
Abstract
Due to the rapid increase of new information on the multiple roles of Toll-like receptors (TLRs), this paper reviews several main properties of TLRs and their ligands and signaling pathways. The investigation of pathogen infections in knockout mice suggests that specific TLRs play a key role in the activation of immune responses. Although the investigation of TLR biology is just beginning, a number of important findings are emerging. This review focuses on the following seven aspects of this emerging field: (a) a history of TLR and ligand studies; (b) the molecular basis of recognition by TLRs: TLR structures, pathogen-associated molecular pattern binding sites, TLR locations and functional responses; (c) cell types in TLR expression; (d) an overview of TLRs and their ligands: expression and ligands of cell-surface TLRs and of intracellular TLRs; (e) TLR-signaling pathways; (f) discussion: TLRs control of innate and adaptive systems; the trafficking of intracellular TLRs to endolysosomes; investigation of TLRs in regulating microRNA; investigation of crystal structure of TLRs with ligand binding; incidence of infectious diseases associated with single nucleotide polymorphisms (SNPs) in TLR genes; risk of cancer related to SNPs in TLR genes; TLR-ligand mediated anti-cancer effects; and TLR-ligand induced chronic inflammation and tumorigenesis; and (g) conclusions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During the last 10 years, extensive investigation has identified Toll-like receptors (TLRs) as a major class of pattern-recognition receptors (PRRs) in Drosophila and mammalians, including humans. Recognition of pathogen-associated molecular patterns (PAMPs) through TLRs, either alone or in heterodimerization with other TLR or non-TLR receptors, triggers signals responsible for the activation of innate and adaptive immune responses. Most classes of TLRs are found in innate immune cells such as polymorphonuclear neutrophils (PMN), monocytes/macrophages, dendritic cells [1], NK cells [2], and mast cells [3], where they trigger an immediate response against pathogens. However, recent studies indicate that a number of TLRs are widely expressed in T and B cells [1, 4–6] as well as others, such as endothelial cells [7, 8], epithelial cells [9], skin keratinocytes [10–12], fibroblasts [13] and cancer cells [14, 15], showing that TLRs also directly regulate innate and adaptive immunity, inflammation reaction, inflammatory disease, cancer cells, and other cell responses [1, 4–16].
There is increasing evidence to show that TLRs are receptors for endogenous ligands and damaged tissue, suggesting pathogen-derived molecules and products of damaged tissue can be ligands to mediate inflammatory responses in the host [17, 18].
The discovery and characterization of the TLR family and their ligands have facilitated our understanding of TLR function and host defense against infection, disease and injury [19, 20]. Each TLR shows a differential expression pattern, extra- or intracellular localization and signaling pathway, resulting in the pathogenesis of sepsis and autoimmune disease [21]. Results of studies of the ligands or agonists and antagonists for TLRs and inhibitors of TLR-signaling molecules are currently being used in a variety of therapeutic applications. Thus this review highlights recent advances related to TLR recognition of their ligands and their signaling pathways, discussing the most advanced data in TLR fields to provide a new understanding and a basis for new therapeutic targeting and future considerations.
A history of TLRs and ligands
A century ago, Elie Metchnikoff was first to recognize that organisms possess an inherent ability to detect foreign matter through a cell-based defensive system. He observed in lower organisms “phagocytes” (e.g. macrophages) that could attack foreign substrates, ranging from carmine granules and rose thorns to fungal spores. He pointed out that a similar cell-based defensive system (e.g. white blood cells) also exists in humans [22]. Metchnikoff received the Nobel Prize in Medicine and Physiology in 1908 for his work on the cellular theory of immunology. Two key findings in innate immunity have promoted immunological development in recent years [23]. One was the discovery in 1996 of the Drosophila transmembrane protein, Toll, mediating recognition for fungal response and embryo dorsal–ventral polarity formation [24, 25]. The other was the discovery in 1998 of Toll-like receptor 4 (TLR4), which was demonstrated to be the receptor for lipopolysaccharide (LPS) of Gram-negative bacteria in mice [26]. These discoveries showed that the pathogen-sensing apparatuses in insects and mammals are closely related.
The molecular basis of recognition by TLRs
The protein Toll, German slang for “fantastic,” was first derived from the Toll gene of Drosophila, a maternal-effect gene that plays a central role in the establishment of dorsal–ventral patterning during embryonic development [24, 27]. At almost the same time, it was discovered that Toll and the Toll-signaling pathways are instrumental in the resistance of Drosophila to infection by several peptides [25]. Two peptides are involved in mediating this resistance. One is a powerful antifungal agent, drosomycin. The other, attacin, is responsible for protection against Gram-negative bacteria. The Toll protein acts as the receptor to mediate production of drosomycin to respond to fungal infection, whereas related receptor, the “18-wheeler,” is related to production of attacin. Receptors and signaling pathways quite similar to those of the Toll family in Drosophila have been indentified in a variety of other organisms ranging from plants to humans [28]. Owing to these receptors’ similarity to the Drosophila Toll, they are called Toll-like receptors, or TLRs. So far, 11 mammalian and 13 murine TLRs have been identified [6, 20].
TLR family members are expressed differentially in a variety of cells and tissues, including innate immune response cells, specific T and B lymphocytes and other non-immune cells. These receptors are localized both on the cellular membrane and within different cellular compartments, where they recognize and respond to various molecules related to a broad class of pathogens. Because the recognition system of mammalian TLRs is based on a strategy for a limited number of conserved microbial molecules, TLRs derive from an evolutionarily ancient germline encoded as and consisting of a “recognition” domain and a protein–protein-interacting region for activating downstream signalings [29]. The mechanism of recognition is inherently different from that of the adaptive immune system since varieties in the recognition of T and B cell antigen receptors are based on the recombination of V, D, and J gene segments [30]. Although the innate immune system does not have the specificity of adaptive immunity that is essential in triggering immunological memory, it can distinguish self from non-self and other danger signals. TLR receptors were first found on monocytes/macrophages, neutrophils and dendritic cells, and TLRs can recognize specific molecular structural motifs on pathogens, called pathogen-associated molecular patterns, or PAMPs. A number of investigations have demonstrated that TLRs and their homologs play an important role in initiating the innate immune response against pathogens in organisms, including plants [31], insects and mammals [32].
TLR ligands are varied, consisting of bacterial cell wall components; bacterial genome DNA; viral, fungal and parasitic products; and synthetic analogs of natural products. However, TLRs can also bind with autologous (self) molecules such as heat shock proteins (HSPs), intercellular matrix products, and mammalian genomic DNA, revealing that the TLR immune system is concerned with damage signals from injured tissue, including endogenous ligands such as high mobility group box 1 protein (HMGB1), HSPs (HSP22, HSP60, HSP70, HSP96), hyaluronan, type III repeat extra domain of fibronectin, uric acid crystals, mouse mammary tumor virus envelope proteins and respiratory syncytial virus fusion protein, β-defensin and plant ligands (paclitaxel) [17, 18, 33–36]. It is suggested that TLRs recognize not only PAMPs, but also stress- or damage-associated molecular patterns (DAMPs) [37]. The key to understanding exactly how a TLR is able to discriminate each ligand has been a frontier field.
TLR structures
To understand the structural basis of ligand interaction, analysis of TLRs and their interactions with ligands is important. Studies of the crystal structure of TLR receptors or complexes bound to their ligands have suggested that the ligands bridge two TLR molecules, forming a dimer between the ectodomains that have a similar overall architecture and serve to dimerize the cytoplasmic TIR (Toll/IL-1R) domains [38] or TIR (Toll/IL-1R and IL-18R) domains [32].
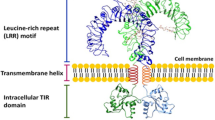
All TLRs belong to the type I transmembrane glycoprotein receptor family, with molecular weights ranging from 90 to 115 kDa and containing 16–28 extracellular leucine-rich repeat (LRR) domains [39]. The signal transmembrane α-helix portion and the conserved intracellular domain are homologous to those of the human interleukin-1 receptor (IL-1R) and human interleukin-18 receptor (IL-18R) [40]. The presence of the LRR domain in plant and animal proteins suggests an evolutionarily conserved role as a molecular PRR. Both humans and mice express TLR1 to TLR9. In addition, humans, but not mice, express TLR10. In contrast, mice express TLR11, 12, and 13, which humans lack [41]. TLRs are germline-encoded receptors that recognize highly conserved motifs present in microbes, including viruses, bacteria, fungi and protozoans, referred to as PAMPs. Some TLRs (e.g. TLR1, TLR2, TLR4, TLR5 and TLR6) are located on the cell surface, whereas others (e.g. TLR3, TLR7, TLR8, TLR9) remain sequestered in intracellular compartments. Clearly, TLRs are distinguished by LRRs that deviate markedly in both sequence and length from the TLR consensus [20, 42].
Each LRR contains 24–29 amino acids, the leucine-rich sequence XLXXLXLXX, and another conserved sequence XΦXXΦX4FXXLX, where X denotes any amino acid and Φ a hydrophobic amino acid. The repeats comprise a β-strand and an α-helix connected by loops [32].
The LRR domains of TLRs form a horseshoe-like structure, and it is through this structure that their concave surface is involved directly in the recognition of various pathogen molecules. Binding of ligands to the extracellular domains of TLRs causes rearrangement of the receptor complexes and triggers the recruitment of specific adaptor proteins to the intracellular TIR domains [43].
PAMP binding sites
Study shows that the potency of TLR ligands relies on their ability to induce homo- or heterodimerization and conformational change of the TLR [44]. It is widely believed that most TLRs are homodimeric but others (TLR1/TLR2 and TLR2/TLR6) are heterodimeric. Further, all TLRs are single-spanning transmembrane proteins with ectodomains largely composed of LRRs and cytoplasmic domains largely composed of a TIR (Toll/IL-1R or IL-18R) [40].
As mentioned above, some TLRs are clearly intracellular, residing predominantly or entirely within the endosomes. These include TLR3, TLR7, TLR8, and TLR9 [41]. The structure of a representative TLR9 has been solved through X-ray crystallography, revealing that TLR9 is a dimeric protein composed of two horseshoe-shaped subunits that stack together side by side. The repeats in TLR ectodomains frequently contain insertions following positions 10 and 15 in the TLR consensus sequence. At those positions, they can interact with a larger PAMP or with an accessory molecule, such as MD-2, an assistant molecule of TLR4. Importantly, TLR9 recognizes and responds to bacterial DNA and oligodeoxynucleotides (ODNs) that contain the unmethylated CpG sequence. Although TLR7 and TLR8 are highly homologous to TLR9, their natural ligands have not yet been found [32]. As Fig. 1 indicates, the LRR of the TLR9 receptor contains four insertions at position 10 (shown in red) that could create bacterial DNA and an unmethylated CpG binding site formed in the LRR loops [32].

A Toll-like receptor: All TLRs are integral membrane glycoproteins with an N-terminal ectodomain and a signal transmembrane domain. The ectodomain of TLR7, TLR8 and TLR9 is depicted, with the LRR solenoid shown with a gray molecular surface, and the N- and C-terminal flanking regions shown in green and purple. Insertions at position 10, indicated in red, might contribute to the information of the PAMP binding site. The insert at position 15, indicated in yellow, is expected to originate on the convex face of the TLR. Also shown is a cartoon of the transmembrane domain (presumed to be a single α-helix) followed by a molecular representation of the Toll-IL-1 and IL-18R (TIR) domain. This figure is from [32]
Recent site-directed mutagenesis and reporter gene experiments have indicated that the extracellular domain of TLR9 contains 25 LRR motifs. LRR 2, 5, and 8 may be receptors for CpG DNA activation in RAW264.7 cells [45].
TLR locations and functional responses
TLRs constitute critical components functioning to sense the presence of pathogens and initiate inflammatory responses. However, a pathogen can divide itself into microbes that replicate outside of host cells and others that replicate inside cells. Similarly, TLRs that recognize PAMPs on extracellular microbes are expressed on the cell surface, and those that recognize PAMPs on intracellular microbes are expressed inside the cell. Based on the specificity of PAMPs, correspondingly, TLRs can be broadly divided into two groups. The first group is TLRs expressed on the cell surface recognizing PAMPs on extracellular pathogens. In humans, TLR1, TLR2, TLR4, and TLR6 are out-membrane and respond mainly to bacterial surface-associated PAMPs. The second group, TLR3, TLR7, TLR8 and TLR9, is found on the surfaces of endosomes, where they bind mainly to the nuclear acid of PAMPs from viruses and bacteria. Upon binding with their cognates, TLRs activate their major signaling pathways. The first group, for example TLR4-MD-2-CD14, mediates the activation of the transcription factor NF-κB (nuclear factor κB). The second group, such as TLR3, triggers transcription factor IRF (IFN regulatory factor) to induce type I IFN (IFN-α and IFN-β) gene expressions [46, 47].
TLRs 1, 2 and 6 are a phylogenetically related subfamily. High sequence similarity and tandem arrangement of TLR1 and TLR6 in the human genome show that these two receptors arose from a recent gene duplication event [48]. TLR2 demands either TLR1 or TLR6 as a co-receptor for recognizing bacterial lipoproteins and lipopeptides. These lipoproteins are anchored to the cell membrane via conserved N-termini modified by a lipid chain and induce strong pro-inflammatory signals by macrophages [49].
It has been demonstrated that TLR2 can form heterodimers with TLR1 or TLR6, which recognize a wide variety of acylated cell wall components, such as lipoproteins and lipopeptides derived from Gram-positive bacteria, Gram-negative bacteria, mycoplasma, spirochetes and fungi [50].
Because microbial ligands and an increasing number of endogenous ligands can stimulate TLR activation, the functions of TLRs include (1) distribution on endothelial cells and epithelial cells and as the first line of defense at skin (including vascular endothelial cells and keratinocytes) and mucosal sites such as the respiratory, gastrointestinal and genitourinary tracts; (2) induction of cellular surface adhesion molecule expression, such as E-selectin and intercellular adhesion molecule 1 (ICAM1), that are important for leukocyte rolling and adhesion, as well as the secretion of chemokines, resulting in the adhesion of leukocytes to endothelium; (3) increasing phagocytosis of microorganisms by phagocyte cells, killing through the LPS- or other ligand-induced intracellular production of reactive oxygen and nitrogen intermediates; (4) regulating the differentiation and maturation of immune cells, and stimulating T and B cells to mediate adaptive immune responses. (For example, TLRs can upregulate the expression of co-stimulatory molecules CD80 and CD86 and production of IL-12, IL-23 and IL-27 to regulate the differentiation and maintenance of T and B cells to promote a specific immune response.); and (5) playing a critical role in tissue injury and tissue repair and regeneration, including liver regeneration and the protective response to injury of the brain and spinal cord by a TLR-dictated complex regulatory process of cell proliferation, survival and apoptosis that involves angiogenesis and tissue remodeling by the production of cyclooxygenases, chemokines, VEGF and matrixmetalloproteinases, as well as by the activation of mesenchymal stem cells (MSCs) [18, 51–54].
Cell types in TLR expression
Accumulating evidence indicates that TLRs are expressed in a variety of cell types, including immune and non-immune cells. For example, macrophages and myeloid DC preferentially express TLR2, TLR3, TLR4 and TLR 8, whereas plasmacytoid dendritic cells (also called natural interferon producing cells, IPC) express TLR 7 and TLR9 [55, 56].
To identify more accurately potential sites of action, real-time quantitative PCR analyses of the expression of TLRs in human and mouse tissues have been performed [5, 57, 58]. These studies have demonstrated that TLRs mainly express on all peripheral blood leukocytes, including CD4+ and CD8+ T cells and B cell populations and spleen tissue cells involved in immune function, but also on other tissues, such as ovaries, prostate, pancreas, placenta and testis [57].
In the past, non-immune cells have primarily been considered as targets of immune cell activities. For example, the importance of the barrier maintained by the gut epithelium has long been known. Epithelial cells also play additional key roles through TLR expression responding to pathogenic molecules. Thus, epithelial cells are more than just a barrier located at potential sites of entry, such as the skin, respiratory, intestinal and genitourinary tracts [59, 60].
Interestingly, a receptor of innate immunity, TLR3, is also predominantly expressed in brain, heart, lung and muscle [61], suggesting that TLR3 may play a role in anti-infection or anti-inflammatory reaction in these tissues. For example, TLR3 expresses in the central nervous system (CNS), where control of herpes simplex virus (HSV-1) spreading is required [62]. Also, TLR3 on neurons has been found to be a potent negative regulator of axonal growth in mammals [63], and inhibiting some human breast cell lines after binding with its ligand [64]. In addition, the TLR3 ligand with type I interferon can directly mediate the inhibition of proliferation and induce cell apoptosis in human melanoma cell lines [65].
Recent study has identified that more primitive cord-blood-derived MSCs express low RNA levels of TLR1, TLR3, TLR5 and TLR9 and high levels of TLR4 and TLR6. Stimulation of these MSCs with LPS or flagellin mediates MSC differentiation and production of cytokine, depending on the cell type that expresses them. TLR cell-expression-pattern study has been suggested that TLR networks regulate innate and adaptive immunity, but also active, integrate and select cell differentiation [60, 63]. However, it has been reported that many cancer cells express TLRs, suggesting that these cells also take TLRs and ligands to mediate their signal response (see “TLR-ligand induced tumorigenesis”).
An overview of TLRs, their ligands and signalling
Expression and ligands of cell-surface TLRs
TLR2 recognizes PAMPs derived from various pathogens, ranging from viruses, bacteria and fungi to parasites [50]. These ligands include tri-acyl lipopeptides from microplasma; peptidoglycan (PGN) and lipoteichoic acid (LTA) from Gram-positive bacteria; porin from Neisseria; lipoarabinomanan from mycobacteria; zymosan (containing β-glycan, mannans, chitin, lipid and protein) from fungi; and hemagglutinin protein from measles virus.
Figure 2 shows how TLR2 usually forms a heterodimer with TLR1, TLR6 or non-TLR molecules such as CD36, CD14 and dectin-1 to discriminate the molecular structure of the ligands. For example, TLR1–TLR2 recognizes the bacterial tri-acylated lipopeptide, whereas TLR2–TLR6 recognizes mycobacterial LTA and zymosan. The LPS receptor TLR4 depends even more strongly on CD14 than does the TLR2/TLR6 heterodimer. Without CD14, TLR4 cannot mobilize all of the adaptor proteins that it requires for signaling activity (Fig. 2). It has been determined that four adaptor proteins (MyD88, Mal, TRIF and TRAM) are essential for signal transduction by many TLRs and for full signaling by TLR4. If these proteins are absent, mice are even more susceptible to infection [40]. In addition, individual mutations of TLR also induce the compromising of the immune response.

Shared and unique components of the TLR complex. Germline mutations have proven the participation of CD14, CD36 and MD-2 in signaling by TLR2 and TLR4 complexes. This figure is from [40]. Current studies show that TLR1/TLR2 is activated by a ligand, a synthetic diacylated lipopeptide, PAM2 CSK4 [66]. As indicated above left, TLR2 can be homodimer or TLRX (candidates include TLR11, TLR12, and TLR13 in mice) [40]. MALP-2 (a lipopeptide from Maycoplasma fermentans) and LTA require CD36 for full signaling efficacy. CD14 is partly required by all TLR1/TLR2 and TLR2/TLR6 ligands. Ligand LPS are divided in two categories based on the morphology of bacteria colonies: smooth or rough (S-LPS or R-LPS, respectively). S-LPS contains O-polysaccharide chains which are absent from R-LPS. Wild-type Gram-negative bacteria synthesize S-LPS which needs CD14 to signal through TLR4 [44]. VSV-G vesicular stomatitis virus glycoprotein G, MMTV-G mouse mammary tumor virus surface glycoprotein
TLR4 was first demonstrated to be the receptor for LPS of Gram-negative bacteria in mice in 1998 [26]. TLR4 mainly expresses on myeloid cells (such as monocytes/macrophages, myeloid dendrite cells) and mast cells, NK cells, T and B lymphocytes, endothelial, epithelial cells, keratinocytes (Table 1) as well as fibroblasts [13], and even tumor cells (Table 2). Ligands of human TLR4 include exogenous LPS, PTX (paclitaxel), and several endogenous HSPs, fibronectin, heparin sulfate, and HMGB1 (Table 1).
TLR5 is mainly expressed on the surface of monocytes and epithelial cells. It is localized to the basolateral surface of intestinal epithelial cells, where it is capable of recognizing flagellin from bacteria that have invaded the epithelia [67]. Flagellin is a 55 kDa monomeric component of bacterial flagella [68]. A study on TLR5 knockout mice highlighted redundancy within TLRs. It appears that TLR4 can function to induce an antimicrobial response in TLR5-deficient mice challenged by Salmonella typhimurium and Pseudomonas aeruginosa [69].
It has been shown that some bacteria, such as Helicobacter pylori and Campylobacter jejuni, are capable of evading recognition by TLR5 by possessing flagellin with no immune stimulatory properties. However, a recent study indicated that TLR5 knockout mice are susceptible to E. coli-induced urinary tract infection, indicating TLR5 is host protection in the urinary tract [70].
TLR6 is described above and in Fig. 2. TLR6 is required to form a heterodimer with TLR2 for ligand recognition and NF-κB activation in mice and in humans. For example, TLR6 mediates the recognition of diacylated lipoproteins [or a synthetic diacylated lipopeptide, macrophage-activating lipopeptide-2 (MALP-2)] from mycoplasma, and PGN from Gram-positive bacteria that require cooperation with TLR2. If TLR2 mutates (Cys 30 and Cys 36 in TLR2 were substituted with Ser), it cannot mediate a MALP-2-induced NF-κB activation in the presence of TLR6 in human cells [71].
Human TLR10 is an orphan receptor that can be homodimerized and heterodimerized with TLR1 and TLR2 [72]. TLR10 is expressed on myeloid cells, T cells, B cells, endothelial cells and epithelial cells (Table 1). TLR10 may potentially act as a TLR2 co-receptor [72].
TLR11 is functional only in mice because the human TLR11 gene has a stop codon that prevents expression of the full-length version of the open reading frame (ORF) [73]. TLR11 knockout mice are highly susceptible to infection by uropathogenic E. coli. TLR11 also responds to profilin-like proteins from Toxoplasma gondii [74]. Human TLR11 may be non-functional against uropathogenic E. coli; instead, TLR5 may perform a function within the urinary tract against uropathogenic E. coli [75].
Expression and ligands of intracellular TLRs
As described above, TLR3, TLR7, TLR8 and TLR9 are expressed by intracellular compartments such as the endosome, lysome or ER [76, 77]. These intracellular TLRs act as sensors of foreign nucleic acids and trigger the anti-viral immune response by producing type I IFN and inflammatory cytokines.
TLR3 recognizes a synthetic analogue of double-strand RNA (dsRNA), polyinosinic–polycytidylic acid (polyI:C) (which structurally mimics dsRNA of viral origin), genomic RNA purified from dsRNA viruses such as retrovirus, and dsRNA produced in the course of replication of single-strand RNA (ssRNA) viruses such as RSV, encephalomyocarditis virus (EMCV) and West Nile virus (WNV) [78, 79]. TLR3-deficient mice died earlier than wild-type mice following infection with murine cytomegalovirus (MCMV). Accordingly, TLR3 deficiency is associated with susceptibility to herpes simplex virus (HSV)-1 infection in humans [62, 80]. TLR3 is also implicated in the recognition of small interfering RNA (siRNA) in a sequence-independent manner and induces the production of IL-12 and IFN-γ, which efficiently suppress angiogenesis in a mouse model, suggesting that genetic siRNA might treat angiogenic disorders, which affect 8% of the world’s population [81]. It has been suggested that WNV crosses the blood–brain barrier through TLR3, which can lead to lethal encephalitis [79].
TLR7, TLR8 and TLR9, like TLR3, are all located within the endosomal membrane. TLR7 has been reported to play an important role in resistance to fluenza infection [82]. In addition, TLR7 and TLR8 have been demonstrated to recognize numerous ssRNA viruses for anti-viral defensive responses through type I IFN and cytokine IL-12 and TNF and chemokine IP-10 production [83]. TLR7 and TLR8 both confer responsiveness to the anti-viral compound R848 [84].
TLR9 is the receptor for both bacterial and viral DNA, in contrast with TLR3, TLR7 and TLR8 for detecting RNA. As Fig. 1 indicates, TLR9 is a potent activator of immune cells in the recognition of bacterial CpG. TLR9-deficient mice did not show any response to CpG DNA [85]. Experiments done by gene knockdown and gain of function have demonstrated that TLR9 recognizes unmethylated CpG motifs commonly present in the genomes of bacteria and viruses [85, 86]. However, in vertebrates, the frequency of CpG motifs is severely suppressed and the cytosine residues of the CpG motifs are highly methylated, which leads to abrogation of immunostimulatory activity. Synthetic ODNs containing unmethylated CpG motifs also activate immune cells and elicit Th1-like immune responses. Therefore, CpG DNA is now expected to be useful as an adjuvant for a variety of vaccines against infectious disease, cancer and allergy [6, 20].
The ligands for mouse TLR12 and TLR13 are currently unknown. Table 1 shows known human ligands, and TLRs that are mainly expressed in immune cells, endothelial cells, epithelial cells, keratinocytes and fibroblasts (although not in Table 1). They play an important role in normal and pathological inflammation reaction and tumor environments. Recently, increased evidence indicates that TLRs are also expressed in human cancer cells to mediate cancer cell responses (Table 2).
TLR-signaling pathways: from TLR4 to other TLRs
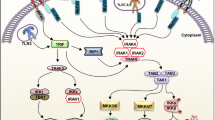
Before the last two decades, little was known about signal transduction by LPS-mediated modulation of murine peritoneal macrophages, including LPS-dependent production of IL-12 and prostaglandins [110]. However, as a result of the first demonstration of TLR family homology with the Drosophila Toll protein [111] and evidence that the human homologue of the Drosophila protein can induce NF-κB signals for activation of both innate and adaptive immunity [112], as well as the discovery of LPS sensitivity associated with the TLR4 gene in mice [26], LPS- and other ligand-mediated TLR-signaling, research has made tremendous progress (Fig. 3).

TLR4 signaling pathways in macrophages. Exposing macrophages to a TLR4 ligand, such as LPS, induces at least two pathways; an MyD88 dependent cascade, recruiting the IRAK family of proteins (IRAK1, 4 are active isoforms, but IRAK2 and IRAK-M are negatively regulating isoforms controlled by PI3K/Akt [118, 119]), that regulate TRAF6 and TAK1 leading to activation of NF-κB and MAPKs and transcriptions of a number of genes, including pro-inflammatory cytokines, chemokines, cytosolic PLA2, Cox-2, iNOS, etc. The other is an MyD88 independent pathway, through TRAM, TRIF TKB/1/IKKi, activate IRF3 and IRF7, which controls the gene expression of type I IFNs. In addition, TRIF can also interact with RIP1 (receptor-interacting protein, not shown in this figure) and activate TAK1, inducing IKK complex and MAPK activation. LPS-mediated activation of PKC, PI3K and phospholipases may involve the activation of G protein coupled receptors [120]
LPS, potentiallythe most immunostimulatory glycolipids, consist of the out-membrane of Gram-negative bacteria recognized by the TLR4 and MD-2 receptor complexes [113]. MD-2 is associated with the extracellular domain, LRR of TLR4, triggering TLR4 clustering [113]. LPS also binds another CD14 molecule existing in the serum or cell surface to enhance LPS function [114]. However, the precise relationship between CD14 and TLR4 remains unclear [82]. Currently, two main signaling pathways have been identified by an alkylating agent, N-ethyl-N-nitrosourea (ENU), that induces point mutations on haploid DNA [82]. The first pathway requires two adaptor proteins, myeloid differentiation factor 88 (MyD88) and TIR domain-containing protein (TIRAP)/MyD88 adaptor-like (Mal), recruiting the serine/thionine kinases (IRAK4, 1) to trigger the activation of TRAK6/IKK complex and MAPKK, respectively, resulting in quick or early activation of the transcription factor NF-κB and MAPK [17, 114]. The second pathway needs TIR domain-containing adaptor inducing interferon-β (TRIF)/TIR, domain-containing adaptor molecule-1 (TICAM-1), and TRIF-related adaptor molecule (TRAM)/TIR domain-containing adaptor molecule-2 (TICAM-2) that induce late NF-κB activation and LPS-mediated phosphorylation and dimerization of the transcription factor interferon regulatory factors (IRF), resulting in IFN-α and IFN-β production and activation of acquired immunity [47, 114] (Fig. 3).
All TLRs belong to the class I transmembrane receptor family [112, 115]. Ligand binding to its receptor results in the formation of a homo- or heterodimer. TIR associates with different adaptor proteins. Currently, four positive adaptors—MyD88, Mal (TIRAP), TRIF (TICAM-1) and TRAM (TICAM-2)—and one negative adaptor—SARM, which blocks TRIF-dependent signaling—have been demonstrated [43, 116].
The adaptor protein MyD88, which is recruited by the Mal adaptor protein, is utilized by all kinds of TLR receptors, but not by TLR3. However, TLR1, TLR2, TLR4 and TLR6 require using the TIRAP molecule as a linker adaptor for contacting MyD88, resulting in a cascade along the signaling pathway including IL-1 receptor associated kinases (IRAKs) and TNF-receptor associated factor 6 (TRAF6). TRAF6 triggers the activation of transforming growth factor β-activated kinases (TAKs), which initiate a kinase cascade involved in activation of the IKKα/IKKβ/NEMO complex and phosphorylation of inhibitory protein IκB. In turn, phosphorylated IκB dissociates from the complex and is rapidly targeted for ubiquitination and degradation by proteasomes, causing the activation of transcription factor NF-κB, which translocates to the nucleus [112], mediating a number of gene transcriptions [117]. In addition, TAK1 activates IKK complex and MAP kinases such as ERK1/2, JNK and p38MAPK, leading to AP-1, c-Jun and c-fos activation to trigger inflammatory cytokine gene transcriptions. Recent investigation indicates that the MyD88-independent TRIF pathway mediates IRF3 and IRF7 activation and the subsequent induction of IFN-α and IFN-β [17, 43, 46, 47]. A comprehensive figure including all TLR signal pathways has been published [47].
Discussion
TLRs control the innate and adaptive systems
The immune system is divided into two parts, innate and adaptive. The innate immune system is immediately available to combat threats. There is no memory of the same threat molecules upon the second or third exposure. However, the innate immune system responds to the common structures or ligands shared by a vast majority of threats. These common PAMP structures are recognized by TLRs. In addition to the cellular TLRs, an important part of the innate immune system is the humoral complement system that opsonizes and kills pathogens through the PAMP recognition mechanism. Furthermore, highly conserved soluble and membrane-bound proteins, collectively called recognition receptors (PRRs), interact with PAMPs to trigger innate immunity. Thus TLRs belong to one of the PRRs. Research has shown that cytosolic PRRs, including retinoic acid-inducible gene-1 (PIG-1)-like receptors (RLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), also play a role in pathogen recognition [19]. The adaptive immune response consists of T and B lymphocytes. They are specific for particular antigens and depend on clonal selection for foreign antigens and antibody production that takes from 4 to 7 days to ramp up to kill pathogens escaping from the innate immune system [29, 30, 121, 122].
Recently, much research in microbiology and immunology has revealed the pivotal role of TLRs in sensing microbial infections and regulating the subsequent adaptive immune system [20, 60]. Growing evidence shows that TLRs are not only expressed on innate immune cells, but also on T and B lymphocytes, indicating that TLRs play an important role indirectly and directly in regulating T and B lymphocytes [1, 6].
Additionally, much evidence has emphasized that TLRs play a key role in resistance to microbes. Three aspects show TLRs performing their remarkable function in resistance to infection. One, TLR4 mutation causes enhanced susceptibility to S. typhimurium [123] and E. coli [124]. Two, TLR2 deletion causes enhanced susceptibility to Staphylococcus aureus [125]. Three, TLR3 gene knockout (Tlr3 −/−) mice and TLR9 immunodeficiency phenotype mice (TLR9 Tlr9 CpG1/CpG1) expressed a significant immune impairment to cytomegalovirus infection in vivo [125]. In short, the importance of TLRs in resistance to microbes has been strongly identified.
The trafficking of intracellular TLRs to endolysosomes
Recent investigation shows that before intracellular TLRs (TLR3, TLR7, TLR8 and TLR9) respond to viral infection, all may need to interact with a 598 amino acid protein with 12 transmembrane domains, UNC-93B, located on the endoplasmic reticulum (ER) [126]. This interaction regulates the trafficking of intracellular TLRs to the endolysosomes when stimulated with ssRNA or DNA [41] (Fig. 4), indicating a role of the NC-93B protein in the induction of intracellular TLRs to bind pathogen DNA and RNA molecules.

Trafficking of intracellular TLRs to endolysosomes. UNC-93B physically interacts with the transmembrane domain of intracellular nucleotide-sensing TLR3, TLR7 and TLR9 in the endoplasmic reticulum (ER). UNC-93B regulates the trafficking of TLR3, TLR7 and TLR9 to the endolysosomes, gp96 mediates all TLR maturation and PPAT4A involves the trafficking of specific TLR7 and TLR9 upon stimulation with dsRNA, ssRNA and DNA, respectively [41, 127–129]
A recent report has indicated that the ectodomain of TLR9 must cleave proteolytically to generate a functional TLR9. Although both the full-length and processed forms of TLR9 can bind with the ligand, it is suggested that the processed form is essential to lead to its signal activity [127].
Furthermore, ER membrane accessory molecule UNC-93B has been shown to play an important role in the interaction between ER membrane trafficking and signaling with TLR3, TLR7 and TLR9 [128]. It will be interesting to know whether UNC-93B also works for TLR8 and how the trafficking and recognizing of ligands are controlled by ER membrane accessory molecules, including PPAT4A and gp96, since these accessory molecules could be promising treatment targets in infectious diseases and immune disorders [129].
Investigation of TLRs in regulating microRNAs (miRNAs)
The first miRNA4, Lin-4, was discovered as a regulator that turns off lin14 gene expression, controlling the timing of larval development in the nematode C. elegans. In 1997, Tam et al. found a new gene, BIC, which was transcriptionally activated at a common retroviral integration site in B cell lymphomas. The BIC gene, as a proto-oncogene, lacks an extensive ORF, leading to short BIC RNA (about 1.7 kb spliced and polyadenylated) that can function as a non-protein-coding RNA. By now, it is known that miR-155 is processed from the BIC gene through a spliced and polyadenylated BIC RNA [130–133]. Mice deficient for bic/miR-155 have increased lung airway remodeling (such as increased bronchiolar subepithelial collagen deposition and increased cell mass of sub-bronchiolar myofibroblast) and defective adaptive immunity, including BIC-deficient CD4+ T cells that are intrinsically biased toward Th2 differentiation [134]. In addition, miR-155 can also be induced during the macrophage inflammatory response [135].
Currently, one of most important aspects in TLR biology is the study of miRNAs, a class of single-stranded, non-coding small RNA consisting of approximately 22 nucleotides. Recently, miRNAs have been shown to possess a critical role in regulating gene function by directing 3′UTR (the 3′untranslated region) interaction of target mRNA to repress the translation of specific target genes [136, 137]. miRNA genes are encoded in genomic DNA and arise mostly from intergenic regions or introns or exons of miRNA-coding genes [138, 139]. By analyzing the proximity of miRNA genes, research indicates that many miRNA genes exist as clusters [140, 141]. miRNA genes are transcribed by polymerase II or III into long transcripts, called pri-miRNA (primary miRNA), which are cleaved into an intermediate pre-miRNA (precursor miRNA) by RNase III endonucleases (Drosha) in the nucleus, creating an approximately 70-nucleotide-hairpin-shaped pre-miRNA. Following export from the nucleus to the cytosol, the pre-miRNA undergoes further processing by Dicer (another RNase III enzyme), leading to the production of a functional miRNA with about 22 nucleotides that is incorporated into RISC (RNA-induced silencing complex) and unwound into a mature single strand to mediate translational repression and mRNA degradation. Over 700 human miRNAs have been cloned [142]; some are highly conserved, and others are lineage specific [137].
As described above, most miRNAs control gene expression by sequence-specific translational repression or degradation of the target mRNA. However, under particular conditions, for example during cell cycle arrest, some miRNAs can switch from repression to activation [143].
Recent reports further demonstrate that miRNAs play an important role in immune cell development, differentiation and response, including TLR-mediated inflammatory responses [119, 144–146]. The expression of miRNAs themselves is directly controlled by transcriptional factors through both the NF-κB and MAPK signaling pathways. LPS stimulation can up-regulate the expression of miRNAs in human innate immune phagocytes, such as monocytes and PMN, involving the induction of a number of miRNAs, such as let-7e, miR-187, miR-9, miR-99b, miR-135a, miR-132, miR-146a, miR-155, miR-222 [147], suggesting these miRNAs are inducted by inflammatory stimuli and play an important role in the immune system.
The induction of miR-155 is not only essential in activated T and B cells, but also is crucial in the maturation of macrophages and DCs and in antigen presentation [134, 148]. Lu et al. [149] and Androulidaki et al. [119] found that miR-155 represses the expression of SOCS1/suppressor of cytokine signaling 1 in murine Foxp3-dependent regulatory T (TR) cells and macrophages. Another study further indicates the miR-155 mediated repression of inositol phosphatase SHIP mRNA expression in murine macrophage RAW264.7 cells in response to LPS, suggesting that miRNAs not only are novel regulators of gene expression, but also possess distant target molecules within TLR-mediated signal loops. For example, it has become increasingly clear that Akt1 mediates cell survival, proliferation, and differentiation, positively regulating let-7e (targeting TLR4) and miR-181 (targeting TNSF11/TNF superfamily member 11) and negatively regulating miR-155 (targeting TNF-α, PU.1, SOCS1/suppressor of cytokine signaling 1 and SHIP1/Sac homology 2 containing inositol phosphatase-1) and miR-125b (targeting TNF-α) to LPS. These results have been identified through a PI3K/Akt1 signaling pathway, since by using Akt1 /− macrophages, LPS leads to increased amounts of TLR4 and reduced amounts of SOCS1 in macrophages. It further suggests that Akt1 acts as a negative regulator controlling LPS response through regulating the expression of miRNAs [119]. These findings may have a significant role in the treatment of inflammatory diseases and cancers.
Investigation of crystal structures of TLRs with ligand binding
What is the most exciting progress being made among the investigations of TLRs and their ligands? One answer should be the study of the crystal structures of four TLR complexes bound to their ligands. They are (1) TLR1/TLR2 heterodimer bound to tri-acylated lipopeptide [150]; (2) TLR3 dimer complexed to dsRNA [38]; (3) TLR4-MD-2-LPS complex, including the crystal structure of LPS Lipid A, binding to human MD-2; and (4) CpG DNA binding to N-terminal LRR of TLR9 [17, 45, 151–153]. These elegant results have given us the first high-resolution view of LPS-TLR4-MD-2, showing the mechanisms for innate recognition of pathogens, also reflected in investigations involving the single nucleotide polymorphisms (SNPs) in the human TLR4 molecule, such as common, co-segregating missense mutations, Asp299Gly (or D299G, a Gly substitution at Asp 299 due to a point mutation, an A–G substitute, at nt 896 from the start codon of the TLR4 cDNA) and Thr399Ile (or T399I, an Ile substitution at Thr399), affecting the extracellular domain for LPS binding [17, 154].
Incidence of infectious disease associated with SNPs in TLRs
SNPs in TLRs have revealed a single nucleotide change leading to a subtle distortion. It suggests that host genetic factors are of key importance [155]. Alternatively, TLRs, in particular TLR4, can respond to host endogenous molecules such as ox-LDL, Aβ amyloid peptide and HSPs, mediating tissue injury. Thus the investigation of SNP-affected TLR function has been critical. For example, the TLR4 gene residing on chromosome 9 in 9q32–q33 region and its two SNPs encoding Asp299Gly and Thr399Ile substitutions in the TLR4 ectodomain increase susceptibility to infections [156]. People with C1625G in their MD-2 promoter may have increased susceptibility to infection, organ dysfunction and sepsis after major trauma [157]. In addition, people with polymorphism (C558T) of the Mal adaptor protein are linked to increased susceptibility to meningeal tuberculosis [158]. Another recent report also indicates that susceptibility to viral-mediated pneumonia is significantly associated with T1237C polymorphism in the TLR9 gene [159].
Risk of cancer related to SNPs in TLRs
Chronic inflammation can induce most common cancers (in about 15% of global cancer patients) [35]. The etiology of inflammation involves multiple factors, including ROS (reactive oxygen species) mediated non-specific DNA-damage and specific activation of oncogenes through hydroxyl ions as well as polymorphisms in TLRs genes. A specific example of SNPs with cancer risk is H. pylori infection and induction of gastric cancer. A recent report identifies that defective signaling through the TLR4 may result in an exaggerated inflammatory reaction with severe tissue destruction in Asp299Gly people [160, 161].
Sequence variants in several TLR genes have been linked to prostate cancer involving TLR4 and the TLR1–TLR6–TLR10 gene cluster in Sweden. This report indicates that the populations from Swedes having an SNP in the 3′UTR region of TLR4 (11381G/C) genotypes had a 39% increased risk of early-onset prostate cancer when compared with a wild-type GG genotype [162]. Additionally, polymorphisms in the TLR6–TLR1–TLR10 gene cluster residing within a 54-kb region on 4p14 (TLR6 in promoter −1401 A/G, promoter −673 C/T; TLR1 in promoter −7202 A/G, promoter −6399 C/T, intron3 −833 C/T; and TLR10 in promoter −3260 C/T, promoter −1692 C/T, Exon2 −260 A/G, Exon3 720 A/G, Exon3 1104 A/C, Exon3 2322 A/G) were associated with prostate cancer, suggesting that sequence variants in TLR genes may disrupt the inflammation, thereby contributing to the onset of cancer [163].
TLR-ligand mediated anti-cancer effects
Since the eighteenth and nineteenth centuries, microbes have been known to possess anti-cancer properties [18, 164], suggesting a positive association between the infection and remission of malignant disease. That TLRs express on immune cells and trigger immune or inflammatory responses has led to a huge body of effort in anti-tumor therapy using TLR ligands. For example, Bacillus Calmette-Guerin (BCG) has been used to treat bladder cancer for three decades, and OK-432, a lyophilized production of group A streptococcus, has been utilized to treat uterine, cervical, gastric and oral squamous cell carcinoma [18]. OK-432 induced IFN-γ-mediated antitumor immune response may utilize TLR4 signaling pathways [165, 166].
TLR7 ligand, imiquimod, molecular formula, C14-H16-N4, also called R-837, S26308, is an immune response modification (IRM) licensed in 1997 by the US Food and Drug Administration to treat human papilloma virus infection-induced genital warts [105] and basal cell carcinoma of the skin [105, 106].
In addition, increased reports indicate that ligands of selected TLR7 (imidazoquinoline, S28690 and 852A) were used to treat chronic lymphocytic leukemia (CLL) [167] and tested in clinical phases I/II [168].
A TLR9 ligand, ODN containing unmethylated CpG dinucleotide (CpG ODN), has been used in a phase I trial of non-Hodgkin’s lymphoma (NHL) for anti-B cell malignancies [169].
Cutaneous T cell lymphoma (CTCL) is a clonally derived skin-invasive CD4+ T cell lymphoma markedly damaging the Th1-type cell immune response and reducing the production of Th1-type cytokines IFN-γ and IL-2. In contrast, the production of the Th2-type cytokines IL-4, IL-5 and IL-10 was increased. The TLR9 ligand CpG-A (ODN2216) can stimulate PBMC-induced IFN-γ production in healthy volunteers, but not in CTCL patients. However, following CpG-A stimulation with IL-15, the production of IFN-γ was significantly higher in both healthy individuals and CTCL patients [170].
CpG ODN has also been used to treat renal cell carcinoma, melanoma and non-small-cell lung cancer alone and in combination with other agents in phase I and II clinical trials [171].
Based on the principle that TLRs play a key role in inducing adaptive immune responses through antigen-present cells, DCs and macrophages, and a ‘danger signal,’ a nuclear protein—high-mobility group box 1 protein (HMGB1)—from dying cancer cells can trigger an antigen-specific immune response. HMGB1 can trigger an adaptive immune response against different mouse cancer cell models through TLR4 activation [172]. Very recently, Krishnamachari et al. [173] further discussed several recent and innovative strategies for co-delivering antigens and CpG oligonucleotides for antigen-specific immune therapy. Using HMGB1, an endogenous ligand or alarmin protein, to mediate endogenous TLR2 activation leads to TLR2-dependent brain tumor regression from within a microenvironment in mice [174].
TLR-ligand induced chronic inflammation and tumorigenesis
Chronic inflammation and tumor-created microenvironments
Inflammation plays a key role in preventing microbial infection and tissue injury. However, chronic inflammation is linked with tumorigenesis. For example, patients with ulcerative colitis, a chronic colorectal inflammation, have an increased (tenfold) possibility of developing colorectal carcinoma. Additionally, patients with chronic hepatitis and cirrhosis are at risk for development of liver cancer [175].
With the release of endogenous molecules from injured tissue, TLRs recognize these molecules, triggering an inflammatory response to increase the risk of cancer [18].
Solid tumors are set up from an initially tumor-induced microenvironment consisting of tumor cells, stromal cells and migratory immune cells, including tumor-associated macrophages (TAMs) and regulatory T cells (Treg) [176, 177]. The tumor-created microenvironment mediates a complex interaction, including aborting the activation of immune cells and promoting tumor growth and metastasis [176, 177].
Macrophages can be regulated by a number of signals from a tumor microenvironment. Under different conditions, peripheral blood monocyte-derived macrophages can be polarized into M1 and M2 phenotypes that refer to the Th1/Th2 paradigm. Immune infiltration of host leukocytes by neutrophils, TAMs, dendritic cells, eosinophils, mast cells and lymphocytes is a remarkable phenomenon in tumor development [178]. TAMs mainly show an IL-12low and IL-10high phenotype including the expression (MR) and scavenger receptor of mannose that probably can be considered an M2 phenotype [179].
Increasing evidence indicates that a large number of myeloid macrophages are present in epithelial cancers, mediating tumor development and spread, showing that a variety of factors in the tumor microenvironment result in TAM [177].
TLRs expressed on cancer cells and their signaling
Currently, a number of reports indicate that functional TLRs are widely expressed on cancer cells or cancer cell lines in mice and humans [14, 15, 96, 166, 180] (Table 2). Although we largely do not know why and how tumor cells control or utilize TLR activation, the opposite effects of the NF-κB protein in normal tissues and cancer cells have been suggested. Normally, NF-κB activation of acute inflammation can regulate a short term expression of pro-inflammatory mediators as well as cell death before infection is resolved or injured tissue is repaired. In contrast, in cancers, chronic inflammatory-induced precancerous epithelial cells utilize NF-κB activation to elevate expression of pro-inflammatory and cell-survival genes, inhibiting the cell death pathways to promote the growth of malignant cells [181]. A normal immune host mediates an important NF-κB activation and cytokine and chemokine production, forming an acute inflammatory reaction beneficial against pathogens. However, if it is not ordered and timely, it can result in cancer or other diseases such as arthritis, heart attack and Alzheimer’s disease [181].
Chen et al. [182] have drawn a link between TLRs, inflammatory disease, and carcinogenesis, pointing out that chronic inflammation mediated by TLR stimulation through their exogenous or endogenous ligands can induce precancerous cells, increasing neoplastic transformation of normal cells, through active NF-κB pathways, into cancer cells.
Tumor cells and cell lines utilize and devise a TLR4-triggered MyD88/NF-κB and c-Jun signaling pathway to mediate proliferation and growth, suggesting the tumor cells have developed a mechanism by seizing or usurping the host TLR-signaling pathway to proliferate and grow, escaping host-mediated immune surveillance [14, 166, 183].
TLR expression on human cancer cells can enhance the NF-κB cascade, leading to cell proliferation and pro-apoptosis. For example, TLR4 expresses in human lung cancer cells, HNSCC, ovarian cancer (OvCa) cells and OvCa cell lines [14, 97], prostate cancer cell lines (where TLR9 also expresses and up-regulates NF-κB activity) [101] and colorectal cancer [183]. In particular, in OvCa cells and OvCa cell lines, TLR4 signaling triggered NF-κB and MyD88 signaling that was related OvCa progression and chemoresistance, promoting immune escape [14, 183]. Furthermore, high expression of TLR4 and MyD88 protein by colorectal cancer cells is associated with liver metastasis and poor prognosis [183].
In addition, several cases have reported that TLR activation on cancer cells mediates positively to inhibit tumor proliferation and apoptosis. The TLR3 ligand directly mediates anti-human breast cancer cells, TLR5 can inhibit human colorectal cancer cell lines and TLR7/8 and TLR9 ligands are used to treat CLL (Table 2). These inconsistent results are not completely understood. On the one hand, they reflect the complexity of TLRs in cancer cells, which remains a target for future research. On the other hand, the inconsistency may be due in part to the differences in cells, TLRs and test conditions. However, chronic infection or inflammatory disease mediated TLR abnormal expression, aberrant response, oncogenesis and tumor microenvironments may lead to cancer cells increasing NF-κB activity and taking TLR4/MyD88 signaling to drive cancer growth [94, 178, 183].
Thus, TLR-related tumors have been one of the important aspects in TLR investigation. Currently, stimulating “good” inflammation in the tumor microenvironment has been mentioned [184], indicating that TAMs can be modulated from a suppressive “alternative” phenotype (requiring IκB kinase β-mediated NF-κB activation) to a “classical” phenotype (IL-12 high and IL-10 low). These re-educated “classical macrophages” have been shown to possess a capacity to kill cancer cells while TLR2/TLR4 were activated and NF-κB was inhibited [185, 186].
In addition, TLR7 and TLR8 ligands may also provide a path for cancer immunotherapy in the tumor microenvironment [105].
With the use of curcumin (diferuloylmethane, also called turmeric), it has been revealed that curcumin potentiates the antitumor effect of BCG adjuvant through inactivation of NF-κB and up-regulation of TRAIL (TNF-related apoptosis-inducing ligand) receptors that mediate anti-bladder cancer cells in mice [187, 188]. TRAIL, also known as Apo2 ligand, is a type II transmembrane of the TNF family that induces apoptosis in a variety of tumor cells.
This review has summarized the growing body of evidence showing that TLRs play a key role in host defense against infection by recognizing and killing microbes. This review has also discussed the crucial role of TLR-mediated inflammation and cancer, showing that TLR inappropriate regulation results in chronic inflammation and cancer. Better knowledge of inflammation, cancer and tumor microenvironments and new strategies against cancer cells and tumor microenvironments may provide us new treatments targeting inflammatory disease and cancer [17, 176, 189, 190].
Conclusions
This review describes the important aspects and advances of TLR studies within the last two decades. Through several exciting inroads of TLR biology, we are learning that TLRs play diverse roles in the detection of invading pathogens and the induction of innate and adaptive immune responses that are necessary for the up-regulation of cytokines, MHC molecules, including miRNA expression in macrophages and DCs, through complicated signal pathways. This review article also describes the incidence with which infections and cancers are linked to SNPs in TLRs. Importantly, TLRs and TLR ligands as well as TLR antagonism can be used distinctly as vaccine adjuvants or targets for treatment of inflammatory diseases and cancers [17]. Furthermore, several reports indicate that cancer cells utilize TLR receptors or their signal pathways to escape immune surveillance, which has become a new task for TLR biology research.
References
Chang ZL. Role of Toll-like receptors in regulatory functions of T and B cells. Chin Sci Bull. 2008;53:1121–7.
Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50.
Yang H, Wei J, Zhang H, Lin L, Zhang W, He S. Upregulation of Toll-like receptor (TLR) expression and release of cytokines from P815 mast cells by GM-CSF. BMC Cell Biol. 2009;10:37.
Liu G, Zhang L, Zhao Y. Modulation of immune responses through direct activation of Toll-like receptors to T cells. Clin Exp Immunol. 2010 [Epub ahead of print].
Gururajan M, Jacob J, Pulendran B. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS One. 2007;2:e863.
McGettrick AF, O’Neill LA. Toll-like receptors: key activators of leucocytes and regulator of haematopoiesis. Br J Haematol. 2007;139:185–93.
Pegu A, Qin S, Fallert Junecko BA, Nisato RE, Pepper MS, Reinhart TA. Human lymphatic endothelial cells express multiple functional TLRs. J Immunol. 2008;180:3399–405.
Fitzner N, Clauberg S, Essmann F, Liebmann J, Kolb-Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vaccine Immunol. 2008;15:138–46.
Palladino MA, Savarese MA, Chapman JL, Dughi MK, Plaska D. Localization of Toll-like receptors on epididymal epithelial cells and spermatozoa. Am J Reprod Immunol. 2008;60:541–55.
Miller LS, Modlin RL. Human keratinocyte Toll-like receptors promote distinct immune responses. J Invest Dermatol. 2007;127:262–3.
Miller LS. Toll-like receptors in skin. Adv Dermatol. 2008;24:71–87.
Kurokawa I, Danby FW, Ju Q, Wang X, Xiang LF, Xia L, et al. New developments in our understanding of acne pathogenesis and treatment. Exp Dermatol. 2009;18:821–32.
Ospelt C, Gay S. TLRs and chronic inflammation. Int J Biochem Cell Biol. 2010;42:495–505.
Szajnik M, Szczepanski MJ, Czystowska M, Elishaev E, Mandapathil M, Nowak-Markwitz E, et al. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene. 2009;28:4353–63.
Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, et al. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009;69:3105–13.
Pasare C, Medzhitov R. Toll-like receptors and acquired immunity. Semin Immunol. 2004;16:23–6.
O’Neill LA, Bryant CE, Doyle SL. Therapeutic targeting of toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev. 2009;61:177–97.
Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63.
Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–37.
Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe. 2008;3:352–63.
Cristofaro P, Opal SM. Role of Toll-like receptors in infection and immunity: clinical implications. Drugs. 2006;66:15–29.
Hoebe K, Beutler B. TLRs as bacterial sensors. In: O’Neil L, Brint E, editors. Toll-like receptors inflammation. Berlin: Birkauser Verlag; 2005. p. 1–17.
Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–63.
Belvin MP, Anderson KV. A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol. 1996;12:393–416.
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–83.
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8.
Bassett EH, Rich T. Toll receptors and the renaissance of innate immunity. New York: Kluwer Academic/Plenum Publisher; 2005. p. 1–17.
Ligoxygakis P, Bulet P, Reichhart JM. Critical evaluation of the role of the Toll-like receptor 18-Wheeler in the host defense of Drosophila. EMBO Rep. 2002;3:666–73.
Murphy K, Travers P, Walport M. Janeway’s immuno biology. New York: Garland Science; 2008. p. 39–108.
Murphy K, Travers P, Walport M. Janeway’s immuno biology. New York: Garland Science; 2008. p. 143–79.
Fluhr R, Kaplan-Levy RN. Plant disease resistance: commonality and novelty in multicellular innate immunity. Curr Top Microbiol Immunol. 2002;270:23–46.
Bell JK, Mullen GE, Leifer CA, Mazzoni A, Davies DR, Segal DM. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 2003;24:528–33.
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5.
Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–9.
Hold GL, El-Omar EM. Genetic aspects of inflammation and cancer. Biochem J. 2008;410:225–35.
Roelofs MF, Boelens WC, Joosten LA, Abdollahi-Roodsaz S, Geurts J, Wunderink LU, et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol. 2006;176:7021–7.
Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–78.
Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, et al. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–81.
Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, et al. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics. 2007;8:124.
Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, et al. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–89.
Chaturvedi A, Pierce SK. How location governs Toll-like receptor signaling. Traffic. 2009;10:621–8.
Ishii KJ, Uematsu S, Akira S. ‘Toll’ gates for future immunotherapy. Curr Pharm Des. 2006;12:4135–42.
O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64.
Dellacasagrande J. Ligands, cell-based models, and readouts required for toll-like receptor action. Methods Mol Biol. 2009;517:15–32.
Peter ME, Kubarenko AV, Weber AN, Dalpke AH. Identification of an N-terminal recognition site in TLR9 that contributes to CpG-DNA-mediated receptor activation. J Immunol. 2009;182:7690–7.
Uematsu S, Akira S. Toll-like receptors and Type I interferons. J Biol Chem. 2007;282:15319–23.
Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–5.
Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, et al. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci USA. 2005;102:9577–82.
Henderson B, Poole S, Wilson M. Bacterial modulins: a novel class of virulence factors which cause host tissue pathology by inducing cytokine synthesis. Microbiol Rev. 1996;60:316–41.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801.
Fukata M, Chen AL, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862–77.
Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF, et al. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J Clin Invest. 2007;117:258–69.
Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282:14975–83.
Rakoff-Nahoum S, Medzhitov R. Role of toll-like receptors in tissue repair and tumorigenesis. Biochemistry (Mosc). 2008;73:555–61.
Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7.
Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–9.
Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–61.
Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J Immunol. 2006;176:3885–9.
Danese S. Nonimmune cells in inflammatory bowel disease: from victim to villain. Trends Immunol. 2008;29:555–64.
Parker LC, Prince LR, Sabroe I. Translational mini-review series on Toll-like receptors: networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin Exp Immunol. 2007;147:199–207.
Carpenter S, O’Neill LA. How important are Toll-like receptors for antimicrobial responses? Cell Microbiol. 2007;9:1891–901.
Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7.
Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, et al. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007;27:13033–41.
Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894–901.
Salaun B, Lebecque S, Matikainen S, Rimoldi D, Romero P. Toll-like receptor 3 expressed by melanoma cells as a target for therapy? Clin Cancer Res. 2007;13:4565–74.
van den Berk LC, Jansen BJ, Siebers-Vermeulen KG, Netea MG, Latuhihin T, Bergevoet S, et al. Toll-like receptor triggering in cord blood mesenchymal stem cells. J Cell Mol Med. 2009;13:3415–26.
Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–5.
Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–103.
Feuillet V, Medjane S, Mondor I, Demaria O, Pagni PP, Galan JE, et al. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc Natl Acad Sci USA. 2006;103:12487–92.
Andersen-Nissen E, Hawn TR, Smith KD, Nachman A, Lampano AE, Uematsu S, et al. Cutting edge: Tlr5−/− mice are more susceptible to Escherichia coli urinary tract infection. J Immunol. 2007;178:4717–20.
Nakao Y, Funami K, Kikkawa S, Taniguchi M, Nishiguchi M, Fukumori Y, et al. Surface-expressed TLR6 participates in the recognition of diacylated lipopeptide and peptidoglycan in human cells. J Immunol. 2005;174:1566–73.
Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, et al. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol. 2005;174:2942–50.
Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, et al. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–6.
Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–9.
Lauw FN, Caffrey DR, Golenbock DT. Of mice and man: TLR11 (finally) finds profilin. Trends Immunol. 2005;26:509–11.
Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–8.
Nishiya T, Kajita E, Miwa S, Defranco AL. TLR3 and TLR7 are targeted to the same intracellular compartments by distinct regulatory elements. J Biol Chem. 2005;280:37107–17.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8.
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–73.
Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004;101:3516–21.
Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–7.
Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–407.
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–9.
Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499.
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5.
Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–7.
Prakken AB, van Hoeij MJ, Kuis W, Kavelaars A, Heynen CJ, Scholtens E, et al. T-cell reactivity to human HSP60 in oligo-articular juvenile chronic arthritis is associated with a favorable prognosis and the generation of regulatory cytokines in the inflamed joint. Immunol Lett. 1997;57:139–42.
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34.
Huang QQ, Sobkoviak R, Jockheck-Clark AR, Shi B, Mandelin AM 2nd, Tak PP, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182:4965–73.
Holm CK, Petersen CC, Hvid M, Petersen L, Paludan SR, Deleuran B, et al. TLR3 ligand polyinosinic:polycytidylic acid induces IL-17A and IL-21 synthesis in human Th cells. J Immunol. 2009;183:4422–31.
Girart MV, Fuertes MB, Domaica CI, Rossi LE, Zwirner NW. Engagement of TLR3, TLR7, and NKG2D regulate IFN-gamma secretion but not NKG2D-mediated cytotoxicity by human NK cells stimulated with suboptimal doses of IL-12. J Immunol. 2007;179:3472–9.
MacRedmond R, Greene C, Taggart CC, McElvaney N, O’Neill S. Respiratory epithelial cells require Toll-like receptor 4 for induction of human beta-defensin 2 by lipopolysaccharide. Respir Res. 2005;6:116.
Hart OM, Athie-Morales V, O’Connor GM, Gardiner CM. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol. 2005;175:1636–42.
Sato Y, Goto Y, Narita N, Hoon DS. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron. 2009;2(Suppl 1):205–14.
Szczepanski M, Stelmachowska M, Stryczynski L, Golusinski W, Samara H, Mozer-Lisewska I, et al. Assessment of expression of toll-like receptors 2, 3 and 4 in laryngeal carcinoma. Eur Arch Otorhinolaryngol. 2007;264:525–30.
He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol. 2007;44:2850–9.
Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66:3859–68.
Schmausser B, Andrulis M, Endrich S, Muller-Hermelink HK, Eck M. Toll-like receptors TLR4, TLR5 and TLR9 on gastric carcinoma cells: an implication for interaction with Helicobacter pylori. Int J Med Microbiol. 2005;295:179–85.
Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, et al. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer. 2010;102:908–15.
Molteni M, Marabella D, Orlandi C, Rossetti C. Melanoma cell lines are responsive in vitro to lipopolysaccharide and express TLR-4. Cancer Lett. 2006;235:75–83.
Kundu SD, Lee C, Billips BK, Habermacher GM, Zhang Q, Liu V, et al. The toll-like receptor pathway: a novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate. 2008;68:223–9.
Rhee SH, Im E, Pothoulakis C. Toll-like receptor 5 engagement modulates tumor development and growth in a mouse xenograft model of human colon cancer. Gastroenterology. 2008;135:518–28.
Chang S, Kodys K, Szabo G. Impaired expression and function of toll-like receptor 7 in hepatitis C virus infection in human hepatoma cells. Hepatology. 2010;51:35–42.
Spaner DE, Masellis A. Toll-like receptor agonists in the treatment of chronic lymphocytic leukemia. Leukemia. 2007;21:53–60.
Smits EL, Ponsaerts P, Berneman ZN, Van Tendeloo VF. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist. 2008;13:859–75.
Schon M, Bong AB, Drewniok C, Herz J, Geilen CC, Reifenberger J, et al. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. J Natl Cancer Inst. 2003;95:1138–49.
Lee JW, Choi JJ, Seo ES, Kim MJ, Kim WY, Choi CH, et al. Increased toll-like receptor 9 expression in cervical neoplasia. Mol Carcinog. 2007;46:941–7.
Ilvesaro JM, Merrell MA, Swain TM, Davidson J, Zayzafoon M, Harris KW, et al. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate. 2007;67:774–81.
Droemann D, Albrecht D, Gerdes J, Ulmer AJ, Branscheid D, Vollmer E, et al. Human lung cancer cells express functionally active Toll-like receptor 9. Respir Res. 2005;6:1.
Zhang ZL, Song QB, Lin MQ, Ding YM, Kang XW, Yao Z. Immunomodulated signaling in macrophages: studies on activation of Raf-1, MAPK, cPLA(2) and secretion of IL-12. Sci China Ser C Life Sci. 1997;40:583–92.
Gay NJ, Kubota K. The signal transduction pathway leading from the toll receptor to nuclear localization of dorsal transcription factor. Biochem Soc Trans. 1996;24:35–8.
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7.
Miyake K, Ogata H, Nagai Y, Akashi S, Kimoto M. Innate recognition of lipopolysaccharide by Toll-like receptor 4/MD-2 and RP105/MD-1. J Endotoxin Res. 2000;6:389–91.
Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun. 2008;368:94–9.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80.
Jiang Z, Georgel P, Li C, Choe J, Crozat K, Rutschmann S, et al. Details of Toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc Natl Acad Sci USA. 2006;103:10961–6.
Zhang JS, Feng WG, Li CL, Wang XY, Chang ZL. NF-kappa B regulates the LPS-induced expression of interleukin 12 p40 in murine peritoneal macrophages: roles of PKC, PKA, ERK, p38 MAPK, and proteasome. Cell Immunol. 2000;204:38–45.
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202.
Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–31.
Hamilton T. Molecular basis of macrophage activation: from gene expression to phenotype diversity. In: Lewis BBCE, editor. The macrophage. Oxford: Oxford University Press; 2002. p. 73–102.
Murphy K, Travers P, Walport M. Janeway’s immuno biology. New York: Garland Science; 2008. p. 111–42.
Murphy K, Travers P, Walport M. Janeway’s immuno biology. New York: Garland Science; 2008. p. 1–38.
O’Brien AD, Rosenstreich DL, Scher I, Campbell GH, MacDermott RP, Formal SB. Genetic control of susceptibility to Salmonella typhimurium in mice: role of the LPS gene. J Immunol. 1980;124:20–4.
Hagberg L, Hull R, Hull S, McGhee JR, Michalek SM, Svanborg Eden C. Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect Immun. 1984;46:839–44.
Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–6.
Conley ME. Immunodeficiency: UNC-93B gets a toll call. Trends Immunol. 2007;28:99–101.
Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–62.
Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177:265–75.
Akashi-Takamura S, Miyake K. TLR accessory molecules. Curr Opin Immunol. 2008;20:420–5.
Tam W, Ben-Yehuda D, Hayward WS. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol Cell Biol. 1997;17:1490–502.
Tam W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene. 2001;274:157–67.
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–32.
Tam W, Dahlberg JE. miR-155/BIC as an oncogenic microRNA. Genes Chromosom Cancer. 2006;45:211–2.
Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11.
O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007;104:1604–9.
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5.
Faller M, Guo F. MicroRNA biogenesis: there’s more than one way to skin a cat. Biochim Biophys Acta. 2008;1779:663–7.
Alexiou P, Vergoulis T, Gleditzsch M, Prekas G, Dalamagas T, Megraw M, et al. miRGen 2.0: a database of microRNA genomic information and regulation. Nucleic Acids Res. 2009;38D:137–41.
Sonkoly E, Pivarcsi A. Advances in microRNAs: implications for immunity and inflammatory diseases. J Cell Mol Med. 2009;13:24–38.
Sewer A, Paul N, Landgraf P, Aravin A, Pfeffer S, Brownstein MJ, et al. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinform. 2005;6:267.
Ro S, Song R, Park C, Zheng H, Sanders KM, Yan W. Cloning and expression profiling of small RNAs expressed in the mouse ovary. RNA. 2007;13:2366–80.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–8.
Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–4.
Lu LF, Liston A. MicroRNA in the immune system, microRNA as an immune system. Immunology. 2009;127:291–8.
Taganov KD, Boldin MP, Baltimore D. MicroRNAs and immunity: tiny players in a big field. Immunity. 2007;26:133–7.
Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–45.
Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA. 2009;106:5282–7.
Sheedy FJ, O’Neill LA. Adding fuel to fire: microRNAs as a new class of mediators of inflammation. Ann Rheum Dis. 2008;67(Suppl 3):iii50–5.
Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91.
Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–82.
Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–17.
Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5.
Vasl J, Prohinar P, Gioannini TL, Weiss JP, Jerala R. Functional activity of MD-2 polymorphic variant is significantly different in soluble and TLR4-bound forms: decreased endotoxin binding by G56R MD-2 and its rescue by TLR4 ectodomain. J Immunol. 2008;180:6107–15.
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–91.
Turvey SE, Hawn TR. Towards subtlety: understanding the role of Toll-like receptor signaling in susceptibility to human infections. Clin Immunol. 2006;120:1–9.
Awomoyi AA, Rallabhandi P, Pollin TI, Lorenz E, Sztein MB, Boukhvalova MS, et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol. 2007;179:3171–7.
Gu W, Shan YA, Zhou J, Jiang DP, Zhang L, Du DY, et al. Functional significance of gene polymorphisms in the promoter of myeloid differentiation-2. Ann Surg. 2007;246:151–8.
Hawn TR, Dunstan SJ, Thwaites GE, Simmons CP, Thuong NT, Lan NT, et al. A polymorphism in Toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J Infect Dis. 2006;194:1127–34.
Carvalho A, Cunha C, Carotti A, Aloisi T, Guarrera O, Di Ianni M, et al. Polymorphisms in Toll-like receptor genes and susceptibility to infections in allogeneic stem cell transplantation. Exp Hematol. 2009;37:1022–9.
Higgins SC, Lavelle EC, McCann C, Keogh B, McNeela E, Byrne P, et al. Toll-like receptor 4-mediated innate IL-10 activates antigen-specific regulatory T cells and confers resistance to Bordetella pertussis by inhibiting inflammatory pathology. J Immunol. 2003;171:3119–27.
El-Omar EM, Ng MT, Hold GL. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene. 2008;27:244–52.
Zheng SL, Augustsson-Balter K, Chang B, Hedelin M, Li L, Adami HO, et al. Sequence variants of toll-like receptor 4 are associated with prostate cancer risk: results from the CAncer Prostate in Sweden Study. Cancer Res. 2004;64:2918–22.
Sun J, Wiklund F, Zheng SL, Chang B, Balter K, Li L, et al. Sequence variants in Toll-like receptor gene cluster (TLR6–TLR1–TLR10) and prostate cancer risk. J Natl Cancer Inst. 2005;97:525–32.
Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991;262:3–11.
Okamoto M, Oshikawa T, Tano T, Ahmed SU, Kan S, Sasai A, et al. Mechanism of anticancer host response induced by OK-432, a streptococcal preparation, mediated by phagocytosis and Toll-like receptor 4 signaling. J Immunother. 2006;29:78–86.
Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene. 2008;27:218–24.
Shi Y, White D, He L, Miller RL, Spaner DE. Toll-like receptor-7 tolerizes malignant B cells and enhances killing by cytotoxic agents. Cancer Res. 2007;67:1823–31.
Spaner DE, Shi Y, White D, Shaha S, He L, Masellis A, et al. A phase I/II trial of TLR-7 agonist immunotherapy in chronic lymphocytic leukemia. Leukemia. 2010;24:222–6.
Leonard JP, Link BK, Emmanouilides C, Gregory SA, Weisdorf D, Andrey J, et al. Phase I trial of toll-like receptor 9 agonist PF-3512676 with and following rituximab in patients with recurrent indolent and aggressive non Hodgkin’s lymphoma. Clin Cancer Res. 2007;13:6168–74.
Wysocka M, Benoit BM, Newton S, Azzoni L, Montaner LJ, Rook AH. Enhancement of the host immune responses in cutaneous T-cell lymphoma by CpG oligodeoxynucleotides and IL-15. Blood. 2004;104:4142–9.
Murad YM, Clay TM. CpG oligodeoxynucleotides as TLR9 agonists: therapeutic applications in cancer. BioDrugs. 2009;23:361–75.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Krishnamachari Y, Salem AK. Innovative strategies for co-delivering antigens and CpG oligonucleotides. Adv Drug Deliv Rev. 2009;61:205–17.
Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10.
Hagemann T, Balkwill F, Lawrence T. Inflammation and cancer: a double-edged sword. Cancer Cell. 2007;12:300–1.
Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–12.
Hallam S, Escorcio-Correia M, Soper R, Schultheiss A, Hagemann T. Activated macrophages in the tumour microenvironment-dancing to the tune of TLR and NF-kappaB. J Pathol. 2009;219:143–52.
Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their Toll—the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27:225–33.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Huang B, Zhao J, Li H, He KL, Chen Y, Chen SH, et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005;65:5009–14.
Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431:405–6.
Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107.
Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, et al. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer. 2010;102:908–15.
Balkwill F, Mantovani A. Cancer and inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Ther. 2010;87:401–6.
Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–8.
Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE. Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood. 2009;113:3139–46.
Jagetia GC, Aggarwal BB. “Spicing up” of the immune system by curcumin. J Clin Immunol. 2007;27:19–35.
Kamat AM, Tharakan ST, Sung B, Aggarwal BB. Curcumin potentiates the antitumor effects of Bacillus Calmette-Guerin against bladder cancer through the downregulation of NF-{kappa}B and upregulation of TRAIL receptors. Cancer Res. 2009;69:8958–66.
Herberman RB, Whiteside TL. Summary of the international cancer microenvironment from meeting in Pittsburgh, Pennsylvania on October 3–6, 1999. Cancer Res. 2000;60:1465–9.
Whiteside TL. Immune responses to malignancies. J Allergy Clin Immunol. 2010;125:S272–83.
Acknowledgments
Author thanks Dr. K. Hayden, Dr. C. Tittiger, Dr. W. Yan and Dr. R. Song (University of Nevada, Reno, NV, USA) for helpful discussion and critical review. I also thank Dr. David Segal (Immune Targeting Section, Experimental Immunology Branch, NCI, NIH, Bethesda, USA), and Dr. Bruce Beutler (Department of Genetics, Professor and Chairman at the Scripps Research Institute, CA, USA) for generously providing Figs. 1 and 2, respectively.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Liwu Li.
Rights and permissions
About this article
Cite this article
Chang, Z.L. Important aspects of Toll-like receptors, ligands and their signaling pathways. Inflamm. Res. 59, 791–808 (2010). https://doi.org/10.1007/s00011-010-0208-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-010-0208-2