
Abstract
Idiopathic nephrotic syndrome (INS) is a multifactorial disease, characterized by proteinuria, hypoalbuminemia, edema and hyperlipidemia. Studies in humans and animal models have associated INS with changes in the immune response. The purpose of this article is to review clinical and experimental findings showing the involvement of the immune response in the pathogenesis of INS. The role of the immune system in INS has been shown by clinical and experimental studies. However, the pattern of immune response in patients with INS is still not clearly defined. Many studies show changes in the dynamics of T lymphocytes, especially the regulatory T cells. Alternatively, there are other reports regarding the involvement of the complement system and B lymphocytes in the pathophysiology of INS. Indeed, none of the immunological biomarkers evaluated were undeniably linked to changes in glomerular permeability and proteinuria. On the other hand, some studies suggest a link between urinary chemokines, such as IL-8/CXCL8 and MCP-1/CCL2, and changes in glomerular permeability and/or the deterioration of glomerulopathies. To understand the pathophysiology of INS, longitudinal studies are clearly needed. The characterization of the profile of the immune response might help the development of specific and individualized therapies, leading to clinical improvement and better prognosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nephrotic syndrome (NS) is a fairly common glomerulopathy in children and adults [1, 2], characterized by massive proteinuria, hypoalbuminemia, generalized edema and hyperlipidemia. NS can be caused by a primary renal lesion or can be associated with systemic diseases [1]. The term idiopathic nephrotic syndrome (INS) refers to the condition caused by a primary renal lesion, in which kidney histology can reveal minimal podocyte changes (MCNS) or focal and segmental glomerulosclerosis (FSGS) [1, 3]. Due to the clinical relevance of the response to corticosteroid treatment, INS can also be classified as steroid-sensitive nephrotic syndrome (SSNS), steroid-dependent nephrotic syndrome and steroid-resistant nephrotic syndrome [1].
Some studies suggest an important role of the immune system in triggering or maintaining INS, such as the abnormal response of T lymphocytes [4, 5] and increased local cytokine release [6]. In addition, the existence of a circulating factor that increases glomerular permeability factor was postulated many years ago [7]. This hypothetical factor might be related to recurrent disease after renal transplantation [8] and the clinical improvement observed after treatment with steroids and immunosuppressant drugs [1, 2].
Despite advances in INS studies in recent decades, especially in the field of immunology, the pathophysiology of this disease remains unknown [3]. The aim of this paper is to review clinical and experimental studies showing the participation of the immune system in INS pathophysiology.
Immune system and nephrotic syndrome
Historical aspects
The first report of immune dysfunction in patients with NS was probably in 1956 by Gitlin et al. [9], who showed an increased synthesis of gamma-globulins in children with INS. Some years later, in the 70s and 80s, several researchers focused studies of NS on the role of the immune system, especially searching for changes in T lymphocytes and immunoglobulins (Ig) [4, 9–13]. Some studies reported increases in serum IgM [12] and reduction of IgG in patients with NS [11, 12, 14]. These findings were subsequently confirmed in an animal model of NS induced by puromycin aminonucleoside [15].
In 1974, Shalboub et al. [7] hypothesized that a change in T cells would result in the production of toxic circulating lymphokines, being responsible for the disorder known as lipoid nephrosis. In 1976, Moorthy et al. [16] demonstrated that plasma from patients with lipoid nephrosis inhibited lymphocyte blastogenic response to the phytohemagglutinin mitogen. Lipoid nephrosis was later named MCNS and the toxic lymphokine as a vascular permeability factor [17, 18]. In initial studies, the culture supernatant of leukocytes from patients with INS induced proteinuria in rats [10, 13] and the substance present in this supernatant, described as a lymphokine and called skin reactive factor [19], caused a reduction in anionic sites of the glomerular basal membrane leading to proteinuria [13, 20].
Renal inflammatory infiltrate in the pathogenesis of INS
In NS, an early tubulointerstitial inflammatory infiltrate of mononuclear cells, predominantly monocytes/macrophages and T lymphocytes, was observed [21–23]. The intensity of the inflammatory infiltrate is associated with a reduction in glomerular filtration, protein deposition in the extracellular matrix, scar tissue formation and subsequent interstitial sclerosis [21, 24]. Children with FSGS had more lymphocytes and macrophages in renal tissue than those with MCNS [25]. Tubular epithelial cells can act as antigen-presenting cells and stimulate T lymphocytes, thereby perpetuating the inflammatory process [21].
The glomeruli of rats with experimentally induced NS showed higher expression of B7-1 (CD80), a co-stimulatory molecule generally present on the surface of B lymphocytes and antigen-presenting cells [26]. The renal tissue of animals with NS induced by doxorubicin revealed initial interstitial accumulation of macrophages [27–29], with subsequent reduction of them [28, 30] and an increase in the number of TCD4+ and TCD8+ cells [30], with a predominance of TCD4+ [31]. The infiltration of monocytes/macrophages in the renal tissue was related to the increased expression of adhesion molecules like ICAM-1 [32]. CD25+ lymphocytes were also observed in the renal interstitium of animals with NS induced by doxorubicin [29]. Although there are reports of the presence of macrophages also in the glomerulus of animals with NS [32], most studies have not detected glomerular macrophage infiltration [23, 28, 30, 33].
Tissue factors determine the phenotype of monocytes/macrophages recruited into the renal tissue, whereas the profile of locally released cytokines regulates the differentiation of mononuclear cells. Th1-type cytokines induce differentiation into classical macrophages, denominated M-1, that produce cytotoxic and proinflammatory cytokines. In contrast, Th2-type cytokines induce alternative macrophages, denominated M-2, responsible for the synthesis of anti-inflammatory cytokines [23, 34–36]. Macrophages from control mice cultured in the presence of interleukin (IL)-4 and IL-13 induced the M-2 phenotype, while the addition of lipopolysaccharides resulted in the M-1 phenotype. When M-2 cells were injected into mice with NS, the amount of native renal macrophages (M-1) reduced, as well as the biochemical and histological alterations of the disease. Alternatively, injection of M-1 cells worsened the experimental NS [36]. More recently, it was shown that M-2 macrophages originating from the action of IL-10 and TGF-β also inhibited M-1 macrophages and TCD4+ and TCD8+ lymphocytes. In addition, this cell line also induced the differentiation of regulatory T cells at the renal interstitium of rats with NS induced by doxorubicin, with consequent improvement of the disease [23].
T lymphocytes in the pathogenesis of INS
Initial studies suggested the existence of a serum factor responsible for the reduction in T lymphocyte function in individuals with INS [4, 16, 37, 38], as well as changes in the percentages of TCD4+ and TCD8+ lymphocytes [38]. In 1985, Schnaper and Aune [37] postulated the existence of a soluble factor in urine and serum of children with MCNS, called soluble immune response suppressor. Other studies showed changes in the relationship between subpopulations of T lymphocytes TCD4+/TCD8+ in the peripheral blood of patients with NS, with lower TCD4+ cell activity [12, 39, 40] and increased amounts of TCD8+ lymphocytes [39] and natural killer (NK) cells [40]. In an animal model of NS, the depletion of TCD4+ cells induced an increase in TCD8+ lymphocytes and macrophages in renal tissue and worsened the disease, thus suggesting a protective function for TCD4+ cells [41]. In a parallel study, the depletion of TCD8+ lymphocytes, also in an NS animal model, significantly reduced renal injury and the number of tissue macrophages, pointing to a deleterious role for this cell line [42].
Tejani et al. [43] provided the initial evidence for the participation of T lymphocytes in the pathogenesis of INS [43]. These authors showed that the INS remission obtained with cyclosporin-A treatment was associated with a reduction in IL-2 levels, a cytokine mainly produced by T lymphocytes [43]. Indeed, cyclosporin A is the first choice for the treatment of SSNS [7].
More recent reports also showed changes in the response of T lymphocytes in INS [44, 45]. For instance, Okuyama et al. [44] evaluated peripheral blood mononuclear cells (PBMC) from patients with INS and detected high mRNA expression for the apoptotic protein TRAIL, also suggesting an altered function of T lymphocytes. Musial et al. [45] detected a reduction in zeta chain expression on NK cells in the peripheral blood of children with INS and also in TCD8+ lymphocytes stimulated in culture with anti-CD3/IL-2r. The zeta chain is a complex component, important for the activation and proliferation of NK cells by co-stimulation. The decrease in zeta chain expression may be due to increased degradation of this component in response to hyperactivity of NK cells [45].
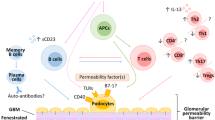
INS severity is associated with decreased activity of regulatory T cells [6, 18, 23, 46–48]. The primary cell with immunosuppressive capacity is the natural regulator T lymphocyte, a TCD4+ lymphocyte differentiated in the thymus and with a constitutive expression of CD25 at high concentrations (CD4+CD25+) [49]. The induction of regulatory T cells occurs in the peripheral circulation from TCD4+ naïves under the action of antigenic stimuli [49] or immunosuppressive cytokines, such as TGF-β [50] (Fig. 1). Regulatory cells are subdivided into Th3 (TGF-β-producing cells) and Tr1 (IL-10- and IFN-α-producing cells) [51]. The transcription factor FOXP3 acts in the conversion of naïve TCD4 cells into regulatory TCD4 [50, 51]. Other cell lines such as regulatory TCD8+ lymphocytes and regulatory NK may also exert immunosuppressive effects (for details, see review of Le and Chao [51]). Figure 1 shows the immunosuppression exerted by regulatory T cells.

T regulatory cells’ suppressive function, modified from Wang et al. [47]. Mechanisms and pathways elicited by T-regulatory cells; Th1 and Th2 response related to renal inflammation, cell infiltration, proteinuria, glomerulosclerosis and tissue injury. IL interleukin, LTCD4 + lymphocytes TCD4+, Th T-helper response, TGF-β transforming growth factor beta
Wang et al. [41] first described the role of TCD4+ cells in the pathophysiology of experimental NS. These authors observed that the depletion of TCD4+ cells worsened the disease, thus suggesting a protective function for these lymphocytes. However, this study did not define which subclass of TCD4+ lymphocytes exerts protective actions [41]. Subsequently, the same research group reported that viral transfection of FOXP3 in TCD4+CD25+ cells generated regulatory T cells with high levels of FOXP3, TGF-β and CTLA-4 (TCD4+CD25+ cells), which inhibited in vitro proliferation. The injection of these regulatory cells in mice with NS induced by doxorubicin improved biochemical alterations and kidney histological injury [52]. Furthermore, TCD4+CD25+ lymphocytes inhibited the production of TNF-α, IL-12, MIP-1α/CCL3 and iNOS in culture of macrophages culture [53]. This action was dependent on TGF-β release, but not on IL-10 [53]. TCD4+CD25+ lymphocytes reduced the amount of macrophages in the kidney interstitium of animals with NS induced by doxorubicin and increased local expression of FOXP3 [53]. The improvement of renal tissue injury was positively correlated with the levels of FOXP3 and dependent on the presence of TGF-β [53]. Pediatric patients with INS also showed a reduction in the amount of FOXP3 in renal tissue [25].
In doxorubicin-induced NS, the animals presented an increase in renal expression of TGF-β by T cells γδ, a T-cell subset whose receptor is formed by γ and δ subunits. The depletion of these cells worsened the disease, thus suggesting that production of TGF-β by T cells γδ would be related to the activation of regulatory T cells [46]. Regulatory T cells (CD4+CD25+FOXP3+) of patients with MCNS had a poorer suppressive capacity on the proliferation of effector T cells (CD4+FOXP3+CD25−), when cultured together. Although there was no reduction in the number of regulatory T cells, or IL-2 or TGF-β levels, IL-10 was reduced in these patients [5]. In the study by Shao et al. [54], mononuclear cells in peripheral blood of children with INS exhibited a reduction in regulatory T cells parallel to an increase in Th17. The elevated Th17/regulatory-T ratio might induce local tissue inflammation and contribute to renal injury, proteinuria and progression of INS [54]. Recently, the increased Th17 response was also demonstrated in experimental NS induced by doxorubicin [55].
Some parallel observations have supported the hypothesis that changes in the balance and/or the functional profile of regulatory T cells may contribute to INS. As an example, NS is very frequent in patients with X-linked syndrome (IPEX syndrome), a genetic disorder caused by FOXP3 gene mutation, which in turn impairs the production of regulatory T lymphocytes (CD4+CD25+) [18, 48].
According to Sellier-Leclerc et al. [56], the pathogenesis of INS is linked more closely with the performance of immature cells than with the peripheral differentiation of T cells. These authors reported that the injection of immature stem cells (CD34+) and peripheral blood mononuclear cells (CD34−), collected from patients with INS, in immunodeficient mice induced the appearance of CD45+ cells (human leukocytes) in peripheral blood of these animals. However, only those animals that received stem cells (CD34+) developed proteinuria.
B lymphocytes and the complement system in the pathogenesis of INS
Few studies have investigated the role of B lymphocytes in INS. Pioneering studies showed an increase in the number of these cells in the active phase of the disease [14, 38, 67]. Recent studies have shown the participation of B lymphocytes in the INS, either directly by histological [25] and hematological analysis [40], or indirectly by the therapeutic effect of the inhibition of B lymphocytes by the administration of rituximab in patients with INS [25, 68–70]. In patients with FSGS, treatment with rituximab reduced proteinuria and the number of B lymphocytes in peripheral blood [68]. Elevated serum levels of soluble IgE receptor (sCD23) and high serum and urinary levels of the soluble receptor of IL-2 (sCD25) during relapse in SSNS suggest simultaneous abnormalities in T and B lymphocytes [60]. On the other hand, in experimental NS induced by doxorubicin, B lymphocytes were not detected in renal tissue [30].
The complement system consists of a group of over 30 plasma proteins, which are soluble or attached to surface proteins. The activation of each complement component promotes a cascade of sequential reactions with anaphytoxin production (C3a and C5a), potent chemotaxis for neutrophils and monocytes. The final result of complement activation is the formation of the membrane attack complex (MAC; C5b-9), also called terminal complement complex, responsible for pore formation in the membrane, resulting in cell lysis [71].
The complement system is related to inflammatory changes in renal tissue. Activation of this system in vitro induced MAC formation in human proximal-tubule epithelial cell surface in addition to the production of IL-6 and TNF-α cytokines, an effect dependent on C6 component [72]. Glomerular ultrafiltration of complement system components and intratubular MAC formation are related to peritubular myofibroblast accumulation in rats with NS induced by doxorubicin [74]. These events depended on the action of C6 component of the complement system. In this experimental model, the blockade of MAC formation improved the animals [74].
The proximal tubule cell surface exhibits C3 convertase activity [72]. Therefore, alterations in the glomerular filtration barrier and the presence of complement system proteins in the tubular filtrate induce activation of these proteins and subsequent MAC formation [72]. In an animal model of non-proteinuric chronic renal disease, MAC did not influence the development of tubulointerstitial injury, thus confirming that the presence of proteins in the tubular lumen is critical for renal injury [75]. In general, the reports concerning the activation of the complement system in NS refer to components located at tubular and peritubular sites [73, 76].
Different animal models of NS or glomerulonephritis have clearly shown the pathophysiological role of the complement system [73, 76–78]. In the NS model induced by protein overload, C3 component deposits and MAC formation were detected on the proximal-tubule cell luminal surface [76]. The absence of the C6 component protected the animal against peritubular myofibroblast accumulation, but did not alter the renal function decline. This finding suggests the participation of other factors in tubule-interstitial injury [73]. In animal models of glomerulonephritis, the blockade of CD59, a regulatory protein that inhibits the ultimate MAC formation, caused glomerular macrophage accumulation, increased MAC formation, increased adhesion molecule expression (ICAM-1) and increased fibrin deposition in glomeruli [79]. Similar effects have been obtained by the induction of glomerulonephritis in mice with genetic deletion of the CD59a gene [78, 80]. The induction of immune-mediated glomerulonephritis in knockout mice for another regulatory factor of the complement system, CD55, increased renal sclerosis and produced biochemical changes typical of NS [77]. In doxorubicin-induced nephropathy, the alternative pathway of the complement system is activated [78].
Cytokines, chemokines and circulating factors in the pathogenesis of INS
Cytokines are soluble proteins, with low molecular weight, secreted by leukocytes and other cells of the organism, which act as messengers of the immune system. Chemokines, or chemoattractant cytokines, are a class of cytokines responsible for leukocyte basal and inflammatory traffic control by chemotaxis [81]. To date, approximately 50 chemokines and 20 receptors have been described in humans [81]. Chemokines are divided into four families based on differences in structure and function. The largest family comprises CC chemokines, so named because the first two cysteine residues are adjacent to each other, which are primarily involved in the attraction of mononuclear cells to sites of chronic inflammation. The CXC family, in which the first two cysteine residues are separated by a single amino acid, consists of two subfamilies based on the presence of a characteristic glutamic acid–leucine–arginine (ELR) motif near the N terminal of the molecule. ELR(+) CXC chemokines, of which CXCL8/IL-8 (IUPHAR nomenclature/original name) is the prototype molecule, attract polymorphonuclear leukocytes to sites of acute inflammation [81]. Conversely ELR(−) CXC chemokines, like CXCL9/MIG, CXCL10/IP-10 and CXCL11/I-TAC, are IFN-γ-inducible chemokines, being involved in the recruitment of Th1 lymphocytes, among other cell types. The remaining chemokine families comprise CX3C (with three amino acids separating the first two cysteine residues) with a single member CX3CL1/fractalkine, and XC (with a single cysteine residue) with XCL1/lymphotactin and XCL2 [81].
Some studies have shown the involvement of cytokines/chemokines in the inflammatory process responsible for the progression of chronic renal disease and renal transplant rejection (for review, see refs. [82] and [83]). It was also suggested that the cytokines/chemokines might act as biomarkers of renal disease progression [84] and as predictors of graft function in renal transplantation [85].
Different cytokines/chemokines were measured in patients with INS or in animal models of the disease (Tables 1, 2). Some of these cytokines/chemokines were correlated with proteinuria and suggested as candidates for glomerular permeability factors responsible for proteinuria in patients with INS or in animal models of the disease, such as IL-1 [63], IL-6 [66] and IL-8/CXCL8 [86, 87]. Another candidate for glomerular permeability factor is vascular endothelial growth factor (VEGF). Since VEGF was able to induce fenestrations in capillaries not normally fenestrated [88] and was constitutively present in peritubular and glomerular capillaries [89], it was hypothesized that this mediator could be the permeability factor related to proteinuria in INS [90, 91]. However, in patients with INS, VEGF levels did not differ between relapse and remission of the disease. Furthermore, the injection of VEGF obtained from the serum of patients with INS was not able to induce proteinuria in mice [90, 91].
In an animal model of NS induced by doxorubicin, IL-1 secreted by glomerular macrophage residents was associated with proteinuria [63]. Increased concentrations of IL-6 were detected in the urine and renal tissue of rats with NS induced by doxorubicin and showed positive correlation with proteinuria [66]. Several studies have also demonstrated high concentrations of IL-8/CXCL-8 in serum [58, 62, 86] and urine [87] of patients with INS, as well as increased levels of mRNA for IL-8/CXCL-8 in peripheral blood mononuclear cell (PBMC) culture of these patients [86]. The IL-8/CXCL-8 present in the PBMC culture supernatant from patients with INS alters the sulfated component metabolism at the glomerular basement membrane in rats [57, 86]. Urinary concentrations of IL-8/CXCL-8 showed positive correlation with 24-h proteinuria in children with INS [87]. Increased concentrations of IL-2 and IL-4 in PBMC-stimulated culture were detected in patients with INS [58]. High levels of plasma TGF-β were also found in corticoid-resistant patients [87], and increased expression of TGF-β genes and cytotoxic T lymphocytes effectors in renal tissue [92]. In animal models of NS, increased renal concentrations of many cytokines such as IL-4, IL-1, TNF-α, IL-12p40 and IL-17 [55] and of the chemokines RANTES/CCL5, eotaxin/CCL11 [28, 55], TCA-3/CCL1 (T cell activation-3), MCP-1/CCL2, MIP-1α/CCL3 (macrophage inflammatory protein-1 alpha) and MIP-1β (macrophage inflammatory protein-1 beta) have been reported [28]. Blockade of the CCR1 chemokine receptor reduced the infiltration of macrophages, lymphocytes and fibroblasts in renal tissue [28]. Wang et al. [64] showed that high concentrations of albumin stimulated proximal tubular cells in culture to increase the production and secretion of MCP-1/CCL2. Rats immunized by MCP-1/CCL2 and RANTES/CCL5 DNA before the induction of NS by doxorubicin presented low chemotaxis of monocytes/macrophages and improvement in the biochemical changes and in renal histopathology [29]. Table 3 summarizes recent studies related to cytokines and chemokines in INS.
On the other hand, some studies did not detect changes in the expression or in the levels of cytokines in patients or animal models of NS. For instance, Strehlau et al. [92] found no significant changes in renal mRNA expression for IL-2, IL-4, IL-7, IL-15, IL-8/CXCL-8, IL-17 or RANTES/CCL5 in children with INS. Neuhaus et al. [58] found no correlation between serum levels of IL-4, IL-8/CXCL-8 and IFN-γ and proteinuria in children with INS despite the increased levels of these mediators.
Another important factor that probably has a role in the pathogenesis of INS is the nuclear transcription factor called NF-κB. According to Valanciuté et al. [61], there was an increase in the activity of NF-κB in patients with INS. Indeed, this factor controls the expression of several cytokines and cellular adhesion molecules [31, 93]. In NS induced by doxorubicin, the increased activity of NF-κB was also positively correlated with renal damage [32, 65, 93].
More recently, Wei et al. [94] proposed soluble urokinase-type plasminogen activator receptor (su-PAR) as a circulating factor responsible for FSGS. These authors reported that su-PAR is elevated in two-thirds of subjects with primary FSGS, but not in people with other glomerular diseases, and that a higher concentration of su-PAR before transplantation underlies an increased risk for recurrence of FSGS after transplantation [94]. In addition, the authors showed that circulating su-PAR activates podocyte β(3) integrin in both native and grafted kidneys, causing foot process effacement, proteinuria and FSGS-like glomerulopathy in three different mouse models [94]. Although the results obtained with animal models of FSGS-like glomerulopathies are very suggestive, at present there is no proof that any known human su-PAR fragment causes FSGS in humans [see ref. 95 for review]. Therefore, at the present time the measurement of su-PAR using currently available assays has no value in decision-making in routine clinical practice [95].
Immune response pattern in nephrotic syndrome
T-helper lymphocyte classification and cytokine response pattern
In 1986, Mosmann et al. [96] classified CD4+ T lymphocytes of mice into Th1 lymphocytes, which produce IL-2, IL-3 and IFN-γ, and Th2 lymphocytes, which synthesize IL-3 as well as two other cytokines, initially described as growth factors for T cells (TCGF-2) and for mast cells (MCGF-2). Afterwards the same classification was proposed for humans [97]. However, many T cells cannot be classified into Th1 or Th2 based on these original criteria, due to the complex mixture of cytokines released by them, and are therefore classified as Th0 [97, 98].
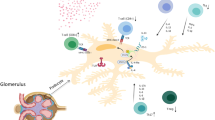
Th0 cells are possibly intermediate in the differentiation of naïve T cells (naïves) into Th1 or Th2 cells [95]. A mixed pattern of cytokines (Th0) occurs early after lymphocyte activation and differentiation into Th1 or Th2 usually occurs at the chronic phase of the disease [96]. In general, Th1 cells produce IL-2, IFN-γ and TNF-β, whereas Th2 cells produce IL-4, IL-5, IL-6, IL-9, IL-10 and IL-13. However, the main markers for these patterns of immune system response are, respectively, IFN-γ (Th1) and IL-4 (Th2) (Fig. 2) [6, 98]. Naïve CD4+ T cells under IL-12 or IFN-γ stimuli differentiate into Th1, whereas under IL-4 influence the cells differentiate into Th2. IL-4, the main inducer of Th2 response, results from the activation of lymphocytes. Since lymphocyte activation is considered a later event in the immune response, this fact might explain the difficulties in defining a specific immune response pattern in INS [98].

Th1 and Th2 lymphocyte functions. (a) Th1 cells induce phagocytosis and defense mediated by T cells against microorganisms. (b) Th2 cells induce IgE production and subsequent mast cell degranulation and activation of eosinophils—modified from Abbas, Murphy and Sher [98]
Th1 versus Th2 response pattern in INS
Some types of human glomerulonephritis, including crescentic and membranoproliferative glomerulonephritis, have a predominantly Th1 immune response pattern, while others, such as membranous nephropathy, IgA nephropathy and MCNS, show a predominantly Th2 response [97]. Some studies have considered that INS presents an imbalance between Th1/Th2 responses [6, 22], with a trend toward greater Th2 response [22, 26, 55, 59, 99].
The Buffalo/Mna rat, an animal strain with spontaneous NS, exhibited early changes in the balance between Th1 and Th2 with predominance of Th2 (IL-10 and IL-13) and inhibition of Th1 (IL-2 and IFN-γ) before the onset of proteinuria [22]. In addition, TCD4+ and TCD8+ lymphocytes of children with INS presented high expression of mRNA for IL-13 [26]. Mice transfected with IL-13 developed NS with overexpression of receptors for IL-4 and IL-13 in glomeruli [26]. Serum levels of IL-13 were correlated with the glomerular expression of B7-1 (CD80) in these animals [26]. CD80 is a co-stimulatory molecule generally present on the surface of B lymphocytes and of antigen-presenting cells that is associated with decreased apoptosis and induction of proliferation of TCD4+ cells [100].
The susceptibility to renal injury by doxorubicin may be related to T-helper response, since C57BL/6 mice, which present a predominance of Th1 response, are resistant to nephropathy induced by doxorubicin, while BALB/c mice, in which Th2 response predominates, are susceptible to the disease [101]. Recently, high concentrations of IL-4 and of eoxtaxin/CCL11, both mediators related to Th2 response, were detected in renal tissue of animals with NS induced by doxorubicin [55].
In sharp contrast, several studies showed the predominance of Th2 response in INS. As an example, Lama et al. detected high levels of IL-2 and of IFN-γ in children with steroid-sensitive INS. According to Araya et al. [6] there is no compelling evidence to define a predominance of Th2 response in INS. The absence of standardization in the selection of patients with INS and the various techniques used to measure the levels of cytokines may explain the different results [6, 62].
Some limitations should be taken into account when analyzing data on cytokines in INS. First, the measurement of cytokines in plasma might not always reflect the local production of these mediators in renal tissue. Moreover, plasma cytokines can bind to soluble receptors or antagonistic molecules and their production may vary depending on the stage of the disease [58]. External factors can also affect the cytokine pattern. For example, changes in the type or amount of protein in the diet were associated with modifications in the expression of cytokines in renal tissue and in lymphoid organs in animal models of NS [102, 103].
Final considerations and perspectives
In recent decades, much has been discovered about the role of the immune system in INS; however, there remain many questions about the immunological mechanisms associated with this disease. Because of the diversity of methods used, the results of clinical and experimental studies are quite variable, preventing the definition of a pattern of immune response.
Many findings point to an altered response of regulatory T lymphocytes, while other studies suggest an increase in cytotoxicity, dependent on CD8+ lymphocytes, NK cells and complement molecules. Furthermore, the participation of macrophages should be considered, especially at early stages of renal injury. Surveys have failed to identify the permeability factor related to proteinuria in INS, although there is some evidence for the participation of cytokines/chemokines, such as IL-1, Il-6 and IL-8/CXCL8, in increasing glomerular permeability.
Due to the intricate relationship between different cytokines and cell components of the immune system, we believe that INS does not have a single causal factor, but results from simultaneous or successive changes in different cell subtypes and many cytokines, depending on individual immune response, stage of the disease, and response to therapy. Thus, we consider that longitudinal studies with patients and animal models may help in understanding the immune response in INS and in the detection of biomarkers for the evolution and prognosis of the disease.
References
Schachter AD. The pediatric nephrotic syndrome spectrum: clinical homogeneity and molecular heterogeneity. Pediatr Transpl. 2004;8:344–8.
D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398–411.
Souto MFO, Teixeira MM, Penido MGMG, Simões e Silva AC. Fisiopatologia da Síndrome Nefrótica em crianças e adolescentes. Arch Latin Nefr Ped. 2008;8:1–10.
Fodor P, Saitúa MT, Rodriguez E, González B, Schlesinger L. T-cell dysfunction in minimal-change nephrotic syndrome of childhood. Am J Dis Child. 1982;136:713–7.
Araya C, Diaz L, Wasserfall C, Atkinson M, Mu W, Johnson R, et al. T regulatory cell function in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol. 2009;24:1691–8.
Araya CE, Wasserfall CH, Brusko TM, Mu W, Segal MS, Johnson RJ, et al. A case of unfulfilled expectations cytokines in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol. 2006;21(603–610):3.
Shalboub RJ. Pathogenesis of lipoid nephritis: a disorder of T cell function. Lancet. 1974;304:556–60.
Savin VJ, Sharma R, Sharma M, McCarthy ET, Swan SK, Ellis E, et al. Circulating fator associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med. 1996;334:878–83.
Gitlin D, Janeway CA, Farr LE. Studies of the metabolism of plasma proteins in the nephrotic syndrome. I. Albumin, γ-globulin and immunoglobulin. J Clin Invest. 1956;35:44–56.
Lagrue G, Branellec A, Blanc C, Xheneumont S, Beaudoux F, Sobel A, et al. A vascular permeability factor in lymphocyte culture supernatants from patients with nephrotic syndrome II: pharmacological and physicochemical properties. Biomedicine. 1975;23:73–5.
Giangiacomo J, Cleary TG, Cole BR, Hoffstein P, Robson AM. Serum immunoglobulins in the nephrotic syndrome. A possible cause of minimal change nephrotic syndrome. N Engl J Med. 1975;293:08–12.
Herrod HG, Stapleton FB, Trouy RL, Roy S. Evaluation of T lymphocyte subpopulations in children with nephrotic syndrome. Clin Exp Immunol. 1983;52:581–5.
Boulton JJM, Tulloch I, Dore B, Mclay A. Changes in the glomerular capillary wall induced by lymphocyte products and serum of nephrotic patients. Clin Nephrol. 1983;20:72–7.
Yokoyama H, Kida H, Tani Y, Abe T, Tomosugi N, Koshino Y, et al. Immunodynamics of minimal change nephrotic syndrome in adults T and B lymphocyte subsets and serum immunoglobulin levels. Clin Exp Immunol. 1985;61:601–7.
Beaman M, Oldfield S, Maclennan ICM, Michael J, Adu D. Hypogammaglobulinaemia in nephrotic rats is attributable to hypercatabolism of IgG. Clin Exp Immunol. 1988;74:425–30.
Moorthy AV, Zimmerman SW, Burkholder PM. Inhibition of lymphocyte blastogenesis by plasma of patients with minimal change nephrotic syndrome. Lancet. 1976;1:1160–2.
Kausman JY, Kitching AR. A new approach to idiopathic nephrotic syndrome. J Am Soc Nephrol. 2007;18:2621–2.
Hashimura Y, Nozu K, Kanegane H, Miyawaki T, Hayakawa A, Yoshikawa N, et al. Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Nephrol. 2009;24:1181–6.
Lagrue G, Branellec A, Xheneumont S, Weil B. Lymphokine ‘skin reactive factor’ (SRF) and the nephrotic syndrome. Bibl Anat. 1975;13:331–4.
Tanaka R, Yoshikawa N, Nakamura H, Ito H. Infusion of peripheral blood mononuclear cell products from nephrotic children increases albuminuria in rats. Nephron. 1992;60:35–41.
Riyuzo MC, Soares V. Revisão: Papel do infiltrado inflamatório na fibrose túbulo-intersticial e evolução das glomerulopatias. J Bras Nefrol. 2002;24:40–7.
Le Berre L, Herve C, Buzelin F, Usal C, Soulillou JP, Dantal J. Renal macrophage activation and Th2 polarization precedes the development of nephrotic syndrome in Buffalo/Mna rats. Kidney Int. 2005;68:2079–90.
Cao Q, Wang Y, Zheng D, Sun Y, Wang YA, et al. IL-10/TGF-β-modified macrophages induce regulatory t cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21:933–42.
Kuncio GS, Neilson EG, Haverty T. Mechanisms of tubulointerstitial fibrosis. Kidney Int. 1991;39:550–6.
Benz K, Büttner M, Dittrich K, Campean V, Dötsch J, Amann K. Characterization of renal immune cell infiltrates in children with nephrotic syndrome. Pediatr Nephrol. 2010;25:1291–8.
Lai K-W, Wei Ch-L, Tan L-K, Tan P-H, Chiang GSC, Lee CGL, et al. Overexpression of interleukin 13 induces minimal-change-like nephropathy in rats. J Am Soc Nephrol. 2007;18:1476–85.
Lee VWS, Wang Y, Qin X, Wang Y, Zheng G, Mahajan D, et al. Adriamycin nephropathy in severe combined immunodeficient (SCID) mice. Nephrol Dial Transpl. 2006;21:3293–8.
Vielhauer V, Berning E, Eis V, Kretzler M, Segerer S, Strutz F, et al. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int. 2004;66:2264–78.
Wu H, Wang Y, Tay Y-C, Zheng G, Shang C, Alexander S, et al. DNA vaccination with naked DNA encoding MCP-1 and RANTES protects against renal injury in Adriamycin nephropathy. Kidney Int. 2005;67:2178–86.
Wang Y, Wang YP, Tay Y-C, Harris DCH. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 2000;58:1797–804.
Rossmann P, Matousovic K, Bohdanecká M. Experimental adriamycin nephropathy: fine structure, morphometry, glomerular polyanion and cell membrane antigens. J Pathol. 1993;169:99–108.
Muñoz M, Rincón J, Pedreañez A, Viera N, Hernández-Fonseca JP, Mosquera J. Proinflammatory role of angiotensin II in a rat nephrosis model induced by Adriamycin. J Renin Angiotensin Aldosterone Syst. 2001; doi:10.1177/14703203114100922011.
Van Goor H, Van Der Horst MLC, Atmosoerodjo J, Joles JA, Van To A, Grond J. Renal apolipoproteins in nephrotic rats. Am J Pathol. 1993;142:1804–12.
Erwig LP, Kluth DC, Rees AJ. Macrophage heterogeneity in renal inflammation. Nephrol Dial Transpl. 2003;18:1962–5.
Eardley KS, Cockwell P. Macrophages and progressive tubulointerstitial disease. Kidney Int. 2005;68:437–55.
Wang Y, Wang YP, Zheng G, Lee VWS, Ouyang L, Chang DHH, et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007;72:290–9.
Schnaper HW, Aune TM. Identification of the lymphokine soluble immune response suppressor in urine of nephrotic children. J Clin Invest. 1985;76:341–9.
Sasdelli M, Rovinetti C, Cagnoli L, Beltrandi E, Barboni F, Zucchelli P. Lymphocyte subpopulations in minimal-change nephropathy. Nephron. 1980;25:72–6.
Fiser RT, Arnold WC, Charlton RK, Steele RW, Childress SH, Shirkey B. T-lymphocyte subsets in nephrotic syndrome. Kidney Int. 1991;40:913–6.
Lama G, Luongo I, Tirino G, Borriello A, Carangio C, Salsano ME. T-lymphocyte populations and cytokines in childhood nephrotic syndrome. Am J Kidney Dis. 2002;39:958–65.
Wang Y, Wang Y, Feng X, Bao S, Yi S, Kairaitis L, et al. Depletion of CD4 T cells aggravates glomerular and interstitial injury in murine adriamycin nephropathy. Kidney Int. 2001;59:975–84.
Wang Y, Wang YP, Tay Y-C, Harris DCH. Role of CD8 cells in the progression of murine adriamycin nephropathy. Kidney Int. 2001;59:941–9.
Tejani AT, Butt K, Trachtman H, Suthanthiran M, Rosenthal CJ, Khawar MR. Cyclosporine-A induced remission of relapsing nephrotic syndrome in children. Kidney Int. 1988;33:729–34.
Okuyama S, Komatsuda A, Wakui H, Aiba N, Fujishima N, Iwamoto K, et al. Up-regulation of TRAIL mRNA expression in peripheral blood mononuclear cells from patients with minimal-change nephrotic syndrome. Nephrol Dial Transpl. 2005;20:539–44.
Musial K, Ciszak L, Kosmaczewska A, Szteblich A, Frydecka I, Zwolińska D. Zeta chain expression in T and NK cells in peripheral blood of children with nephrotic syndrome. Pediatr Nephrol. 2010;25:119–27.
Wu H, Wang YM, Wang Y, Hu M, Zhang GY, Knight JF, et al. Depletion of gama–delta T cells exacerbates murine adriamycin nephropathy. J Am Soc Nephrol. 2007;18:1180–9.
Wang YM, Hu M, Wang Y, Polhill T, Zhang GY, Wang Y, et al. Regulatory T cells in renal disease. Int J Clin Exp Med. 2008;1:294–304.
Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, et al. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. 2009;32:111–6.
Apostolou I, Von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199:1401–8.
Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− naïve T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor FOXP3. J Exp Med. 2003;198:1875–86.
Le NT, Chao N. Regulating regulatory T cells. Bone Marrow Transpl. 2007;39:01–9.
Wang YM, Zhang GY, Wang Y, Hu M, Wu H, Watson D, et al. Foxp3-transduced polyclonal regulatory T cells protect against chronic renal injury from adriamycin. J Am Soc Nephrol. 2006;17:697–706.
Mahajan D, Wang Y, Qin X, Wang Y, Zheng G, Wang YM, et al. CD4+CD25+ regulatory T cells protect against injury in an innate murine model of chronic kidney disease. J Am Soc Nephrol. 2006;17:2731–41.
Shao XS, Yang XQ, Zhao XD, Li Q, Xie YY, Wang XG, et al. The prevalence of Th17 cells and FOXP3 regulate T cells (Treg) in children with primary nephrotic syndrome. Pediatr Nephrol. 2009;24:1683–90.
Pereira RL, Reis VO, Semedo P, Buscariollo BN, Donizetti-Oliveira C, Cenedeze MA, et al. Invariant natural killer T cell agonist modulates experimental focal and segmental glomerulosclerosis. PLoS One. 2012;. doi:10.1371/journal.pone.0032454.
Sellier-Leclerc AL, Duval A, Riveron S, Macher MA, Deschenes G, Loirat C, et al. A humanized mouse model of idiopathic nephrotic syndrome suggests a pathogenic role for immature cells. J Am Soc Nephrol. 2007;18:2732–9.
Garin EH, Blanchard DK, Matsushima K, Djeu JY. IL-8 production by peripheral blood mononuclear cells in nephrotic patients. Kidney Int. 1994;45:1311–7.
Neuhaus TJ, Wadhwa M, Callard R, Barratt TM. Increased IL-2, IL-4 and interferon-gamma (IFN-γ) in steroid-sensitive nephrotic syndrome. Clin Exp Immunol. 1995;100:475–9.
Yap HK, Cheung W, Murugasu B, Sim SK, Seah CC, Jordan SC. Th1 and Th2 cytokine mRNA profiles in childhood nephrotic syndrome: evidence for increased IL-13 mRNA expression in relapse. J Am Soc Nephrol. 1999;10:529–37.
Kemper MJ, Meyer-Jark T, Lilova M, Müller-Wiefel DE. Combined T- and B-cell activation in childhood steroid-sensitive nephrotic syndrome. Clin Nephrol. 2003;60:242–7.
Valanciuté A, Le SG, Solhonne B, Pawlak A, Grimbert P, Lyonnet L, et al. NF-kappa-B p65 antagonizes IL-4 induction by c-maf in minimal change nephrotic syndrome. J Immunol. 2004;172:688–98.
Kanai T, Yamagata T, Momoi MY. Macrophage inflammatory protein-1b and interleukin-8 associated with idiopathic steroid sensitive nephrotic syndrome. Pediatr Int. 2009;51:443–7.
Bricio T, Molina A, Egido J, Gonzalez E, Mampaso F. IL-1-like production in adriamycin-induced nephrotic syndrome in the rat. Clin Exp Immunol. 1992;87:117–21.
Wang Y, Chen J, Chen L, Tay Y-C, Rangan GK, Harris DCH. Induction of monocyte chemoattractant protein-1 in proximal tubule cells by urinary protein. J Am Soc Nephrol. 1997;8:1537–45.
Rangan GK, Wang Y, Tay YC, Harris DCH. Inhibition of nuclear factor-κB activation reduces cortical tubulointerstitial injury in proteinuric rats. Kidney Int. 1999;56:118–34.
Wang LM, Chi HJ, Wang LN, Nie L, Zou YH, Zhao TN, et al. Expression of interleukin 6 in rat model of doxorubicin induced nephropathy. Chin J Contemp Pediatr. 2010;12:912–4.
Kerpen HO, Bhat JG, Kantor R, Gauthier B, Rai KR, Schacht RG, et al. Lymphocyte subpopulations in minimal change nephrotic syndrome. Clin Immunol Immunopathol. 1979;14:130–6.
Pescovitz MD, Book BK, Sidner RA. Resolution of recurrent focal segmental glomerulosclerosis proteinuria after rituximab treatment. N Engl J Med. 2006;354:1961–3.
Ahmed MS, Wong CF. Rituximab and nephrotic syndrome: a new therapeutic hope? Nephrol Dial Transpl. 2008;23:11–7.
Takei T, Nitta K. Rituximab and minimal change nephrotic syndrome: a therapeutic option. Clin Exp Nephrol. 2011;15:641–7.
Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011; doi:10.1007/s00441-010-1034-0.
David S, Biancone L, Caserta C, Bussolati B, Cambi V, Camussi G. Alternative pathway complement activation induces proinflammatory activity in human proximal tubular epithelial cells. Nephrol Dial Transpl. 1997;12:51–6.
Rangan GK, Pippin JW, Couser WG. C5b-9 regulates peritubular myofibroblast accumulation in experimental focal segmental glomerulosclerosis. Kidney Int. 2004;66:1838–48.
He C, Imai M, Song H, Quigg RJ, Tomlinson S. Complement inhibitors targeted to the proximal tubule prevent injury in experimental nephrotic syndrome and demonstrate a key role for C5b-91. J Immunol. 2005;174:5750–7.
Rangan GK, Pippin JW, Coombes JD, Couser WG. C5b-9 does not mediate chronic tubulointerstitial disease in the absence of proteinuria. Kidney Int. 2005;67:492–503.
Eddy AA. Interstitial nephritis induced by protein overload proteinuria. Am J Pathol. 1989;135:719–73.
Bao L, Haas M, Pippin J, Wang Y, Miwa T, Chang A, et al. Focal and segmental glomerulosclerosis induced in mice lacking decay-accelerating factor in T cells. J Clin Invest. 2009;119:1264–74.
Turnberg D, Lewis M, Moss J, Xu Y, Botto MH, Cook T. Complement activation contributes to both glomerular and tubulointerstitial damage in adriamycin nephropathy in mice. J Immunol. 2006;177:4094–102.
Matsuo S, Nishikage H, Yoshida F, Nomura A, Piddlesden SJ, Morgan BP. Role of CD59 in experimental glomerulonephritis in rats. Kidney Int. 1994;46:191–200.
Turnberg D, Botto M, Warren J, Morgan BP, Walport MJ, Cook HT. CD59a deficiency exacerbates accelerated nephrotoxic nephritis in mice. J Am Soc Nephrol. 2003;14:2271–9.
Ransohoff RM. Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity. 2009;31(5):711–21.
Vianna HR, Bouissou CMMS, Tavares MS, Teixeira MM, Simões e Silva AC. Inflamação na doença renal crônica: papel de citocinas. J Bras Nefrol. 2011;33:351–64.
Pereira AB, Rezende NA, Teixeira Junior AL, Teixeira MM, Simões e Silva AC. Citocinas e quimiocinas no transplante renal. J Bras Nefrol. 2009;31:286–96.
Vianna HR, Soares CMBM, Silveira KD, Elmiro GS, Mendes PM, Tavares MS, et al. Cytokines in chronic kidney disease: potential link of MCP-1 and dyslipidemia in glomerular diseases. Pediatr Nephrol. 2013;28:463–9.
Pereira AB, Teixeira AL, Rezende NA, Pereira RM, Miranda DM, Oliveira EA, et al. Urinary chemokines and anti-inflammatory molecules in renal transplanted patients as potential biomarkers of graft function: a prospective study. Int Urol Nephrol. 2012;44:1539–48.
Garin EH, Laflam P, Chandler L. Anti-interleukin 8 antibody abolishes effects of lipoid nephrosis cytokine. Pediatr Nephrol. 1998;12:381–5.
Souto MFO, Teixeira AL, Russo RC, Penido M-GMG, Silveira KD, Teixeira MM, Simões e Silva AC. Immune mediators in idiopathic nephrotic syndrome: evidence for a relation between interleukin 8 and proteinuria. Pediatr Res. 2008;64:637–42.
Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108:2369–79.
Simon M, Gröne HJ, Jöhren O, Kullmer J, Plate KH, Risau W. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol. 1995;268:240–50.
Webb NJA, Watson CJ, Roberts ISD, Bottomley MJ, Jones CA, Lewis MA, et al. Circulating vascular endothelial growth factor is not increased during relapses of steroid-sensitive nephrotic syndrome. Kidney Int. 1999;55:1063–71.
Laflam PF, Garin EH. Effect of tumor necrosis factor-α and vascular permeability growth factor on albuminuria in rats. Pediatr Nephrol. 2005;21:177–81.
Strehlau J, Schachter AD, Pavlakis M, Singh A, Tejani A, Strom TB. Activated intrarenal transcription of CTL-effectors and TGF-1 in children with focal segmental glomerulosclerosis. Kidney Int. 2002;61:90–5.
Ruiz-Ortega M, Lorenzo Ó, Rupérez M, Blanco J, Egido J. Systemic infusion of angiotensin II into normal rats activates nuclear factor-κB and AP-1 in the kidney role of AT1 and AT2 receptors. Am J Pathol. 2001;158:1743–56.
Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor (suPAR) as a cause of focal segmenter glomerulosclerosis. Nat Med. 2011;17:952–60.
Maas RJ, Deegens JK, Wetzels JF. Serum suPAR in patients with FSGS: trash or treasure? Pediatr Nephrol. 2013;28:1041–8.
Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;175:05–14.
Del Prete GF, De Carli M, Mastromauro C, Biagiotti R, Macchia D, Falagiani P, Ricci M, Romagnani S. Purified protein derivative of Mycobacterium tuberculosis and excretory-secretory antigen(s) of Toxocara canis expand in vitro human T cells with stable and opposite (type 1 T helper or type 2 T helper) profile of cytokine production. J Clin Invest. 1991;88:346–50.
Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1986;383:787–93.
Hurtado A, Johnson RJ. Hygiene hypothesis and prevalence of glomerulonephritis. Kidney Int. 2005;68 (Supplement):62–7.
Odobasic D, Kitching AR, Tipping PG, Holdsworth SR. CD80 and CD86 costimulatory molecules regulate crescentic glomerulonephritis by different mechanisms. Kidney Int. 2005;68:584–94.
Lee VWS, Harris DCH. Adriamycin nephropathy: a model of focal segmental glomerulosclerosis. Nephrology. 2011;16:30–8.
Tovar AR, Murguía F, Cruz C, Hernández-Pando R, Aguilar-Salinas CA, Pedraza-Chaverri J, et al. A soy protein diet alters hepatic lipid metabolism gene expression and reduces serum lipids and renal fibrogenic cytokines in rats with chronic nephrotic syndrome. J Nutr. 2002;132:2562–9.
Kim SY, Lim AY, Jeon SK, Lee IS, Choue R. Effects of dietary protein and fat contents on renal function and inflammatory cytokines in rats with adriamycin-induced nephrotic syndrome. Mediat Inflamm. 2011; doi:10.1155/2011/945123.
Acknowledgments
This study was partially supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil) and FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais, Brazil) by the Grant INCT-MM (Instituto Nacional de Ciência e Tecnologia—Medicina Molecular: FAPEMIG: CBB-APQ-00075-09/CNPq 573646/2008-2). Dr. AC Simões e Silva received a research productivity grant from CNPq.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: John Di Battista.
Rights and permissions
About this article
Cite this article
de Fátima Pereira, W., Brito-Melo, G.E.A., Guimarães, F.T.L. et al. The role of the immune system in idiopathic nephrotic syndrome: a review of clinical and experimental studies. Inflamm. Res. 63, 1–12 (2014). https://doi.org/10.1007/s00011-013-0672-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-013-0672-6