
Abstract
Objective and design
Toll-like receptor 4 (TLR4) plays important roles in the recognition of lipopolysaccharide (LPS) and the activation of inflammatory cascade. In this study, we evaluated the effect of TAK-242, a selective TLR4 signal transduction inhibitor, on acute lung injury (ALI).
Materials and methods
C57BL/6J mice were intravenously treated with TAK-242 15 min before the intratracheal administration of LPS or Pam3CSK4, a synthetic lipopeptide. Six hours after the challenge, bronchoalveolar lavage fluid was obtained for a differential cell count and the measurement of cytokine and myeloperoxidase levels. Lung permeability and nuclear factor-κB (NF-κB) DNA binding activity were also evaluated.
Results
TAK-242 effectively attenuated the neutrophil accumulation and activation in the lungs, the increase in lung permeability, production of inflammatory mediators, and NF-κB DNA-binding activity induced by the LPS challenge. In contrast, TAK-242 did not suppress inflammatory changes induced by Pam3CSK4.
Conclusion
TAK-242 may be a promising therapeutic agent for ALI, especially injuries associated with pneumonia caused by Gram-negative bacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the last few decades, several studies have indicated that the survival rate for patients with acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) has improved, although the mortality rate remains high, ranging between 25 and 40% [1]. ALI/ARDS may occur in association with direct lung injury, including pneumonia, aspiration of gastric contents, and inhalation of noxious gas, or indirect lung injury, such as sepsis, blood transfusions and shock. Among the various predisposing factors, severe pneumonia is one of the most common causes [2]. Experimental endotoxin (lipopolysaccharide; LPS) administration via the tracheal route has been extensively used to study the pathogenesis of ALI/ARDS following severe pneumonia [3].
Toll-like receptors (TLRs) have been shown to play an essential role in the activation of innate immunity by recognizing specific patterns of microbial components [4]. Among the TLRs, TLR4 recognizes LPS, triggers the activation of an intracellular signaling pathway involving nuclear factor-κB (NF-κB), and results in the upregulation of adhesion molecules and inflammatory mediators, such as cytokines and chemokines. TLR4 mutant (C3H/HeJ) mice have been shown to be hyporesponsive to LPS [5] and do not develop ALI after being challenged with aerosolized LPS [6]. Conversely, the overexpression of TLR4 in transgenic mice augmented the responses to inhaled LPS, including bronchoconstriction, the production of tumor necrosis factor (TNF) and keratinocyte-derived chemokine (KC), lung epithelial and endothelial cell damage, and the recruitment of neutrophils in the lung [7].
To date, many clinical trials have been conducted to examine the efficacy of blocking a single inflammatory mediator in patients with ALI/ARDS, but none of them have shown a sufficient efficacy for clinical application [8, 9]. Since these outcomes could be due to the combined contribution of various pathogenic mediators, we hypothesized that a treatment strategy aimed at inhibiting the upstream inflammatory signaling pathway, such as TLR4, might be reasonable. In fact, a synthetic lipid A analogue that functions as a TLR4 antagonist reportedly showed protective effects against septic shock in mice [10].
TAK-242, a selective TLR4 signal transduction inhibitor, has been shown to suppress LPS-induced inflammatory responses in vitro [11] and lethality after systemic LPS challenge in vivo [12]. In this study, we evaluated the effect of TAK-242 using a murine model of ALI induced by an intratracheal LPS challenge. We examined inflammatory cell recruitment; lung permeability; the levels of pro-inflammatory cytokines, chemokines, myeloperoxidase (MPO) in bronchoalveolar lavage (BAL) fluid; and NF-κB DNA-binding activity. In addition, to elucidate whether the effect of TAK-242 is specifically through the inhibition of the TLR4 pathway, we evaluated the effect of TAK-242 treatment on the inflammatory cell accumulation and cytokine production in the lungs following an intratracheal challenge of Pam3CSK4, a potent activator of TLR2/TLR1 pathway.
Materials and methods
Materials
TAK-242, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (molecular weight; 362 Da), was synthesized at Takeda Pharmaceutical Company Limited (Osaka, Japan). TAK-242 was dissolved in a fat emulsion. Pam3CSK4, (S)-[2,3-Bis (palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-(S)-Ser-(S)-Lys4-OH·3HCl] was purchased from InvivoGen (San Diego, CA).
Mice
Eight-week-old male C57BL/6J mice were obtained from Charles River Laboratories Japan (Yokohama, Japan). Mice were given free access to water and standard rodent chow and were housed in pathogen-free cages. All animal experiments were approved by the Animal Care and Use Committee of Keio University School of Medicine.
Murine model of lung injury
Mice were anesthetized using intraperitoneal ketamine (50 mg/kg) and xylazine (5 mg/kg). During the following procedures, mice were spontaneously breathing. The trachea was exposed surgically and punctuated with a 24-gauge angiocatheter to instill 0.3 mg/kg of LPS from Escherichia coli O55:B5 (Sigma-Aldrich, St. Louis, MO) or phosphate-buffered saline (PBS) into the left lung. Fifteen minutes before the intratracheal administration of LPS, the animals received an intravenous injection of 0.3, 1.0, or 3.0 mg/kg of TAK-242 (LPS + TAK groups) or the vehicle alone (LPS control group) via the tail vein. The sham group animals received an intravenous injection of vehicle alone followed by the intratracheal administration of PBS.
In another series, mice were challenged intratracheally with 0.25 mg/kg of Pam3CSK4 15 min after an intravenous injection of 3.0 mg/kg of TAK-242 (Pam3 + TAK group) or the vehicle alone (Pam3 control group) via the tail vein.
Bronchoalveolar lavage
Six hours after intratracheal instillation, the mice were euthanized with deep anesthesia using intraperitoneal pentobarbital sodium (50 mg/kg). The trachea was exposed and cannulated with a 20-gauge angiocatheter. Both lungs were lavaged with two separate 0.7-mL volumes of ice-cold PBS. The BAL fluid was centrifuged at 400×g for 10 min at 4°C to pellet the cell fraction, and the supernatant was stored at −80°C until the measurements of the cytokines, chemokines, MPO, and human serum albumin (HSA) levels to calculate the permeability index. The cell pellet was resuspended in 400 μL of cold saline, and the total cell counts were determined using a hemacytometer. Differential cell counts were performed using cytocentrifuge smears stained with Diff-Quik (Sysmex, Kobe, Japan).
Measurement of proinflammatory mediators in bronchoalveolar lavage fluid
BAL fluid was assayed for TNF-α, interleukin (IL)-1β, IL-6, macrophage inflammatory protein (MIP)-2 and KC using a multiplex cytokine bead array system (Bio-Plex™; Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. The reaction mixture was read using the Bio-Plex protein array reader, and the data were analyzed using the Bio-Plex Manager software program. Interferon-gamma inducible protein (IP)-10 (CXCL10) in BAL fluid was quantified using an ELISA kit (R&D Systems, Minneapolis, MN). MPO in the BAL fluid was assayed using an ELISA kit (Hycult Biotechnology, Uden, The Netherlands).
Lung permeability index
Mice were given 10 mg/kg of HSA dissolved in 100 μL of saline intravenously, 1 h before euthanasia. At the time of sacrifice, the blood was drawn from the inferior vena cava. The permeability index was defined as the ratio of the HSA concentration in the BAL fluid to that in the plasma, presented as a percentage. The HSA concentration was measured using an immunoassay with a Human Albumin ELISA Quantitation Kit (Bethyl Laboratories, Montgomery, TX). The lower limit of detection was 5 ng/mL.
NF-κB (p65) DNA-binding activity in the lung
Nuclear protein extraction
After performing BAL, the left lungs were harvested and snap frozen in liquid nitrogen and then stored at −80°C until analysis. The lungs were homogenized in 2 mL of ice-cold Buffer A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.5 mM PMSF) with a 0.1% volume of Nonidet P-40 and a protease inhibitor cocktail (1 mg/mL leupeptin, 1 mg/mL aprotinin, 10 mg/mL soy bean trypsin inhibitor, 1 mg/mL pepstatin). Following 10 min of incubation on ice, the homogenates were centrifuged at 850×g for 10 min at 4°C. The pellets were resuspended in 2 mL of Buffer A and centrifuged at 1,200×g for 10 min at 4°C. The crude nuclear pellets were resuspended in 40 mL of Buffer B (20 mM HEPES, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% vol/vol glycerol, 0.5 mM DTT, 0.5 mM PMSF) with a protease inhibitor cocktail (as described above) and incubated for 30 min on ice. Nuclear extracts were recovered following centrifugation at 20,000×g for 15 min at 4°C and stored at −80°C. The protein concentration of the nuclear extracts was determined using a BCA Protein Assay Kit (Pierce, Rockford, IL) with bovine serum albumin used as a standard.
NF-κB (p65) DNA-binding activity assay
NF-κB (p65) DNA-binding activity was examined using the TransAM™ ELISA kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol. In brief, 0.5 μg of nuclear extract was subjected to the binding of NF-κB to an immobilized consensus sequence (5′-GGGACTTTCC-3′) in a 96-well plate, and the primary and secondary antibodies were added. After the colorimetric reaction, the samples were measured in a spectrophotometer at the wavelength of 450 nm. Recombinant NF-κB p65 (Active Motif) was used as a protein standard. The DNA binding specificity was assessed using wild-type or mutated oligonucleotides.
Statistical analysis
Data are presented as the mean ± SEM. All statistical analyses were carried out using SAS software (version 6.1, SAS Institute, Cary, NC). Differences in the differential cell counts, permeability index and mediator levels in the BAL fluid in vehicle-treated versus TAK-242-treated groups were analyzed using a one-tailed Williams or one-tailed Shirley–Williams test. We chose these tests for analysis because Williams’ test is more powerful than Dunnett’s test for dose-related data [13, 14]. Differences between the sham and vehicle-treated groups were analyzed using a Student or Welch t test and were considered statistically significant when P < 0.05.
Results
Inhibitory effect of TAK-242 on inflammatory cell recruitment to the lung
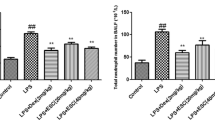
Mice were treated with 0.3 mg/kg of LPS administered into the left lung; 6 h later, BAL fluid was obtained and used to perform a differential cell count. The total cell and neutrophil counts are shown in Fig. 1a, b. LPS instillation induced a significant increase in inflammatory cell recruitment into the alveolar space, compared with the instillation of PBS. Treatment with 3.0 mg/kg of TAK-242 effectively suppressed the total cell count in BAL fluid, compared with the control group (P < 0.01; Fig. 1a). It also markedly reduced the recruitment of neutrophils into the alveolar space (P < 0.01; Fig. 1b).

Inhibitory effect of TLR4 signaling blockade on inflammatory cell recruitment into the lung. Mice received an intravenous injection of 0.3, 1.0, or 3.0 mg/kg of TAK-242 (treatment groups, n = 6 for each group) or vehicle (control group, n = 6), followed by the administration of 0.3 mg/kg of LPS into the left lung; 6 h later, BAL fluid was obtained and used to perform a differential cell count. a TAK-242 (3.0 mg/kg) effectively suppressed the total cell number, compared with the control group. b TAK-242 (3.0 mg/kg) dramatically reduced the recruitment of neutrophils into the alveolar space. Values are the mean ± SD. *P < 0.025 (one-tailed Williams test) compared with the control group (gray bar)
Inhibitory effects of TAK-242 on cytokine, chemokine and MPO levels in BAL fluid
Six hours after LPS instillation, BAL fluid was recovered and the concentration of TNF-α, IL-1β, IL-6, KC, MIP2, IP-10, and MPO in the BAL fluid was measured. LPS instillation markedly increased the levels of these inflammatory cytokines and chemokines in the alveolar space (Fig. 2a–f). The MPO level in the BAL fluid was also significantly enhanced after LPS instillation, indicating neutrophil degranulation in the alveolar space (Fig. 2g). TAK-242 markedly inhibited the LPS-induced increase in TNF-α in a dose-dependent manner, compared with the control group (Fig. 2a). Especially, 3.0 mg/kg of TAK-242 suppressed the elevation of TNF-α by 95%. Lower doses of TAK-242 (0.3 and 1.0 mg/kg) also suppressed the elevation in the TNF-α level by 55 and 80%, respectively, compared with the control group (Fig. 2a). In the mice treated with 3.0 mg/kg of TAK-242, the LPS-induced elevation of the IL-1β level in the BAL fluid was reduced by 60% (Fig. 2b). Lower doses of TAK-242 also reduced the level of IL-1β significantly (Fig. 2b). Treatment with 1.0 and 3.0 mg/kg of TAK-242 inhibited the elevation of IL-6 by 66 and 83%, respectively, compared with the control group (Fig. 2c). KC and MIP-2, murine homologs of the CXC chemokine family, are known to function as neutrophil chemoattractants and activators [15]. Treatment with 3.0 mg/kg of TAK-242 significantly inhibited the upregulation of KC and MIP-2 by 70 and 60%, respectively, compared with the control group (Fig. 2d, e). The level of IP-10, another CXC chemokine, was significantly decreased by TAK-242 treatment in a dose-dependent manner (Fig. 2f). Especially, 3.0 mg/kg of TAK-242 decreased the level of IP-10 to a level similar to that in the sham group. Treatment with 1.0 mg/kg of TAK-242 also inhibited the LPS-induced elevation of IP-10 by 85%, compared with the control group. A dose of 0.3 mg/kg of TAK-242 suppressed the upregulation of IP-10 by 50%, but the difference did not reach statistical significance. The administration of 3.0 mg/kg of TAK-242 markedly reduced the MPO level in the BAL fluid (P < 0.01), suggesting that TAK-242 might inhibit the degranulation of neutrophils (Fig. 2g). Although 0.3 and 1.0 mg/kg of TAK-242 suppressed the level of MPO in the BAL fluid by 30 and 50% respectively, compared with the control group, the difference did not reach statistical significance.

Inhibitory effect of TLR4 signaling blockade on cytokine, chemokine and MPO levels in BAL fluid. Six hours after LPS instillation. BAL fluid was recovered and the amounts of cytokines, chemokines and MPO were measured. a TAK-242 dramatically inhibited the LPS-induced increase in TNF-α in a dose-dependent manner, compared with the control group. b TAK-242 also reduced the level of IL-1β significantly in a dose-dependent manner. c The elevation of IL-6 was effectively inhibited by treatment with 1.0 and 3.0 mg/kg of TAK-242. d TAK-242 (3.0 mg/kg) suppressed the level of MIP-2 significantly. e The level of KC was also suppressed by treatment with 3.0 mg/kg of TAK-242. f The level of IP-10 was strongly suppressed by treatment with TAK-242 in a dose-dependent manner. g Administration of TAK-242 markedly reduced the level of MPO in the BAL fluid. Values are the mean ± SD; n = 6 for each treatment group and vehicle. *P < 0.025 (one-tailed Williams test) compared with the control group (gray bar)
Effect of TAK-242 on LPS-induced increase in lung permeability
The permeability index was calculated as the BAL fluid-to-plasma ratio of the concentration of human albumin that was injected intravenously 1 h before sacrifice. Thus, this index reflects pulmonary endothelial and alveolar septal permeability. The permeability index in the control group was significantly higher than that in the sham group (0.048 ± 0.011 vs. 0.002 ± 0.001%; P < 0.01), and treatment with TAK-242 (3.0 mg/kg) significantly suppressed the elevation of this index, compared with the control group (0.015 ± 0.005 vs. 0.048 ± 0.011%; P < 0.01) (Fig. 3). Although 1.0 mg/kg of TAK-242 decreased the permeability index by 40%, no significant differences in this index were seen between the control group and those treated with 0.3 or 1.0 mg/kg of TAK-242. We observed the exhibition of the animals carefully, but no difference was observed after the instillation.

Effect of TLR4 signaling blockade on LPS-induced increase in lung permeability. The permeability index was calculated as the BAL fluid-to-plasma ratio of the concentration of human serum albumin injected intravenously 1 h before sacrifice. Treatment with 3.0 mg/kg of TAK-242 significantly suppressed the elevation of this index, compared with the control group. Values are the mean ± SD; n = 6 for each treatment group and vehicle. *P < 0.025 (one-tailed Shirley–Williams test) compared with the control group (gray bar)
Inhibitory effect of TAK-242 on NF-κB DNA-binding activity in the lung
To evaluate the effect of TAK-242 on the LPS-induced upregulation of the NF-κB signaling pathway in the lung, nuclear extracts of lung homogenates were analyzed using the TransAM™ ELISA kit. Because the most frequently activated form of NF-κB in TLR signaling is a heterodimer composed of Rel A(p65)-p50 [16] and p50 lacks the transcription activation domain, we used p65 as a marker of NF-κB activation. As shown in Fig. 4, LPS stimulation induced high levels of NF-κB DNA-binding activity. Treatment with 1.0 and 3.0 mg/kg of TAK-242 showed a marked reduction in LPS-induced DNA–protein complex binding activity (Fig. 4). A lower dose (0.3 mg/kg) of TAK-242 did not significantly change the NF-κB DNA-binding activity. Additionally, the binding was specific, since the wild-type consensus oligonucleotide prevented NF-κB binding to the probe immobilized on the plate; conversely, the mutated oligonucleotide had no effects on NF-κB binding (data not shown).

Inhibitory effect of TLR4 signaling blockade on NF-κB DNA-binding activity in the lung. To evaluate the effect of TAK-242 on the LPS-induced upregulation of the NF-κB signal pathway in the lung, nuclear extracts of lung homogenates were analyzed using the TransAM™ ELISA kit. TAK-242-treatment (1.0 and 3.0 mg/kg) produced a marked reduction in LPS-induced DNA–protein complex binding activity. Values are the mean ± SD; n = 6–7 for each treatment group and vehicle. *P < 0.025 (one-tailed Williams test) compared with the control group (gray bar)
Effects of TAK-242 in Pam3CSK4-induced lung inflammation
To elucidate whether the effect of TAK-242 is specifically through the inhibition of TLR4 pathway, we evaluated the effect of TAK-242 on the inflammatory cell accumulation and cytokine production in the lungs following an intratracheal challenge of Pam3CSK4, a potent activator of TLR2/TLR1 pathway. Pam3CSK4 induced marked infiltration of inflammatory cells into the alveolar space (Fig. 5a, b) as well as inflammatory mediator production including IL-1β, IL-6 and KC (Fig. 6a–c). Treatment with TAK-242 did not inhibit the infiltration of inflammatory cells and the release of inflammatory mediators into the alveolar space elicited by Pam3CSK4.

Pam3CSK4-induced inflammatory cell accumulations into the alveolar space. Mice received an intravenous injection of 3.0 mg/kg of TAK-242 or vehicle, followed by intratracheal administration of 2.5 mg/kg of Pam3CSK4 into the left lung; 6 h later, BAL fluid was obtained for a differential cell count. a Total cell counts in BAL fluid were not changed by treatment with TAK-242. b The recruitment of neutrophils into the alveolar space after Pam3CSK4 challenge did not differ between the groups. Values are the mean ± SE; n = 4–5 for each group. *P < 0.05 compared with the sham group (open bar)

Cytokine and chemokine levels in BAL fluid following Pam3CSK4 challenge. Six hours after Pam3CSK4 instillation, BAL fluid was recovered and the levels of inflammatory mediators were measured. Pam3CSK4 induced significant upregulation of a IL-1β, b IL-6, and c KC. The elevation of these mediators was not inhibited by treatment with 3.0 mg/kg of TAK-242. Values are the mean ± SE; n = 4–5 for each group. *P < 0.05 compared with the sham group (open bar)
Discussion
In this study, we demonstrated that TAK-242 inhibited LPS-induced neutrophil recruitment and activation in the lung and lessened the LPS-induced increase in lung permeability. In addition, it attenuated the release of pro-inflammatory mediators into the alveolar space and inhibited the activation of the NF-κB signaling pathway. These results suggest that TAK-242, a specific inhibitor of TLR4 signaling, might be effective for the alleviation of LPS-induced lung injury. To the best of our knowledge, this is the first report on the effect of a synthesized TLR4 signaling inhibitor with a small molecular size on the production of inflammatory mediators and tissue injury after a locally administered inflammatory stimulus.
TLR4 has been shown to play a critical role in LPS recognition and subsequent signal transduction [17]. The activation of signaling requires CD14-bound LPS to adhere to and to transactivate the TLR4-myeloid differentiation protein (MD)-2 complex on the cell membrane [18–20]. TLR4 recruits four adaptor proteins, including myeloid differentiation factor 88 (MyD88), Toll/IL-1 receptor (TIR)-associated protein (TIRAP), TIR-domain-containing adaptor protein-inducing IFN-γ (TRIF) and TRIF-related adaptor molecule (TRAM), with TIR domains. These interactions trigger downstream signaling cascades leading to the activation of NF-κB, which controls the induction of pro-inflammatory cytokines and chemokines such as TNF-α, IL-1β, IL-6, IL-8, and IFN-γ [21]. Therefore, to suppress the inflammation cascade effectively, we hypothesized that blocking a point upstream of TLR4 signaling might be superior to blocking each mediator.
In the present study, TAK-242 effectively inhibited the nuclear translocation of NF-κB and the release of TNF-α, IL-1β, IL-6, KC, MIP-2 and IP-10 in the alveolar space in a dose-dependent manner. Although TAK-242 was reported to selectively suppress the TLR4 signal, commercial preparations of LPS may have contamination that could function as a TLR2 agonist [22, 23]. Therefore, we evaluate whether the inhibitory effects of TAK-242 were truly through inhibition of TLR4 signaling, using Pam3CSK4 that represents the N-terminal part of bacterial lipopeptide. Pam3CSK4, a potent activator of TLR2/TLR1, is known as an important tool for studying the TLR2-specific immune recognition mechanism because it is free of other contaminating bacterial components [24]. We observed that intratracheal challenge of Pam3CSK4 induced marked inflammatory cell accumulation and release of inflammatory mediators into the alveolar space. Since the TAK-242 treatment made no change in the Pam3CSK4-induced inflammatory response, we considered that the inhibitory effect of TAK-242 on LPS-induced lung injury could be through selective blockade of TLR4 signaling pathway.
To date, the beneficial effects of inhibiting TLR4 signaling have been shown in some experimental acute injury models using TLR4 mutant mice [6] and a lipid A analogue [10]. Since previous investigations have revealed no effect of TAK-242 on the function of the extracellular components (MD2 and CD14) and intracellular adaptor proteins (MyD88, TIRAP, TRIF and TRAM), the inhibitory effects of TAK-242 are considered to be due to the inhibition of signaling mediated by the intracellular domain of TLR4, such as the TIR domain [11, 25]. Mutational analysis using TLR4 mutants indicated that TAK-242 inhibits TLR4 signaling by binding to Cys747 in the intracellular domain of TLR4 [26]. Because of the small molecular size of 362 Da, TAK-242 can penetrate into tissues and cells and act directly on intracellular signaling pathways. TAK-242 inhibited MyD88-independent pathways as well as MyD88-dependent pathways and its inhibitory effect was largely unaffected by LPS concentration and types of TLR4 ligands [26]. TAK-242 had no effect on the LPS-induced conformational change of TLR4-MD-2 and TLR4 homodimerization. Therefore, TAK-242 might be effective in a variety of clinical settings.
The inflammatory cascade in ALI/ARDS is initiated by several inflammatory mediators, including pro-inflammatory cytokines and chemokines [27]. For example, TNF-α and IL-1 are early response cytokines that are produced in response to inflammatory stimuli, such as LPS or other microbial products [28]. In this study, TAK-242 strongly inhibited the production of these cytokines that promote the production of other mediators by macrophage, endothelial cells, fibroblasts, and epithelial cells.
In this study, we administered TAK-242 15 min before the intratracheal instillation. We previously showed that TAK-242 inhibited LPS lethality when administered 1 h before or simultaneous with intravenous LPS [12]. In this study, we examined whether TAK-242 is similarly effective on intratracheal stimuli. The time point of treatment was chosen because TAK-242 is supposed to be distributed in the whole body within 15 min.
Neutrophils have been recognized as important contributors to the pathogenesis of ALI/ARDS [29–31]. In addition, LPS is known to induce a large influx of neutrophils into the alveolar space [32]. In rodents, the two most important chemokines for neutrophil recruitment into the lung are KC and MIP-2 [15]. Since the production of these chemokines was significantly inhibited in the groups treated with TAK-242, we speculated that TAK-242 might attenuate neutrophil accumulation mainly via this inhibitory effect. In addition, TAK-242 treatment reduced the level of MPO, a parameter of neutrophil activation, in BAL fluid. It was indicated that TAK-242 might inhibit not only neutrophil accumulation, but also activation or degranulation in the alveolar space.
Since TAK-242 reduced the level of MPO in BAL fluid, treatment with TAK-242 has been suggested to inhibit not only neutrophil infiltration, but also activation or degranulation in the alveolar space. However, whether the effect of TAK-242 on neutrophil activation occurs through a direct effect on neutrophils or via an inhibitory effect on pro-inflammatory cytokines and chemokines remains to be determined.
Another limitation of our study was that we used LPS, not live bacteria, as an insult to induce lung injury. The TLR4 signaling pathway is important for host defense, especially against Gram-negative bacteria, as shown by the impaired defense of TLR4 mutant mice with pneumonia arising from infection with Klebsiella pneumoniae [33] and Bordetella bronchiseptica [34]. However, not all studies have shown that TLR4 is essential for adequate pulmonary host defense. For example, TLR4 mutant mice showed no difference in bacterial clearance following intranasal inoculation with Legionella pneumophilia, compared with a related substrain wild type for TLR4 [35]. TLR4 mutant mice also showed reduced inflammatory responses with no impairment in their ability to eliminate E. coli from the lungs [36]. Therefore, TLR4 may not be necessary for lung host defense against all Gram-negative bacteria, and we think that the inhibitory effects of TAK-242 on TLR4 signaling observed in the present study were not overestimated by our use of a lung injury model induced by LPS, and not live bacteria. In addition, we did not examine the effect of TAK-242 at later time points, because we focused on its effect on acute phase of lung injury. A preliminary experiment showed, however, a significant difference in the cell count in BAL fluid that was collected 12 h after LPS challenge [mean ± 1 SEM for the mice without TAK-242 treatment: 22.0 ± 1.1 (×104 per mL), whereas for those treated with 3.0 mg/kg of TAK-242: 8.5 ± 3.2 (×104 per mL); P < 0.01]. It was suggested that TAK-242 treatment might exert a beneficial effect on LPS-induced lung injury at later time points, which will be a subject of future investigation.
In conclusion, TAK-242 suppressed the LPS-induced production of inflammatory mediators, neutrophil recruitment in the lung, and the development of lung injury in a murine lung injury model. Neutrophils are responsible for both host defense against bacterial pathogens and tissue injury by releasing elastase and reactive oxygen species. In the clinical practice, most of the patients with ALI are treated with antibiotics. We think that, at least when bacterial activity is controlled by antibiotic therapy, blockade of the TLR4 signaling pathway might attenuate neutrophil-mediated lung injury rather than worsen bacterial infection.
References
McIntyre RC Jr, Pulido EJ, Bensard DD, Shames BD, Abraham E. Thirty years of clinical trials in acute respiratory distress syndrome. Crit Care Med. 2000;28:3314–31.
Bersten AD, Edibam C, Hunt T, Moran J. Incidence and mortality of acute lung injury and the acute respiratory distress syndrome in three Australian states. Am J Respir Crit Care Med. 2002;165:443–8.
Brigham KL, Meyrick B. Endotoxin and lung injury. Am Rev Respir Dis. 1986;133:913–27.
Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9.
Morrison DC, Ryan JL. Bacterial endotoxins and host immune responses. Adv Immunol. 1979;28:293–450.
Jeyaseelan S, Chu HW, Young SK, Freeman MW, Worthen GS. Distinct roles of pattern recognition receptors CD14 and Toll-like receptor 4 in acute lung injury. Infect Immun. 2005;73:1754–63.
Togbe D, Schnyder-Candrian S, Schnyder B, Couillin I, Maillet I, Bihl F, et al. TLR4 gene dosage contributes to endotoxin-induced acute respiratory inflammation. J Leukoc Biol. 2006;80:451–7.
Abraham E, Laterre PF, Garbino J, Pingleton S, Butler T, Dugernier T, et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase iii trial with 1,342 patients. Crit Care Med. 2001;29:503–10.
Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The interleukin-1 receptor antagonist sepsis investigator group. Crit Care Med. 1997;25:1115–24.
Shiozaki M, Doi H, Tanaka D, Shimozato T, Kurakata S. Syntheses of glucose analogues of E5564 as a highly potent anti-sepsis drug candidate. Bioorg Med Chem. 2006;14:3011–6.
Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, et al. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits Toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol Pharmacol. 2006;69:1288–95.
Sha T, Sunamoto M, Kitazaki T, Sato J, Ii M, Iizawa Y. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur J Pharmacol. 2007;571:231–9.
Shirley E. A non-parametric equivalent of Williams’ test for contrasting increasing dose levels of a treatment. Biometrics. 1977;33:386–9.
Williams DA. A note on Shirley’s nonparametric test for comparing several dose levels with a zero-dose control. Biometrics. 1986;42:183–6.
Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28.
Hayden MS, West AP, Ghosh S. NF-κB and the immune response. Oncogene. 2006;25:6758–80.
Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274:10689–92.
Ulevitch RJ, Tobias PS. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–57.
Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–82.
Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–3.
Christman JW, Lancaster LH, Blackwell TS. Nuclear factor kappa B: a pivotal role in the systemic inflammatory response syndrome and new target for therapy. Intensive Care Med. 1998;24:1131–8.
Xu WY, Wang L, Wang HM, Wang YQ, Liang YF, Zhao TT, et al. TLR2 and TLR4 agonists synergistically up-regulate SR-A in RAW264.7 through p38. Mol Immunol. 2007;44:2315–23.
Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–22.
Xue Y, Yun D, Esmon A, Zou P, Zuo S, Yu Y, et al. Proteomic dissection of agonist-specific TLR-mediated inflammatory responses on macrophages at subcellular resolution. J Proteome Res. 2008;7:3180–93.
Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. Tak-242 selectively suppresses toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40–8.
Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, et al. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol. 2009;157(7):1250–62.
Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–56.
Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–26.
Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150:113–22.
Parsons PE, Fowler AA, Hyers TM, Henson PM. Chemotactic activity in bronchoalveolar lavage fluid from patients with adult respiratory distress syndrome. Am Rev Respir Dis. 1985;132:490–3.
Matthay MA. Conference summary: acute lung injury. Chest. 1999;116:119S–26S.
Reutershan J, Ley K. Bench-to-bedside review: acute respiratory distress syndrome—how neutrophils migrate into the lung. Crit Care. 2004;8:453–61.
Schurr JR, Young E, Byrne P, Steele C, Shellito JE, Kolls JK. Central role of Toll-like receptor 4 signaling and host defense in experimental pneumonia caused by gram-negative bacteria. Infect Immun. 2005;73:532–45.
Mann PB, Kennett MJ, Harvill ET. Toll-like receptor 4 is critical to innate host defense in a murine model of bordetellosis. J Infect Dis. 2004;189:833–6.
Lettinga KD, Florquin S, Speelman P, van Ketel R, van der Poll T, Verbon A. Toll-like receptor 4 is not involved in host defense against pulmonary Legionella pneumophila infection in a mouse model. J Infect Dis. 2002;186:570–3.
Lee JS, Frevert CW, Matute-Bello G, Wurfel MM, Wong VA, Lin SM, et al. TLR-4 pathway mediates the inflammatory response but not bacterial elimination in E. coli pneumonia. Am J Physiol Lung Cell Mol Physiol. 2005;289:L731–8.
Acknowledgments
We thank Miyuki Yamamoto of Keio University School of Medicine and Masumi Uekata of Takeda Pharmaceutical Co. Ltd. for their technical assistance. This study was supported in part by a grant-in-aid for scientific research from the Ministry of Health, Labour and Welfare of Japan (no. 19590913) (S. T.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: K. Visvanathan.
Rights and permissions
About this article
Cite this article
Seki, H., Tasaka, S., Fukunaga, K. et al. Effect of Toll-like receptor 4 inhibitor on LPS-induced lung injury. Inflamm. Res. 59, 837–845 (2010). https://doi.org/10.1007/s00011-010-0195-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-010-0195-3