
Abstract
Endoplasmic reticulum stress (ER stress) has been increasingly recognized as an important mechanism in various liver diseases. However, its intrinsic physiological role in acute liver failure (ALF) remains largely undetermined. This study aimed to examine how ER stress orchestrates glycogen synthase kinase 3β (GSK3β) and inflammation to affect ALF. In a murine ALF model induced by d-galactosamine (d-GalN) and lipopolysaccharide (LPS), 4-phenylbutyric acid (4-PBA) is to be administered to relieve ER stress. The lethality rate, liver damage, cytokine expression, and the activity of GSK3β were evaluated. How to regulate LPS-induced inflammation and TNF-α-induced hepatocyte apoptosis by ER stress was investigated in vitro. In vivo, ER stress was triggered in the liver with the progression of mice ALF model. ER stress was essential for the development of ALF because ER stress inhibition by 4-PBA ameliorated the liver damage through decreasing liver inflammation and hepatocyte apoptosis. 4-PBA also decreased GSK3β activity in the livers of ALF mice. In vitro, ER stress induced by tunicamycin synergistically increased LPS-triggered pro-inflammatory cytokine induction and promoted the activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathway in bone marrow-derived macrophages; moreover, tunicamycin also cooperated with TNF-α to increase hepatocyte apoptosis. ER stress promoted LPS-triggered inflammation depending on GSK3β activation because inhibition of GSK3β by SB216763, the specific inhibitor of GSK3β, resulted in downregulation of pro-inflammatory genes. ER stress contributes to liver inflammation and hepatotoxicity in ALF, particularly by regulating GSK3β, and is therefore a potential therapeutic target for ALF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Acute liver failure (ALF), an inflammation-mediated hepatocellular injury process, is a dramatic clinical syndrome that results from hepatocellular apoptosis and hemorrhagic necrosis [1]. It frequently results from hepatitis virus infection, induction of drugs and toxins, or hepatic ischemia-reperfusion injury. The prognosis of ALF is extremely ominous, and there is no effective therapy for the disease other than liver transplantation [2]. Although liver inflammation is an important determinant in the physiopathology of ALF, its mechanisms are still not well understood.
Endoplasmic reticulum stress (ER stress) as a result of accumulation of unfolded or misfolded proteins in the ER leads to the activation of three ER-localized transmembrane protein sensors, including inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6), which in turn initiate unfolded protein response (UPR) [3, 4]. The chaperone protein glucose-regulated protein 78 (Grp78) and glucose-regulated protein 94 (Grp94) are a master regulator of ER homeostasis which interact with PERK, IRE1α, and ATF6, and they are hallmarks for ER stress responses [5–7]. The transient and low-grade ER stress can be overcome by the UPR; if the UPR fails to promote cell survival, persistent and severe ER stress results in cell apoptosis, the c-jun N-terminal kinase (JNK), C/EBP-homologous protein (CHOP), and caspase 12 in rodents (caspase 4 in humans) are recruited to participate in ERS-induced apoptosis [8, 9]. At present, a growing body of evidence suggests that the signaling pathways in ER stress and inflammation are interconnected through various mechanisms [10], and impairment of endoplasmic reticulum in the liver is an early consequence of the systemic inflammatory response [11]. Distinctive effects of ER stress on inflammatory response have been reported; ER stress was shown to increase logfold LPS-mediated inflammatory cytokine induction in the macrophage cell [12–15].
Glycogen synthase kinases 3 (GSK3) are a group of ubiquitously expressed serine/threonine kinases that are initially found to regulate glycogen synthesis. There are two highly homologous isoforms, designated as GSK3α and GSK3β, respectively. Among the diverse functions that are regulated by GSK3β, inflammation has recently emerged as one of the major interesting focuses. Studies showed that GSK3β is an important positive regulator in inflammatory process [16]. Additionally, GSK3β promotes cell apoptosis caused by the intrinsic apoptotic pathway, while inhibits the death receptor-mediated extrinsic apoptotic signaling pathway [17]. Our study has shown that the activity of GSK3β is promoted in the progression of ALF, and inhibition of GSK3β ameliorates ALF model of mice [18, 19].
Despite the rapid growth of ER stress research in liver diseases, the exact contribution of the ER stress response to ALF is still unclear; moreover, the mechanistic focus has been limited mostly to cell apoptosis [20]. The ultimate result of ALF is the apoptosis of liver parenchymal cells, but the full development of injury is critically dependent on the inflammatory immune response of the liver. Thus, the questions of whether and how ER stress regulates liver inflammation are of high interest to further our understanding of the ALF mechanism. The model of hepatic injury induced by simultaneous injection of d-galactosamine (d-GalN) and lipopolysaccharide (LPS) has been widely used to examine the mechanisms of ALF, which is primarily a model of TNF-induced hepatotoxicity and produces typical hepatic apoptosis and necrosis [21, 22], though it is hardly a specific model for a common human condition that results in ALF. In the present study, we found that ER stress was triggered in the progression of ALF model induced by d-GalN/LPS; inhibition of ER stress by chemical chaperone protected mice from liver injury and significantly suppressed the inflammation, meanwhile decreased the activity of GSK3β. Therefore, we first explored a pivotal role of ER stress-GSK3β pathway in regulating inflammation in the mechanism of ALF.
MATERIALS AND METHODS
Animal Experiments
Male wild-type (WT; C57BL/6) mice (8–12 weeks of age) were purchased from the Capital Medical University (Beijing, China) and housed in the Capital Medical University animal facility under specific pathogen-free conditions and received humane care according to the Capital Medical University Animal Care Committee guidelines. The animal protocol had been approved by the Institutional Animal Care and Use Committee (IACUC) of the Capital Medical University.
To induce ALF, the mice (except for the control) were injected intraperitoneally with d-GalN (700 mg/kg; Sigma, St Luis, MO) and LPS (10 μg/kg; InvivoGen, San Diego, CA). 4-Phenylbutyrate (4-PBA; 100 mg/kg, Sigma, St Luis, MO), which is a chemical chaperone to relieve ER stress, was suspended in PBS and administered intraperitoneally 6 h prior to d-GalN/LPS exposure. At selected time points after d-GalN/LPS treatment, mice were anesthetized and blood was collected. The liver was harvested and used immediately to prepare messenger RNA (mRNA). Both the mRNA and liver tissues were stored at −75 °C for later analysis.
Serum Aminotransferase Activities and Histopathological Analysis
Serum samples were taken from the mice at 6 h after d-GalN/LPS injection. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) as markers of hepatic damage, were measured by using a multiparametric analyzer (AU5400; Olympus, Japan), according to an automated procedure.
Liver tissues were fixed in formalin and embedded in paraffin wax, and sections in 5 μm were stained with hematoxylin and eosin (H&E) using a standard protocol and then analyzed by light microscopy. Histological severity of liver injury was graded using Suzuki’s criteria on a scale from 0 to 4. No necrosis, congestion/centrilobular ballooning is given a score of 0, whereas severe congestion/degeneration and >60 % lobular necrosis are given a value of 4 [23].
Quantitative Reverse-Transcription Polymerase Chain Reaction
Total RNA was isolated from hepatic samples using TRIzol reagent according to manufacturer’s protocol. Using SuperScript™ III First-Strand Synthesis System (Invitrogen, Carlsbad, CA), 2.5 μg of RNA was reverse transcribed into complementary DNA (cDNA). Quantitative RT-PCR was performed using the DNA Engine with Chromo4 Detector (MJ Research, Waltham, MA). In a final reaction volume of 25 μl, the following were added: 1× SuperMix (Platinum SYBR Green qPCR Kit; Invitrogen, Carlsbad, CA), cDNA (2 μl) and 0.5 μM of each primer. Amplification conditions were as follows: 50 °C (2 min), 95 °C (5 min) followed by 50 cycles of 95 °C (15 s), 60 °C (30 s). Primers used to amplify a specific mouse gene fragments were listed in Table 1.
Western Blot Analysis
Protein was extracted from the liver tissue or cell in RIPA buffer together with phosphatase and protease inhibitors. Proteins in sodium dodecyl sulfate (SDS)-loading buffer were subjected to SDS-12 % polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membrane (Bio-Rad, Hercules, CA). Antibodies against target proteins pan-IP3R, (Calbiochem, Frankfort, KY, USA), p-GSK3β, total GSK3β, Grp78, Grp94, cleaved PARP, β-actin (Cell Signaling Technology Inc., Santa Cruz, CA), XBP-1 (Abcam, Cambridge, MA), ATF-6 (Imgenex, San Diego, CA), p-PERK, and PERK (Santa Cruz Biotechnology, Inc., Dallas, Texas) were used for Western blot analysis. Membranes were probed with primary antibody (1:500–1000) in 10 ml blocking buffer overnight at 4 °C. After washing, membranes were further probed with appropriate horseradish peroxidase-conjugated secondary antibody (1:2000) in 10 ml of blocking buffer for 1 h at room temperature. SuperSignal West Pico Chemiluminescent Substrates (Thermo Fisher Scientific, Rockford, IL) were used for chemiluminescence development.
Myeloperoxidase Activity Assay
Presence of myeloperoxidase (MPO) was used as an index for neutrophil accumulation in the liver. Briefly, frozen tissue was thawed and weighed, and 100 mg tissue was placed in 4 ml of iced 0.5 % hexadecyltrimethyl-ammonium bromide and 50 mM of potassium phosphate buffer solution with the pH adjusted to 5. Each sample was then homogenized for 30 s and centrifuged at 15,000 rpm for 15 min at 4 °C. Supernatants were mixed with hydrogen peroxide-sodium acetate and tetramethylbenzidine solutions. The change in absorbance was measured by spectrophotometry at 655 nm. This absorbance was then corrected for the weight of the tissue sample, and results are expressed as specific enzyme activity.
Determination of Hepatic GSK3β Kinase Activity
To determine the GSK3β kinase activity in the liver tissue, liver homogenates were made in lysis buffer and analyzed using a colorimetric GSK3β kinase assay kit (Jianglaibio Co, Shanghai) according to manufacturer’s instruction. In the presence of inhibitor GSK3α, peptide substrates are phosphorylated by GSK3β, and further make a catalytic reaction by pyruvate kinase and lactate dehydrogenase, and measure the ADP formed from a kinase reaction which is concomitant with the oxidation reaction of reduced nicotinamide adenine dinucleotide (NADH). The determination of the peak changes after oxidation using spectroscopy to analyze activity of GSK3β kinase.
TUNEL Assay
Apoptosis in liver sections was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL; red fluorescence) using the In Situ Cell Death Detection Kit (Roche, Indianapolis, IN). Negative control was prepared through omission of terminal transferase. Positive controls were generated by treatment with DNase. Nuclei were stained with 49,6-diamino-2-phenylindole (DAPI; 1 μg/ml) for 10 min. Images were performed on a Nikon Eclipse E800 fluorescent microscope.
Isolation and Treatment of Murine Bone Marrow-Derived Macrophages
Murine bone marrow-derived macrophages (BMM) were differentiated from bone marrow from 6- to 10-week-old C57BL/6 mice by culturing in DMEM containing 10 % fetal bovine serum, 1 % penicillin/streptomycin, and 20 % L929-conditioned medium for 6 days. BMMs were stimulated with tunicamycin (TM; 10 μg/ml, Sigma) or SB216763 (10 μM; Sigma, St Louis, MO) and TM for 6 h, followed by 6 or 24 h LPS stimulation.
Isolation and Treatment of Primary Mouse Hepatocytes
The mouse livers were perfused with collagenase-containing Hanks’ solution at 7 weeks of age, and viable hepatocytes were isolated by Percoll isodensity centrifugation as described [24]. To study the effects of ER stress on hepatocyte apoptosis induced by TNF-α (20 ng/ml; Sigma, St Louis, MO) and actinomycin D (ActD; 1 nM, Sigma, St Louis, MO), TM (20 μg/ml) was added 2 h prior to the TNF-α and ActD treatment. Furthermore, the primary hepatocytes also stimulated with TNF-α (20 ng/ml)/ActD (1 nM) in the absence or presence of 4-PBA (1 mM) for 12 h. Cell apoptosis was evaluated at 12 h LDH assays (BioChain Institute, Hayward, CA) and Western blot for cleaved caspase-3 expression.
ELISA for Cytokine Measurements
Mouse TNF-α (eBioscience, San Diego, CA) in the BMM culture supernatants was measured using ELISA kits from eBioscience based on the manufacturer instructions.
Statistical Analysis
The results are shown as the mean ± standard error of the mean (SEM). Kaplan-Meier curves present mouse survival rates; the statistical analyses were performed using an unpaired Student’s t test, and p < 0.05 (two tailed) was defined as significant.
RESULTS
ER Stress Is Triggered in the Liver in Progression of ALF
We first determined whether ALF triggers ER stress in mouse livers followed by various lengths of d-GalN/LPS stimulation. In treatment of mice with d-GalN/LPS for 2, 4, and 6 h, we found that the liver tissue of mice appears spotty hemorrhage at 2 h; the increased inflammation, hepatic lobules disorder, a large number of inflammatory cell infiltration, and visible hepatocyte apoptosis or confluent necrosis are shown at 4 h; there are a lot of visible large necrosis areas in the liver tissue, the entire liver congestion at 6 h (Fig. 1a). There are increased ALT and AST enzyme levels (Fig. 1b). Meanwhile, the levels of Grp78 and Grp94 were increased as the liver injury progressed (Fig. 1c, d). ER stress leads to the activation of IRE1α, PERK, and ATF6 pathways, which in turn initiate UPR, so we tested the activation of UPR in the development of d-GalN/LPS-induced liver injury. The results showed that the levels of p-PERK, p50ATF6, and XBP-1 increased from 2 to 4 h and then restored after 6 h (Fig. 1e). Disruption of ER Ca2+ homeostasis plays an important role in the induction of ER stress and calcium, and ER stress mediates hepatic apoptosis after severe injury [25, 26]. There are multiple different plasma-membrane channels that control Ca2+ influx in response to various stimuli, including inositol 1,4,5-trisphosphate receptors (IP3Rs). The Western blot results showed that the expression of pan-IP3R was increased along with the liver injury progressed (Fig. 1f). Taken together, these results indicated that ER stress is triggered in the progression of d-GalN/LPS-induced liver injury.

ER stress is triggered in the progression of ALF. The mice were injected intraperitoneally with d-GalN (700 mg/kg) and LPS (10 μg/kg) for 2, 4, and 6 h (10 mice/groups); the mice of the control group (n = 8) were injected only PBS and sacrificed at 6 h after PBS injection. a Representative livers and H&E staining of livers from 2, 4, and 6 h control groups and group averages of liver Suzuki scores. b Serum AST and ALT enzyme levels from 2, 4, and 6 h control groups. c Gene expression of Grp78 and Grp94 was measured by qRT-PCR in livers. Average target gene/HPRT ratios for each experimental group were plotted. d Protein expression of Grp78, Grp94, and β-actin was measured by Western blot in livers. A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample. e Protein expression of p-PERK, PERK, XBP-1, p50ATF6, ATF6, and β-actin was measured by Western blot in livers. A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample. f Protein expression of pan-IP3R and β-actin was measured by Western blot in livers. A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample.
Inhibition of ER Stress Ameliorates Hepatotoxicity Induced by d-GalN/LPS
To evaluate the role of ER stress in the pathogenesis of d-GalN/LPS-induced liver injury, we evaluated whether relieving ER stress could rescue hepatotoxicity by applying 4-phenylbutyric acid (4-PBA), which is a small chemical chaperone that has been shown to reduce ER stress both in vivo and in vitro [27, 28]. In our results, inhibition of ER stress in vivo by 4-PBA was confirmed by the reduced levels of Grp78, Grp94, CHOP, and by the increased levels of XBP-1 (Fig. 2a). In the mortality analysis, compared with d-GalN/LPS-treated group, 4 -PBA treatment significantly increased the survival rate from 40 (survival in 6 of 15 mice) to 80 % at 48 h (survival in 12 of 15 mice) (Fig. 2b). For gross morphology of the liver, 4-PBA-treated mice almost appeared substantially the normal liver morphology (Fig. 2c). With respect to liver damage, there was a better preserve of the liver architecture, decreased inflammation, and less necrosis areas (only little spotty necrosis) after 4-PBA treatment (Fig. 2d), and sALT and sAST levels were significantly lower (Fig. 2e). These results indicated that ER stress is critical for d-GalN/LPS-induced liver injury and that its inhibition can protect the liver from injury.

ER stress inhibition by 4-PBA protects against d-GalN/LPS-induced liver injury. 4-PBA/d-GalN/LPS-treated mice were administered with 4-PBA (100 mg/kg) via intraperitoneal injection at 6 h prior to d-GalN/LPS exposure (n = 12); d-GalN/LPS-treated mice were pretreated with vehicle (PBS) at 6 h prior to d-GalN/LPS exposure (n = 12); control mice were pretreated with vehicle (PBS) at 6 h prior to PBS injection (n = 8). a Protein expression of Grp78, Grp94, XBP-1, CHOP, and β-actin was measured by Western blot. A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample. b The survival rate of mice was measured in different groups (15 mice/groups). c Representative livers from the different groups. d Representative H&E staining of livers from the different groups (black arrows indicate confluent necrosis; black triangles indicate spotty necrosis) and group averages of liver Suzuki scores. e Serum AST and ALT enzyme levels from the different groups.
ER Stress Facilitates Cytokine Programs, Neutrophil Infiltration, and Hepatocyte Apoptosis
LPS can trigger inflammatory cascades involving the induction of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6, which are essential for inflammation and consequent liver damage in d-GalN/LPS-treated mice. In order to determine the impact of ER stress inhibition on the induction of these cytokines, we further investigated the inflammatory cytokine profiles in the liver. Indeed, 4-PBA treatment decreased the expression of TNF-α, IL-1β, IL-6 (Fig. 3a), and the injury-related MPO activity (Fig. 3b). These results indicated that 4-PBA can suppress the expression of pro-inflammatory cytokines and reduce neutrophil infiltration in the liver.

ER stress inhibition by 4-PBA decreases pro-inflammatory cytokine expression, neutrophil infiltration, and hepatocyte apoptosis. 4-PBA/d-GalN/LPS-treated mice were administered with 4-PBA (100 mg/kg) via intraperitoneal injection at 6 h prior to d-GalN/LPS exposure (n = 12); d-GalN/LPS-treated mice were pretreated with vehicle (PBS) at 6 h prior to d-GalN/LPS exposure (n = 12); control mice were pretreated with vehicle (PBS) at 6 h prior to PBS injection (n = 8). a Gene expression of TNF-α, IL-6, and IL-1β at 6 h after d-GalN/LPS injection. Average target gene/HPRT ratios for each experimental group were plotted. b Liver MPO levels at 6 h after d-GalN/LPS injection. c Protein expression of caspase-3, cleaved caspase-3, and cleaved PARP was measured in the liver tissue by Western blot. A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample. d TUNEL staining (red) liver tissue at 6 h after d-GalN/LPS administration. Representative of one experiment is shown. Original magnification ×200.
Because of the critical role of hepatocyte apoptosis in the mechanism of ALF, the impact of ER stress inhibition on hepatocyte apoptosis was examined. In animal model, the cleavage of caspase-3 was markedly increased in d-GalN/LPS-induced liver failure, and treatment with 4-PBA significantly inhibited the expression of the cleavage of caspase-3 and the cleavage of PARP (Fig. 3c). Similar results were also observed in liver tissue samples by applying TUNEL assay (Fig. 3d). Therefore, ER stress inhibition is capable of inhibiting hepatocyte apoptosis in hepatic failure induced by d-GalN/LPS.
Inhibition of ER Stress Decreases GSK3β Activity and Regulates the NF-κB, MAPK Pathways in the Liver
Previous studies have shown that the GSK3β participates in the regulation of inflammation in macrophages [16]; the activity of GSK3β is triggered in the progression which promotes the hepatotoxicity induced by d-GalN/LPS [18, 19], so we evaluated whether 4-PBA is able to decrease the activity of GSK3β in the liver of d-GalN/LPS-treated mice. Western blot results showed that 4-PBA increased the phosphorylated GSK3β (at serine 9) level (Fig. 4a), which means that the activity of GSK3β is decreased; furthermore, it was also directly confirmed by detecting the activity of GSK3β in the liver tissues (Fig. 4b). These results indicated that the activity of GSK3β is inhibited by 4-PBA in d-GalN/LPS-induced ALF model.

ER stress inhibition by 4-PBA decreases the activity of GSK3β in the liver. 4-PBA/d-GalN/LPS-treated mice were administered with 4-PBA (100 mg/kg) via intraperitoneal injection at 6 h prior to d-GalN/LPS exposure (n = 12); d-GalN/LPS-treated mice were pretreated with vehicle (PBS) at 6 h prior to d-GalN/LPS exposure (n = 12); control mice were pretreated with vehicle (PBS) at 6 h prior to PBS injection (n = 8). a The expression of p-GSK3β, total GSK3β, and β-actin was measured by Western blots in liver. A representative blot from two samples of every group is shown. After quantification of the signals, results were normalized relative to the total GSK3β expression. Data are presented as the means ± SEM. b The activity of GSK3β in the liver tissue was measured. c The relative proteins of NF-κB and MAPK pathway were measured in the liver tissue by Western blot. A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample.
Nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) are the most important transcription factors involved in inflammatory pathways [29], and thus, we assessed the impact of 4-PBA on these two pathways in the liver. The level of IkB-α was decreased at 6 h after d-GalN/LPS treatment, and 4-PBA treatment increased the expression of IkB-α. Moreover, the levels of the phosphorylation of NF-κBp65, ERK, p38, and JNK were increased in mice treated by d-GalN/LPS, and they were all inhibited by 4-PBA (Fig. 4c). These results indicated that ER stress inhibition protects the liver from inflammatory injury by regulating the NF-κB and MAPK pathways.
ER Stress Synergistically Increases Inflammation in Macrophage Induced by LPS Through GSK3β Pathway In Vitro
To investigate the cellular mechanism of our in vivo findings, we analyzed the effects of ER stress on macrophage response to TLR4 stimulation by LPS in vitro. Bone marrow-derived macrophages (BMM) were stimulated with LPS in the absence or presence of tunicamycin. Remarkably, compared with only LPS treatment, the gene expression of TNF-α, IL-6, and IL-1β was synergistically increased when the cells were simultaneously induced by LPS and tunicamycin (Fig. 5a). We also measured TNF-α expression in cell culture supernatants using ELISA, which gave results similar to the gene transcription assays for TNF-α (Fig. 5b). These results further demonstrated that ER stress synergistically promotes the production of pro-inflammatory cytokines.

ER stress synergistically increases inflammation in macrophage induced by LPS through activating GSK3β. a BMMs were stimulated with tunicamycin (TM; 10 μg/ml) or SB216763 (10 μM) and TM for 6 h, followed by 4 h LPS stimulation. Gene expression of TNF-α, IL-1β, and IL-6 was measured by qRT-PCR. Average target gene/HPRT ratios for each experimental group were plotted. b BMMs were stimulated with TM (10 μg/ml) or SB216763 (10 μM) and TM for 6 h, followed by 12 h LPS stimulation. TNF-α levels were measured by ELISA. c BMMs were stimulated by TM (10 μg/ml) for different times. The cell lysates were analyzed for the phosphorylated (serine 9) and total GSK3β levels by Western blot quantified by densitometry (mean ± SD, n = 3). d BMMs were stimulated by TM with different concentrations for 6 h. The cell lysates were analyzed for the phosphorylated (serine 9) and total GSK3β levels by Western blot quantified by densitometry (mean ± SD, n = 3). e BMMs were stimulated with or without TM for 6 h, followed by LPS for 15 min, 30 min, 1 h, or 2 h stimulation. For only TM stimulation, BMMs were also stimulated by TM for 15 min, 30 min, 1 h, or 2 h stimulation. The cell lysates were analyzed by Western blot in different times.
Next, we evaluated whether ER stress regulates GSK3β activity in macrophages. Western blot results showed that tunicamycin-induced ER stress promoted GSK3β activity (dephosphorylation at serine 9) in a time-dependent and does-dependent manner (Fig. 5c, d). We also examined whether GSK3β was involved in the regulation of the inflammation induced by tunicamycin and LPS. Western blot results showed that, compared with only tunicamycin stimulation, the activity of GSK3β was further upregulated by tunicamycin and LPS (Fig. 5e). Moreover, inhibition of GSK3β by SB216763 attenuated the expression of TNF-α, IL-1β, and IL-6 induced by tunicamycin and LPS (Fig. 5a, b).
Next, in vitro, we assessed the impact of tunicamycin on NF-κB and MAPK pathways in macrophages stimulated by LPS. In the NF-κB pathway, the level of IkB-α was decreased at 30 min after LPS stimulation in macrophages, and tunicamycin cooperated with LPS to significantly reduce the level of IkB-α. In contrast, the phosphorylation of NF-κBp65 increased in tunicamycin/LPS treatment (Fig. 5e). In the MAPK pathway, under LPS signaling, the phosphorylation of ERK, p38, and JNK increased after 30 min and maintained at a significantly high level for 1 h and restored at 2 h; they were significantly magnified by tunicamycin/LPS (Fig. 5e). These results indicated that ER stress and LPS can interact to regulate the NF-κB and MAPK pathways.
ER Stress Synergistically Promotes TNF-α-induced Hepatocyte Apoptosis In Vitro
Given the central role of TNF-α in the mechanisms of ALF [30, 31], we investigated the impact of ER stress on TNF-α-induced hepatocyte apoptosis. Preincubation of Hepa 1 cells with tunicamycin significantly increased cell apoptosis in response to TNF-α/ActD. There was a significant increase in LDH activity in the culture supernatant of hepatocytes pretreated with tunicamycin after incubation with TNF-α/ActD (Fig. 6a). Western blot analysis showed that tunicamycin additionally promoted the cleavage of caspase-3 upon TNF-α/ActD challenge compared with tunicamycin or TNF-α/ActD treatment (Fig. 6b). Moreover, we further investigated the impact of ER stress inhibition on TNF-α-induced hepatocyte apoptosis. Preincubation of primary hepatocytes with 4-PBA significantly decreased cell apoptosis in response to TNF-α/ActD. There was a significant decrease in LDH activity in the culture supernatant of hepatocytes pretreated with 4-PBA after incubation with TNF-α/ActD (Fig. 6c). Western blot analysis showed that 4-PBA downregulated the level of the cleavage of caspase-3, Grp78, and CHOP and decreased the activity of GSK3β upon TNF-α/ActD challenge (Fig. 6d). Therefore, these results indicated that ER stress may sensitize hepatocytes to TNF-α-induced cell apoptosis by mediating the activity of GSK3β.

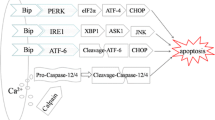
ER stress synergistically promotes TNF-α-induced hepatocyte apoptosis in vitro. a TM (20 μg/ml) was added 2 h before TNF-α (20 ng/ml) and ActD (1 nM) which is added into the 48-well culture plate for 24 h (n = 3 wells). LDH activity assays in the hepatocyte culture supernatant. b TM (20 μg/ml) was added 2 h before TNF-α (20 ng/ml) and ActD (1 nM) which is added into culture plate (diameter 60 mm) for 12 h; cells were harvested and analyzed for expression of caspase-3, cleaved caspase-3, and β-actin by Western blots and quantified by densitometry (mean ± SD, n = 3). c 4-PBA (1 mM) was added 2 h before TNF-α (20 ng/ml) and ActD (1 nM) which is added into the 48-well culture plate for 24 h (n = 3 wells). LDH activity assays in the hepatocyte culture supernatant. d 4-PBA (1 mM) was added 2 h before TNF-α (20 ng/ml) and ActD (1 nM) which is added into culture plate (diameter 60 mm) for 12 h; cells were harvested and analyzed for expression of caspase-3, cleaved caspase-3, Grp78, CHOP, p-GSK3β, total GSK3β, and β-actin by Western blots and quantified by densitometry (mean ± SD, n = 3). e In the progression of d-GalN/LPS-induced ALF mice model, ER stress is triggered which increases the activity of GSK3β, promotes the expression of pro-inflammatory cytokines and incremental neutrophil infiltration in the liver. These events ultimately induce liver inflammation, further leading to induce hepatocyte apoptosis and accelerate the development of ALF. Red arrows indicate 4-phenylbutyric acid (4-PBA), which is a small chemical chaperone to relieve ER stress, and induced changes in ALF mice relative to their DMSO-injected counterparts.
DISCUSSION
Our current study first demonstrated that ER stress plays a critical role in the mechanism of ALF induced by d-GalN/LPS. Furthermore, we showed that ER stress is required for the development of hepatocellular damage through synergistic promotion of local pro-inflammatory responses. Importantly, the present study highlights a novel role of GSK3β in modulating the ER stress to regulate the inflammatory mechanism in ALF. Therefore, our findings suggest that ER stress exacerbates liver inflammation and hepatotoxicity induced by d-GalN/LPS via activating GSK3β.
ALF is associated with various inflammatory cells, including Kupffer cells (macrophage in the liver), neutrophils, and monocytes, which are recruited and activated in the stressed liver [32, 33]. Kupffer cell not only contributes to the early phase of the disease but also results in sustained inflammation [34, 35]. Moreover, inflammatory immune-mediated liver injury is a consequence of the recruitment of effector leukocytes to the liver where they mediate tissue damage; leukocyte migration from the vascular lumen into the surrounding extravascular tissue makes a significant contribution to liver injury in ALF [36, 37]. Our study showed evidences that ER stress promotes the inflammatory response to aggravate liver damage in ALF. In vivo, ER stress inhibition significantly suppressed liver inflammation; in vitro, ER stress sensitized macrophages to LPS, triggering a hundredfold increase in pro-inflammatory cytokine expression. We also showed that ER stress significantly enhanced the LPS-mediated activation of MAPK and NF-κB in macrophages, which fully illustrated the molecular mechanism about how ER stress synergistically promotes inflammatory response. Moreover, ER stress significantly increased expression of TNF-α in vivo and in vitro, and ER stress synergistically promoted the hepatocyte apoptosis induced by TNF-α. Therefore, we concluded that, on the one hand, the sustained presence of ER stress results in abundant expression of inflammatory cytokines in the liver, further promoting the infiltration of neutrophils into the liver and aggravating the liver inflammation; on the other hand, ER stress cooperates with TNF-α to synergistically promote hepatocyte apoptosis, which eventually leads to further trigger aggravation of liver injury. Therefore, ER stress appears to play the role of a catalyst to amplify the inflammatory response and promote hepatocyte apoptosis in ALF.
Originally identified in mammals as a cytoplasmic modulator of glycogen metabolism, GSK3β is now recognized as a central regulator of cellular events, including cell fate determination, microtubule function, cell cycle regulation, apoptosis, and inflammatory responses [38–42]. Recently, GSK3β plays an important role in the inflammation. Martin et al. firstly demonstrated that toll-like receptor-mediated cytokine production is differentially regulated by GSK3β: GSK3β regulates a multitude of transcription factors, including NF-κB, AP-1, NF-AT, and CREB; inhibiting GSK3β allows CREB to sequester CBP from NF-κB, promoting CREB-dependent IL-10 production and suppressing NF-κB-dependent inflammatory cytokine expression [16]. Next to that, Wang et al. further tested that IFN-β production by TLR4-stimulated innate immune cells is negatively regulated by GSK3β [43]. So, GSK3β is an important positive regulator in inflammatory process. Furthermore, accumulating evidences has suggested that GSK3β participates in the ER stress-induced signaling pathway and is involved in ER stress-induced apoptosis [44–46], and our previous study has shown that the increased activity of GSK3β promotes acute liver injury and GSK3β inhibition relieves ER stress in ALF mice induced by d-GalN/LPS [18]. Here, our results directly proved that ER stress promotes GSK3β activity in the progression of ALF in vivo and in the primary macrophage in vitro; furthermore, inhibition of GSK3β significantly attenuates the expression of pro-inflammatory cytokines triggered by ER stress/LPS in macrophage. Thus, we can conclude that, in the progression of ALF, d-GalN/LPS treatment triggers ER stress which can increase the activity of GSK3β and promote the expression of pro-inflammatory cytokines and incremental neutrophil infiltration in the liver; these events ultimately induce liver inflammation, further leading to induce hepatocyte apoptosis and accelerate the development of ALF (as depicted in Fig 6e).
Our prior work demonstrated that the activity of GSK3β is increased and promotes the hepatotoxicity in ALF induced by d-GalN/LPS [18], but Hoeflich et al. showed that GSK3β protects hepatocytes from TNF-α induced-apoptotic cell death in mid gestation [39]. How then would that activity be reconciled with the proposed pro-apoptotic role of GSK3β in ALF model? As we know, GSK3β, constitutively active (in dephosphorylated form) in resting cells, has a broad range of substrates regulating cell activation, differentiation, and survival. In the paper of Hoeflich, compared to normal mice, the GSK3β activity is depressed by disruption of the murine GSK3β gene or lithium treatment on the basis of resting cells, which made the fibroblasts from GSK3β-deficient embryos hypersensitive to TNF-α, so GSK3β protects hepatocytes from TNF-α-induced apoptotic cell death. In our paper [18], compared to normal mice, d-GalN/LPS increased the GSK3β activity during ALF, so inhibition of GSK3β ameliorates liver injury. So, we speculated that the GSK3β activity of resting cells is a balance adjustment points; above or below the constitutive activity of resting cells, GSK3β might play differential roles. The study of Peiyong et al. showed the differential roles of GSK3β during myocardial ischemia and ischemia/reperfusion by mediating autophagy [47]. Autophagy is a protective mechanism against injury, and our study also showed that autophagy can regulate the inflammatory response to protect the liver from injury in d-GalN/LPS-induced ALF [48]. Taken together, we proposed the hypothesis that below the constitutive activity of resting cells, GSK3β can increase autophagy to protect hepatocytes from apoptotic cell death; otherwise, above the constitutive activity of resting cells, GSK3β can decrease autophagy to promote apoptotic cell death. At present, this hypothesis is under exploration.
In summary, ER stress plays a key role in the inflammatory immune modulatory mechanism of ALF, especially through the regulation of GSK3β. ER stress inhibition represents a potent strategy to ameliorate the liver pathology of ALF through immunomodulation to reduce local inflammation. Further preclinical studies with ER stress inhibitors are warranted for the development of a clinically applicable therapeutic strategy against ALF.
Abbreviations
- ALF:
-
Acute liver failure
- ER:
-
Endoplasmic reticulum
- ER stress:
-
Endoplasmic reticulum stress
- GSK3β:
-
Glycogen synthase kinase 3β
- d-GalN:
-
d-galactosamine
- LPS:
-
Lipopolysaccharide
- UPR:
-
Unfolded protein response
- Grp78:
-
Glucose-regulated protein 78
- Grp94:
-
Glucose-regulated protein 94
- HPRT:
-
Hypoxanthine-guanine phosphoribosyltransferase
- IP3R: inositol 1:
-
4, 5-trisphosphate receptors
- PARP:
-
Poly ADP ribose polymerase
- NF-κB:
-
Nuclear factor-κB
- MAPK:
-
Mitogen-activated protein kinase
- IkB-α:
-
IkappaB-alpha
- TNF-α:
-
Tumor necrosis factor-α
- IL-1β:
-
Interleukin1β
- IL-6:
-
Interleukin6
- 4-PBA:
-
4-phenylbutyrate
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- MPO:
-
Myeloperoxidase
- ELISA:
-
Enzyme-linked immunosorbent assay
- TLR4:
-
Toll-like receptor 4
- BMM:
-
Bone marrow-derived macrophage
- PAGE:
-
Polyacrylamide gel electrophoresis
- TM:
-
Tunicamycin
References
Hoofnagle, J.H., R.L.J. Carithers, C. Shapiro, and N. Ascher. 1995. Acute hepatic failure: summary of a workshop. Hepatology 21: 240–252.
Riordan, S.M., and R. Williams. 2003. Mechanisms of hepatocyte injury, multiorgan failure, and prognostic criteria in acute liver failure. Seminars in Liver Disease 23: 203–215.
Kaufman, R.J. 1999. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes and Development 13: 1211–1233.
Ron, D., and P. Walter. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology 8: 519–529.
Todd, D.J., A.H. Lee, and L.H. Glimcher. 2008. The ERS response in immunity and autoimmunity. Nature Reviews Immunology 8: 663–674.
Lin, J.H., P. Walter, and T.S. Yen. 2008. Endoplasmic reticulum stress in disease pathogenesis. Annual Review of Pathology 3: 399–425.
Little, E., M. Ramakrishnan, B. Roy, G. Gazit, and A.S. Lee. 1994. The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications. Critical Reviews in Eukaryotic Gene Expression 4: 1–18.
Xu, C., M.B. Bailly, and J.C. Reed. 2005. Endoplasmic reticulum stress: cell life and death decisions. Journal of Clinical Investigation 115: 2656–2664.
Inki, K., J.X. Wen, and C.R. John. 2008. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature Reviews Drug Discovery 7: 1013–30.
Zhang, K., and R.J. Kaufman. 2008. From endoplasmic-reticulum stress to the inflammatory response. Nature 24: 455–62.
Nürnberger, S., I. Miller, J.C. Duvigneau, E.T. Kavanagh, S. Gupta, R.T. Hartl, et al. 2012. Impairment of endoplasmic reticulum in liver as an early consequence of the systemic inflammatory response in rats. American Journal of Physiology - Gastrointestinal and Liver Physiology 303: G1373–1383.
Liu, J., R. Feng, Q. Cheng, L. Bai, X. Shen, F. Gao, et al. 2012. Endoplasmic reticulum stress modulates liver inflammatory immune response in the pathogenesis of liver ischemia and reperfusion injury. Transplantation 94: 211–217.
Martinon, F., X. Chen, A.H. Lee, and L.H. Glimcher. 2010. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature Immunology 11: 411–418.
Smith, J.A., M.J. Turner, M.L. DeLay, E.I. Klenk, D.P. Sowders, and R.A. Colbert. 2008. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. European Journal of Immunology 38: 1194–1203.
Zeng, L., Y.P. Liu, H. Sha, H. Chen, L. Qi, and J.A. Smith. 2010. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. Journal of Immunology 185: 2324–2330.
Martin, M., K. Rehani, R.S. Jope, and S.M. Michalek. 2005. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nature Immunology 6: 777–784.
Beurel, Eléonore, and Richard S. Jope. 2006. The paradoxical pro- and anti-apoptotic actions of GSK-3 in the intrinsic and extrinsic apoptosis signaling pathways. Progress in Neurobiology 79: 173–189.
Chen, L., F. Ren, H. Zhang, T. Wen, Z. Piao, L. Zhou, et al. 2012. Inhibition of GSK3β ameliorates D-GalN/LPS-induced liver injury by reducing ERS-triggered apoptosis. Plos One 7: e45202.
Linlin, W., R. Feng, Z. Xiangying, Wen Tao, Shi Hongbo, Zheng Sujun, et al. 2014. Oxidative stress promotes D-GalN/LPS-induced acute hepatotoxicity by increasing glycogen synthase kinase 3β activity. Inflammation Research 63: 485–494.
Tabas, I., and D. Ron. 2011. Integrating the mechanisms of apoptosis induced by ERS. Nature Cell Biology 13: 184–190.
Mignon, A., N. Rouquet, M. Fabre, S. Martin, J.C. Pagès, J.F. Dhainaut, et al. 1999. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. American Journal of Respiratory and Critical Care Medicine 159: 1308–1315.
Nakama, T., S. Hirono, A. Moriuchi, S. Hasuike, K. Nagata, T. Hori, et al. 2001. Etoposide prevents apoptosis in mouse liver with D-GalN/LPS-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology 33: 1441–1450.
Suzuki, S., L.H. Toledo-Pereyra, F.J. Rodriguez, and D. Cejalvo. 1993. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation 55: 1265–1272.
Klaunig, J.E., P.J. Goldblatt, D.E. Hinton, M.M. Lipsky, J. Chacko, and B.F. Trump. 1981. Mouse liver cell culture. I. Hepatocyte isolation. In Vitro 17: 913–925.
Jeschke, M.G., C.C. Finnerty, D.N. Herndon, J. Song, D. Boehning, R.G. Tompkins, et al. 2012. Severe injury is associated with insulin resistance, endoplasmic reticulum stress response, and unfolded protein response. Annals of Surgery 255: 370–378.
Jeschke, M.G., G.G. Gauglitz, J. Song, G.A. Kulp, C.C. Finnerty, R.A. Cox, et al. 2009. Calcium and ER stress mediate hepatic apoptosis after burn injury. Journal of Cellular and Molecular Medicine 13: 1857–1865.
Ozcan, U., E. Yilmaz, L. Ozcan, M. Furuhashi, E. Vaillancourt, R.O. Smith, et al. 2006. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313: 1137–1140.
Zode, G.S., M.H. Kuehn, D.Y. Nishimura, C.C. Searby, K. Mohan, S.D. Grozdanic, et al. 2011. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. Journal of Clinical Investigation 121: 3542–3553.
Gyongyi, S., M. Pranoti, and D. Angela. 2007. Innate immune response and hepatic inflammation. Seminars in Liver Disease 27: 339–350.
Streetz, K., L. Leifeld, D. Grundmann, J. Ramakers, K. Eckert, U. Spengler, et al. 2000. Tumor necrosis factor-alpha in the pathogenesis of human and murine fulminant hepatic failure. Gastroenterology 119: 446–460.
Bradham, C.A., J. Plümpe, M.P. Manns, D.A. Brenner, and C. Trautwein. 1998. Mechanisms of hepatic toxicity I: TNF-induced liver injury. American Journal of Physiology 275: G387–392.
Zimmermann, H.W., C. Trautwein, and F. Tacke. 2012. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Frontiers in Physiology 3: 56. doi:10.3389/fphys.2012.00056.
Wagner, J.G., and R.A. Roth. 1999. Neutrophil migration during endotoxemia. Journal of Leukocyte Biology 66: 10–24.
Su, G.L. 2002. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. American Journal of Physiology - Gastrointestinal and Liver Physiology 283: G256–265.
Matsuno, K., H. Nomiyama, H. Yoneyama, and R. Uwatoku. 2002. Kupffer cell-mediated recruitment of dendritic cells to the liver crucial for a host defense. Developmental Immunology 9: 143–149.
Klugewitz, K., D.H. Adams, M. Emoto, K. Eulenburg, and A. Hamann. 2004. The composition of intrahepatic lymphocytes: shaped by selective recruitment? Trends in Immunology 25: 590–594.
Bertus, E., C.A. Simon, J.W. Stephen, A.P. Holt, and D.H. Adams. 2007. Immune-mediated liver injury. Seminars in Liver Disease 27: 351–366.
Beurel, E., S.M. Michalek, and R.S. Jope. 2010. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends in Immunology 31: 24–31.
Hoeflich, K.P., J. Luo, E.A. Rubie, M.S. Tsao, O. Jin, and J.R. Woodgett. 2000. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406: 86–90.
Cao, J., X.X. Feng, L. Yao, B. Ning, Z.X. Yang, D.L. Fang, et al. 2014. Saturated free fatty acid sodium palmitate-induced lipoapoptosis by targeting glycogen synthase kinase-3β activation in human liver cells. Digestive Diseases and Sciences 59: 346–357.
Choi, S.E., Y. Kang, H.J. Jang, H.C. Shin, H.E. Kim, H.S. Kim, et al. 2007. Involvement of glycogen synthase kinase-3beta in palmitate-induced human umbilical vein endothelial cell apoptosis. Journal of Vascular Research 44: 365–374.
Shinohara, M., M.D. Ybanez, S. Win, T.A. Than, S. Jain, W.A. Gaarde, et al. 2010. Silencing glycogen synthase kinase-3beta inhibits acetaminophen hepatotoxicity and attenuates JNK activation and loss of glutamate cysteine ligase and myeloid cell leukemia sequence 1. Journal of Biological Chemistry 285: 8244–8255.
Wang, H., C.A. Garcia, K. Rehani, C. Cekic, P. Alard, D.F. Kinane, et al. 2008. IFN-beta production by TLR4-stimulated innate immune cells is negatively regulated by GSK3β. Journal of Immunology 181: 6797–6802.
Kim, A.J., Y. Shi, R.C. Austin, and G.H. Werstuck. 2005. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. Journal of Cell Science 118: 89–99.
Srinivasan, S., M. Ohsugi, Z. Liu, S. Fatrai, E. Bernal-Mizrachi, and M.A. Permutt. 2005. ERS-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-Kinase/Akt and increased GSK3β in mouse insulinoma cells. Diabetes 54: 968–975.
Takadera, T., R. Yoshikawa, and T. Ohyashiki. 2006. Thapsigargin-induced apoptosis was prevented by GSK3 inhibitors in PC12 cells. Neuroscience Letters 408: 124–128.
Zhai, P., S. Sciarretta, J. Galeotti, M. Volpe, and J. Sadoshima. 2011. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circulation Research 109: 502–511.
Mingjing, J., R. Feng, Z. Li, Z. Xiangying, Z. Li, W. Tao, et al. 2014. Peroxisome proliferator-activated receptor α activation attenuates inflammatory response to protect liver from acute failure by promoting autophagy pathway. Cell Death & Disease 5: e1397. doi:10.1038/cddis.2014.361.
Acknowledgments
This work was supported by the China National Key Project of the Twelfth Five-year Plan (2012ZX10002004-006, 2012ZX10004904-003-001, 2013ZX10002002-006-001, 2012ZX10002005-003-003), the National Natural Science Foundation of China (81270532, 81372094,81300349), the Beijing Excellent Talents Training Funding (2011D003034000022), the Technology Foundation for Selected Overseas Chinese Scholar, the Ministry of Personnel of Beijing (2012), the Applied Research for the Clinical Characteristics of Capital (Z1211070010112167), and the Cooperation Research Project of CMU and Clinical (13JL33).
Conflict of Interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Feng Ren and Li Zhou have contributed equally to this study.
Rights and permissions
About this article

Cite this article
Ren, F., Zhou, L., Zhang, X. et al. Endoplasmic Reticulum Stress-Activated Glycogen Synthase Kinase 3β Aggravates Liver Inflammation and Hepatotoxicity in Mice with Acute Liver Failure. Inflammation 38, 1151–1165 (2015). https://doi.org/10.1007/s10753-014-0080-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-014-0080-2