
Abstract
Objective
We questioned whether infection with murine gammaherpesvirus 68 (HV-68) might exacerbate inflammatory bowel disease using mice deficient in IL-10 (IL-10−/−) as a model of developing colitis.
Methods
Groups of C57BL/6 mice and IL-10−/− mice were mock-treated or infected with HV-68. Two months following infection, mice were euthanized and a variety of parameters were measured to quantify the extent of inflammation and the presence of virus. Measurements included survival, body weight, splenomegaly, colonic disease scores, liver histopathology, viable bacteria in the liver, and splenic viral burden.
Results
IL-10−/− mice infected with HV-68 displayed reduced survival, lower body weights, increased splenomegaly, exacerbated colonic disease scores, increased numbers of viable bacteria in the liver, and increased leukocyte liver infiltration when compared to mock-treated IL-10−/− mice or HV-68 infected C57BL/6 mice. Surprisingly, levels of infectious or latent virus were not significantly different between the groups of mice exposed to HV-68.
Conclusions
The presence of HV-68 in IL-10−/− mice exacerbates the developing clinical disease in this animal model of colitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Infection with human gammaherpesviruses has been associated with the exacerbation of a variety of inflammatory diseases. For example, Epstein Barr virus (EBV) infection has been implicated as a contributing factor for the severity of a variety of autoimmune disorders including multiple sclerosis [1–3], systemic lupus erythematosus [2, 4], and rheumatoid arthritis [2, 5]. It is important to note that investigators are not suggesting that EBV is the direct cause of any of these autoimmune diseases. Rather, the immune dysregulation caused by this virus during a lifetime of reactivation [6] has been postulated to contribute to the exacerbation of autoimmunity.
EBV can also contribute to the severity of bacterial co-infections. The most studied example of this relationship comes from investigations of periodontal disease [7–9]. Numerous studies have clearly demonstrated the presence of EBV along with multiple bacterial species in this clinical disease, and the presence of EBV in oral tissues correlates with severity. In addition, bacterial infections of tonsilar tissue [10, 11] and nasopharynx [12] associated with EBV infection have been reported. There have also been a few reported cases where EBV infection has been associated with sepsis [13–15].
In light of the association of EBV with inflammatory diseases, it is not surprising that the presence of this virus has been linked to inflammatory bowel diseases. In several studies, investigators have concluded that co-infection with EBV correlates with the presence or severity of inflammatory bowel disease in some patients [16–22]. Clearly EBV infection does not cause colitis, however, the presence of EBV in intestinal lesions, and increases in EBV-specific antibodies, have led some investigators to suggest that one environmental factor that could be responsible for exacerbating this inflammatory disease might be co-infection with this virus.
Unfortunately, a definitive association between EBV and its role in exacerbating inflammatory bowel disease will be difficult to prove in patients for several reasons. It has been estimated that 95% of adults have been exposed to the virus [23]. However, despite exposure, recent studies demonstrate that EBV viral loads differ greatly from individual to individual [24], making some researchers suggest that more recent and sensitive methods of viral quantification need to be utilized to assess a patient’s predeposition for EBV-associated diseases. Stated simply, some individuals pay more of a price for harboring this latent herpesvirus than do others [25, 26]. Furthermore, it is highly likely that other factors such as genetics, diet, and other preexisting medical conditions will influence the impact that this viral infection has on immune dysregulation. Thus, even though EBV is ubiquitous, susceptibility to virus-exacerbated diseases will vary greatly within the population based on multiple factors. Such a complexity of influences, however, does not diminish the potential for EBV to contribute to the development and/or increased severity of inflammatory bowel diseases.
Fortunately, a rodent model of gammaherpesvirus infection exists that can be used to investigate viral-induced exacerbation of inflammatory bowel disease. Murine gammaherpesvirus 68 (HV-68) has been used as a mouse model for EBV and human herpesvirus 8 infections [27–30] and is the only well-characterized rodent model for studying gammaherpesviruses. Upon intranasal or oral inoculation in mice [31–36], there is a productive infection of epithelial cells, followed by infection of B lymphocytes, and also macrophages and dendritic cells. A marked leukocytosis (i.e., mononucleosis) and splenomegaly occur, which peak around days 15–20 postinfection. This leukocytosis results from expansions of CD4+ T lymphocytes, CD8+ T lymphocytes, and B lymphocytes, the majority of which are not specific for viral antigens. Within the first week following infection, latent herpesvirus can be detected in B lymphocytes, as is the case following infections of humans with EBV. Given an appropriate stimulus, gammaherpesviruses can re-emerge from latency, resulting in a productive infection and re-establishment of latency. This virus cycle ensures that the host remains infected for life [6]. The pathophysiology of HV-68 closely mimics that observed for EBV infections [27–30], making it an excellent model to investigate gammaherpesvirus pathogenesis.
In the present study, we studied whether infection with HV-68 might exacerbate inflammatory bowel disease using mice deficient in IL-10 (IL-10−/−) as a model of developing colitis [37–39]. We report here that IL-10−/− mice infected with HV-68 developed more rapid and more severe inflammatory bowel disease.
Materials and methods
Murine gammaherpesvirus 68 (HV-68) propagation and isolation
HV-68 was kindly provided by Dr. Anthony Nash (University of Edinburgh, Edinburgh, UK) and Dr. Peter Doherty (St. Jude’s Hospital, Memphis, TN). Virus stock was prepared by infecting BHK-21 cells [American Type Culture Collection (ATCC), Rockville, MD; CCL-10) with HV-68 at a low multiplicity of infection, followed by isolation of virus as previously described [31–36]. Replicating virus present in viral stocks was quantified using serial dilutions on NIH-3T3 cell (ATCC CRL 1658) monolayers.
Intranasal inoculation with HV-68
Groups of 6-week-old C57BL/6 mice and B6.129P2-IL10tm1cgn (IL-10−/−) mice (Jackson Laboratory, Bar Harbor, ME) were bred and housed under specific pathogen-free conditions. Intranasal inoculations with HV-68 were performed as previously described [31–36]. Briefly, mice were anesthetized and allowed to aspirate, via the nasal passages, 20 ml of inoculum containing 6,000 plaque-forming units of HV-68. Groups of control mice were mock-treated using intranasal exposure with a similar amount of UV-killed virus. Mice were allowed to recover from the anesthesia prior to being returned to their cages. Mice were given food and water ad libitum and were housed in isolation cages throughout the experimental period.
Survival following HV-68 infection
Groups of IL-10−/− mice (n = 6) were weighed and monitored for activity following mock treatment or infection with HV-68. Following weighing, an assessment of the activity–inactivity continuum was made. Continual locomotion, rearing, or grooming movements for 15 s were scored as 4. Movement for 10 of 15 s was scored as 3. Movement for 5 of 15 s was scored as 2. No movement for 15 s was scored as 1. Mice were considered moribund and euthanized as a humane endpoint when they had lost 20% of their body weight when compared to controls and also had an activity score of less than 2.0. Statistical significance in survival analyses (i.e., moribund as defined above) were performed using Kaplan–Meier survival curves.
Colonic disease scores, cholangitis, and liver histopathology
Intestine and liver tissue were removed for histological analyses 2 months following mock treatment or HV-68 infection. Tissues were fixed in 10% neutral buffered formalin, imbedded, sectioned at 5 μm, and stained with hematoxylin and eosin for light microscopic examination.
Colon sections were scored for colonic disease similar to that previously described [40]. For scoring, grade 0 represented no change from normal tissue, grade 1 had a few foci of mononuclear infiltrates with limited epithelial cell hyperplasia, grade 2 lesions were frequent with mild epithelial cell hyperplasia, grade 3 lesions involved frequent large areas of mucosa with submucosa involvement, grade 4 lesions had severe inflammation including ulcers with marked epithelial cell hyperplasia. At least three colon segments were scored for each mouse, and the average colonic score was determined for each group.
Cholangitis was defined as clear obstruction of bile flow out of the gallbladder as observed following euthanasia.
Bacterial colony counts present in liver tissue
Liver tissue (25 mg) was taken from each mouse following euthanasia and homogenized in 0.5% Triton X-100. Dilutions of each homogenate were plated onto Luria agar plates, and the plates incubated in reduced O2 for 48 h. Bacterial colonies were counted to quantify viable bacteria present in liver tissue.
Several bacterial colonies were selected from each plate for identification using API-20 strips (Fisher, Fair Lawn, NJ).
Leukocytosis, splenomegaly, and isolation of splenic leukocytes for quantification of viral burden
Groups of mice (n = 4–6) were killed 2 months following mock treatment or infection with HV-68. Blood was collected, red blood cells lysed, and total white blood cell counts performed using a Coulter Counter (Coulter, Miami, FL).
Spleens were removed, weighed, and single-cell suspensions made for quantification of infectious and latent virus burdens. Single-cell suspensions were made by pressing tissue through a 30-gauge wire mesh screen, followed by hypotonic lysis. Splenocytes were washed, and the pellet resuspended in RPMI-1640 with 1% fetal calf serum. Total leukocytes were counted using a Coulter Counter.
Quantification of infectious virus was performed as previously described [31–36]. Briefly, isolated splenic leukocytes were pulse sonicated (Vibra Cell, Newtown, CT) to release intracellular virus. After sonication, lysates were centrifuged at 2,500g to remove cellular debris. Limiting dilutions of the lysates were then placed on NIH-3T3 monolayers for 1 h followed by washing and overlaying with 0.15% agar (Difco, Detroit, MI) in RPMI-1640 with 30% fetal calf serum (FCS). After 5 days, overlays were removed, and cell monolayers were stained with crystal violet. Plaque-forming units were quantified in duplicate at several serial dilutions of lysate to assure accuracy.
Quantification of latent virus was performed as previously described [31–36] using an infectious-centers assay. Briefly, limiting dilutions of isolated splenic leukocytes were placed onto monolayers of NIH-3T3 cells. After 24 h, an agar overlay supplemented with medium and calf serum was added and allowed to incubate in culture for 5 days. The monolayers were then fixed and stained with crystal violet, and the number of infectious centers was counted in duplicate for several dilutions of cells for each experimental condition.
FACS analysis of splenic leukocyte populations
The phenotypic characterization of splenocytes was performed using a FACSCalibur instrument (BD, Franklin Lakes, NJ) and CellQuest software as previously described [33]. Briefly, splenocytes were isolated as described above, washed with PBS and resuspended in 10% nonimmune rabbit serum (Zymed, San Francisco, CA), and FcBlock (anti-mouse CD16/CD32, BD Biosciences, San Jose, CA) for 20 min at 4°C. FITC-conjugated anti-mouse CD3, anti-mouse B220, anti-mouse CD11b, or anti-mouse CD11c antibodies (BD Biosciences) were added to separate aliquots of cells, and incubated for 30 min at 4°C in the dark. Control samples were prepared using isotype-matched, irrelevant antibodies that were fluorochrome-conjugated (BD Biosciences). Cells were washed three times, resuspended in PBS, and kept at 4°C in the dark until FACS analyses were performed. For each sample, data from 10,000 volume-gated viable cells were collected.
Statistical analyses
Results were tested statistically by one-way ANOVA or Student’s t test whenever appropriate and were determined to be statistically significantly when P < 0.05 was obtained.
Results
IL-10−/− mice succumb to HV-68 infection
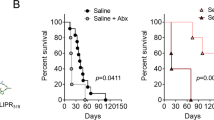
To investigate the ability of HV-68 infection to exacerbate inflammatory bowel disease in IL-10−/− mice, groups of 6-week-old deficient mice (n = 6) were mock-treated or infected with HV-68. To our surprise, approximately 2–3 months following treatment, the infected IL-10−/− mice began to succumb (Fig. 1) to what appeared to be a lethal wasting disease. Conversely, the mock-treated IL-10−/− mice appeared relatively normal. This result was consistent with previous reports of IL-10−/− mice on the C57BL/6 background, which are relatively resistant to developing colitis and have little overt disease at approximately 4 months of age [40].

Survival in HV-68-infected IL-10−/− mice. Groups (n = 6) of 6-week-old IL-10−/− mice were mock-treated (Mock) or infected with murine gammaherpsesvirus 68 (HV-68). A moribund nature as a humane survival endpoint was determined over time
Exaggerated clinical disease in IL-10−/− mice infected with HV-68
To begin to understand the significant differences in survival observed in our initial studies, we documented differences in the inflammatory responses of these mice using a variety of parameters. For these studies, groups of 6-week-old C57BL/6 or IL-10−/− mice (n = 4–6) were mock-treated or infected with HV-68. At 2 months post-treatment, mice were weighed and then euthanized to evaluate the extent of disease. Figure 2 shows that, at the time of euthanasia, IL-10−/− mice had significant reductions in total body weight when compared to all other groups of mice. This was consistent with our initial observation of a lethal wasting disease.

Loss of body weight in HV-68-infected IL-10−/− mice. Groups of 6-week-old normal C57BL/6 mice (C57) or IL-10-deficient mice (IL-10−/−) were mock-treated (Mock) or infected with murine gammaherpesvirus 68 (HV-68) as indicated. Two months after infection, body weights were determined. Results are presented as mean values (±SD). The asterisk indicates a statistically significant difference when compared to all other groups of mice. This study was repeated twice with similar results
A gross splenomegaly and leukocytosis were also observed in IL-10−/− mice infected with HV-68. Figure 3a shows significant increases in spleen weights in these mice compared to all other groups. Representative photomicrographs (Fig. 3b) show the dramatic increase in the size of spleens isolated from IL-10−/− mice infected with HV-68. This splenomegaly was also accompanied by a marked leukocytosis as mock-treated IL-10−/− mice had white blood cell counts of 2.2 (± 0.09) × 106/ml compared to 6.9 (± 0.65) × 106/ml.

Splenomegaly in HV-68-infected IL-10−/− mice. Groups of 6-week-old normal C57BL/6 mice (C57) or IL-10-deficient mice (IL-10−/−) were mock-treated (Mock) or infected with murine gammaherpesvirus 68 (HV-68) as indicated. Two months following infection, mice were euthanized and spleen weights were determined. Results in a are presented as mean values (±SD). The asterisk indicates a statistically significant difference when compared to all other groups of mice. b Representative photomicrographs of the size of spleens isolated from mice treated as indicated. This study was repeated three times with similar results
FACS analysis of CD3+, B220+, CD11b+, and CD11c+ cells in the spleens of IL-10−/− mice infected with HV-68 showed significant increases in the number of each of these subpopulations when compared to all other groups (data not shown). However there were no significant differences in the percentage of cells in each subpopulation when comparing groups, suggesting that all subpopulations were expanded proportionally.
A significant leukocytic infiltration into the livers of IL-10−/− mice infected with HV-68 was also observed. Figure 4 shows representative histology from the liver sections of treated mice, demonstrating increased mononuclear cell infiltration in infected IL-10−/− mice. These mice also had a significantly higher incidence of cholangitis than control groups. In one experiment, all six IL-10−/− mice infected with HV-68 had obvious gallbladder obstruction, while only one of six mock-treated IL-10−/− mice had such an obstruction.

Leukocytic infiltration in the liver of HV-68-infected IL-10−/− mice. Groups of 6-week-old normal C57BL/6 mice (C57) or IL-10-deficient mice (IL-10−/−) were mock-treated (Mock) or infected with murine gammaherpesvirus 68 (HV-68) as indicated. Two months following infection, mice were euthanized and liver tissue taken for histology. Results are presented as representative photomicrographs of liver tissue following staining with hematoxylin and eosin
Finally, significant increases in colonic disease were observed in IL-10−/− mice infected with HV-68 when compared to mock-treated mice (Fig. 5a). Microscopic inspection of colon sections typically showed grade 3 to grade 4 lesions [40], with large areas of the mucosa and submucosa showing inflammation and epithelial cell hyperplasia. While some inflammation could be seen in mock-treated IL-10−/− mice at this age (i.e., 3.5 months), it was much less severe than virally infected mice. Figure 5b shows representative photomicrographs of the gross intestinal pathology observed in these mice. Thickening of the bowel wall in response to the inflammatory response was readily visible in IL-10−/− mice infected with HV-68.

Colonic disease in HV-68-infected IL-10−/− mice. Groups of 6-week-old, normal C57BL/6 mice (C57) or IL-10-deficient mice (IL-10−/−) were mock-treated (Mock) or infected with murine gammaherpesvirus 68 (HV-68) as indicated. Two months following infection, mice were euthanized and colonic disease scores were determined (a). Results are presented as mean values (±SD). The asterisk indicates a statistically significant difference when compared to all other groups of mice. b Representative photomicrographs of colon and cecum segments from mice treated as indicated
Infectious and latent viral burden in IL-10−/− mice infected with HV-68
One possible explanation for the exacerbated inflammatory response in IL-10−/− mice infected with HV-68 might be the reactivation of latent virus during developing disease. The persistent presence of infectious virus can result in systemic inflammation and death if uncontrolled, as occurs in interferon alpha/beta receptor-deficient mice [41]. Therefore we questioned whether the levels of infectious virus or latent viral burden were significantly different in these groups of mice. As illustrated in Fig. 6, no significant differences in infectious virus (Fig. 6a) or latent viral burden (Fig. 6b) were observed when comparing IL-10−/− mice and C57BL/6 mice. Therefore differences in viral burden could not explain the exacerbated inflammation observed.

Quantification of infectious and latent viral burdens in HV-68-infected mice. Groups of 6-week-old normal C57BL/6 mice (C57) or IL-10-deficient mice (IL-10−/−) were mock-treated (Mock) or infected with murine gammaherpesvirus 68 (HV-68) as indicated. Two months following infection, mice were euthanized and spleens were removed for quantification of infectious (a) and latent (b) viral burdens. To quantify infectious virus, splenic tissue was homogenized and subjected to a plaque-forming cell assay (a). To quantify latent virus, splenic leukocytes were isolated and subjected to an infectious-centers assay (b). Results are presented as mean values (±SD). This study was repeated twice with similar results
Increased viable bacteria in livers of IL-10−/− mice infected with HV-68
As colitis develops, translocation of bacteria from the inflamed gut into the portal circulation has been used to explain some aspects of liver and gall bladder disease in these patients [42–45]. Therefore we questioned whether the increased incidence of cholangitis and liver disease in IL-10−/− mice infected with HV-68 might be associated with the presence of viable bacteria in liver tissue. For these studies, liver was taken from mice, homogenized, and plated onto Luria agar to detect the presence of viable bacteria. Figure 7 shows the results of one such study where six of six IL-10−/− mice infected with HV-68 had viable bacteria in liver, whereas two of six mock-treated IL-10−/− mice had significantly lower numbers of viable bacteria.

Quantification of viable bacteria in liver homogenates. Groups of 6-week-old normal C57BL/6 mice (C57) (n = 4) or IL-10-deficient mice (IL-10−/−) (n = 6) were mock-treated (Mock) or infected with murine gammaherpesvirus 68 (HV-68) as indicated. Two months following infection, mice were euthanized and liver tissue homogenized in 0.5% Triton X-100. Dilutions of tissue homogenates were plated onto Luria agar plates and incubated for 48 h. Numbers of bacterial colonies per 25 mg of liver tissue were counted, and results are presented as colony counts in each individual animal. Bars represent mean values of these individual determinations
Identification of selected bacterial colonies present on Luria plates was performed using API-20 test strips. E. coli, Enterococcus sp., Bacillus sp., and a gram-negative, non-lactose-fermenting bacterium whose identity could not be determined were present in the liver homogenates from IL-10−/− mice and from IL-10−/− mice infected with HV-68. There were no significant differences in the species of bacteria present in the liver of the two groups. However this result could be due to colony selection or to the singular use of Luria agar plates for bacterial growth.
Discussion
In previous work, we discovered that while IL-10−/− mice could be readily infected by HV-68, they had less latent virus than wild-type C57BL/6 mice [36]. We postulated, based on this [36] and other work [31], that this result was due to an enhanced, protective IL-12 response in IL-10−/− mice. Surprisingly, however we also discovered increased pathophysiology following this acute viral infection of IL-10−/− mice, suggesting that the presence of this cytokine helped to limit a destructive viral-induced inflammation [36]. The increased inflammation following this acute viral infection caused us to question whether a sustained inflammatory response accompanied latent viral infection with HV-68 in this animal model of colitis.
To address this possibility, we infected young, 6-week-old IL-10−/− mice with HV-68. By 2 months following infection, viral latency was established in these mice (Fig. 6b), and it was difficult to detect the presence of infectious virus in most animals (Fig. 6a). Therefore we were surprised to find that IL-10−/− mice harboring this latent gammaherpesvirus infection succumbed (Fig. 1) to an inflammatory wasting-like disease characterized by weight loss (Fig. 2), splenomegaly (Fig. 3), leukocytosis and leukocytic infiltration of the liver (Fig. 4), cholangitis, and exacerbated inflammation of the colon (Fig. 5). Increased colitis correlated with the presence of viable bacteria in the liver (Fig. 7), suggesting that translocation of gut flora into the portal circulation may be responsible for the inflammatory wasting disease observed in the IL-10−/− mice infected with HV-68.
Based on these results, we suggest that IL-10−/− mice harboring latent HV-68 represent a chronic model of bacterial and viral co-infection that, over time, develops into an exacerbated, fatal inflammatory response. The relevance for such a model can be found in patients who experience gammaherpesvirus-exacerbated diseases whose etiology is bacterial, including periodontal diseases [7–9] and inflammatory bowel diseases [16–22]. While it is clear from these studies in patients that gammaherpesviruses, like EBV, have been associated with exacerbated inflammation, it is not altogether clear how such a ubiquitous viral infection can contribute to disease severity.
Previous studies performed in our laboratory suggest a possible mechanism to explain HV-68-induced exacerbated disease in IL-10−/− mice. IL-10−/− mice have reduced viral burdens [36]. This is likely due to the presence of an increased IL-12-induced T helper type 1 response [36], which can contribute to the protective host response [31]. While somewhat protective, the host’s IL-12-mediated T helper type 1 anti-viral response is unable to prevent acute HV-68 infection from establishing latency [31]. In addition, this IL-12-mediated host response contributes to the pathophysiology seen during the mononucleosis phase of the disease [36]. Therefore while an IL-12-induced T helper type 1 response can contribute to the protective host response, it contributes to host-induced pathophysiology as well. Therefore it appears that infection with this gammaherpesvirus results in a dysregulated T helper type 1 response that is only partially protective, but also contributes to viral-induced pathology.
It has been proposed that the IL-10−/− model of colitis represents a T helper type 1-mediated disease [46–48]. Furthermore, it has also been suggested that the host response directed against normal bacterial flora translocating from the gut is likely one stimulus for activating the T helper type 1 response in animal models of colitis [49–52]. In the present work, IL-10−/− mice infected with HV-68 had an increased translocation of bacteria from the gut into peripheral tissues (Fig. 7). Therefore if infection with HV-68 can dysregulate T helper type 1 responses [31, 36], an exacerbated inflammatory response may occur as IL-10−/− mice respond to bacteria translocating from the gut.
Based on the work presented here, it will be possible to utilize this unique model of latent gammaherpesvirus and bacterial co-infection to address T helper type 1 exacerbated inflammatory diseases. Such studies should provide unique insights into the mechanisms used by gammaherpesviruses to dysregulate normal host immune responses in an effort to maintain viral latency throughout the life of the host [6].
References
Lunemann JD, Munz C. Epstein–Barr virus and multiple sclerosis. Curr Neurol Neurosci Rep. 2007;7(3):253–8.
Niller HH, Wolf H, Minarovits J. Regulation and dysregulation of Epstein–Barr virus latency: implications for the development of autoimmune diseases. Autoimmunity. 2008;41(4):298–328.
Peacock JW, Elsawa SF, Petty CC, Hickey WF, Bost KL. Exacerbation of experimental autoimmune encephalomyelitis in rodents infected with murine gammaherpesvirus-68. Eur J Immunol. 2003;33(7):1849–58.
Harley JB, Harley IT, Guthridge JM, James JA. The curiously suspicious: a role for Epstein–Barr virus in lupus. Lupus. 2006;15(11):768–77.
Costenbader KH, Karlson EW. Epstein–Barr virus and rheumatoid arthritis: is there a link? Arthritis Res Ther. 2006;8(1):204.
Nikolich-Zugich J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nature Rev. 2008;8(7):512–22.
Saygun I, Kubar A, Sahin S, Sener K, Slots J. Quantitative analysis of association between herpesviruses and bacterial pathogens in periodontitis. J Periodontal Res. 2008;43(3):352–9.
Slots J. Herpesviruses, the missing link between gingivitis and periodontitis? J Int Acad Periodontol. 2004;6(4):113–9.
Slots J. Herpesviral-bacterial synergy in the pathogenesis of human periodontitis. Curr Opin Infect Dis. 2007;20(3):278–83.
Brook I. The association of anaerobic bacteria with infectious mononucleosis. Anaerobe. 2005;11(6):308–11.
Stenfors LE, Bye HM, Raisanen S, Myklebust R. Bacterial penetration into tonsillar surface epithelium during infectious mononucleosis. J Laryngol Otol. 2000;114(11):848–52.
Stenfors LE, Bye HM, Raisanen S. Causes for massive bacterial colonization on mucosal membranes during infectious mononucleosis: implications for acute otitis media. Int J Pediatr Otorhinolaryngol. 2002;65(3):233–40.
Garimorth K, Kountchev J, Bellmann R, Semenitz B, Weiss G, Joannidis M. Lemierre’s syndrome following infectious mononucleosis. Wien Klin Wochenschr. 2008;120(5–6):181–3.
Givner LB. Arcanobacterium haemolyticum sepsis and Epstein–Barr virus infection. Pediatr Infect Dis J. 1992;11(5):417–8.
Yoon TY, Yang TH, Hahn YS, Huh JR, Soo Y. Epstein–Barr virus-associated recurrent necrotic papulovesicles with repeated bacterial infections ending in sepsis and death: consideration of the relationship between Epstein–Barr virus infection and immune defect. J Dermatol. 2001;28(8):442–7.
Bertalot G, Villanacci V, Gramegna M, Orvieto E, Negrini R, Saleri A, et al. Evidence of Epstein–Barr virus infection in ulcerative colitis. Dig Liver Dis. 2001;33(7):551–8.
Kangro HO, Chong SK, Hardiman A, Heath RB, Walker-Smith JA. A prospective study of viral and mycoplasma infections in chronic inflammatory bowel disease. Gastroenterology. 1990;98(3):549–53.
Spieker T, Herbst H. Distribution and phenotype of Epstein–Barr virus-infected cells in inflammatory bowel disease. Am J Pathol. 2000;157(1):51–7.
Takeda Y, Takada K, Togashi H, Takeda H, Sakano M, Osada Y, et al. Demonstration of Epstein–Barr virus localized in the colonic and ileal mucosa of a patient with ulcerative colitis. Gastrointest Endosc. 2000;51(2):205–9.
Van Kruiningen HJ, Poulin M, Garmendia AE, Desreumaux P, Colombel JF, De Hertogh G, et al. Search for evidence of recurring or persistent viruses in Crohn’s disease. Apmis. 2007;115(8):962–8.
Wakefield AJ, Fox JD, Sawyerr AM, Taylor JE, Sweenie CH, Smith M, et al. Detection of herpesvirus DNA in the large intestine of patients with ulcerative colitis and Crohn’s disease using the nested polymerase chain reaction. J Med Virol. 1992;38(3):183–90.
Yanai H, Shimizu N, Nagasaki S, Mitani N, Okita K. Epstein–Barr virus infection of the colon with inflammatory bowel disease. Am J Gastroenterol. 1999;94(6):1582–6.
Cohen JI. Epstein–Barr virus infection. N Engl J Med. 2000;343(7):481–92.
Compston LI, Sarkobie F, Li C, Candotti D, Opare-Sem O, Allain JP. Multiplex real-time PCR for the detection and quantification of latent and persistent viral genomes in cellular or plasma blood fractions. J Virol Methods. 2008;151(1):47–54.
Cameron B, Bharadwaj M, Burrows J, Fazou C, Wakefield D, Hickie I, et al. Prolonged illness after infectious mononucleosis is associated with altered immunity but not with increased viral load. J Infect Dis. 2006;193(5):664–71.
Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein–Barr virus. Annu Rev Immunol. 2007;25:587–617.
Doherty PC, Tripp RA, Hamilton-Easton AM, Cardin RD, Woodland DL, Blackman MA. Tuning into immunological dissonance: an experimental model for infectious mononucleosis [see comments]. Curr Opin Immunol. 1997;9(4):477–83.
Flano E, Woodland DL, Blackman MA. A mouse model for infectious mononucleosis. Immunol Res. 2002;25(3):201–17.
Mistrikova J, Raslova H, Mrmusova M, Kudelova M. A murine gammaherpesvirus. Acta Virol. 2000;44(3):211–26.
Simas JP, Efstathiou S. Murine gammaherpesvirus 68: a model for the study of gammaherpesvirus pathogenesis. Trends Microbiol. 1998;6(7):276–82.
Elsawa SF, Bost KL. Murine gamma-herpesvirus-68-induced IL-12 contributes to the control of latent viral burden, but also contributes to viral-mediated leukocytosis. J Immunol. 2004;172(1):516–24.
Elsawa SF, Taylor W, Petty CC, Marriott I, Weinstock JV, Bost KL. Reduced CTL response and increased viral burden in substance P receptor-deficient mice infected with murine gamma-herpesvirus 68. J Immunol. 2003;170(5):2605–12.
Gasper-Smith N, Marriott I, Bost KL. Murine gamma-herpesvirus 68 limits naturally occurring CD4+CD25+T regulatory cell activity following infection. J Immunol. 2006;177(7):4670–8.
Gasper-Smith N, Singh S, Bost KL. Limited IL-6 production following infection with murine gammaherpesvirus 68. Arch Virol. 2006;151(7):1423–9.
Peacock JW, Bost KL. Infection of intestinal epithelial cells and development of systemic disease following gastric instillation of murine gammaherpesvirus-68. J Gen Virol. 2000;81(Pt 2):421–9.
Peacock JW, Bost KL. Murine gammaherpesvirus-68-induced interleukin-10 increases viral burden, but limits virus-induced splenomegaly and leukocytosis. Immunology. 2001;104(1):109–17.
Bhan AK, Mizoguchi E, Smith RN, Mizoguchi A. Colitis in transgenic and knockout animals as models of human inflammatory bowel disease. Immunol Rev. 1999;169:195–207.
Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–74.
Powrie F, Uhlig H. Animal models of intestinal inflammation: clues to the pathogenesis of inflammatory bowel disease. Novartis Found Symp. 2004;263:164–74. discussion 74-8, 211-8.
Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98(4):1010–20.
Dutia BM, Allen DJ, Dyson H, Nash AA. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology. 1999;261(2):173–9.
DeMeo MT, Mutlu EA, Keshavarzian A, Tobin MC. Intestinal permeation and gastrointestinal disease. J Clin Gastroenterol. 2002;34(4):385–96.
Karlinger K, Gyorke T, Mako E, Mester A, Tarjan Z. The epidemiology and the pathogenesis of inflammatory bowel disease. Eur J Radiol. 2000;35(3):154–67.
Swidsinski A, Lee SP. The role of bacteria in gallstone pathogenesis. Front Biosci. 2001;6:E93–103.
Zeuzem S. Gut-liver axis. Int J Colorectal Dis. 2000;15(2):59–82.
Kennedy RJ, Hoper M, Deodhar K, Erwin PJ, Kirk SJ, Gardiner KR. Interleukin 10-deficient colitis: new similarities to human inflammatory bowel disease. Br J Surg. 2000;87(10):1346–51.
Madsen KL. Inflammatory bowel disease: lessons from the IL-10 gene-deficient mouse. Clin Invest Med. 2001;24(5):250–7.
Rennick DM, Fort MM. Lessons from genetically engineered animal models. XII. IL-10-deficient (IL-10(−/−) mice and intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2000;278(6):G829–33.
Brimnes J, Reimann J, Nissen M, Claesson M. Enteric bacterial antigens activate CD4(+) T cells from scid mice with inflammatory bowel disease. Eur J Immunol. 2001;31(1):23–31.
Madsen KL, Doyle JS, Tavernini MM, Jewell LD, Rennie RP, Fedorak RN. Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology. 2000;118(6):1094–105.
Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66(11):5224–31.
Takahashi I, Matsuda J, Gapin L, DeWinter H, Kai Y, Tamagawa H, et al. Colitis-related public T cells are selected in the colonic lamina propria of IL-10-deficient mice. Clin Immunol. 2002;102(3):237–48.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: S. Stimpson.
Rights and permissions
About this article
Cite this article
Nelson, D.A., Petty, C.C. & Bost, K.L. Infection with murine gammaherpesvirus 68 exacerbates inflammatory bowel disease in IL-10-deficient mice. Inflamm. Res. 58, 881–889 (2009). https://doi.org/10.1007/s00011-009-0059-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-009-0059-x