) inducing several inflammatory events, including lung edema and recruitment and activation of leukocytes [91–93]. Immune cells, such as monocytes, macrophages, neutrophils and lymphocytes can produce, by hydrolyzing membrane phospholipids, and be activated by PAF [94, 95]. During IAV infection, the expression of the enzyme responsible for PAF synthesis in inflammatory conditions (LPAFAT/LPAFAT2) is increased after 1 day of infection in the lungs [96] and it coincides with the first wave of leukocyte recruitment [97] revealing the possible role of PAF during the inflammatory response. Our group showed that absence of PAFR (as seen in PAFR-deficient mice) or administration of a PAFR antagonist prevented the intense inflammatory response, lung injury and death associated with infection by IAV without affecting viral titers in the lungs [96]. Of interest, the PAFR antagonist was effective even when drug treatment was started 3 days after infection, suggesting there was a therapeutic window for administration of PAFR antagonists. Our studies clearly reinforce the pivotal role of inflammation at inducing mortality and morbidity during IAV infection and suggest immunomodulation as a strategy to control the symptoms and death related to infection.
LTB4 is another member of the family of lipid mediators that are produced mainly by leukocytes of the myeloid-lineage [98]. It acts through two G protein-coupled receptors: a high affinity receptor called BLT1R (predominantly expressed in leukocytes) [99] and a low-affinity receptor named BLT2R (expressed in different cell types) [100, 101]. On contrary to PAF, LTB4 has a protective role in the context of IAV infection. It was shown that administration of LTB4 increased the expression of antimicrobial peptides in the lungs and improved viral clearance [102]. In agreement with that, another study showed that LTB4-treated human neutrophils increased levels of myeloperoxidase and the antimicrobial peptide α-defensin enhancing the antiviral state of the cells against IAV infection [103].
The complement system is an important component of innate responses against pathogen infections [104]. Its activation cascade is a multistep cleavage process leading to opsonization or/and lysis of the target cell and can also produce the anaphylatoxins C3a and C5a that are worth to mention in this section [105–107]. C5a is a potent neutrophil chemoattractant and activates neutrophils to generate reactive oxygen species and release of enzymes contributing to the inflammatory response during IAV pneumonia [108, 109]. Indeed, extensive deposition of cleavage products of the complement cascade was found in the lungs of fatal cases of IAV pandemic strains, 2009 H1N1 and also1957 H2N2 [110]. Therefore, C5a plays a detrimental role during IAV infection leading to increased lung injury and death. Our group and others showed that during IAV infection, the levels of C3a and C5a are increased in bronchoalveolar lavage fluid (BALF) of mice and it is associated with intense inflammation and lung damage [109, 111]. Furthermore, the inhibition of C5 cleavage or use of anti-C5a during IAV infection in mice or green monkeys could prevent lung damage and decreased several inflammatory parameters that are associated with poor outcome from disease [109, 112].
Lastly but not less important are the chemokines. These molecules are chemotactic cytokines that control the migration and homing of immune cells during homeostasis or disease [77]. During infections, the recruitment of leukocytes is essential for proper setting of the immune response, pathogen clearance and subsequent reestablishment of tissue homeostasis. Chemokines are a large family of 50 small molecular weight proteins (8–12 kDa) that are subdivided into four groups based on the positioning of their N-terminal cysteine residues [77]. The C–X–C group is defined by the presence of a variable amino acid separating the first two cysteines; C–C chemokines are characterized by the presence of two cysteines adjacent to each other; the C group contains only a single cysteine residue in the conserved position and in the CX3C group the last two cysteine residues are separated by three amino acids. The CXC and CC groups are the most studied type of chemokines and are produced and act on a variety of immune cells [113].
During IAV infection there is marked and early production of chemokines and consequently recruitment of macrophages and neutrophils to the lungs and airways [114]. The production of chemokines such as CCL2 induces the recruitment of natural killer cells and inflammatory monocytes that are important for the early control of virus replication [115]. As discussed for the other cytokines in this section, the chemokine production must be finely tuned to generate effective adaptive immune responses and viral control but to avoid inflammatory injury. In fact, several studies showed that the increased production of CCL2 and consequent recruitment of inflammatory monocytes can increase the morbidity due to influenza infection and the absence of CCL2 receptor (CCR2) also decreased mice susceptibility to a bacterial secondary infection [116–118]. On the other hand, CCL2 seems to be important for an efficient immune response during IAV infection as CCL2 knockout mice presented increased weight loss, viral loads and levels of pro-inflammatory cytokines regardless of the reduced number of macrophages and neutrophils in the lungs [119], in addition, treatment with neutralizing CCL2 antibodies enhanced epithelial damage following IAV infection in mice [120, 121]. The differences presented by the distinct studies might be explained by the strategies used and the affected cells. CCL2 is considered protective when knockout mice or depleting antibodies are used against the chemokine, which causes a great reduction in lymphocytes and thus turns the immunity down [119]. On the other hand, a detrimental role is associated to CCR2, when receptor knockout or blockage is used, resulting in reduced lung inflammatory monocytes.
The neutrophils-associated chemokines such as CXCL1 and CXCL2 are mostly associated with a harmful role during IAV infection. The activation of receptor for both chemokines, CXCR2, was shown to be important for the intense recruitment of neutrophils and contributes to the lung injury and mortality of mice during IAV infection, without affecting viral clearance [122]. In fact, production of CXCL2 by the pulmonary epithelial cells is associated with an increased recruitment of inflammatory cells and is crucial for the acute lung damage during flu [123]. Moreover, the overproduction of CXCL2 is associated with increased lung damage, weight loss and death of mice in a model of IAV and bacteria coinfection [56].
In summary, different pro-inflammatory cytokines can contribute to the generation of an effective antiviral response during IAV infection, but can also be harmful as they can exacerbate the inflammatory response and contribute to lung injury. Therefore, understanding the dynamics of these molecules can be helpful in the development of new therapeutic targets in the context of IAV infection.
Leukocytes and influenza
After IAV infection, resident cells orchestrate the sequential recruitment of different leukocyte populations to the lung parenchyma and airway space. The recruitment of these cells is dependent on virus sensing and release of inflammatory mediators by the infected cells. Innate and adaptive immune cells play an important role in the clearance of the virus but, similar to cytokines production, the activation of leukocytes has to be finely controlled to insure effective killing of virus, minimal lung damage and restoration to organ homeostasis.
Alveolar macrophages (AMs) are resident lung cells that present variable susceptibility to influenza virus infection depending on the viral strain; strains that do not infect AMs, like PR8, are more virulent to mice [124]. Upon infection with different IAV strains, as shown for strains ST169 (H1N1), ST602 (H3N2) and HKG9 (H9N2), AMs polarize early into the M1 phenotype, with high expression of pro-inflammatory cytokines and iNOS, enhanced endocytic functions and enhanced ability to kill intracellular pathogens. Later, macrophages became M2b activated, with reduced IL-6 and IL-12 expression, but increased IL-10 and STAT1 expression. AMs become immunosuppressed with downregulation of most markers of immune response. This polarization occurs via the PI3 K/Akt signaling pathway [125]. AMs are very important to keep lung homeostasis and gas exchange function upon IAV infection. Two recent studies in which depletion of AMs was achieved by different genetic approaches showed increased morbidity and pathology following IAV infection and this was associated with increased respiratory failure with loss of gas exchange. The depletion of AMs also results in increased viral loads in early time points after infection, but no changes in cellular CD8+ T cell responses [126]. A previous study has shown that AM depletion before, but not after infection with recombinant Influenza virus bearing HA and NA from the 1918 IAV, resulted in increased lethality and virus levels coincident with reduced IFN-α, IFN-γ, TNF-α and CCL3 [127].
Contrasting conclusions about the role of alveolar macrophages in alveolar epithelial damage have been published. As mentioned before, the protective role of alveolar macrophages was shown to be mediated by the preservation of lung tissue and the epithelial barrier [126]. On the other hand, an ex vivo study pointed that the production of IFN-β by AM during IAV infection might contribute to apoptosis of alveolar epithelial cell and lung injury [128]. The differences in the studies may be due to the different experimental strategies and the more complex system during in vivo conditions. Another important feature of AMs biology during influenza infection is their ability to phagocyte, together with neutrophils, apoptotic infected cells, probably via TLR4 activation contributing to cross-presentation and an adequate adaptative response [129]. Therefore, AM are essential for the development of an effective innate response against the virus and might contribute to both epithelial injury and maintenance of lung homeostasis. The conflicting results highlight the need of more kinetic studies for understanding the contribution of AM and the various macrophage subtypes during infection with different influenza subtypes and strains.
Besides alveolar macrophage polarization upon influenza infection, inflammatory macrophages (Ly6Chigh) are recruited early to lungs and airways during lethal IAV infection and accumulation is observed until late time points [130]. CD11b+ cells upregulate CD11c and MHCII and mature to exudate macrophages or monocyte-derived DC and contribute to tissue damage and mortality via NOS2 expression, CCR2 activation and production of inflammatory cytokines during IAV infection in mice [130].
Neutrophils are recruited early to the lungs after influenza infection and can be infected and produce viable virus progeny, at least for the A(H1N1)pdm09 strain [131]. These cells have an important role in killing the virus [132], especially by phagocytosing infected apoptotic cells [129]. In addition to their role in innate control of the virus, neutrophils are associated with immunopathology. A direct correlation between neutrophil recruitment and severity of lung disease associated to highly pathogenic avian influenza has been shown [133]. Aiming to investigate the role of neutrophils during influenza infection, different studies have performed neutrophil depletion protocols in experimental infection in mice. If neutrophils are depleted before infection using the antibody RB6, the disease is more severe and lethality is enhanced [134]. If these cells are depleted after 3 or 5 days of infection, there was no change in disease severity and lethality [127]. Using different schedules and doses of the antibody 1A8, it has been shown that limiting antibody concentrations protected mice, without changes in virus levels. In contrast, high doses of the antibody, and therefore neutrophil depletion, enhanced disease severity [130]. Although the distinct strategies of neutrophil depletion differs in specificity—RB6 depletes both Gr1+ neutrophils and non-neutrophils Gr1+ cells like monocytes, whereas 1A8 depletes only neutrophils [135]—the two strategies showed that a finely tuned equilibrium in the number of neutrophils is important to control infection and prevent lung damage and lethality.
Distinct mechanisms of neutrophil activation have also been investigated in the context of influenza infection. Release of myeloperoxidase and neutrophil extracellular traps (NETs) by activated neutrophils has been shown to contribute to disease severity [136]. Reactive oxygen species released by activated neutrophils and macrophages might be a potential target to limit disease severity. Indeed, antioxidant agents such as catalase, N-acetyl-l-cysteine or Nox2 inhibitor result in reduced disease severity and also virus levels in murine models of influenza infection [137–140].
NK cells, a group 1 innate lymphoid cell (ILC) population, are normally present in the airways in homeostatic states. During influenza infection, more NK cells are recruited and become activated by type I IFN to produce IFN-γ, granzyme B and IL-12 [141]. The involvement of NK cells in virus control or influenza-related pathology is not completely understood. It has been shown that in the absence of NCR1 (NKp46), the main NK cell receptor, influenza infection becomes more lethal in two mouse strains [142]. Indeed, the human NK receptors NKp46 and NKp44 are capable of recognizing the IAV hemagglutinin and neuraminidase and lead to the death of the infected cell and control of virus proliferation [143]. Together with the direct recognition of infected cells, NK cells were recently shown to be capable of mediating antibody-dependent cellular cytotoxicity in IAV infected patients [144]. Furthermore, another study showed a correlation between high NK activation and low pathogenic avian influenza, whereas highly pathogenic avian influenza induced weak NK activation [145]. However, NK cells have also a contrasting role during IAV infection: it can mediate part of the injury observed after infection. It was shown that depletion of NK cells using anti-GM1 or anti-NK1.1 reduced lethality in influenza infected mice and this deleterious role of NK cells was observed only in high inoculums of influenza infection [146]. In addition, the absence of IL-15, the cytokine responsible for the proliferation and maintenance of NK cells, or depletion of these cells protected mice from lethality and morbidity caused by influenza infection. It was associated with a reduction of neutrophils and mononuclear cells recruitment as well as reduced IL-6 and IL-12 levels and increased IL-10 levels, but no changes in viral control [147].
Innate lymphoid cells 2 (ILC2) have been reported to play distinct roles during influenza A infection. This cell population is activated by IL-33 production and is a source of IL-5 and IL-13, important for eosinophil recruitment and epithelial proliferation. During experimental murine infection with H3N1 [148] or H1N1—PR8 [149, 150] or pdmH1N1 [151]—increased lung ILC2 populations as well as higher IL-33, IL-5 and IL-13 levels were found. Experimental H3N1 infection causes an intense airway hyper-reactivity (AHR) and inflammation. The AHR induced by H3N1 infection was shown to be an innate and acute mechanism, dependent on ST2 (IL-33 receptor), stimulated by the induction of IL-33 production, mainly by AM, which, in turn, results in greater ILC2 abundance in the lungs, and therefore, intense IL-13 production [148]. Using a different virus strain, PR8, Monticelli and colleagues demonstrated that this IAV also induces ILC2 population. On the contrary to the H3N1 infection, PR8 induces intense epithelial lung injury and do not induce AHR; in this scenario, ILC2 induction promotes the activation of genes involved in wound healing and amphiregulin production, which restores lung function epithelial integrity and airway remodeling [150]. Later, another study also investigated another role of ILC2 during influenza infection. Using PR8 mouse infection, it was shown that NKT cells, together with AM, are also an important source of IL-33 that will result in greater infiltration of ILC2 that in turns produce IL-5 and stimulate eosinophils recruitment in the recovery phase of the infection [149]. Lastly, a recent study showed that among children presenting acute asthmatic symptoms after pandemic H1N1 virus infection, most of them (81 %) did not report previous history of asthma. Mice infected with the pandemic virus presented AHR manifestations associated with ILC2, IL-4, IL-5 and IL-33 presence [151]. To sum up, ILC2 induction during IAV infection might induce acute AHR (pathogenic), or late tissue epithelial repair (protective), depending on the virus strain and the microenvironment [152].
Lipid reactive lymphocytes with reduced variability and expression of NK cell markers, the so-called invariant NKT cells, are important for innate responses to influenza infection. The activation of NKT favors virus clearance and reduces disease severity [153], whereas NKT depletion using CD1d KO or Jα18 KO mice succumb to influenza infection [154]. iNKT activation is also important to control airway inflammation and to stimulate CD8+ T cell response [155]. Also, the role of iNKT cells mediating the protection during a secondary pneumococcal infection was recently demonstrated [156].
Adaptive immune responses during influenza infection are performed by CD4+ and CD8+ T cells and B cells, that become activated, expand clonally and migrate to the lungs around seven days after infection. The latter cells, respectively, produce cytokines, lyses virally infected cells or produce great amounts of antibodies that can directly neutralize the virus (IgM) or confer protection to a secondary homosubtypic influenza infection (IgA and IgG) (reviewed in [157]). CD4+ and CD8+ T cells have also been related to influenza immunopathology (reviewed by [158]). One example is that, in the absence of the immunoadaptor DAP12 expressed in innate immune cells, there is marked infiltration of CD4+ T cells presenting enhanced FasL expression and cytotoxicity that may cause uncontrolled lung injury and death [159]. Antigen-specific CD8+ T cells may also contribute to immunopathology by producing TNF-α after recognizing IAV antigen in infected alveolar cells [160]. Indeed, classical CD8+ T cells (“Tc1”) can produce large amounts of IFN-γ, TNF-α and IL-2, activating themselves and other cell types during IAV infection [161]. Transferring IAV-specific CD8+ T cells to a transgenic mice with expression of HA in alveolar epithelial cells leads to intense lung injury and progressive weight loss, reinforcing the harmful role of overactivation of those cells during IAV infection [162]. The balanced response and activation of CD8+ T cells present several regulation mechanisms including inhibitory and costimulatory signals, cytokine production and signaling and also cell–cell interactions to assure effective viral clearance and less cytotoxic damage. CD4+ regulatory T cells (Treg) are induced after influenza infection and are important in limiting antigen-specific CD4+ and CD8+ T cell activation [163]. During avian H5N1 influenza infection CD8+, but not CD4+, Tregs are induced and suppress CD8+ T cell response by IL-10 production, and therefore, limit control of virus infection [164].
Secondary bacterial infections
It is estimated that one quarter of deaths during IAV epidemics [165] and 50–95 % of deaths during IAV pandemics [166, 167] are caused by secondary bacterial pneumonia. This lethal synergism between IAV and bacteria such as Streptococcus pneumoniae, Staphylococcus aureus, Streptococcus pyogens or Haemophilus influenzae is only partially understood. The increased susceptibility to bacterial infections during or shortly after IAV infection is multifactorial but there are two main theories. Enhanced susceptibility could be due to the intrinsic virulence of the virus, i.e., IAV infection directly causes lung damage and, thus, facilitates adherence and spread of bacteria [168]. Indeed, data from autopsy studies from patients of IAV pandemics showed increased adherence of bacteria on damaged epithelium [169]. Therefore, the cytotoxic potential of the virus, which may cause tissue damage, is important for the increase in secondary bacterial infection susceptibility. As an example, the protein PB1-F2, expressed by some influenza virus, is a cytotoxin known to increase cell death and inflammation thus contributing for the severity of pneumonia during coinfection [170]. Natural barriers such as the mucus layer and the tracheal mucociliary epithelium are important for host protection against respiratory pathogens. The neuraminidase activity of IAV can expose attachment sites for bacteria by the cleavage of sialic acids from the pulmonary mucus layer [171, 172] and provide sialylated substrates as nutrients for the proliferation of bacteria [173]. In addition, IAV infection decreases the mucociliary velocity and, as a consequence, also decreases the clearance of bacteria during co-infection [174].
The other theory for the increased susceptibility to secondary bacterial infection after IAV infection is the impairment of host defense mechanisms. It is suggested that IAV infection might compromise the local immunity of ears or lungs then predisposing the host to secondary infections. It has been shown that the production of type I IFNs after IAV infection in mice can impair the effective response against bacteria by decreasing the Th17 responses which are important to bacteria clearance [56, 175]. IFN-γ production during IAV infection is also associated with an impairment of phagocytosis [176]. Moreover, IAV infection can decrease intracellular production of oxygen reactive species and phagocytosis in neutrophils [177] and increase the production of the anti-inflammatory mediator IL-10 [178]. In addition, some studies have shown that IAV infection can lead to increased apoptosis of macrophages and neutrophils and that the resulting leukopenia could predispose the host to a sequential bacterial infection [172, 179, 180]. However, a large number of studies demonstrate that in most cases of secondary bacterial infection leukocytosis rather than leukopenia occurs [181].
Regardless of the mechanisms explaining enhanced susceptibility to secondary infection, it is clear that the synergistic effects of viral and bacterial infection in stimulating inflammation contribute to the severity of pneumonia [172]. Indeed, the intense inflammatory response observed in co-infected cases is associated with increased mortality as the persistent activation of macrophages and neutrophils in the lungs contributes to intense tissue damage [172]. In a murine model of secondary bacterial infection post-Influenza, there is massive influx of neutrophils and macrophages into the lungs and higher levels of pro-inflammatory cytokines in the lungs and blood [182]. It was also reported that human dendritic cells exposed to IAV and then infected with Streptococcus pneumoniae change their phenotypes and become more pro-inflammatory, producing higher levels of TNF-α, IL-6 and IL-12 [183, 184]. These data in conjunction with postmortem findings of patients that died with secondary bacterial infection after IAV infection [185] support the idea of tissue damage due to inflammation as an important contributor to the severity of pneumonia and morbidity.
Anti-inflammatory cytokines, such as IL-10, are also increased in the lungs after IAV and during bacteria co-infection [178, 182]. IL-10 has been suggested to play a role in the impairment of host defenses that follows IAV infection because treatment with anti-IL10 during a model of secondary bacterial infection restored the host ability to deal with infection [178]. Therefore, despite the increased expression of pro-inflammatory cytokines and striking influx of leukocytes into the lungs, the host defenses are impaired during post-influenza bacterial infections in part because of the markedly increased levels of IL-10. Another cytokine group that may contribute to the hyporesponsiveness of the immune system after an IAV infection is the type I and II interferons. Type I interferons may be important for the antiviral response during IAV infection but can also suppress IL-17 production by γδ T cells [175]. The inhibition of the Th17 pathway attenuates the production of antimicrobial peptides necessary for bacterial clearance in the lung [186], thus contributing to the increased susceptibility to secondary bacterial infection. Type II interferon (IFN-γ) produced after viral infections leads to a defect in alveolar macrophage-mediated phagocytosis of bacteria during a secondary infection and neutrophil dysfunction has also been observed after IAV infection [176, 177, 187, 188]. Associated with the dysfunction of phagocytes, it has been suggested that impairment of NK cells responses after IAV infection increases susceptibility to secondary S. aureus infection.
IAV infection can also lead to downregulation and desensitization of receptors such as the class A scavenger receptor MARCO and TLRs in alveolar macrophages thus impairing detection, phagocytosis and killing of bacteria [176, 189]. The desensitization of TLRs can persist for weeks or months after IAV infection contributing to the sustained susceptibility to secondary bacterial infections [189]. In addition, the increased expression of the receptor for the negative regulatory ligand CD200 during the recovery phase of IAV infection augments the threshold for innate immune activation enabling bacterial outgrowth [190].
Lastly, the increased severity of pneumonia observed during a co-infection can also be related with the impairment of the lung repair responses after IAV infection. A murine model of infection with a pandemic strain of IAV showed that the expression of epithelium cell proliferation and lung repair genes were decreased after virus infection and pro-inflammatory genes were strongly increased during the secondary pneumococcal infection [191]. In agreement with that, a most recent study showed that during co-infection with IAV and Legionella pneumophila genes involved in tissue protection and repair were specifically downregulated and it was determinant for the high morbidity and mortality observed [192]. Therefore, the lack of appropriate lung repair responses after IAV infection can also play a role in enhancing disease severity during bacterial co-infection.
The intense inflammatory response in the lungs that is caused by IAV infection is followed by a resolution state characterized by enhanced susceptibility to bacterial infection. A few molecular players of enhanced susceptibility have been found but there is much more research to be conducted to understand how IAV predisposes to subsequent infection.
Anti-inflammatory therapies
As discussed above, inflammatory responses are essential for the clearance of the virus and return to lung homeostasis during IAV infection. However, excessive, altered (different mediators) or misplaced (systemic) activation of inflammatory responses is clearly associated with intense lung damage and death. In principle, it is possible that we may develop immunomodulatory therapies aiming to modifying the unwanted inflammatory response triggered by IAV infection without interfering with the inflammatory response necessary to clear the virus [193]. The idea is that administration of such anti-inflammatory drugs during severe cases of flu may reduce patient symptoms related with the increased levels of pro-inflammatory mediators [43] and potentially modify favorably the prognosis of the infection. In this respect, one would expect that anti-inflammatory drugs decreased the number of hospitalizations and complications associated with IAV infection. Additional benefits of this kind of therapy are that therapeutic strategies aiming at host responses would be expected to decrease inflammatory responses regardless of the virus strain [194] and that they may be synergic with antiviral drugs.
Table 2 summarizes some of the immunomodulatory strategies that have been investigated for IAV infection treatment. Four important aspects deserve consideration in the context of IAV infection. First, timing of administration should be such that any anti-inflammatory drug would need to be effective even after infection had initiated and symptoms were present (in infected patients) or to prevent occurrence of severe disease in patients with contact with other infected patients. In this regard, pre-clinical studies do need to take into account this delayed administration of drugs and test potential anti-inflammatory compound in a more therapeutic setting: i.e., treatment should start and be effective several days after the infection of experimental animals. Second, anti-inflammatory therapies against IAV should work in association with and provide additional benefit to currently used antiviral drugs. Third, anti-inflammatory therapies should preferentially decrease the risk and/or severity of secondary bacterial infections that follow IAV infection. Fourth and most importantly, anti-inflammatory therapies should not interfere with the ability of the human host to deal with infection, even mild infection, i.e., the generation of adaptive immune responses should not be altered by anti-inflammatory drugs. An example of detrimental effects of anti-inflammatory/immunomodulatory drugs during IAV infection was observed during the 2009 IAV pandemic. Systemic treatment with corticosteroids during early or mild stages of pH1N1 infection increased the risk of secondary bacterial or fungal infections, critical illness and death [195, 196]. However, a Japanese study found no association between corticosteroid therapy during IAV infection and worse outcome [197]. Regardless of the conflicting available evidence, the data do reflect the necessary caution for the use of systemic corticosteroids to treat acute infections and put a word of caution to any potential anti-inflammatory strategy against severe IAV infection.
Statins are an example of drugs used as anti-inflammatory and immunomodulatory in the context of IAV infection. Statins are drugs used to lower lipids levels in blood, but are known also to decrease several features of inflammation, including neutrophil recruitment and secretion of pro-inflammatory cytokines [198]. There are a few studies that suggest that statins are beneficial in the context of IAV infection [199, 200]. A relevant study of 3043 patients hospitalized with IAV infection showed that administration of statins prior to or during the hospitalization period could reduce mortality by 41 % [201]. However, other groups have not observed the same effects of statin administration during IAV infection [202]. Studies in murine models of IAV infection are also controversial. Kumaki et al. in 2012 showed that oral or systemic administration of different statins could not improve the outcome of mice after infection with the highly pathogenic avian influenza H5N1, seasonal or H1N1pdm09 virus [203]. On the other hand, Liu et al. 2009 showed that statin treatment ameliorated lung damage and inhibited viral replication of influenza H5N1, H3N2 or H1N1, especially when given before infection [204]. A more recent study, also demonstrated that administration of sinvastatin in vitro could decrease the levels of pro-inflammatory cytokines and reduce virus replication by inhibiting proteins related to cytoskeleton function [205]. Therefore, given the safety profile of statins in the context of IAV infection, the use of these drugs may be useful patients at high risk of severe disease secondary to IAV infection, a tenet that deserves further investigation.
Another class of anti-inflammatory drugs that have been suggested to improve patient outcome during IAV infection is the cyclooxygenase-2inhibitors. Cyclooxygenase-2 (COX-2) are enzymes that catalyze the conversion of arachidonic acid in prostaglandins and, thus, may contribute to inflammation [206]. COX inhibitors are largely used as analgesic, anti-inflammatory and anti-pyretic drugs and have been suggested to be of therapeutic benefit during IAV infection [207]. Cyclooxygenase-2 is up-regulated during infection with the pandemic H5N1 influenza virus in vitro and increased expression of this enzyme is also observed in lung tissue samples obtained during autopsy of patients who died of H5N1 disease [208]. Inhibition of COX-2 during a lethal H5N1 virus challenge was demonstrated to be beneficial when associated with a neuraminidase inhibitor, even when given as a delayed treatment [209]. In addition, inhibition of COX-2 by paracetamol or a selective inhibitor (celecoxib) during H1N1 and H3N2 infection prevented lung immunopathology without affecting virus clearance in mice [210]. Associated with these findings, COX-2 deficiency is protective in a murine model of IAV H3N2 infection and it is associated with a decrease of the inflammatory responses [211]. Therefore, animal studies suggest that the use of COX-2 inhibitors during IAV infection could be beneficial. However, clinical studies with human patients are still lacking to reinforce the proof of concept of using this class of drugs during IAV infection. Currently, a clinical trial is recruiting patients to test the efficacy of celecoxib during influenza infection (ClinicalTrials.gov Identifier: NCT02108366).
Several anti-inflammatory strategies have been studied in the context of experimental IAV infection (see Table 2). Many of these have been investigated in some detail and will be described below. Peroxisome proliferator-activated receptor (PPAR) agonists provide beneficial effects in treatment of metabolic disorders, such as diabetes, and may also have anti-inflammatory effects [194]. PPAR activation leads to down regulation of NF-kB, AP1 and STAT signaling resulting in reduction of several pro-inflammatory cytokines [212]. In murine models of IAV infection, administration of Gemfibrozil, a synthetic PPAR-α agonist, was shown to protect mice from severe H2N2 infection possible due to its anti-inflammatory effect [213]. The same protection could not be seen in a model of H5N1 infection [209]. PPAR-γ agonists such as pioglitazone and rosiglitazone were also suggested as immunomodulatory drugs to treat IAV infection. Interestingly, prophylactic treatment with both of these drugs reduced weight loss and lethality induced in a murine model of seasonal H1N1 infection. In addition, rosiglitazone pre-treatment could also partially prevent lethality of animals infected with the 2009 pandemic H1N1 strain [214].
Pro-resolving mediators are also listed as a potential strategy for controlling the overwhelming inflammation during IAV infection. Lipoxins and protectins are lipid mediators that have anti-inflammatory and pro-resolving effects and are shown to be protective during several models of infection [215]. During IAV H5N1 infection, there was down regulation of genes related to lipoxin pro-resolving effects and up regulation of pro-inflammatory cytokines genes and these changes associated with dissemination of the virus to other organs. Therefore, it seems that lipoxin may have protective roles during IAV infection, acting as a modulator of the inflammatory responses. Levels of another endogenous resolution-associated molecule, protectin D1, was decreased during a model of severe IAV H5N1 infection and correlated inversely with immunopathology [216]. Associated with this, protectin D1 treatment improved survival of infected mice and could also inhibit IAV replication [216].
We have previously shown that blockade of the PAFR could improve survival of IAV infected mice as it decreased several parameters of the inflammatory response [96]. Effects of PAFR antagonists were observed even when the drug was initiated 3 days after infection. Importantly, association of PAFR antagonist and oseltamivir (Tamiflu®) provided cooperative inhibition of lethality in infected mice (World Intellectual Property Organization, International publication number: WO 2009/05938 A1).
CCR2 is chemokine receptor expressed on a variety of leukocyte and has been shown to contribute to recruitment of mononuclear phagocytes [217]. Monocyte-derived mononuclear cells are thought to contribute to the immunopathology observed during IAV infection [118]. Indeed, pharmacological blockage of CCR2 reduced pathology, morbidity, and mortality during IAV infection when given as a prophylactic treatment without affecting the virus clearance or aggravating secondary bacterial infections [218].
Another receptor that has been described as a therapeutic target during IAV infection is the sphingosine-1-phosphate receptor (S1P). Sphingosine 1-phosphate is a metabolite of sphingolipid that binds to five G protein-coupled receptors (S1P1-5) and controls different processes, including the immune response [219]. Administration of S1P ligands reduces the levels of cytokines and chemokines, and the tissue injury associated with infection with a human IAV strain [32] and a mouse-adapted influenza virus [220]. This effect was mostly due to the binding of the S1P agonist on S1P1 expressed by lung endothelium, suggesting that endothelial cells have an important role in inflammation induction after IAV infection [221]. Of note, specific S1P1 agonists did not impair the generation of neutralizing antibodies and did not alter viral clearance [220].
In summary, different studies have been suggested modulation of inflammatory response as a potential strategy for a better outcome during severe cases of flu [222–227].
Concluding remarks
There is now no doubt that severe influenza cases are associated with intense lung inflammation and injury. However, the inflammatory response is also necessary to control infection acutely and drive adaptive immune responses that will ultimately control viral replication. As described above, several molecules or cell types which are necessary for protective immune responses may also contribute to tissue injury and death when present in large quantities. In addition, there are molecules and pathways which are preferentially associated with tissue injury and death rather than protection. Defining pathways of disease in clinical samples and pre-clinical studies is crucial if we are to identify novel targets for IAV infection. There are several pre-clinical studies which have suggested potential targets and these clearly provided the necessary proof of concept that it is possible to develop anti-inflammatory drugs for influenza. The difficult task ahead will be to translate these pre-clinical findings into therapies for humans. A great advantage of anti-inflammatory drugs is that, on the contrary of antivirals, they are not susceptible to virus resistance. Therefore, it is very important to consider combined treatment of antiviral and anti-inflammatory drugs to reduce virus burden and disease severity.
Ideal anti-inflammatory drugs should be effective when administered after symptoms onset, be used in combination with approved antivirals such as oseltamivir or zanamivir, be able to reduce the risk of secondary bacterial infection and not to interfere with virus clearance mechanisms. Novel targets are needed and it is central that pre-clinical experiments model all of these parameters during drug development. Clinical studies with statins have provided mixed results and available murine studies with COX-2 inhibitors suggest these drugs may have potential benefit to patients.
The principle that infectious diseases are amenable to treatment with anti-inflammatory drugs appears to be a valid one. Ahead, we need to move into clinical development potential anti-inflammatory strategies that will decrease the suffering and death caused by severe IAV infection and secondary bacterial infections.
References
WHO. Influenza (Seasonal) - Fact sheet N°211. http://www.who.int/mediacentre/factsheets/fs211/en/ (2014). Accessed 02 Feb 2016.
Berri F, et al. Switch from protective to adverse inflammation during influenza: viral determinants and hemostasis are caught as culprits. Cell Mol Life Sci. 2014;71(5):885–98.
Hutchinson EC, Fodor E. Transport of the influenza virus genome from nucleus to nucleus. Viruses. 2013;5(10):2424–46.
Yoon SW, Webby RJ, Webster RG. Evolution and ecology of influenza A viruses. Curr Top Microbiol Immunol. 2014;385:359–75.
Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14(5):315–28.
Kuiken T, et al. Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol. 2012;2(3):276–86.
WHO. Influenza update N°258. http://www.who.int/influenza/surveillance_monitoring/updates/2016_03_07_surveillance_update_258.pdf?ua=1 (2016). Accessed 08 Mar 2016.
Edinger TO, Pohl MO, Stertz S. Entry of influenza A virus: host factors and antiviral targets. J Gen Virol. 2014;95(Pt 2):263–77.
Kohlmeier JE, Woodland DL. Immunity to respiratory viruses. Annu Rev Immunol. 2009;27:61–82.
Hendrickson CM, Matthay MA. Viral pathogens and acute lung injury: investigations inspired by the SARS epidemic and the 2009 H1N1 influenza pandemic. Semin Respir Crit Care Med. 2013;34(4):475–86.
Bruder D, Srikiatkhachorn A, Enelow RI. Cellular immunity and lung injury in respiratory virus infection. Viral Immunol. 2006;19(2):147–55.
de Jong MD, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12(10):1203–7.
Peiris JS, et al. Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol. 2009;30(12):574–84.
Baskin CR, et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci USA. 2009;106(9):3455–60.
Perrone LA, et al. Mice lacking both TNF and IL-1 receptors exhibit reduced lung inflammation and delay in onset of death following infection with a highly virulent H5N1 virus. J Infect Dis. 2010;202(8):1161–70.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
Xagorari A, Chlichlia K. Toll-like receptors and viruses: induction of innate antiviral immune responses. Open Microbiol J. 2008;2:49–59.
Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34(5):637–50.
Imai Y, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–49.
Le Goffic R, et al. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol. 2007;178(6):3368–72.
Le Goffic R, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2(6):e53.
Guillot L, et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280(7):5571–80.
Diebold SS, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–31.
Lee SM, et al. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci USA. 2014;111(10):3793–8.
Owen DM, Gale M Jr. Fighting the flu with inflammasome signaling. Immunity. 2009;30(4):476–8.
Allen IC, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30(4):556–65.
McAuley JL, et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013;9(5):e1003392.
Rehwinkel J, et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell. 2010;140(3):397–408.
Kato H, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23(1):19–28.
Kandasamy M, et al. RIG-I signaling Is critical for efficient polyfunctional T cell responses during influenza virus infection. PLoS Pathog. 2016;12(7):e1005754.
Tisoncik JR, et al. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76(1):16–32.
Walsh KB, et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA. 2011;108(29):12018–23.
Damjanovic D, et al. Immunopathology in influenza virus infection: uncoupling the friend from foe. Clin Immunol. 2012;144(1):57–69.
Tscherne DM, Garcia-Sastre A. Virulence determinants of pandemic influenza viruses. J Clin Invest. 2011;121(1):6–13.
Arankalle VA, et al. Role of host immune response and viral load in the differential outcome of pandemic H1N1 (2009) influenza virus infection in Indian patients. PLoS One. 2010;5(10) pii: e13099.
Cheng XW, et al. Three fatal cases of pandemic 2009 influenza A virus infection in Shenzhen are associated with cytokine storm. Respir Physiol Neurobiol. 2011;175(1):185–7.
Li N, et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell Mol Life Sci. 2015.
Ng HH, et al. Doxycycline treatment attenuates acute lung injury in mice infected with virulent influenza H3N2 virus: involvement of matrix metalloproteinases. Exp Mol Pathol. 2012;92(3):287–95.
Reshi ML, Su YC, Hong JR. RNA viruses: ROS-mediated cell death. Int J Cell Biol. 2014;2014:467452.
Kash JC, Taubenberger JK. Infectious disease theme issue: the role of viral, host, and secondary bacterial factors in influenza pathogenesis. Am J Pathol. 2015;185(6):1528–36.
La Gruta NL, et al. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol. 2007;85(2):85–92.
Hsieh YC, et al. Influenza pandemics: past, present and future. J Formos Med Assoc. 2006;105(1):1–6.
Kaiser L, et al. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol. 2001;64(3):262–8.
Hayden FG, et al. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest. 1998;101(3):643–9.
Julkunen I, et al. Molecular pathogenesis of influenza A virus infection and virus-induced regulation of cytokine gene expression. Cytokine Growth Factor Rev. 2001;12(2–3):171–80.
Jewell NA, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol. 2010;84(21):11515–22.
Wareing MD, et al. Chemokine expression during the development and resolution of a pulmonary leukocyte response to influenza A virus infection in mice. J Leukoc Biol. 2004;76(4):886–95.
To KF, et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J Med Virol. 2001;63(3):242–6.
Lee N, et al. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS One. 2011;6(10):e26050.
Fensterl V, Sen GC. Interferons and viral infections. BioFactors. 2009;35(1):14–20.
Chelbi-Alix MK, Wietzerbin J. Interferon, a growing cytokine family: 50 years of interferon research. Biochimie. 2007;89(6–7):713–8.
Price GE, Gaszewska-Mastarlarz A, Moskophidis D. The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J Virol. 2000;74(9):3996–4003.
Szretter KJ, et al. Early control of H5N1 influenza virus replication by the type I interferon response in mice. J Virol. 2009;83(11):5825–34.
Davidson S, et al. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun. 2014;5:3864.
Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89(Pt 1):1–47.
Shahangian A, et al. Type I IFNs mediate development of post influenza bacterial pneumonia in mice. J Clin Invest. 2009;119(7):1910–20.
Stifter SA, et al. Functional interplay between type I and II interferons is essential to limit influenza A virus-induced tissue inflammation. PLoS Pathog. 2016;12(1):e1005378.
Heise MT, Virgin HWT. The T-cell-independent role of gamma interferon and tumor necrosis factor alpha in macrophage activation during murine cytomegalovirus and herpes simplex virus infections. J Virol. 1995;69(2):904–9.
Stanton GJ, et al. Nondetectable levels of interferon gamma is a critical host defense during the first day of herpes simplex virus infection. Microb Pathog. 1987;3(3):179–83.
Boehm U, et al. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–95.
Verhoeven D, Perry S, Pryharski K. Control of influenza infection is impaired by diminished interferon-gamma secretion by CD4 T cell in the lungs of toddlers. J Leukoc Biol. 2016;100(1):203–12.
Turner SJ, et al. Disregulated influenza A virus-specific CD8+ T cell homeostasis in the absence of IFN-gamma signaling. J Immunol. 2007;178(12):7616–22.
Ramana CV, et al. Inflammatory impact of IFN-gamma in CD8+ T cell-mediated lung injury is mediated by both Stat1-dependent and -independent pathways. Am J Physiol Lung Cell Mol Physiol. 2015;308(7):L650–7.
Baumgarth N, Kelso A. In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J Virol. 1996;70(7):4411–8.
Sommereyns C, et al. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4(3):e1000017.
Mordstein M, et al. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008;4(9):e1000151.
van de Sandt CE, Kreijtz JH, Rimmelzwaan GF. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses. 2012;4(9):1438–76.
Crotta S, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 2013;9(11):e1003773.
Acosta-Rodriguez EV, et al. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8(9):942–9.
Luft T, et al. IL-1 beta enhances CD40 ligand-mediated cytokine secretion by human dendritic cells (DC): a mechanism for T cell-independent DC activation. J Immunol. 2002;168(2):713–22.
Ichinohe T, et al. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87.
Schmitz N, et al. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol. 2005;79(10):6441–8.
Liu Y, et al. Genetic variants in IL1A and IL1B contribute to the susceptibility to 2009 pandemic H1N1 influenza A virus. BMC Immunol. 2013;14:37.
Robinson KM, et al. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J Immunol. 2013;191(10):5153–9.
Guarda G, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34(2):213–23.
Chiaretti A, et al. IL-1 beta and IL-6 upregulation in children with H1N1 influenza virus infection. Mediat Inflamm. 2013;2013:495848.
Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702.
Yamaya M, et al. Magnitude of influenza virus replication and cell damage is associated with interleukin-6 production in primary cultures of human tracheal epithelium. Respir Physiol Neurobiol. 2014;202:16–23.
Shi X, et al. Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice. Crit Care. 2013;17(6):R301.
Szretter KJ, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. 2007;81(6):2736–44.
Hamada H, et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol. 2009;182(6):3469–81.
McKinstry KK, et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J Immunol. 2009;182(12):7353–63.
Wang X, et al. A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection. Cell Mol Immunol. 2011;8(6):462–8.
Crowe CR, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. 2009;183(8):5301–10.
Bermejo-Martin JF, et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care. 2009;13(6):R201.
Liu FD, et al. Timed action of IL-27 protects from immunopathology while preserving defense in influenza. PLoS Pathog. 2014;10(5):e1004110.
Mayer KD, et al. Cutting edge: T-bet and IL-27R are critical for in vivo IFN-gamma production by CD8 T cells during infection. J Immunol. 2008;180(2):693–7.
Liu L, et al. Influenza A virus induces interleukin-27 through cyclooxygenase-2 and protein kinase A signaling. J Biol Chem. 2012;287(15):11899–910.
Ivanov S, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol. 2013;87(12):6911–24.
Pociask DA, et al. IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol. 2013;182(4):1286–96.
Uhlig S, Goggel R, Engel S. Mechanisms of platelet-activating factor (PAF)-mediated responses in the lung. Pharmacol Rep. 2005;57(Suppl):206–21.
Ishii S, Shimizu T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res. 2000;39(1):41–82.
Weijer S, et al. Host response of platelet-activating factor receptor-deficient mice during pulmonary tuberculosis. Immunology. 2003;109(4):552–6.
Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000;80(4):1669–99.
Chao W, Olson MS. Platelet-activating factor: receptors and signal transduction. Biochem J. 1993;292(Pt 3):617–29.
Garcia CC, et al. Platelet-activating factor receptor plays a role in lung injury and death caused by influenza A in mice. plos Pathog. 2010;6(11):e1001171.
van der Sluijs KF, et al. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006;290(1):L194–9.
McCarthy MK, Weinberg JB. Eicosanoids and respiratory viral infection: coordinators of inflammation and potential therapeutic targets. Mediat Inflamm. 2012;2012:236345.
Yokomizo T, et al. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387(6633):620–4.
Yokomizo T, et al. A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. J Exp Med. 2000;192(3):421–32.
Tager AM, Luster AD. BLT1 and BLT2: the leukotriene B(4) receptors. Prostaglandins Leukot Essent Fatty Acids. 2003;69(2–3):123–34.
Gaudreault E, Gosselin J. Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. J Immunol. 2008;180(9):6211–21.
Widegren H, et al. LTB4 increases nasal neutrophil activity and conditions neutrophils to exert antiviral effects. Respir Med. 2011;105(7):997–1006.
Ricklin D, et al. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–97.
Sun S, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol. 2013;49(2):221–30.
Tong HH, et al. Deletion of the complement C5a receptor alleviates the severity of acute pneumococcal otitis media following influenza A virus infection in mice. PLoS One. 2014;9(4):e95160.
Xu GL, et al. C5a/C5aR pathway is essential for the pathogenesis of murine viral fulminant hepatitis by way of potentiating Fgl2/fibroleukin expression. Hepatology. 2014;60(1):114–24.
Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52.
Garcia CC, et al. Complement C5 activation during influenza A infection in mice contributes to neutrophil recruitment and lung injury. PLoS One. 2013;8(5):e64443.
Monsalvo AC, et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. 2011;17(2):195–9.
O’Brien KB, et al. A protective role for complement C3 protein during pandemic 2009 H1N1 and H5N1 influenza A virus infection. PLoS ONE. 2011;6(3):e17377.
Sun S, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis. 2015;60(4):586–95.
Turner MD, et al. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. 2014;1843(11):2563–82.
Gao R, et al. Cytokine and chemokine profiles in lung tissues from fatal cases of 2009 pandemic influenza A (H1N1): role of the host immune response in pathogenesis. Am J Pathol. 2013;183(4):1258–68.
van Helden MJ, Zaiss DM, Sijts AJ. CCR2 defines a distinct population of NK cells and mediates their migration during influenza virus infection in mice. PLoS One. 2012;7(12):e52027.
Maelfait J, et al. A20 deficiency in lung epithelial cells protects against influenza A virus infection. PLoS Pathog. 2016;12(1):e1005410.
Ellis GT, et al. TRAIL + monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep. 2015;16(9):1203–18.
Lin KL, et al. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. 2008;180(4):2562–72.
Dessing MC, et al. Monocyte chemoattractant protein 1 contributes to an adequate immune response in influenza pneumonia. Clin Immunol. 2007;125(3):328–36.
Narasaraju T, et al. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am J Respir Cell Mol Biol. 2010;42(6):732–43.
Sakai S, et al. Therapeutic effect of anti-macrophage inflammatory protein 2 antibody on influenza virus-induced pneumonia in mice. J Virol. 2000;74(5):2472–6.
Wareing MD, et al. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral Immunol. 2007;20(3):369–78.
DeBerge MP, et al. ADAM17-mediated processing of TNF-alpha expressed by antiviral effector CD8+ T cells is required for severe T-cell-mediated lung injury. PLoS One. 2013;8(11):e79340.
Tate MD, et al. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol. 2010;84(15):7569–80.
Zhao X, et al. PI3 K/Akt signaling pathway modulates influenza virus induced mouse alveolar macrophage polarization to M1/M2b. PLoS One. 2014;9(8):e104506.
Schneider C, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 2014;10(4):e1004053.
Tumpey TM, et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol. 2005;79(23):14933–44.
Hogner K, et al. Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 2013;9(2):e1003188.
Hashimoto Y, et al. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol. 2007;178(4):2448–57.
Brandes M, et al. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154(1):197–212.
Zhang Z, et al. Infectious progeny of 2009 A (H1N1) influenza virus replicated in and released from human neutrophils. Sci Rep. 2015;5:17809.
Fujisawa H. Inhibitory role of neutrophils on influenza virus multiplication in the lungs of mice. Microbiol Immunol. 2001;45(10):679–88.
Perrone LA, et al. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4(8):e1000115.
Fujisawa H. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J Virol. 2008;82(6):2772–83.
Daley JM, et al. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83(1):64–70.
Narasaraju T, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210.
Zhang RH, et al. N-acetyl-l-cystine (NAC) protects against H9N2 swine influenza virus-induced acute lung injury. Int Immunopharmacol. 2014;22(1):1–8.
Vlahos R, et al. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011;7(2):e1001271.
Shi X, et al. PEGylated human catalase elicits potent therapeutic effects on H1N1 influenza-induced pneumonia in mice. Appl Microbiol Biotechnol. 2013;97(23):10025–33.
Geiler J, et al. N-acetyl-l-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem Pharmacol. 2010;79(3):413–20.
Hwang I, et al. Activation mechanisms of natural killer cells during influenza virus infection. PLoS One. 2012;7(12):e51858.
Gazit R, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7(5):517–23.
Ishikawa H, et al. IFN-gamma production downstream of NKT cell activation in mice infected with influenza virus enhances the cytolytic activities of both NK cells and viral antigen-specific CD8+ T cells. Virology. 2010;407(2):325–32.
Vanderven HA, et al. What lies beneath: antibody dependent natural killer cell activation by antibodies to internal influenza virus proteins. EBioMedicine. 2016;8:277–90.
Jansen CA, et al. Differential lung NK cell responses in avian influenza virus infected chickens correlate with pathogenicity. Sci Rep. 2013;3:2478.
Zhou G, Juang SW, Kane KP. NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol. 2013;43(4):929–38.
Abdul-Careem MF, et al. Critical role of natural killer cells in lung immunopathology during influenza infection in mice. J Infect Dis. 2012;206(2):167–77.
Chang YJ, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8.
Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. 2013;9(9):e1003615.
Monticelli LA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12(11):1045–54.
Shim DH, et al. Pandemic influenza virus, pH1N1, induces asthmatic symptoms via activation of innate lymphoid cells. Pediatr Allergy Immunol. 2015;26(8):780–8.
Monticelli LA, Sonnenberg GF, Artis D. Innate lymphoid cells: critical regulators of allergic inflammation and tissue repair in the lung. Curr Opin Immunol. 2012;24(3):284–9.
Ho LP, et al. Activation of invariant NKT cells enhances the innate immune response and improves the disease course in influenza A virus infection. Eur J Immunol. 2008;38(7):1913–22.
De Santo C, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. 2008;118(12):4036–48.
Paget C, et al. Potential role of invariant NKT cells in the control of pulmonary inflammation and CD8+ T cell response during acute influenza A virus H3N2 pneumonia. J Immunol. 2011;186(10):5590–602.
Barthelemy A, et al. Influenza A virus-induced release of interleukin-10 inhibits the anti-microbial activities of invariant natural killer T cells during invasive pneumococcal superinfection. Mucosal Immunol. 2016; doi:10.1038/mi.2016.49.
Kreijtz JH, Fouchier RA, Rimmelzwaan GF. Immune responses to influenza virus infection. Virus Res. 2011;162(1–2):19–30.
Hillaire ML, Rimmelzwaan GF, Kreijtz JH. Clearance of influenza virus infections by T cells: risk of collateral damage? Curr Opin Virol. 2013;3(4):430–7.
McCormick S, et al. Control of pathogenic CD4 T cells and lethal immunopathology by signaling immunoadaptor DAP12 during influenza infection. J Immunol. 2011;187(8):4280–92.
Xu L, et al. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-alpha by antiviral CD8+ T cells. J Immunol. 2004;173(2):721–5.
Duan S, Thomas PG. Balancing immune protection and immune pathology by CD8(+) T-cell responses to influenza infection. Front Immunol. 2016;7:25.
Enelow RI, et al. Structural and functional consequences of alveolar cell recognition by CD8(+) T lymphocytes in experimental lung disease. J Clin Invest. 1998;102(9):1653–61.
Betts RJ, et al. Influenza A virus infection results in a robust, antigen-responsive, and widely disseminated Foxp3+ regulatory T cell response. J Virol. 2012;86(5):2817–25.
Zou Q, et al. CD8+ Treg cells suppress CD8+ T cell-responses by IL-10-dependent mechanism during H5N1 influenza virus infection. Eur J Immunol. 2014;44(1):103–14.
Simonsen L. The global impact of influenza on morbidity and mortality. Vaccine. 1999;17(Suppl 1):S3–10.
Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198(7):962–70.
McCullers JA. Do specific virus-bacteria pairings drive clinical outcomes of pneumonia? Clin Microbiol Infect. 2013;19(2):113–8.
Ramphal R, et al. Adherence of Pseudomonas aeruginosa to tracheal cells injured by influenza infection or by endotracheal intubation. Infect Immun. 1980;27(2):614–9.
McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. 2014;12(4):252–62.
McAuley JL, et al. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007;2(4):240–9.
McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186(3):341–50.
McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19(3):571–82.
Siegel SJ, Roche AM, Weiser JN. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe. 2014;16(1):55–67.
Pittet LA, et al. Influenza virus infection decreases tracheal mucociliary velocity and clearance of Streptococcus pneumoniae. Am J Respir Cell Mol Biol. 2010;42(4):450–60.
Kudva A, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 2011;186(3):1666–74.
Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. 2008;14(5):558–64.
McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. 2006;74(12):6707–21.
van der Sluijs KF, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172(12):7603–9.
Kosai K, et al. Increase of apoptosis in a murine model for severe pneumococcal pneumonia during influenza A virus infection. Jpn J Infect Dis. 2011;64(6):451–7.
Wilson HE, et al. Reactions of Monkeys to experimental mixed influenza and Streptococcus Infections: an analysis of the relative roles of humoral and cellular immunity, with the description of an intercurrent nephritic syndrome. J Exp Med. 1947;85(2):199–215.
Speshock JL, et al. Filamentous influenza A virus infection predisposes mice to fatal septicemia following superinfection with Streptococcus pneumoniae serotype 3. Infect Immun. 2007;75(6):3102–11.
Smith MW, et al. Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp Med. 2007;57(1):82–9.
Wu Y, et al. Successive influenza virus infection and Streptococcus pneumoniae stimulation alter human dendritic cell function. BMC Infect Dis. 2011;11:201.
Kuri T, et al. Influenza A virus-mediated priming enhances cytokine secretion by human dendritic cells infected with Streptococcus pneumoniae. Cell Microbiol. 2013;15(8):1385–400.
Centers for Disease C. and Prevention. Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1)—United States, May–August 2009. MMWR Morb Mortal Wkly Rep. 2009;58(38):1071–4.
Robinson KM, et al. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J Infect Dis. 2014;209(6):865–75.
Warshauer D, et al. Effect of influenza viral infection on the ingestion and killing of bacteria by alveolar macrophages. Am Rev Respir Dis. 1977;115(2):269–77.
Martin RR, et al. Effects of infection with influenza virus on the function of polymorphonuclear leukocytes. J Infect Dis. 1981;144(3):279–80.
Didierlaurent A, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205(2):323–9.
Goulding J, et al. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J Infect Dis. 2011;204(7):1086–94.
Kash JC, et al. Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. MBio. 2011; 2(5).
Jamieson AM, et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340(6137):1230–4.
Fedson DS. Confronting the next influenza pandemic with anti-inflammatory and immunomodulatory agents: why they are needed and how they might work. Influenza Other Respir Viruses. 2009;3(4):129–42.
Garcia CC, et al. The development of anti-inflammatory drugs for infectious diseases. Discov Med. 2010;10(55):479–88.
Han K, et al. Early use of glucocorticoids was a risk factor for critical disease and death from pH1N1 infection. Clin Infect Dis. 2011;53(4):326–33.
Kim SH, et al. Corticosteroid treatment in critically ill patients with pandemic influenza A/H1N1 2009 infection: analytic strategy using propensity scores. Am J Respir Crit Care Med. 2011;183(9):1207–14.
Kudo K, et al. Systemic corticosteroids and early administration of antiviral agents for pneumonia with acute wheezing due to influenza A(H1N1)pdm09 in Japan. PLoS ONE. 2012;7(2):e32280.
Abeles AM, Pillinger MH. Statins as antiinflammatory and immunomodulatory agents: a future in rheumatologic therapy? Arthritis Rheum. 2006;54(2):393–407.
Kwong JC, Li P, Redelmeier DA. Influenza morbidity and mortality in elderly patients receiving statins: a cohort study. PLoS One. 2009;4(11):e8087.
Frost FJ, et al. Influenza and COPD mortality protection as pleiotropic, dose-dependent effects of statins. Chest. 2007;131(4):1006–12.
Vandermeer ML, et al. Association between use of statins and mortality among patients hospitalized with laboratory-confirmed influenza virus infections: a multistate study. J Infect Dis. 2012;205(1):13–9.
Brett SJ, et al. Pre-admission statin use and in-hospital severity of 2009 pandemic influenza A(H1N1) disease. PLoS One. 2011;6(4):e18120.
Kumaki Y, Morrey JD, Barnard DL. Effect of statin treatments on highly pathogenic avian influenza H5N1, seasonal and H1N1pdm09 virus infections in BALB/c mice. Future Virol. 2012;7(8):801–18.
Liu Z, et al. Evaluation of the efficacy and safety of a statin/caffeine combination against H5N1, H3N2 and H1N1 virus infection in BALB/c mice. Eur J Pharm Sci. 2009;38(3):215–23.
Mehrbod P, et al. Simvastatin modulates cellular components in influenza A virus-infected cells. Int J Mol Med. 2014;34(1):61–73.
Rocca B, FitzGerald GA. Cyclooxygenases and prostaglandins: shaping up the immune response. Int Immunopharmacol. 2002;2(5):603–30.
Darwish I, Mubareka S, Liles WC. Immunomodulatory therapy for severe influenza. Expert Rev Anti Infect Ther. 2011;9(7):807–22.
Lee SM, et al. Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: a mechanism for the pathogenesis of avian influenza H5N1 infection. J Infect Dis. 2008;198(4):525–35.
Zheng BJ, et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci USA. 2008;105(23):8091–6.
Lauder SN, et al. Paracetamol reduces influenza-induced immunopathology in a mouse model of infection without compromising virus clearance or the generation of protective immunity. Thorax. 2011;66(5):368–74.
Carey MA, et al. Contrasting effects of cyclooxygenase-1 (COX-1) and COX-2 deficiency on the host response to influenza A viral infection. J Immunol. 2005;175(10):6878–84.
Bassaganya-Riera J, et al. PPAR-gamma activation as an anti-inflammatory therapy for respiratory virus infections. Viral Immunol. 2010;23(4):343–52.
Budd A, et al. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob Agents Chemother. 2007;51(8):2965–8.
Moseley CE, Webster RG, Aldridge JR. Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir Viruses. 2010;4(5):307–11.
Russell CD, Schwarze J. The role of pro-resolution lipid mediators in infectious disease. Immunology. 2014;141(2):166–73.
Morita M, et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. 2013;153(1):112–25.
Winter C, et al. Lung-specific overexpression of CC chemokine ligand (CCL) 2 enhances the host defense to Streptococcus pneumoniae infection in mice: role of the CCL2-CCR2 axis. J Immunol. 2007;178(9):5828–38.
Lin KL, et al. CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol. 2011;186(1):508–15.
Iwasaki A, Medzhitov R. A new shield for a cytokine storm. Cell. 2011;146(6):861–2.
Marsolais D, et al. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci USA. 2009;106(5):1560–5.
Teijaro JR, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146(6):980–91.
Sato K, et al. Therapeutic effect of erythromycin on influenza virus-induced lung injury in mice. Am J Respir Crit Care Med. 1998;157(3 Pt 1):853–7.
Karlstrom A, et al. Treatment with protein synthesis inhibitors improves outcomes of secondary bacterial pneumonia after influenza. J Infect Dis. 2009;199(3):311–9.
Higashi F, et al. Additional treatment with clarithromycin reduces fever duration in patients with influenza. Respir Investig. 2014;52(5):302–9.
Planz O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antiviral Res. 2013;98(3):457–68.
Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol. 2014;88(17):10214–27.
Sharma G, et al. Reduction of influenza virus-induced lung inflammation and mortality in animals treated with a phosophodisestrase-4 inhibitor and a selective serotonin reuptake inhibitor. Emerging Microbes Infect 2013;2,e54; doi:10.1038/emi.2013.52.
Acknowledgments
We are grateful to Fundação de Amparo a Pesquisas do Estado de Minas Gerais (FAPEMIG, Brazil), Conselho Nacional do Desenvolvimento Cientifico e Tecnológico (CNPq, Brazil), The Institute of Science and Technology in Dengue (INCT in Dengue, CNPq/Brazil) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Additional information
Responsible Editor: John Di Battista.
Rights and permissions
About this article

Cite this article
Tavares, L.P., Teixeira, M.M. & Garcia, C.C. The inflammatory response triggered by Influenza virus: a two edged sword. Inflamm. Res. 66, 283–302 (2017). https://doi.org/10.1007/s00011-016-0996-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-016-0996-0