
Abstract
Objective
The objective of the review is to examine the crossroads of autophagy, inflammation, and apoptosis signaling pathways and their participation in liver fibrosis.
Introduction
Endoplasmic reticulum (ER) stress was emerged as a common feature relevant to the pathogenesis of diseases associated with organ fibrosis. However, the functional consequences of these alterations on ER stress and the possible involvement in liver fibrosis were currently largely unexplored. Here, we will survey the recent literature in the field and discuss recent insights focusing on some cellular models expressing mutant proteins involved in liver fibrosis.
Methods
A computer-based online search with PubMed, Scopus and Web of Science databases was performed for articles published, concerning ER stress, adaptation, inflammation and apoptosis with relevance to liver fibrosis.
Results and conclusions
Progression of liver fibrosis requires sustained inflammation leading to hepatocytes apoptosis through ER stress, whereas associated with activation of hepatic stellate cells (HSCs) into a fibrogenic and proliferative cell type. Faced with persistent and massive ER stress, HSCs adaptation starts to fail and apoptosis occurs in reversal of liver fibrosis, possibly mediated through calcium perturbations, unfolded protein response, and the pro-apoptotic transcription factor CHOP. Although limited in scope, current studies underscored that ER stress is tightly linked to adaptation, inflammation and apoptosis, and recent evidences suggested that these processes are related to the pathogenesis of liver fibrosis and its recovery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liver fibrosis is caused by a variety of agents, including chronic viral hepatitis, alcohol toxicity, autoimmune disease, hereditary metabolic disorders and the result of the wound-healing response of the liver to repeated injury [1, 2]. After the liver injury, hepatocytes regenerate and replace the necrotic or apoptotic cells, accompanying with an inflammatory response [3, 4]. If liver injury persists, hepatic stellate cells (HSCs) undergo a phenotypic transformation to become myofibroblast-like with expression of α-smooth muscle actin (α-SMA) and deposition of extracellular matrix (ECM) [5–7]. Thus, some studies suggested that the elimination of activated HSCs through cell death has been proposed as a mechanism to attenuate or reverse liver fibrosis [8–10]. However, the pathogenesis of liver fibrosis was so complicated, along with adaptation, inflammation, and apoptosis [11, 12]. Therefore, a better understanding of the pathogenesis of liver fibrosis was very important for the development of new treatment strategies.
The endoplasmic reticulum (ER) is an important organelle whose functions include folding and trafficking of protein, maintaining calcium homeostasis, lipid synthesis, and participating in a number of crucial cellular functions [13–15]. ER stress has emerged as a common feature relevant to the pathogenesis of diseases associated with fibrosis, whereas fibrosis is an integral part of many of the pathological conditions associated with ER stress; the role of these pathways in the fibrotic process is only beginning to emerge [16]. For liver fibrosis, it is assumed that ER stress has two opposing effects, including adaptation and apoptosis. Although ER stress serves a protective role that allows cells to deal with the noxious stimuli, persistent and immense ER stress contributes to cells apoptosis [17–20]. The apoptosis of hepatocytes by death receptors might be profibrogenic, while the apoptosis of HSCs would be expected to be fibrogenic resolution. Recently, a growing body of research has suggested that the ER is involved in the progression of liver fibrosis and its recovery [21–23]. Additional research is required to investigate the roles of the ER and its related signaling in liver fibrosis and to thus help in developing novel therapeutic strategies.
The unfolded protein response and ER stress
ER is the principal cellular organelle that regulates folding and trafficking of secretory proteins and an important storage site for cellular calcium. The dysregulation of protein synthesis or processing within the ER causes the accumulation of unfolded proteins, thereby leading to ER stress and the activation of the unfolded protein response (UPR) [24, 25].
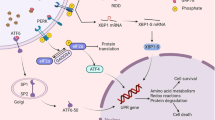
The typical UPR consists of three pathways, which are mediated by three ER-transmembrane transducers: protein kinase RNA (PKR)-like ER kinase (PERK), Inositol requiring 1α (IRE1α), and activating transcription factor-6 (ATF6). The UPR are sensed by the binding immunoglobulin protein (BiP), also called glucose-regulated protein 78 (GRP78) [26, 27]. The accumulation of unfolded proteins sequesters BiP so it dissociates from three ER-transmembrane transducers leading to their activation. The UPR can alleviate ER stress by reducing protein synthesis, promoting protein degradation and producing chaperones to assist with protein folding. Excessive or prolonged ER stress can lead to cell death mediated by apoptosis [28, 29]. CHOP is expressed at a very low level under physiological conditions, but its expression level is significantly increased in the presence of severe or persistent ER stress. The activation of CHOP, a central mediator of ER stress-induced apoptosis, was regulated by all three branches of the UPR; however, activating ATF4 is considered to be a major inducer of CHOP expression [30].
The Ca2+ signaling and ER stress
ER stress occurs when there is an imbalance between protein load and folding capacity, but can also be induced by other mechanisms. The ER was an important storage site for Ca2+ and involved in instigating cytosolic Ca2+ signals and plays a major role in signalling pathways. Many of the ER-resident molecular chaperones are Ca2+-binding proteins, including GRP78, calpain, calnexin and calreticulin, which are relevant to protein folding and protein quality control [31]. When a cell encounters prolonged ER stress or is overloaded with accumulation of proteins in the ER, cell death pathways such as calpain, caspase-12, CHOP, and c-Jun NH2-terminal kinase (JNK) are activated. Calcium release from the ER can activate calpain, which may proteolytically activate caspase-12 to mediate apoptosis. Calpain, the Ca2+-dependent cysteine protease, was ubiquitously expressed in cells and trigger ER stress-induced apoptosis. Caspase-12 was found in the ER membrane, providing a direct link between ER stress and activation of the caspase pathway [32–34]. Disturbance of Ca2+ influences ER function and its role in protein synthesis and folding, including post-translational modifications which lead to ER stress and to the activation of UPR [35–37].
To date, studies investigating the adaptability of ER dysfunction can cause UPR activation, and the UPR and ER stress are linked to Ca2+-signaling pathways and it may play an important role in the pathogenesis of liver fibrosis.
ER stress and autophagy
Cells respond to ER stress through a protective mechanism termed as the unfolded protein response (UPR) to restore normal ER function. As HSCs were activated and generated more ECM, this increased protein synthesis imposed a higher demand on the ER folding capacity that may disturb ER homeostasis [13, 38, 39]. Autophagy plays an important role in maintaining cellular homeostasis through alleviating stress. Although the role of autophagy in normal ER function is not established, there are some studies that have shown that autophagy is associated with the ER and maybe an important part of normal ER function [40].
In this important and well-designed study, Hernandez-Gea et al. [21] reported the molecular mechanism of this response under ER stress conditions. Indeed, blocking the IRE1a pathway in stellate cells decreases their fibrogenic potential. IRE1a is the most conserved branch among the three, normally kept inactive bound to the ER chaperone Bip, and could sense perturbation of ER homeostasis. This study also showed that mouse HSCs with tunicamycin (TN), a classical ER stress inducer, induced a strong increase in mRNA expression of Col1α1, Col1α2, β-Pdgfr, and α-SMA. TN also induced nuclear erythroid 2-related factor 2 (Nrf2) in HSCs, suggesting that Nrf2 may be involved in the ER stress response [41, 42]. Nrf2 is an oxidative stress-mediated transcription factor with a variety of downstream targets aimed at cytoprotection, which has recently been implicated as a new therapeutic target for the treatment of liver fibrosis [43–45]. The aim of this study was to determine the impact of oxidant and ER stress on stellate cell activation, along with the IRE1a branch of the UPR and modulates fibrogenic gene expression through autophagy.
These data implicate mechanisms underlying protein folding quality control in regulating the fibrogenic response in HSCs, together with autophagy. Because both autophagy and the UPR are induced upon perturbation of cellular homeostasis, we have explored the possibility that they contribute to the fibrogenic response in stellate cells [45–47]. These studies bring new insights that ER stress may play an important role in regulating the fibrogenic response in HSCs.
ER stress and inflammation
The ability of ER stress to induce an inflammatory response is considered to play a role in disease pathogenesis. Controlling the inflammatory process can therefore be a potential therapeutic target to inhibit the progress of diseases [48]. ER stress and UPR signaling pathways are linked with some of the major inflammation and stress signaling networks including the IRE1 and the NF-κB–IKK pathways.
In this review, we represented a conceivable mechanism that ER stress plays a pivotal role in the onset of inflammation and injury in the liver. Indeed, ER stress has been linked to several inflammatory response pathways in liver fibrosis, among these are the activation of NF-κB, JNK, ROS, tumor necrosis factor-α (TNF-α) and transforming growth factor-β (TGF-β) [49, 50]. Recently, it was shown that the IRE1 pathway is also linked with the NF-κB–IKK pathway, and it has been suggested that specific inflammatory activators can transmit signals through different UPR pathways. Indeed, recent study found that IRE1, a transmembrane ER protein that contains a cytoplasmic kinase, links ER stress to ERK phosphorylation and activation of ATF4, upregulated inflammatory cytokines through the activation of JNK. Similarly, this study showed that hepatic NF-κB p65 translocation and eIF2α phosphorylation were significantly increased in CCl4-induced mice liver fibrosis [23, 51]. Moreover, CCl4 also increase TNF-α and TGF-β1 in the model of mice liver fibrosis. According to several reports from different laboratories, the IRE1 pathway and the NF-κB–IKK pathway are involved in the regulation of inflammation in the pathogenesis of liver fibrosis [52, 53].
The tight relationship between ER stress and inflammation integrates ER function which plays a crucial role in liver fibrosis. In liver fibrosis, how the ER and its integrated stress signaling system associate with the function of immune cells is unclear. Thus, studies are still needed to clarify the important roles of ER stress and inflammation in liver fibrosis.
ER stress and apoptosis
A short-lasting UPR dampens ER stress and ensures cell survival. However, prolonged and sustained ER stress cannot be alleviated by the UPR, functional homeostasis of the ER cannot be reestablished, and this will induce cell death by apoptosis and activate apoptotic signalling pathways.
ER stress induced hepatocytes apoptosis in the progression of liver fibrosis
The ER stress response is an important homeostatic device for the liver during the onset and progression of chronic liver disease. Indeed, previous studies demonstrated the presence of ER stress during fibrosis, the consequently expected apoptosis of hepatocytes in vivo [54, 55]. There were some studies showing the antiapoptotic protein Bcl-2 and pro-apoptotic proteins, Bak and Bax, control ER calcium and cell death. Bax Inhibitor-1 also regulates ER calcium in cell lines, promoting calcium release under acidic conditions with associated increase in cell death. In addition, some findings suggested the possibility that ER stress induced liver injury through modulation of SERCA and maintaining calcium homeostasis, accompanied by the increased expression of GRP78, CHOP, SREBP1c and phosphorylated JNK2. GRP78, which influences Ca2+ homeostasis in the ER is upregulated in mice models and constitutes a corecomponent of the unfolded protein response [56, 57]. However, the linkage of ER stress to all stages of liver injury, including early and advanced liver disorders, remains poorly understood. In addition, the survival response activates genes that encode ER-residing chaperones such as GRP78, which uses the energy derived from ATP hydrolysis to prevent the aggregation of ER proteins and is considered the classical marker of UPR activation. Recent studies using this novel mouse model also revealed that liver GRP78 was required for neonatal survival, and a loss of GRP78 in the adult liver greater than 50 % caused an ER stress response and dilation of the ER compartment, which was accompanied by the onset of apoptosis.
ER stress induced HSCs apoptosis in the regression of liver fibrosis
The roles of each UPR initiation factor in recognition and response to various types of ER stress are not completely understood. There is also a need for further research to establish how the different UPR pathways function under particular conditions and different cellular environments and to determine whether they participate in different reactions and produce different effects on cells.
GRP78 is responsible for maintaining the inactive state of UPR signaling arms by preventing their dimerization. Elevation of misfolded/unfolded proteins occupies more GRP78 interacting sites and results in dissociation of GRP78 from signaling molecules and their subsequent dimerization. Upon release from GRP78, ATF6 is cleaved and translocated to the nucleus to induce CHOP and XBP1 expression [55–59]. Tashiro et al. [59] revealed that Nogo-B (Reticulon 4B) was a member of the reticulon protein family that is localized primarily to the ER. Nogo-B could accentuate hepatic fibrosis progression though the suppression of HSCs apoptosis. Furthermore, tunicamycin increased cleaved caspase-3 and -8 levels in Nogo-A/B knockout (Nogo-B KO) activated HSCs compared with wild-type (WT) activated HSCs, accompanying with increased GRP-78 expression.
Additionally, there was a study showing that cannabidiol (CBD), a major non-psychoactive component of the plant Cannabis sativa, elicits an endoplasmic reticulum (ER) stress response, characterized by changes in ER morphology and the downstream activation of the pro-apoptotic IRE1/ASK1/c-Jun N-terminal kinase pathway, leading to HSC apoptosis. Furthermore, this study investigated the effect of CBD on the ER in the activated HSCs by examining changes in the expression of calnexin, as well as CHOP, a major marker of prolonged ER stress [60]. Indeed, the expression of CHOP is involved in the pathway of ER stress-induced apoptosis, and while being normally undetectable in proliferating cells, it becomes highly synthesized in cells exposed to conditions that perturb the homeostasis of ER and is linked to the development of apoptosis. These results suggested that the higher degree of apoptosis observed in UPR and ER stress-induced activated HSCs apoptosis.
Although the major function of the ER is protein processing, factors in addition to protein overload can trigger UPR; interruption of ER calcium homeostasis can also induce ER stress and lead to apoptosis. Perturbations in ER calcium are also linked to apoptosis effectors. ER stress-inducing agents led to sustained and prolonged Ca2+ release from the ER, mitochondrial Ca2+ accumulation followed by mitochondrial permeabilization and release of apoptosis effectors from mitochondria into the cytosol [61]. Calpains are calcium-activated proteases that trigger ER stress-induced apoptosis. In addition, calpastatin represents the specific endogenous inhibitor of calpains and is provided with four inhibitory domains (CAST 1, 2, 3 and 4) simultaneously acting in calpains inhibition [62]. De Minicis et al. [22] demonstrated that ER stress sensitized HSCs to apoptosis by increasing in calpain/calpastatin ratio both in bile duct ligation (BDL) and subsequent bile duct diversion (BDD). Moreover, the study also showed that ER stress inducers (tunicamycin and thapsigargin) could induce HSCs apoptosis in vitro, completely reversing the calpain/calpastatin pattern expression compared with quiescent HSCs from normal rats. The experiments also showed that both tunicamycin and thapsigargin incubations in HSCs determine a phosphorylation of JNK and an increased expression of CHOP and caspase-12, as main indices in the process of cell apoptosis. These findings suggested that ER stress may play a crucial role in the regression of hepatic fibrosis, by converting quiescent HSCs to apoptosis-sensible phenotype. Similarly, Lin et al. previously showed that mYGJ (Modified Yi Guan Jian) may induce ER stress in activated HSCs, resulting in an increased cytosolic calcium concentration and activation of caspase-12 concomitant with the increased GRP-78 [63, 64]. Thus, it was concluded that ER stress-induced activated HSCs apoptosis may be used for a therapeutic application of liver fibrosis in the future.
Apoptosis is involved in the progression and regression of liver fibrosis. In the progression of liver fibrosis, ER stress induced hepatocytes apoptosis. While ER stress-induced activated HSCs apoptosis has been proposed as a mechanism to attenuate or reverse liver fibrosis. The role of ER stress in liver fibrosis was a double-edged sword of hepatocytes and HSCs apoptosis (Fig. 1).

Perturbations in ER homeostasis leading to dysfunction and activation of some of the UPR sensors occur in several liver fibrosis
Conclusion and prospective
In recent decades, many studies investigated distinct roles of ER stress in progresssion and regression of liver fibrosis, with the aim of developing novel therapeutic strategies. Yet, the incidence of liver fibrosis is still increasing worldwide, and the need for development of new therapies targeting is therefore urgent. Progression of liver fibrosis requires sustained inflammation leading to hepatocytes apoptosis through ER stress, whereas associated with activation of HSCs into a fibrogenic and proliferative cell type. Besides, the elimination of activated HSCs through ER stress-induced cell apoptosis has been proposed as a mechanism to attenuate or reverse liver fibrosis. As stated above, the ER may be an intersection of integrated multiple stress responses and may be closely related to adaptation, apoptosis, and inflammation in liver fibrosis (Fig. 2). Therefore, aberrant ER stress signalling has been reported in progresssion and regression of liver fibrosis and in addition to revealing pathological insights it may also present potential therapeutic opportunities.

ER stress plays a critical role of in the progression and regression of liver fibrosis
References
Jiang JX, Torok NJ. Liver injury and the activation of the hepatic myofibroblasts. Curr Pathobiol Rep. 2013;1:215–23.
Svegliati-Baroni G, De Minicis S, Marzioni M. Hepatic fibrogenesis in response to chronic liver injury: novel insights on the role of cell-to-cell interaction and transition. Liver Int. 2008;28:1052–64.
Friedman SL. Hepatic fibrosis—overview. Toxicology. 2008;254:120–9.
Wallace K, Burt AD, Wright MC. Liver fibrosis. Biochem J. 2008;411:1–18.
Atzori L, Poli G, Perra A. Hepatic stellate cell: a star cell in the liver. Int J Biochem Cell Biol. 2009;41:1639–42.
Wells RG. The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J Clin Gastroenterol. 2005;39:S158–61.
Safadi R, Friedman SL. Hepatic fibrosis–role of hepatic stellate cell activation. Med General Med. 2002;4:27.
Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S73–8.
Schinoni MI, Parana R. Apoptosis and progression of hepatic fibrosis in liver diseases. Acta Gastroenterol Latinoam. 2006;36:211–7.
Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21(Suppl 3):S84–7.
Leclercq IA, Da SMA, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol. 2007;47:142–56.
Mollica MP, Lionetti L, Putti R, Cavaliere G, Gaita M, Barletta A. From chronic overfeeding to hepatic injury: role of endoplasmic reticulum stress and inflammation. Nutr Metab Cardiovasc Dis. 2011;21:222–30.
Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–90.
Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab. 2010;12(Suppl 2):108–15.
Sovolyova N, Healy S, Samali A, Logue SE. Stressed to death—mechanisms of ER stress-induced cell death. Biol Chem. 2014;395:1–13.
Lenna S, Trojanowska M. The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr Opin Rheumatol. 2012;24:663–8.
Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5:a013169.
Malhotra JD, Kaufman RJ. ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb Perspect Biol. 2011;3:a004424.
Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007.
Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–70.
Hernandez-Gea V, Hilscher M, Rozenfeld R, Lim MP, Nieto N, Werner S, et al. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol. 2013;59:98–104.
De Minicis S, Candelaresi C, Agostinelli L, Taffetani S, Saccomanno S, Rychlicki C, et al. Endoplasmic Reticulum stress induces hepatic stellate cell apoptosis and contributes to fibrosis resolution. Liver Int. 2012;32:1574–84.
Wang JQ, Chen X, Zhang C, Tao L, Zhang ZH, Liu XQ, et al. Phenylbutyric acid protects against carbon tetrachloride-induced hepatic fibrogenesis in mice. Toxicol Appl Pharmacol. 2013;266:307–16.
Foufelle F, Ferre P. Unfolded protein response: its role in physiology and physiopathology. Med Sci (Paris). 2007;23:291–6.
Hollien J. Evolution of the unfolded protein response. Biochim Biophys Acta. 2013;1833:2458–63.
Misra UK, Pizzo SV. Modulation of the unfolded protein response in prostate cancer cells by antibody-directed against the carboxyl-terminal domain of GRP78. Apoptosis. 2010;15:173–82.
Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36:329–37.
Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13.
Feng LJ, Jiang TC, Zhou CY, Yu CL, Shen YJ, Li J, et al. Activated macrophage-like synoviocytes are resistant to endoplasmic reticulum stress-induced apoptosis in antigen-induced arthritis. Inflamm Res. 2014;63(5):335–46.
Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011;23:143–9.
Prell T, Lautenschlager J, Grosskreutz J. Calcium-dependent protein folding in amyotrophic lateral sclerosis. Cell Calcium. 2013;54:132–43.
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103.
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–94.
Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, et al. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36:592–601.
Brostrom MA, Brostrom CO. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium. 2003;34:345–63.
Kooptiwut S, Mahawong P, Hanchang W, Semprasert N, Kaewin S, Limjindaporn T, et al. Estrogen reduces endoplasmic reticulum stress to protect against glucotoxicity induced-pancreatic beta-cell death. J Steroid Biochem Mol Biol. 2014;139:25–32.
Paredes RM, Bollo M, Holstein D, Lechleiter JD. Luminal Ca2+ depletion during the unfolded protein response in Xenopus oocytes: cause and consequence. Cell Calcium. 2013;53:286–96.
Anholt RR, Carbone MA. A molecular mechanism for glaucoma: endoplasmic reticulum stress and the unfolded protein response. Trends Mol Med. 2013;19:586–93.
Kapoor A, Sanyal AJ. Endoplasmic reticulum stress and the unfolded protein response. Clin Liver Dis. 2009;13:581–90.
Carpenter JE, Jackson W, Benetti L, Grose C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J Virol. 2011;85:9414–24.
Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, et al. Toxicogenomics of endoplasmic reticulum stress inducer tunicamycin in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Toxicol Lett. 2007;168:21–39.
Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–22.
Liu J, Wu KC, Lu YF, Ekuase E, Klaassen CD. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid Med Cell Longev. 2013;2013:305861.
Yang JJ, Tao H, Huang C, Li J. Nuclear erythroid 2-related factor 2: a novel potential therapeutic target for liver fibrosis. Food Chem Toxicol. 2013;59:421–7.
Lee BH, Hsu WH, Hsu YW, Pan TM. Suppression of dimerumic acid on hepatic fibrosis caused from carboxymethyl-lysine (CML) by attenuating oxidative stress depends on Nrf2 activation in hepatic stellate cells (HSCs). Food Chem Toxicol. 2013;62:413–9.
Reichard JF, Petersen DR. Hepatic stellate cells lack AP-1 responsiveness to electrophiles and phorbol 12-myristate-13-acetate. Biochem Biophys Res Commun. 2004;322:842–53.
Kohler UA, Kurinna S, Schwitter D, Marti A, Schafer M, Hellerbrand C, et al. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology. 2014;60:670–8.
Tanjore H, Lawson WE, Blackwell TS. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta. 2013;1832:940–7.
Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–94.
Duwaerts CC, Maher JJ. Mechanisms of liver injury in non-alcoholic steatohepatitis. Curr Hepatol Rep. 2014;13:119–29.
Zheng Z, Zhang C, Zhang K. Measurement of ER stress response and inflammation in the mouse model of nonalcoholic fatty liver disease. Methods Enzymol. 2011;489:329–48.
Wang CM, Li SJ, Wu CH, Hu CM, Cheng HW, Chang JS. Transient knock down of Grp78 reveals roles in serum ferritin mediated pro-inflammatory cytokine secretion in rat primary activated hepatic stellate cells. Asian Pac J Cancer Prev. 2014;15:605–10.
Cho HK, Cheong KJ, Kim HY, Cheong J. Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclo-oxygenase 2 expression via activating transcription factor 4. Biochem J. 2011;435:431–9.
Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809.
Ji C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J Gastroenterol Hepatol. 2008;23(Suppl 1):S16–24.
Jeschke MG, Gauglitz GG, Song J, Kulp GA, Finnerty CC, Cox RA, et al. Calcium and ER stress mediate hepatic apoptosis after burn injury. J Cell Mol Med. 2009;13:1857–65.
Zhang J, Li Y, Jiang S, Yu H, An W. Enhanced endoplasmic reticulum SERCA activity by overexpression of hepatic stimulator substance gene prevents hepatic cells from ER stress-induced apoptosis. Am J Physiol Cell Physiol. 2014;306:C279–90.
Gorman AM, Healy SJ, Jager R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther. 2012;134:306–16.
Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda). 2007;22:193–201.
Tashiro K, Satoh A, Utsumi T, Chung C, Iwakiri Y. Absence of Nogo-B (reticulon 4B) facilitates hepatic stellate cell apoptosis and diminishes hepatic fibrosis in mice. Am J Pathol. 2013;182:786–95.
Lim MP, Devi LA, Rozenfeld R. Cannabidiol causes activated hepatic stellate cell death through a mechanism of endoplasmic reticulum stress-induced apoptosis. Cell Death Dis. 2011;2:e170.
Dolai S, Pal S, Yadav RK, Adak S. Endoplasmic reticulum stress-induced apoptosis in Leishmania through Ca2+-dependent and caspase-independent mechanism. J Biol Chem. 2011;286:13638–46.
Raimbourg Q, Perez J, Vandermeersch S, Prignon A, Hanouna G, Haymann JP, et al. The calpain/calpastatin system has opposing roles in growth and metastatic dissemination of melanoma. PLoS One. 2013;8:e60469.
Lin HJ, Tseng CP, Lin CF, Liao MH, Chen CM, Kao ST, et al. A Chinese herbal decoction, modified Yi Guan Jian, induces apoptosis in hepatic stellate cells through an ROS-mediated mitochondrial/caspase pathway. Evid Based Complement Alternat Med. 2011;2011:459531.
Acknowledgments
This study is supported by the Chinese National Natural Science Foundation Project (No. 81102493, 81273526, 81072686) and the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20103420120001).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible Editor: John Di Battista.
Rights and permissions
About this article

Cite this article
Li, X., Wang, Y., Wang, H. et al. Endoplasmic reticulum stress is the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in liver fibrosis. Inflamm. Res. 64, 1–7 (2015). https://doi.org/10.1007/s00011-014-0772-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-014-0772-y