
Abstract
Influenza viruses cause acute respiratory infections, which are highly contagious and occur as seasonal epidemic and sporadic pandemic outbreaks. Innate immune response is activated shortly after infection with influenza A viruses (IAV), affording effective protection of the host. However, this response should be tightly regulated, as insufficient inflammation may result in virus escape from immunosurveillance. In contrast, excessive inflammation may result in bystander lung tissue damage, loss of respiratory capacity, and deterioration of the clinical outcome of IAV infections. In this review, we give a comprehensive overview of the innate immune response to IAV infection and summarize the most important findings on how the host can inappropriately respond to influenza.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza is one of the most important causes of respiratory tract infection and is responsible for widespread morbidity and mortality every winter in moderate climate zones [1, 2]. Worldwide, influenza epidemics result in about 200,000–500,000 deaths each year. In addition to the epidemic outbreaks, a virus of animal origin (usually avian) can also be transmitted to humans and cause a pandemic, which can range from mild (200,000 deaths) to unusual but severe impacts in the population (40 million deaths for the Spanish 1918 pandemic). Thus, influenza is of great concern for human health.
The etiological agents of the disease, the enveloped single-stranded negative-sense RNA influenza viruses, are classified into three types (A, B, and C), of which influenza A virus (IAV) is clinically the most important [2–4]. IAV particles possess two viral surface glycoproteins, hemagglutinin (HA, organized in trimers) and neuraminidase (NA, organized in tetramers) and one matrix-2 protein (M2, organized in tetramers) (Fig. 1). Inside the virion, eight segments of negative-sense RNA are independently encapsidated by the viral nucleoprotein (NP) and a polymerase complex (PB2, PB1, PA), forming the ribonucleoprotein (RNP) complexes. The RNPs are surrounded by a layer of the matrix protein, M1, which line the envelope. In the initial phase of IAV infection, the homotrimer of HA binds to sialic acid on the surface of the host cell, allowing the endocytosis of the virus [4] (Fig. 2). In the endosome, under external acidic pH, the tetrameric channel of M2 proteins conducts protons into the virion, resulting in the dissociation of M1 from the RNP. Fusion of the viral and endosome membranes is mediated by the cleaved HA, which exposes its fusion peptide under acid pH. The vRNPs are then released from the endosome and transported to the nucleus, where replication occurs. The newly synthesized viral RNAs are produced through a complementary positive stranded intermediate RNA (cRNA), which represents a full-length copy of the vRNA. In the nucleus, the polymerase also allows the transcription of the genome into mRNA, which is then transported back to the cytoplasm and translated into viral proteins. Each RNA segment (S) encodes one or two proteins. Proteins NP, PB1, PB2, and PA re-enter the nucleus to form the RNP complex with vRNA. M1 and NEP also re-enter the nucleus and their binding to the vRNPs allows their export to the cytoplasm. Instead, HA, NA, and M2 are transported to the plasma membrane via the reticulum/Golgi route. RNPs bud from the plasma membrane, which expresses the viral HA, NA, and M2 viral proteins to form the newly synthesized IAV virions [4] (Fig. 2). The glycoprotein content of viral proteins on the surface of IAV varies between IAV strains and is dependent on the viral genomic composition of the virus particles [5]. Also, because the envelope is derived from the plasma membrane of the host cell, host cellular proteins such as annexins are also incorporated into the virions [6, 7]. The neuraminidase plays an important role in the last steps of the budding, as it prevents direct re-association of the viral HA with sialic acid of the host cells, so IAV particles can be released [4]. The HA and NA of IAV exhibit a high sequence variability and based on their antigenic differences, IAV are divided into subtypes. To date, 17 HA and ten NA subtypes have been described for IAV [3]. While the bird is the reservoir of all IAV subtypes, only H1, H2, H3, and N1, N2 subtypes have caused infections in humans. Currently, only IAV of the H1N1 and H3N2 strains have established sustained human-to-human transmission. It is noteworthy that in addition to the epidemic outbreaks, a virus of animal origin (usually avian) can also be transmitted to humans and could cause a pandemic if the virus becomes transmissible from human to human. To date, recurrent human infections with IAV of the H5N1 virus subtype and more recently with the newly emerged H7N9 virus has highlighted the important threat caused by influenza [8–10].

Structure of the IAV particle. The virion consists of 8 vRNP (ssRNA, NP, PB1, PB2, PA) surrounded by M1 proteins and an enveloped derived from the plasma membrane of the host cell. The viral HA, NA, and M2 as well as host proteins such as annexins (not shown) are incorporated into the enveloped

Schematic representation of the replication cycle of IAV. The viral HA binds to sialylated glycoprotein receptors (1) and upon binding the virus becomes endocytosed (2). From the endosome, the virus genome is released following a low PH-dependent fusion event mediated by HA (3). The RNPs are transported to the nucleus (4) where the transcription (5) and replication (6) occur. The newly synthesized viral RNAs are produced through a complementary positive-stranded intermediate RNA (cRNA). The mRNA are transported to the cytoplasm and translated into protein (7). HA, NA, and M2 are transported to the plasma membrane through the reticulum/Golgi route (8) while PB1, PB2, PA, NP, NEP, and M1 re-enter the nucleus (9). Association of M1 and NEP with the vRNA complex (vRNA, NP, PA, PB1, PB2) allows the translocation of the vRNPs (10). Budding of the vRNP/M1/NEP from the plasma membrane expressing host proteins and HA, NA, and M2 form the new virions (11)
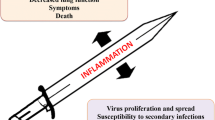
Upon infection with IAV, immune responses are induced that protect the host efficiently [1]. However, when the response to the infection is inappropriately regulated, a deterioration of the respiratory capacity and the clinical outcome of IAV infections can be observed (Fig. 3) [11]. On one hand, if the response is low, the virus can escape immune-surveillance and replicate within the host, leading to a severe infection. On the other hand, hypercytokinemia and excessive recruitment of innate immune cells induce collateral damage of the lungs and increase the immunopathology of influenza. Thus, a better understanding of the mechanism by which inflammation is induced as well as how it fails or turns inappropriate for the host is necessary in order to develop more efficient means of treatment against influenza.

Model of unbalanced inflammation following influenza infection. When the response to influenza infection is low or excessive, immunopathology of influenza develops. Strong interplay may exist between insufficient versus excessive inflammation. Immune escape from immunosurveillance (low response) may increase viral replication, which in turn induces strong release of secretory molecules (intensity of infection). When excessive inflammation is sustained by an uncontrolled host response, collateral lung damage increases IAV pathogeneses
The innate immune response to IAV infection
During the first days of IAV infection, viral replication, particularly in epithelial cells but also in monocytes, macrophages, or dendritic cells, initiates a cascade of signaling pathways involving a myriad of innate immune sensors, called pattern-recognition receptors (PRRs) [12] (Fig. 4). Activation of these receptors results in the release of cytokines and chemokines, which promote a local antiviral state and the recruitment of immune cells to the site of infection. The innate immune response includes both the production of secretory molecules and the recruitment of the cellular components of the immune system. In this paragraph, we will summarize our current knowledge on the innate immune response to influenza.

Pattern-recognition receptors (PRRs) sensing influenza viruses. Three groups of PRRs (TLR, RLR, and NLR) are able to sense influenza viruses. TLR7/8 and TLR3, endosome-expressed receptors, are activated by nucleic acids upon IAV infection. RIG-I, expressed in the cytoplasm recognizes the 5′triphosphate genome of influenza. NLRP3 is activated upon modification of ionic concentration mediated by the viral M2 protein of influenza. Activation of PRRs allows the release of both pro-inflammatory cytokines and IFN
Secretory molecules and pattern-recognition receptors activation
Secretory molecules are key mediators of antiviral immunity. Type I-IFN are the major cytokines that limit viral replication [13]. However, they are not sufficient for effective clearance of the virus, which evolved sophisticated strategies to escape immune-surveillance [13]. Thus, local proinflammatory cytokines are also extremely important for immune cell recruitment to the site of infection and to promote adaptive immune response [1, 14]. Each cytokine is produced in a cell-type-dependent manner. Thus, the nature of the cytokines that are present in the respiratory tract varies as the infection progresses. It is also dependent on the strain of the virus since cell susceptibility is subtype-dependent. Although simplified, the first target of influenza is the epithelial cell and interleukin 6(IL-6), IL-8 and regulated on activation, normal T cell expressed and secreted (RANTES) will be first release. Then, in addition to IL-6 and IL-8, infected alveolar macrophages will release macrophage inflammatory proteins (MIP), IL-1 and tumor necrosis factor-α (TNF-α) while infected dendritic cells will produce additional TNF-α, IL-1, IL-6, and MIP [15]. Each cytokine has specific major functions and thus the relative level of each cytokine will drive the host response (Fig. 4). A high level of IL-1, IL-6, or TNF-α broadly provokes the inflammatory response and causes fever. In contrast, a high level of IL-8 (KC in mouse) or MIP proteins attract and activate neutrophils, while of MCP-1 promote monocytes recruitment [15]. Although the cytokines have specific functions and are released in a cell-type-dependent manner, all of them are produced/activated via a common mechanism involving the activation of PRRs (Fig. 4). Three PRRs detect influenza via pathogen-associated molecular patterns and initiate the release of secretory molecules. Those receptors are the Toll-like receptors (TLR), the RIG-I like receptors (RLR), and the Nod-like receptors (NLR). Thus, TLRs constitute the first group of PRRs that sense influenza and are themselves divided into two groups based on their localization and type of ligand. The first group includes TLR 1, 2, 4, 5, and 6 that are cell surface-expressed and are activated by non-nucleic acid pathogen components. The role of the first group in the defense against IAV infection was poorly investigated and remains controversial [16–18]. The second group includes TLR 3, 7, 8, and 9, which are endosome-localized receptors, recognizing nucleic acids. The intracellular localization of the second group facilitates recognition of IAV, which enter host cells by endocytosis. All TLR, except TLR3 activate NF-κB (proinflammatory) and IRF3/7 (antiviral) through a common signaling adaptor MyD88. Instead, TLR3 recruits TRIF that can also be activated by TLR4. Upon IAV infection, TLR7 or MyD88-deficient dendritic cells are unable to release type-I IFN, in marked contrast to infected wild-type or TLR9-deficient cells [19, 20]. Thus, TLR 7 and 8, which specifically recognize ssRNA, are the main sensors of the ssRNA influenza virus [12, 19–21], while TLR9 does not seem to play a role. In contrast, the antiviral effect of TLR3 (that recognizes dsRNA intermediates) and TRIF remain obscure [22, 23].
The RLRs constitute the second group of PRRs, which sense influenza. RLR are cytoplasm-based receptors that recognize dsRNA and comprise three members; RIG-I, the melanoma differentiation-associated gene 5 (MDA5), and the laboratory of genetics and physiology 2 (LGP2). RLR signal though the mitochondrial antiviral-signaling protein (MAVS) signalosome leading to NF-κB and IRF3 activation. All three receptors contain a helicase domain while RIG-I and MDA 5 also contain a caspase recruitment domain, which allow them to overlap the role of inflammasome for IL1-β release (please see below). Upon IAV infection, RIG-I, which detects 5′ triphosphate RNA [24] and possibly containing short dsRNA structure motifs, but not MDA5, which recognizes stable dsRNA structures, are activated by IAV, while the role of LGP2 is not, so far, well defined. It was indeed demonstrated that RIG-I-deficiency but not MDA5-deficiency affects the release of IFN in response to IAV infection [25]. Finally, the cytosolic NLR receptors form the last group of PRRs that sense IAV. NLR are divided into subfamilies based on their difference in their effectors domains, leading to inflammatory response, autophagy, or cell death. Upon activation, NLR involved in inflammation assemble into platforms called inflammasomes to activate caspase-1 and trigger the maturation and secretion of IL-1 and IL-18, cytokines that play an important role during Flu infections [26]. Those cytokines are synthesized as inactive molecules, which upon enzymatic cleavage by caspase-1 become active and are secreted. So far, four members of the NLR family have been reported to initiate inflammasome multimeric protein platforms: NLRP1, NLRP3, NLRP6, and NLRC4. During macrophages infection by IAV, both the viral RNAs and the viral matrix 2 protein (M2) would be required to produce mature IL1-β via activation of two signals (Fig. 5) [27]. Signal 1 allows pro-IL1 synthesis through TLR7 activation and signal 2 activates the complex NLRP3/ASC/caspase-1 for cleavage of pro-IL-1 into mature IL-1 by active caspase-1. The complex NLRP3/ASC/caspase-1 is activated when ionic concentration is modified by the proton channel activity of the viral M2 protein. In marked contrast to macrophages, IL-1 secretion pathway is different in monocytes, where caspase-1 is constitutively active, and where signal 2 is not necessary [27]. However, the effect of NLRP3 in the experimental model of IAV infection remains controversial. It was initially reported that caspase 1 and IL1-deficient mice (but not NLRP3-deficient mice) are more susceptible to influenza [28]. However, another report showed that NLRP3 deficiency increased influenza-induced mortality [29]. In addition, release of IL1-β by signal 2 may be more complex than a simple NLRP3 activation. Indeed, a recent report has provided evidence that a strong interplay between NLR, TLR, and RLR is necessary to ensure efficient IL1-β release upon IAV infections, at least in epithelial cells [30].

Signals required for IL1 and IL18 release in IAV-infected macrophages TLR7/8 senses influenza and initiates pro-IL-1β (and pro-IL18) synthesis (Signal 1). NLRP3 senses modification of ionic concentration mediated by the viral M2 protein upon IAV infection leading to the assembly of the complex NLRP3/ASC and caspase-1, which is then activated. Caspase1 activation cleaves the immature cytokines into mature IL1 and IL18
Altogether, PPRs are the way by which the host responds primarily to influenza. PPRs, however, are differently expressed between cell subtypes, and cellular tropism of IAV differs between virus subtypes. Thus, this adds complexity in the understanding of the regulation of cytokine production upon IAV infections. The most remarkable example of this complexity is that within one cell subtype, such as macrophages, marked differences can be observed as well. Resident macrophages produce fewer pro-inflammatory cytokines compared to blood-derived macrophages and the latter are also more susceptible to highly pathogenic influenza [31]. Altogether, the combination of all these events likely modulates the quality and the quantity of the cytokine response, which will subsequently drive the protective versus disruptive effect of inflammation.
The cellular components of the inflammation
As mentioned above, cytokines and chemokines that are released upon infection contribute to the recruitment and activation of immune cells, thus facilitating the antiviral defense against the infection. Among the cellular components involved against influenza, three major components of the innate immune response stricto sensu can be mentioned; i.e., neutrophils, macrophages, and natural killer (NK) cells. First, (1) neutrophils recruited in large numbers to the respiratory tract upon influenza infections are implicated in the protection of the host [32, 33]. Depletion of these cells, in IAV-infected mice, increases viral replication, pulmonary inflammation, as well as mortality of the mice [32, 33]. Neutrophils eliminate the virus via different pathways, which include the phagocytosis of apoptotic IAV-infected cells and the degranulation and the production of reactive oxygen species, which assist in the clearance of infected cells [34, 35]. Another important additional weapon of neutrophils against pathogens is the release of neutrophil extracellular trap (NET), which arises from their nuclear contents into the extracellular space and are composed of decondensed chromatin and antimicrobial proteins. It was clearly demonstrated that NETs are formed upon IAV infections, although their role remains controversial [36, 37]. Altogether, neutrophils are important players against influenza. However, excessive recruitment of neutrophils to the lungs is also a major contributor of severe IAV infections and is typically observed upon mice infection with highly pathogenic H1N1 and H5N1 viruses [37, 38]. Their over-reaction further contributes to excessive lung inflammation and additional release of secretory molecules and particularly IL-1, TNF, or MIP proteins.
In addition to neutrophils, alveolar macrophages as well as newly recruited monocytes, which differentiate into macrophages, also contribute to innate immunity against influenza [32]. Macrophages eliminate cellular debris and apoptotic infected cells by phagocytosis. They also act as antigen-presenting cells and contribute to the induction of the adaptive immune response. Depletion of these cells increases lung viral replication as well as pathogenesis and death upon IAV infection [32, 39]. However, as for neutrophils, the presence of excessive macrophages in the lungs is a sign of severe IAV infection, suggesting that these cells could also contribute to the immunopathology of influenza [38]. Finally, the third innate cellular component recruited to the lungs upon IAV infection and playing a key role in IAV immune-surveillance are NK cells [40]. Upon activation, NK cells secrete cytokines and chemokines, and kill sensitive target cells by releasing the content of cytolytic granules [41, 42]. NK cell activation is orchestrated through a balance of inhibitory receptors (KIR) versus activatory (KAR) receptors [43]. First, NK cells detect the loss of human-leukocyte antigen (HLA) at the surface of infected cells via absence of engagement of KIRs, an activation known as the missing self-signal. Secondly, NK cells sense infected targets that express ligands for activation receptors, known as the danger signal [44]. When positive signals tend to be dominant, the functional outcome is tilted in favor of NK responsiveness. Surprisingly, while most viruses down-regulate the expression of HLA molecules at the surface of infected cells, IAV does not alter HLA expression on infected target cells [45] or does so only slightly [46]. IAV even augments NK cell inhibition through reorganization of HLA molecules into lipid rafts [45]. Thus, activation of NK during influenza is not due to a missing self-signal. Instead, during influenza, the KARs, NKp44 and NKp46 (but not NKp30), are engaged by the HA of IAV, which leads to NK cell activation [47–49]. In vivo, mice deficient in NKp46 receptor are more susceptible to IAV infection, demonstrating the importance of NK cell function against influenza [50]. However, as for all the components of the innate immune system, NK cells function can turn deleterious for the host. It was indeed demonstrated that NK cells can also contribute to the pathogenesis of IAV infection [51, 52]. Altogether, this illustrates the importance to consider the severity of infection regarding a protective or deleterious role for any component of the immune response. For example, the role of other cell types of the immune system such as the mucosal-associated invariant T cells could be revised. Their function was initially shown to be restricted to bacterial infections [53] but their role during influenza may be crucial, depending on the type of infection and more importantly during IAV coinfection with bacteria.
Viral escape from immunosurveillance
To evade the immune system, IAV has adopted strategies to efficiently replicate within the host [54]. Viral determinants such as the nonstructural protein 1 (NS1) and PB1-F2 block the antiviral IFN response and induce apoptosis of the recruited cellular components of the immune system, which enable them to react [55]. In addition, IAV upregulates the expression of the powerful immunotolerant human leukocyte antigen-G molecule (HLA-G) [56]. We will here highlight our current knowledge on how IAV manipulate these powerful molecules to escape immune-surveillance.
Role of the NS1 viral protein
NS1 is a nonstructural viral protein that antagonizes host immune responses [13]. The segment 8 of influenza vRNA, also known as the NS gene, encodes two proteins. The primary transcript generated encodes the NS1 protein. The second protein, NS2 (or NEP), is generated by alternative splicing of the primary transcript [57]. Recombinant viruses unable to express NS1 are viable but induce robust IFN secretion and show an attenuated phenotype in vitro and in vivo, which demonstrates the role of NS1 in the virulence of IAV [58]. The major function of NS1 is to limit the antiviral effect of IFN via different pathways. On one hand, NS1 blocks signaling by IRF3 and NF-κB [59, 60] and RIG-I activation [61, 62]. On the other hand, NS1 blocks IFN secretion at the posttranscriptional level with strong inhibition of IFN mRNA synthesis and post-transcriptional processing of IFN [63, 64]. In addition, NS1 is a key player in the manipulation of cell apoptotic machinery [65]. It was demonstrated that NS1 interact with tubulin, leading to disruption of normal cell division and apoptosis [66]. The length of NS1 is variable and strain-specific. In particular, at the C-terminus of NS1, truncations or extensions were observed [3]. While NS1 predominantly localizes in the nucleus and cytoplasm, those modifications may have consequences in its localization and most likely in its function [67]. The fact that NS1 is a virulence factor of IAV makes it a good target to attenuate these viruses. Several studies demonstrated that IAV with partial deletions in NS1 proteins are attenuated and do not cause disease, but induce a protective immune response in different species. These IAV variants are excellent live-attenuated influenza vaccine candidates, which could be of high interest in the future [68].
Role of PB1-F2
PB1-F2 is a proapoptotic viral protein that is expressed from an alternative open reading frame in the PB1 gene of IAV [69]. Some influenza strains do not express PB1-F2 and thus it is not required for viral replication. Nevertheless, PB1-F2 has been established as an important factor of virulence of influenza [70, 71]. Recombinant viruses unable to express PB1-F2 protein are less pathogenic in mice [72]. In addition, viruses with a single mutation in PB1-F2 (N66S) are highly pathogenic in mice as a consequence of increased viral replication [71]. The way by which PB1-F2 mediates increased viral replication is through inhibition of RIG-I-mediated type I IFN production at the level of the MAVS pathway [73–75]. The serine at position 66 (66S) in PB1-F2 further enhances IFN antagonism activity. PB1-F2 also induces apoptosis. After phosphorylation by protein kinase C, PB1-F2 interacts with the inner mitochondrial membrane adenine nucleotide translocase 3 and the outer mitochondrial membrane voltage-dependent anion channel 1, leading to permeabilization and destabilization of mitochondrial membrane, which results in cell death [74–76]. Also, another interesting characteristic of PB1-F2 is its contribution to the virulence of subsequent secondary bacterial pneumonia [77].
Role of the nonclassical host HLA-G molecule
The major histocompatibility complex molecule, HLA-G, is a non-classical antigen, which expression is mainly restricted to the cytotrophoblast, during pregnancy [78]. Several isoforms of HLA-G have been described that exhibit immunotolerant properties and are key factors in maternal-fetal tolerance [78–80]. HLA-G inhibits the lytic activity of NK cells [81, 82] as well as antigen-specific cytotoxic T cells directed against influenza (CTL) and allogeneic proliferative responses [83–85]. Recently, HLA-G has emerged as a key molecule in the evasion of immune response to several pathologic situations, such as tumors [86–90] and bacterial and viral infections, including influenza [56, 91–95]. HLA-G is upregulated at the surface of IAV-infected cells in a strain-dependent manner, at both the mRNA and protein levels [96]. These results suggest that the virulence of IAV may be caused by the differential capability of different strains to upregulate HLA-G. In line with this report, elevated HLA-G expression was ectopically observed in pandemic and seasonal IAV-infected patients [97]. HLA-G has been found to play an important role in several other viral infections and its expression has been correlated with increased severity of infection and poor survival of infected patients [92–95]. Given its broad immune-tolerant properties, by upregulating HLA-G, IAV may efficiently escape from immune surveillance and this likely contributes to IAV pathogenesis.
Uncontrolled deleterious inflammation
Resolution of inflammation is an integral component of the program of acute inflammation. It is absolutely required to protect healthy cells from tissue damage and is a prerequisite for the return of tissue homeostasis. When inflammation is inappropriately regulated, it becomes persistent and excessive. This deregulated inflammation, known as a “cytokine storm”, exacerbates the immunopathology of influenza [98]. Compared to uncomplicated patients, abnormal elevated levels of cytokines and chemokines are commonly detected in severe influenza infections [11]. Here, we will discuss the possible mechanism leading to the uncontrolled inflammation associated with severe influenza infections.
Role of the viral determinants
The role of viral replication in the virulence of IAV is still debated. Clinical studies showed that in severe influenza cases, a high level of virus replication and an excessive inflammatory response can be observed [11]. Whether a direct correlation exists between viral replication and the deregulated immune response remains an open question. An emerging idea is that a high level of virus replication likely contributes but is not the only culprit of excessive inflammation during influenza. The so-called “cytokine storm” would result from two components, which are (1) a high intensity of inflammation mediated by increased viral replication and PRR activation and (2) a sustained inflammation that results from an improper host response. Thus, some viral determinants are assumed to be associated with increased viral replication as well as excessive inflammation. Not surprisingly, chimeric viruses expressing strong activity of the polymerase complex (PA, PB1, and PB2) replicate more efficiently and are a potent inducer of proinflammatory cytokines and chemokines [99]. Also, the PA gene of a highly pathogenic H5N1 virus contributes to its virulence through increased viral replication and subsequent induction of an excessive innate immune response [100]. Another viral determinant that could impact viral replication and cytokine/chemokine release is the presence of a multibasic site in the HA of IAV [56]. After entry into the cell, the virus genome is released from the endosome following a low pH-dependent fusion event mediated by HA, and this fusion occurs only when HA is cleaved. The HA of low pathogenic strains contain a monobasic site that can only be cleaved by extracellular trypsin-like proteases, which thus represent a restricted factor for viral replication. In contrast, the HA of highly pathogenic IAV contain a polybasic site that is cleaved by intracellular furin-type proteases that are present ubiquitously, facilitating viral replication [101]. Indeed, the production of excessive proinflammatory molecules was reported for strains with multibasic cleavage site in HA [102].
Also, as described above, viral proteins NS1 and PB1-F2 are also important determinants that can promote the deregulation of inflammation.
Role of host determinants and hemostasis deregulation
As just mentioned, virus replication is unlikely to be solely responsible for deregulation of innate immunity upon IAV infection. In particular, the crosstalk between the pathogen and the host is a crucial factor driving immunopathogenesis of IAV.
Role of PAR1 in the transition between protective versus deleterious inflammation
Proteases and their receptors have recently emerged as a contributor of immunopathogenesis during viral infections [56, 103, 104]. Protease-activated-receptor 1 (PAR1), a G protein-coupled receptor, is activated as a result of proteolytic cleavage by thrombin, a protease central to the coagulation process. At a high concentration of thrombin, PAR1 plays a proinflammatory role, while at low concentration of thrombin, PAR1 mediates anti-inflammatory effects [105]. Using a mild IAV infection (observed by low levels of cytokine release in the broncho-alveolar lavages of infected WT mice), PAR1 was recently proposed to cooperate with TLR for IFN production [106]. Thus, according to the antiviral effect of TLR and IFN during IAV infections, these results are consistent with a potential protective effect of PAR1 during IAV infections (Fig. 6). The role of PAR1 in promoting innate immunity is platelet-independent, which is in favor of the presence of low concentration of thrombin and a moderate activation of endothelial cells [106]. Interestingly, and in marked contrast, we recently reported that during a severe lethal IAV infection, in which activation of the coagulation is likely to occur, resulting in high thrombin concentrations, PAR1 signaling was deleterious for the host [104]. Administration of PAR1 antagonists or PAR1 deficiency protected mice from lethal inflammation of the lungs. In contrast, activating PAR1 with specific agonists increased the cytokine storm and decreased survival. In addition, during severe infections, a cooperation between the activation of PAR1 and of the fibrinolytic system appeared to promote lethal inflammation [107] (Fig. 6 and discussed below). Similar deleterious role of PAR1 was also during meta-pneumovirus infections [108]. Thus, the severity of the infection likely determines the extent of IAV infection (epithelial versus endothelial cells) and the protective versus deleterious role of PAR-1-triggered anti versus pro-inflammatory responses. Accordingly, endothelial cells have recently emerged at the center of the uncontrolled inflammatory response induced by influenza [109]. The S1P1 receptor has a key position in the control of endothelial cell integrity and the routing of PAR1 towards anti-inflammatory versus proinflammatory responses [105]. At a low concentration of thrombin, PAR1 mediates endothelial barrier protection and anti-inflammatory effects through cross-activation of S1P1 receptor [105]. At a high concentration of thrombin, S1P1 is no longer activated and PAR1 signaling turns pro-inflammatory [105]. In fact, several reports showed that administration of S1P1 receptor (S1P1R) agonists blunt influenza-induced cytokine storm in mice and protect them from mortality induced by several IAV strains [109–111]. Altogether, it is tempting to speculate that modulation of the interactions between PAR1 and S1P1 contributes to regulate and orchestrate inflammation during influenza. More complex regulations of PAR1 may also involve cross-activation of (1) PAR2 [112], previously shown to protect against influenza [113] or (2) endothelial protein C receptor (EPCR) [114], although its role remains to be fully demonstrated [115, 116].

Model of protective and destructive inflammation during influenza. Upon IAV infection (non-severe infection), epithelial cells are infected and release secretory molecules promoting activation of the host immune response. Initial immune system activation is protective and aims at the elimination of the invading pathogen. PAR1, expressed at the surface of epithelial cells, cooperates with PRRs for effective activation of innate immunity against influenza. However, if the infection is not controlled (severe infection), endothelial cells are injured (1). Hemostasis is activated (2) and deregulation of fibrinolysis through hyperactivation of plasminogen/plasmin promotes excessive and deleterious inflammation (3). PAR1, which is also expressed at the surface of the endothelium, cooperates with plasminogen and further exacerbates inflammation and injury
Role of plasminogen and hyperfibrinolysis
Plasminogen is a zymogen that is activated into its active form plasmin by urokinase and tissue plasminogen activators (uPA, tPA). The main function of plasmin is to break down blood clots by dissolving fibrin polymers into soluble fragments, a process called fibrinolysis. Pericellular plasmin contributes to the remodeling of the extracellular matrix directly or indirectly via the activation of metalloproteases and could lead to cell anoïkis when excessive [117]. The generation of plasmin activity is a tightly regulated process. However, since ever, pathogens have exploited the function of plasminogen/plasmin for their own benefit. Particularly, activation of plasminogen by bacteria increases extracellular matrix degradation and fibrinolysis, a way by which the pathogen disseminates within the host. At the same time, this dysregulation of plasminogen activation and fibrinolysis has been associated with excessive inflammation [118]. Not only bacteria but also viruses and IAV in particular have evolved several strategies to sequester and activate plasminogen, through viral or cellular proteins [6, 119, 120]. Neuraminidase of the IAV strain A/WSN/33 can bind plasminogen, conferring this strain with the capacity to replicate efficiently in the brain [119, 121]. IAV can also activate plasminogen through the host cellular protein annexin 2 (A2), which is upregulated at the surface of infected cells and which is incorporated into the virions [6, 120]. Recently, we provided the first evidence that plasminogen plays a central role in influenza pathogenesis and cytokine storm [107]. We found that plasminogen-deficient mice or pharmacological inhibition of plasminogen activation in vivo protected mice from influenza infections and cytokine storm. Furthermore, pharmacological depletion of fibrinogen, the main target of plasmin had a profound deleterious effect on the survival of IAV-infected mice and this whether or not plasminogen activation is triggered (WT versus plasminogen-deficient mice), suggesting that fibrin is rather protective. Thus, these results pointed out for the first time that uncontrolled activation of the plasminergic system drives vascular permeability and excessive lung inflammation upon IAV infections. These results are consistent with clinical reports showing that fibrinolysis deregulation could be associated with fatal outcome of IAV infections in humans [122, 123]. In addition to fibrinolysis, it is well known that plasmin also promotes, in a strain-dependent manner, the proteolytic cleavage of the viral hemagglutinin, an essential step for the infectivity of IAV [2]. In vivo, viruses where HA can be cleaved by plasminogen replicate more efficiently in the lungs of plasminogen-competent mice compared to the ones of plasminogen-deficient mice [107]. Likely, this increased plasminogen-dependent virus replication also contributes to more PPR activation, which may further nourish the vicious circle of inflammation. Thus, these results point to a role for plasminergic and hemostasis deregulation in the control of the deleterious inflammation induced by influenza.
Conclusions
Influenza still causes significant morbidity and mortality associated with severe immunopathology of the lungs, related to excessive innate immune response. However, the mechanisms of such immunopathogenesis remain poorly understood. Based on our recent understanding, a model of inflammation in response to influenza can be proposed (Fig. 6). First, infected epithelial cells sense influenza and activate the innate immune response. Cytokines and chemokines are released and immune cells are recruited to the site of infection to clear the virus (protective immunity). In this context and at the epithelial level, some molecules such as PAR1 cooperate with PPR for protective innate immunity activation. A local and limited formation of fibrin could also be protective by limiting the diffusion of the infection. If the protective barriers are overwhelmed by the infection, endothelial cells are injured. Endothelium injury can result from (1) the acute phase of inflammation leading to increased endothelial cell permeability or (2) a direct infection of endothelial cells by IAV. In these conditions, protective molecules turn deleterious for the host. Deregulation of hemostasis, activation of PAR-1, or of the plasminergic system, then feed a vicious circle leading to malignant inflammation. This recent demonstration of the involvement of unbalanced hemostasis in the pathogenesis of influenza has to be replaced in a broader context. Indeed fibrinolysis plays a fundamental role in the clearance of blood clots and the clearance of extravascular fibrin. The major manifestation of plasminogen deficiency is the absence of fibrin resorption leading to the formation of pseudomembranes on inflamed mucosal surfaces in human [124] and impaired wound healing in mice [125]. In the context of sepsis, impairment of fibrin clearance is assumed to be pivotal in the pathogenesis of microvascular thrombosis and disseminated intravascular coagulation (DIC) [126]. Given the dual role of fibrinolysis, which may dependent on the severity of the infection, our results suggest that it will be essential to define in the next future specific markers of non-severe versus severe IAV infections to direct therapeutics against influenza. During non-severe infections, one could use the current and novel antivirals against influenza aiming at slowing down viral growth. In contrast, during severe IAV infections, where the hallmark of pathogenesis is the deleterious inflammation of the lungs, blocking viral replication may have no effect. Instead, targeting hemostasis looks to be a promising novel strategy for the future. Future research will aim at more precisely elucidating the immune mechanism of protection and deregulation in order to design new intervention strategies against influenza. From our current knowledge, PAR1 antagonists, PAR2 agonists, plasminogen inhibitors, or S1P1 agonists might be explored as a new treatment for influenza. By maintaining the inflammatory responses in their protective role against viral replication, these new strategies would provide protection against severe IAV infections, without encouraging the emergence of virus resistance.
References
Kuiken T, Riteau B, Fouchier RA, Rimmelzwaan GF (2012) Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol 2(3):276–286
Horimoto T, Kawaoka Y (2005) Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 3(8):591–600
Palese P, Shaw ML (2007) Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM (eds) Fields virology, vol 2, 5th edn. Lippincott Williams & Wilkins, Philadelphia, pp 1647–1689
Lamb RAKR (2001) Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, Griffin DE (eds) Fields virology. Lippincott Williams and Wilkins, Philadelphia, pp 1487–1531
Moules V, Terrier O, Yver M, Riteau B, Moriscot C, Ferraris O, Julien T, Giudice E, Rolland JP, Erny A, Bouscambert-Duchamp M, Frobert E, Rosa-Calatrava M, Pu Lin Y, Hay A, Thomas D, Schoehn G, Lina B (2011) Importance of viral genomic composition in modulating glycoprotein content on the surface of influenza virus particles. Virology 414(1):51–62
LeBouder F, Morello E, Rimmelzwaan GF, Bosse F, Pechoux C, Delmas B, Riteau B (2008) Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J Virol 82(14):6820–6828
Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, Palese P (2008) Cellular proteins in influenza virus particles. PLoS Pathog 4(6):e1000085
Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y (2013) Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 368(20):1888–1897
Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus AD, Fouchier RA (2012) Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336(6088):1534–1541
Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, Li C, Kawakami E, Yamada S, Kiso M, Suzuki Y, Maher EA, Neumann G, Kawaoka Y (2012) Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486(7403):420–428
de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J (2006) Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12(10):1203–1207
Ichinohe T (2010) Respective roles of TLR, RIG-I and NLRP3 in influenza virus infection and immunity: impact on vaccine design. Expert Rev Vaccines 9(11):1315–1324
Garcia-Sastre A (2011) Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162(1–2):12–18
Pascale F, Contreras V, Bonneau M, Courbet A, Chilmonczyk S, Bevilacqua C, Epardaud M, Niborski V, Riffault S, Balazuc AM, Foulon E, Guzylack-Piriou L, Riteau B, Hope J, Bertho N, Charley B, Schwartz-Cornil I (2008) Plasmacytoid dendritic cells migrate in afferent skin lymph. J Immunol 180(9):5963–5972
La Gruta NL, Kedzierska K, Stambas J, Doherty PC (2007) A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85(2):85–92
Shinya K, Ito M, Makino A, Tanaka M, Miyake K, Eisfeld AJ, Kawaoka Y (2012) The TLR4-TRIF pathway protects against H5N1 influenza virus infection. J Virol 86(1):19–24
Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM (2008) Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133(2):235–249
Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J, Chen WH, Ernst RK, Rossignol DP, Gusovsky F, Blanco JC, Vogel SN (2013) The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497(7450):498–502. doi:10.1038/nature12118
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA (2004) Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA 101(15):5598–5603
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303(5663):1529–1531
Geeraedts F, Goutagny N, Hornung V, Severa M, de Haan A, Pool J, Wilschut J, Fitzgerald KA, Huckriede A (2008) Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by Toll-like receptor signalling. PLoS Pathog 4(8):e1000138
Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M (2006) Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2(6):e53
Zhao J, Wohlford-Lenane C, Zhao J, Fleming E, Lane TE, McCray PB Jr, Perlman S (2012) Intranasal treatment with poly(I*C) protects aged mice from lethal respiratory virus infections. J Virol 86(21):11416–11424
Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C (2010) RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140(3):397–408
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441(7089):101–105
Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, Hornung V (2011) Inflammasomes: current understanding and open questions. Cell Mol Life Sci 68(5):765–783
Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA (2010) IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog 6(2):e1000661
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206(1):79–87
Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30(4):556–565
Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, Vidal SM (2013) Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus-infected cells. PLoS Pathog 9(4):e1003256
van Riel D, Leijten LM, van der Eerden M, Hoogsteden HC, Boven LA, Lambrecht BN, Osterhaus AD, Kuiken T (2011) Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog 7(6):e1002099
Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF (2005) Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79(23):14933–14944
Tate MD, Deng YM, Jones JE, Anderson GP, Brooks AG, Reading PC (2009) Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol 183(11):7441–7450
Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y (2007) Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol 178(4):2448–2457
Peake J, Suzuki K (2004) Neutrophil activation, antioxidant supplements and exercise-induced oxidative stress. Exerc Immunol Rev 10:129–141
Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA (2011) PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 6(7):e22043
Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT (2011) Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179(1):199–210
Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM (2008) H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4(8):e1000115
Kim HM, Lee YW, Lee KJ, Kim HS, Cho SW, van Rooijen N, Guan Y, Seo SH (2008) Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J Virol 82(9):4265–4274
Ennis FA, Meager A, Beare AS, Qi YH, Riley D, Schwarz G, Schild GC, Rook AH (1981) Interferon induction and increased natural killer-cell activity in influenza infections in man. Lancet 2(8252):891–893
Bryceson YT, Long EO (2008) Line of attack: NK cell specificity and integration of signals. Curr Opin Immunol 20(3):344–352
Riteau B, Barber DF, Long EO (2003) Vav1 phosphorylation is induced by beta2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J Exp Med 198(3):469–474
Thielens A, Vivier E, Romagne F (2012) NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol 24(2):239–245
Orr MT, Lanier LL (2010) Natural killer cell education and tolerance. Cell 142(6):847–856
Achdout H, Manaster I, Mandelboim O (2008) Influenza virus infection augments NK cell inhibition through reorganization of major histocompatibility complex class I proteins. J Virol 82(16):8030–8037
Ronni T, Matikainen S, Sareneva T, Melen K, Pirhonen J, Keskinen P, Julkunen I (1997) Regulation of IFN-alpha/beta, MxA, 2′,5′-oligoadenylate synthetase, and HLA gene expression in influenza A-infected human lung epithelial cells. J Immunol 158(5):2363–2374
Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, Strominger JL, Yewdell JW, Porgador A (2001) Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409(6823):1055–1060
Ho JW, Hershkovitz O, Peiris M, Zilka A, Bar-Ilan A, Nal B, Chu K, Kudelko M, Kam YW, Achdout H, Mandelboim M, Altmeyer R, Mandelboim O, Bruzzone R, Porgador A (2008) H5-type influenza virus hemagglutinin is functionally recognized by the natural killer-activating receptor NKp44. J Virol 82(4):2028–2032
Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O (2001) Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol 31(9):2680–2689
Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O (2006) Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol 7(5):517–523
Zhou G, Juang SW, Kane KP (2013) NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol 43(4):929–938
Abdul-Careem MF, Mian MF, Yue G, Gillgrass A, Chenoweth MJ, Barra NG, Chew MV, Chan T, Al-Garawi AA, Jordana M, Ashkar AA (2012) Critical role of natural killer cells in lung immunopathology during influenza infection in mice. J Infect Dis 206(2):167–177
Le Bourhis L, Martin E, Peguillet I, Guihot A, Froux N, Core M, Levy E, Dusseaux M, Meyssonnier V, Premel V, Ngo C, Riteau B, Duban L, Robert D, Rottman M, Soudais C, Lantz O (2010) Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol 11(8):701–708
Garcia-Sastre A, Biron CA (2006) Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312(5775):879–882
Herold S, Ludwig S, Pleschka S, Wolff T (2012) Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J Leukoc Biol 92(1):75–82
Foucault ML, Moules V, Rosa-Calatrava M, Riteau B (2011) Role for proteases and HLA-G in the pathogenicity of influenza A viruses. J Clin Virol 51(3):155–159
Robb NC, Jackson D, Vreede FT, Fodor E (2010) Splicing of influenza A virus NS1 mRNA is independent of the viral NS1 protein. J Gen Virol 91(Pt 9):2331–2340
Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T (1998) Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252(2):324–330
Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, Garcia-Sastre A (2000) Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74(17):7989–7996
Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, Garcia-Sastre A (2000) Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 74(24):11566–11573
Guo Z, Chen LM, Zeng H, Gomez JA, Plowden J, Fujita T, Katz JM, Donis RO, Sambhara S (2007) NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol 36(3):263–269
Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M Jr, Garcia-Sastre A (2007) Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol 81(2):514–524
Kochs G, Garcia-Sastre A, Martinez-Sobrido L (2007) Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol 81(13):7011–7021
Qiu Y, Krug RM (1994) The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J Virol 68(4):2425–2432
Schultz-Cherry S, Dybdahl-Sissoko N, Neumann G, Kawaoka Y, Hinshaw VS (2001) Influenza virus ns1 protein induces apoptosis in cultured cells. J Virol 75(17):7875–7881
Han X, Li Z, Chen H, Wang H, Mei L, Wu S, Zhang T, Liu B, Lin X (2012) Influenza virus A/Beijing/501/2009(H1N1) NS1 interacts with beta-tubulin and induces disruption of the microtubule network and apoptosis on A549 cells. PLoS ONE 7(11):e48340
Melen K, Kinnunen L, Fagerlund R, Ikonen N, Twu KY, Krug RM, Julkunen I (2007) Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J Virol 81(11):5995–6006
Talon J, Salvatore M, O’Neill RE, Nakaya Y, Zheng H, Muster T, Garcia-Sastre A, Palese P (2000) Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci USA 97(8):4309–4314
Conenello GM, Palese P (2007) Influenza A virus PB1-F2: a small protein with a big punch. Cell Host Microbe 2(4):207–209
Schmolke M, Manicassamy B, Pena L, Sutton T, Hai R, Varga ZT, Hale BG, Steel J, Perez DR, Garcia-Sastre A (2011) Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog 7(8):e1002186
Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P (2007) A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog 3(10):1414–1421
Zamarin D, Ortigoza MB, Palese P (2006) Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol 80(16):7976–7983
Varga ZT, Ramos I, Hai R, Schmolke M, Garcia-Sastre A, Fernandez-Sesma A, Palese P (2011) The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog 7(6):e1002067
Zamarin D, Garcia-Sastre A, Xiao X, Wang R, Palese P (2005) Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog 1(1):e4
Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW (2001) A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 7(12):1306–1312
Chanturiya AN, Basanez G, Schubert U, Henklein P, Yewdell JW, Zimmerberg J (2004) PB1-F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J Virol 78(12):6304–6312
McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, Yewdell JW, McCullers JA (2007) Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2(4):240–249
Rouas-Freiss N, Khalil-Daher I, Riteau B, Menier C, Paul P, Dausset J, Carosella ED (1999) The immunotolerance role of HLA-G. Semin Cancer Biol 9(1):3–12
Menier C, Riteau B, Dausset J, Carosella ED, Rouas-Freiss N (2000) HLA-G truncated isoforms can substitute for HLA-G1 in fetal survival. Hum Immunol 61(11):1118–1125
Riteau B, Moreau P, Menier C, Khalil-Daher I, Khosrotehrani K, Bras-Goncalves R, Paul P, Dausset J, Rouas-Freiss N, Carosella ED (2001) Characterization of HLA-G1, -G2, -G3, and -G4 isoforms transfected in a human melanoma cell line. Transplant Proc 33(3):2360–2364
Khalil-Daher I, Riteau B, Menier C, Sedlik C, Paul P, Dausset J, Carosella ED, Rouas-Freiss N (1999) Role of HLA-G versus HLA-E on NK function: HLA-G is able to inhibit NK cytolysis by itself. J Reprod Immunol 43(2):175–182
Riteau B, Menier C, Khalil-Daher I, Martinozzi S, Pla M, Dausset J, Carosella ED, Rouas-Freiss N (2001) HLA-G1 co-expression boosts the HLA class I-mediated NK lysis inhibition. Int Immunol 13(2):193–201
Riteau B, Menier C, Khalil-Daher I, Sedlik C, Dausset J, Rouas-Freiss N, Carosella ED (1999) HLA-G inhibits the allogeneic proliferative response. J Reprod Immunol 43(2):203–211
Le Gal FA, Riteau B, Sedlik C, Khalil-Daher I, Menier C, Dausset J, Guillet JG, Carosella ED, Rouas-Freiss N (1999) HLA-G-mediated inhibition of antigen-specific cytotoxic T lymphocytes. Int Immunol 11(8):1351–1356
Riteau B, Rouas-Freiss N, Menier C, Paul P, Dausset J, Carosella ED (2001) HLA-G2, -G3, and -G4 isoforms expressed as nonmature cell surface glycoproteins inhibit NK and antigen-specific CTL cytolysis. J Immunol 166(8):5018–5026
Paul P, Rouas-Freiss N, Khalil-Daher I, Moreau P, Riteau B, Le Gal FA, Avril MF, Dausset J, Guillet JG, Carosella ED (1998) HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci USA 95(8):4510–4515
Adrian Cabestre F, Moreau P, Riteau B, Ibrahim EC, Le Danff C, Dausset J, Rouas-Freiss N, Carosella ED, Paul P (1999) HLA-G expression in human melanoma cells: protection from NK cytolysis. J Reprod Immunol 43(2):183–193
Riteau B, Faure F, Menier C, Viel S, Carosella ED, Amigorena S, Rouas-Freiss N (2003) Exosomes bearing HLA-G are released by melanoma cells. Hum Immunol 64(11):1064–1072
Menier C, Riteau B, Carosella ED, Rouas-Freiss N (2002) MICA triggering signal for NK cell tumor lysis is counteracted by HLA-G1-mediated inhibitory signal. Int J Cancer 100(1):63–70
Zilberman S, Schenowitz C, Agaugue S, Benoit F, Riteau B, Rouzier R, Carosella ED, Rouas-Freiss N, Menier C (2012) HLA-G1 and HLA-G5 active dimers are present in malignant cells and effusions: the influence of the tumor microenvironment. Eur J Immunol 42(6):1599–1608
Fainardi E, Castellazzi M, Stignani M, Morandi F, Sana G, Gonzalez R, Pistoia V, Baricordi OR, Sokal E, Pena J (2011) Emerging topics and new perspectives on HLA-G. Cell Mol Life Sci 68(3):433–451
Li C, Toth I, Schulze Zur Wiesch J, Pereyra F, Rychert J, Rosenberg ES, van Lunzen J, Lichterfeld M, Yu XG (2013) Functional characterization of HLA-G(+) regulatory T cells in HIV-1 infection. PLoS Pathog 9(1):e1003140
Larsen MH, Zinyama R, Kallestrup P, Gerstoft J, Gomo E, Thorner LW, Berg TB, Erikstrup C, Ullum H (2013) HLA-G 3′ untranslated region 14-base pair deletion: association with poor survival in an HIV-1-infected Zimbabwean population. J Infect Dis 207(6):903–906
Segat L, Catamo E, Fabris A, Morgutti M, D’Agaro P, Campello C, Crovella S (2010) HLA-G*0105N allele is associated with augmented risk for HIV infection in white female patients. AIDS 24(12):1961–1964
Shi WW, Lin A, Xu DP, Bao WG, Zhang JG, Chen SY, Li J, Yan WH (2011) Plasma soluble human leukocyte antigen-G expression is a potential clinical biomarker in patients with hepatitis B virus infection. Hum Immunol 72(11):1068–1073
LeBouder F, Khoufache K, Menier C, Mandouri Y, Keffous M, Lejal N, Krawice-Radanne I, Carosella ED, Rouas-Freiss N, Riteau B (2009) Immunosuppressive HLA-G molecule is upregulated in alveolar epithelial cells after influenza A virus infection. Hum Immunol 70(12):1016–1019
Chen HX, Chen BG, Shi WW, Zhen R, Xu DP, Lin A, Yan WH (2011) Induction of cell surface human leukocyte antigen-G expression in pandemic H1N1 2009 and seasonal H1N1 influenza virus-infected patients. Hum Immunol 72(2):159–165
Tsotsiashvilli M, Levi R, Arnon R, Berke G (1998) Activation of influenza-specific memory cytotoxic T lymphocytes by Concanavalin A stimulation. Immunol Lett 60(2–3):89–95
Li OT, Chan MC, Leung CS, Chan RW, Guan Y, Nicholls JM, Poon LL (2009) Full factorial analysis of mammalian and avian influenza polymerase subunits suggests a role of an efficient polymerase for virus adaptation. PLoS ONE 4(5):e5658
Hu J, Hu Z, Song Q, Gu M, Liu X, Wang X, Hu S, Chen C, Liu H, Liu W, Chen S, Peng D, Liu X (2013) The PA-gene-mediated lethal dissemination and excessive innate immune response contribute to the high virulence of H5N1 avian influenza virus in mice. J Virol 87(5):2660–2672
Zeng H, Pappas C, Belser JA, Houser KV, Zhong W, Wadford DA, Stevens T, Balczon R, Katz JM, Tumpey TM (2012) Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J Virol 86(2):667–678
Suguitan AL Jr, Matsuoka Y, Lau YF, Santos CP, Vogel L, Cheng LI, Orandle M, Subbarao K (2012) The multibasic cleavage site of the hemagglutinin of highly pathogenic A/Vietnam/1203/2004 (H5N1) avian influenza virus acts as a virulence factor in a host-specific manner in mammals. J Virol 86(5):2706–2714
Riteau B, de Vaureix C, Lefevre F (2006) Trypsin increases pseudorabies virus production through activation of the ERK signalling pathway. J Gen Virol 87(Pt 5):1109–1112
Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, Coughlin SR, Carmeliet P, Lina B, Rimmelzwaan GF, Planz O, Ludwig S, Riteau B (2013) PAR1 contributes to influenza A virus pathogenicity in mice. J Clin Invest 123(1):206–214
Feistritzer C, Riewald M (2005) Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood 105(8):3178–3184
Antoniak S, Owens AP 3rd, Baunacke M, Williams JC, Lee RD, Weithauser A, Sheridan PA, Malz R, Luyendyk JP, Esserman DA, Trejo J, Kirchhofer D, Blaxall BC, Pawlinski R, Beck MA, Rauch U, Mackman N (2013) PAR-1 contributes to the innate immune response during viral infection. J Clin Invest 123(3):1310–1322
Berri F, Rimmelzwaan GF, Hanss M, Albina E, Foucault-Grunenwald ML, Le VB, Vogelzang-van Trierum SE, Gil P, Camerer E, Martinez D, Lina B, Lijnen R, Carmeliet P, Riteau B (2013) Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog 9(3):e1003229
Aerts L, Hamelin MÈ, Rhéaume C, Lavigne S, Couture C, Kim W, Susan-Resiga D, Prat A, Seidah NG, Vergnolle N, Riteau B, Boivin G (2013) Modulation of protease activated receptor 1 influences human metapneumovirus disease severity in a mouse model. Plos One 8:e72529
Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H (2011) Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146(6):980–991
Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M, Kawaoka Y, Rosen H, Oldstone MB (2011) Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA 108(29):12018–12023
Marsolais D, Hahm B, Walsh KB, Edelmann KH, McGavern D, Hatta Y, Kawaoka Y, Rosen H, Oldstone MB (2009) A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci USA 106(5):1560–1565
O’Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF (2000) Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem 275(18):13502–13509
Khoufache K, LeBouder F, Morello E, Laurent F, Riffault S, Andrade-Gordon P, Boullier S, Rousset P, Vergnolle N, Riteau B (2009) Protective role for protease-activated receptor-2 against influenza virus pathogenesis via an IFN-gamma-dependent pathway. J Immunol 182(12):7795–7802
Esmon CT (2012) Protein C anticoagulant system–anti-inflammatory effects. Semin Immunopathol 34(1):127–132
Schouten M, Sluijs KF, Gerlitz B, Grinnell BW, Roelofs JJ, Levi MM, van’t Veer C, Poll T (2010) Activated protein C ameliorates coagulopathy but does not influence outcome in lethal H1N1 influenza: a controlled laboratory study. Crit Care 14(2):R65
Schouten M, van’t Veer C, Levi M, Esmon CT, van der Poll T (2011) Endogenous protein C inhibits activation of coagulation and transiently lowers bacterial outgrowth in murine Escherichia coli peritonitis. J Thromb Haemost 9(5):1072–1075
Meilhac O, Ho-Tin-Noe B, Houard X, Philippe M, Michel JB, Angles-Cano E (2003) Pericellular plasmin induces smooth muscle cell anoikis. FASEB J 17(10):1301–1303
Degen JL, Bugge TH, Goguen JD (2007) Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost 5(Suppl 1):24–31
Goto H, Kawaoka Y (1998) A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc Natl Acad Sci USA 95(17):10224–10228
LeBouder F, Lina B, Rimmelzwaan GF, Riteau B (2010) Plasminogen promotes influenza A virus replication through an annexin 2-dependent pathway in the absence of neuraminidase. J Gen Virol 91(Pt 11):2753–2761
Goto H, Wells K, Takada A, Kawaoka Y (2001) Plasminogen-binding activity of neuraminidase determines the pathogenicity of influenza A virus. J Virol 75(19):9297–9301
Wang ZF, Su F, Lin XJ, Dai B, Kong LF, Zhao HW, Kang J (2011) Serum D-dimer changes and prognostic implication in 2009 novel influenza A(H1N1). Thromb Res 127(3):198–201
Soepandi PZ, Burhan E, Mangunnegoro H, Nawas A, Aditama TY, Partakusuma L, Isbaniah F, Ikhsan M, Swidarmoko B, Sutiyoso A, Malik S, Benamore R, Baird JK, Taylor WR (2010) Clinical course of avian influenza A(H5N1) in patients at the Persahabatan Hospital, Jakarta, Indonesia, 2005–2008. Chest 138(3):665–673
Mehta R, Shapiro AD (2008) Plasminogen deficiency. Haemophilia 14(6):1261–1268
Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL (1996) Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 87(4):709–719
Gando S (2013) Role of fibrinolysis in sepsis. Semin Thromb Hemost 39(4):392–399
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Berri, F., Lê, V.B., Jandrot-Perrus, M. et al. Switch from protective to adverse inflammation during influenza: viral determinants and hemostasis are caught as culprits. Cell. Mol. Life Sci. 71, 885–898 (2014). https://doi.org/10.1007/s00018-013-1479-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-013-1479-x