
Abstract
Objective and design
To investigate the effects of sulforaphane on endothelial inflammatory gene expression in endothelial cells.
Materials and methods
Human aortic endothelial cells were used in the study.
Results
One-hour pretreatment of endothelial cells (EC) with sulforaphane (1–4 μM) suppressed TNF-α-induced MCP-1 and VCAM-1 mRNA and protein levels, but had no effect on TNF-α-induced ICAM-1 expression. Sulforaphane also inhibited TNF-α-induced activation of p38 MAP kinase, but not c-Jun-N-terminal kinase. Sulforaphane had no effect on TNF-α-induced NF-κB nuclear binding activity, IκB-α degradation or activation of NF-κB-driven transcriptional activity. Expression of dominant negative Nrf2 inhibited sulforaphane-induced antioxidant response element (ARE)-driven promoter activity, but had no effect on sulforaphane-mediated inhibition of VCAM-1 and MCP-1 expression.
Conclusion
These data suggest that sulforaphane may be useful as a therapeutic agent for the treatment of inflammatory diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sulforaphane (1-isothiocyanato-4-(methylsulfinyl)-butane) is a naturally occurring isothiocyanate isolated from cruciferous vegetables such as broccoli [1]. Sulforaphane is a potent cancer chemo-preventive agent that functions by inducing phase II detoxification enzymes and antioxidant proteins through the activation of antioxidant response element (ARE)-mediated transcriptional activity [2]. Nrf2 (NF-E2-related factor 2) is the transcription factor that is responsible for both constitutive and inducible expression of ARE-mediated genes [3, 4].
Recent studies indicate that sulforaphane also possesses anti-inflammatory effects. Treatment of cultured Raw 264.7 macrophages with sulforaphane suppressed lipopolysaccharide (LPS)-induced nitric oxide generation, PGE2 production, TNF-α secretion and inducible nitric oxide synthase (iNOS) and Cox-2 expression [5]. Sulforaphane also inhibited cell-mediated immune response in B16F-10 melanoma-induced metastasis-bearing C57BL/6 mice and suppressed the serum levels of proinflammatory cytokines such as IL-1β, IL-6, TNF-α and GM-CSF during metastasis [6]. At the molecular level, sulforaphane treatment inhibited activation of NF-κB [5, 7]. Heiss et al. reported that sulforaphane can selectively reduce DNA binding of NF-κB without interfering with LPS-induced degradation of the inhibitor of NF-κB nor with nuclear translocation of NF-κB [5, 7]. In contrast, Xu reported that sulforpahene inhibited NF-κB transcriptional activity, nuclear translocation of p65 and UVC-induced phosphorylation of IκΒα and blocked UVC-induced IκBα degradation in PC-3 C4 cells [8]. sulforaphane treatment also inhibited LPS-induced activation of AP-1 in Raw 264.7 cells and ultraviolet (UV) light-induced activation of AP-1 in human HaCaT keratinocytes [5, 7, 9].
These observed anti-inflammatory effects of sulforaphane may have involved the activation of the Nrf2/ARE pathway. Several recent studies have demonstrated that the Nrf2/ARE pathway is involved in immune and inflammatory processes. It has been shown that Nrf2-deficient mice had increased inflammatory cell infiltration in hyperoxia and bleomycin-induced lung injury [10, 11]. Nrf2-knockout mice also exhibited prolonged inflammation during cutaneous wound healing [12] and displayed enhanced bronchial inflammation and susceptibility to cigarette smoke-induced emphysema [13]. Previously, we reported that adenovirus-mediated over-expression of Nrf2 suppressed expression of inflammatory genes such as vascular cell adhesion molecule-1 (VCAM-1) and monocyte chemoattractant protein-1 (MCP-1) in endothelial cells [14]. In this study, we investigated the effects of sulforaphane on TNF-α-induced inflammatory gene expression in endothelial cells. Our results demonstrated that treatment of endothelial cells with sulforaphane inhibited TNF-α-induced VCAM-1 and MCP-1 expression and p38 MAP kinase activation. However, these effects were independent of Nrf2/ARE pathway activation. Furthermore, sulforaphane did not inhibit NF-κB activation at concentrations that inhibited VCAM-1 and MCP-1 expression.
Materials and methods
Cell culture and DNA plasmids
Human aortic endothelial cells (HAECs) were obtained from Cambrex (Walkersville, MD, USA) and cultured in EGM-2 growth medium. Cells were used between passages 5 and 9. Human microvascular endothelial cells (HMEC) were grown as described previously [15] and were cultured in modified MCDB 131 (Invitrogen, Carsbad, CA, USA), supplemented with 10% fetal bovine serum and EGM single quote (Cambrex). Cells were maintained at 37°C in a 5% CO2 incubator. p3xARE-luc contains 3 tandem copies of ΝQO1 ARE sequences linked to a luciferase reporter gene and has been described previously [16]. 5xNF-κB/Luc was purchased from Promega Corp (Madison, WI, USA). pcDNA3-DN-Nrf2 expressing dominant negative Nrf2 mutant and pcDNA3-Keap1 were described previously [16]. Sulforaphane was obtained from Calbiochem Corp (San Diego, CA, USA). Antibodies for phosphorylated p38 MAP kinase and phosphorylated c-Jun-amino-terminal kinase (JNK) were obtained from Promega Corporation. Antibodies for p38 MAP kinase and JNK were obtained from Cell Signaling Technologies (Danvers, MA, USA).
Preparation of RNA and Quantikine mRNA analysis
Total RNA samples were isolated by the Trizol method (Life Technologies, Grand Island, NY, USA) and quantitatively measured by UV spectrophotometer. VCAM-1, MCP-1 and GAPDH mRNA levels were determined with a Quantikine mRNA colorimetric quantification kit (R&D Systems, Inc, Minneapolis, MN, USA) according to the manufacturer’s instructions.
ELISA for MCP-1 protein
HAECs grown in 24 well plates were treated with TNF-α (100 U/ml) for 4 h. Conditioned media were collected and assayed for MCP-1 protein levels by ELISA using the Quantikine Colorimetric Sandwich ELISA kit (R&D Systems) according to the manufacturer’s instruction.
ELISA for cell surface expression of adhesion molecules
HAECs grown in 96-well plates were pretreated or not with sulforaphane for 1 h and then incubated with TNF-α (100 U/ml) for 4 h. Primary mouse antibodies for VCAM-1 and ICAM-1 were obtained from Southern Biotechnology Associates. Cell surface expression of adhesion molecules was determined by ELISA with primary binding with specific mouse antibodies, followed by secondary binding with a horseradish peroxidase-conjugated goat anti-mouse IgG antibody. Quantification was performed by determination of colorimetric conversion at OD450 nm of 3,3′,5,5′-tetramethylbenzidine.
Transfection and promoter activity assays
Since HAECs are relatively resistant to efficient transient transfection, we used HMECs for these experiments. HMECs were grown to 60–70% confluence in 12-well plates and transfected with various plasmids as indicated in the figure legends using SuperFect transfection reagent according to the manufacturer’s instructions (Qiagen, Inc, Valential CA, USA). Plasmids pRL-TK (renilla luciferase constitutively expressed under the control of the thymidine kinase promoter) was co-transfected in all samples and used to normalize for transfection efficiency. Firefly and renilla luciferase activities were measured using a luciferase reporter assay system according to the manufacturer’s instructions (Promega Corp).
Western blot analysis
HAECs were lysed for 30 min on ice in 1 ml of a lysis buffer as previously described [16]. Protein samples (15 μg) were subjected to electrophoresis on 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. Antibody-bound protein bands were then visualized via HRP-dependent chemiluminescence (Amersham Corp, Piscataway, NJ, USA).
Monocyte adhesion assay
The human monocytic U937 cell line was used in the adhesion assay as previously described [17]. U937 cells were labeled with 5 μM Calcein AM (Invitrogen) and resuspended in protein-free RPMI culture medium at a concentration of 5 × 106 cells/ml. The fluorescently labeled U937 cell suspension was added to the HAEC monolayers in 96-well plates at a concentration of 3 × 104 cells/well, and the mixture was incubated at 37°C for 30 min. The monolayers were rinsed four times with RPMI, and 200 μl of PBS was added to each well. Fluorescence was measured with a fluorescein filter at 485 nm/535 nm using the Perkin Elmer Victor2 V multi-plate reader.
Adenoviruses
The adenovirus encoding murine dominant negative Nrf2 cDNA (Ad.DN-Nrf2) was previously described [18]. Infection was carried out with the indicated multiplicity of infection (MOI) for 24 h, after which the infection media was aspirated and replaced with fresh media. Ad.GFP, an adenovirus encoding the green florescence protein (GFP) gene, was used as a control for adenovirus infection.
Statistical analysis
Values were expressed as the means ± SD of at least three experiments. Statistics were performed by ANOVA with Tukey’s post hoc test or Student’s t test where appropriate. Values were considered significantly different at the 95% confidence level.
Results
Sulforaphane suppresses TNF-α-induced MCP-1 and VCAM-1, but not ICAM-1, gene expression
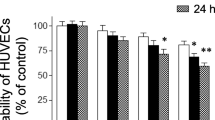
To investigate whether sulforaphane can suppress TNF-α-induced inflammatory gene expression, HAECs were treated with sulforphane (1–4 μM) for 1 h and then exposed to TNF-α (100 U/ml) for 4 h. Treatment of HAECs with TNF-α-induced a marked increase in MCP-1 protein secretion and cell surface expression of VCAM-1 and ICAM-1 (Fig. 1). Pretreatment with sulforaphane suppressed TNF-α-induced MCP-1 protein secretion and cell surface expression of VCAM-1 protein in a concentration-dependent manner (Fig. 1). Similarly, sulforaphane suppressed TNF-α-induced MCP-1 and VCAM-1 mRNA accumulation in HAECs (Fig. 2). However, treatment with sulforaphane had no effect on either TNF-α-induced cell surface expression of ICAM-1 protein (Fig. 1) or ICAM-1 mRNA levels (Fig. 2).

Sulforaphane (SFN) suppressed TNF-α-induced MCP-1 and VCAM-1, but not ICAM-1, protein expression in HAECs. HAECs were pretreated with sulforaphane (1–4 μM) for 1 h and then exposed to TNF-α (100 U/ml) for 4 h. Conditioned medium was collected and MCP-1 protein levels were determined by ELISA. Cell surface expression of VCAM-1 and ICAM-1 protein was determined by ELISA. Values are mean ± SD, n = 4. *P < 0.05 compared with TNF-α-treated group

Sulforaphane suppressed TNF-α-induced MCP-1 and VCAM-1, but not ICAM-1, mRNA accumulation in HAECs. HAECs were pretreated with sulforaphane (1–4 μM) for 1 h and then exposed to TNF-α (100 U/ml) for 4 h. Relative mRNA levels for MCP-1, VCAM-1 and ICAM-1 were determined by Quantikine mRNA kits and normalized to GAPDH levels. Values are mean ± SD, n = 4. *P < 0.05 compared with TNF-α treated group
Sulforaphane suppresses TNF-α-induced monocyte adhesion to endothelial cells
To determine whether inhibition of VCAM-1 gene expression by sulforaphane would suppress TNF-α-induced monocyte adhesion, HAECs were pretreated with sulforaphane (4 μM) for 1 h and then exposed to TNF-α for 4 h. As shown in Fig. 3, TNF-α treatment produced a marked increase in U937 cell adhesion to HAECs. Treatment with sulforphane resulted in approximately 98% inhibition of U937 adhesion to endothelial cells.

Sulforaphane inhibited TNF-α-induced monocyte adhesion. HAECs were pretreated with sulforaphane (4 μM) for 1 h and then exposed to TNF-α for 4 h. Adhesion of monocytic cell line U937 cells was determined as described in “Methods”. Data represent percentage of attached cells from total amount of cells put into wells. Values represent mean ± SD, n = 4
Sulforaphane activates the ARE-driven promoter through an Nrf2-dependent mechanism
Sulforaphane is a well-characterized phase II inducer and activates ARE-mediated transcriptional activity [19–21]. To examine the concentration-dependent responses of sulforaphane on ARE-driven promoter activity, HMECs were transfected with 3xARE-luc and exposed to sulforaphane (1–3 μM) for 4 h. HMECs were used as HAECs are refractory to transient transfection. Treatment with sulforaphane produced a concentration-dependent increase in ARE transcriptional activity (Fig. 4a). To determine if the Nrf2 transcriptional factor is important in sulforaphane-induced ARE promoter activity, HMECs were co-transfected with dominant negative Nrf2 vector. Sulforaphane-induced activation of ARE promoter activity was inhibited by co-transfection with a DN-Nrf2 mutant expression vector or with Nrf2 inhibitory protein Keap1 (Fig. 4b).

Sulforaphane activates ARE-driven transcriptional activity through Nrf2-dependent mechanism in HMECs. a HMECs cultured in 12-well plates were transfected with 0.5 μg of p3xARE/Luc for 24 h and exposed to sulforaphane (1–3 μM) for 4 h. b HMECs cultured in 12-well plates were transfected with 0.5 μg of p3xARE/Luc plus 0.5 μg pcDNA3-DN-Nrf2, pcDNA3-Keap1 or empty vector pcDNA3 and exposed to sulforaphane (4 μM) for 4 h. These cells were also transfected with 0.1 μg of pRL-TK for normalization of transfection efficiency. Cells extracts were harvested and luciferase assays were performed. Values represent mean ± SD, n = 3. *P < 0.05 compared with non-treated treated cells
Sulforaphane suppresses TNF-α-induced VCAM-1 and MCP-1 expression through an Nrf2-independent mechanism
To examine the role of Nrf2 in sulforaphane-mediated inhibition of VCAM-1 and MCP-1 expression, HAECs were infected with Ad.GFP and Ad.DN-Nrf2 for 24 h. Infection with Ad.DN-Nrf2 led to marked expression of truncated Nrf2 protein in HAECs (data not shown). Cells were then pretreated with sulforaphane (4 μM) for 1 h and exposed to TNF-α (100 U/ml) for 4 h. As shown in Fig. 5, sulforaphane inhibited TNF-α-induced cell surface expression of VCAM-1 and secretion of MCP-1 by HAECs. Infection with Ad.DN-Nrf2 did not reverse sulforaphane-mediated inhibition (Fig. 5). These data suggest that the Nrf2/ARE pathway is not involved in sulforaphane-mediated inhibition of VCAM-1 and MCP-1 expression.

Expression of dominant negative Nrf2 has no effect on sulforaphane-mediated inhibition of TNF-α-induced VCAM-1 and MCP-1 expression. HAECs were infected with Ad.GFP or Ad.DN-Nrf2 (MOI of 100) for 24 h and then exposed to TNF-α (100 U/ml) for 4 h. a Cell surface expression of VCAM-1 was determined by ELISA. b Conditioned medium was collected and MCP-1 protein levels were determined by ELISA. Values represent mean ± SD, n = 4. *P < 0.05 compared with TNF-α-treated cells infected with Ad.GFP
Sulforaphane has no effect on TNF-α-induced NF-κB activation
We used three approaches to determine the effect of sulforaphane on NF-κB activation: nuclear NF-κB-binding activity, IκBα degradation, and NF-κB promoter activity. As shown in Fig. 6a and b, TNF-α-induced IκBα degradation and nuclear NF-κB-binding activity were not suppressed by sulforaphane pretreatment in HAECs. Similarly, pretreatment with sulforaphane did not inhibit TNF-α-induced activation of NF-κB-driven promoter activity in HMECs (Fig. 6c). These data suggest that inhibition of MCP-1 and VCAM-1 gene expression by sulforaphane is mediated by an NF-κB-independent mechanism.

Sulforaphane did not inhibit TNF-α-induced activation of NF-κB. a HAECs pretreated with sulforaphane (4 μM) for 1 h were exposed to TNF-α (100 U/ml) for 10 and 20 min. Whole cell lysates were analyzed by immunoblotting with antibody to IκB-α or β-tubulin. b HAECs were pretreated with sulforaphane (4 μM) for 1 h and then exposed to TNF-α (100 U/ml) for 1 h. Nuclear extracts were isolated and nuclear NF-κB-binding activity was determined with TransAM NF-κB p65 Transcription Factor Kit. Values are mean ± SD, n = 4. *P < 0.05 compared to the control group. c HMECs cultured in 12-well plates were transfected with 1 μg 5xNFκB/Luc plus 1 μg LNCX-Nrf2, or empty vector LNCX. After a 24 h recovery, cells were pretreated with sulforaphane (4 μM) for 1 h and then exposed to TNF-α (100 U/ml) for 16 h. Cells extracts were harvested and luciferase assays performed. The firefly luciferase activities were normalized by renilla luciferase activities. Values represent mean ± SD, n = 4
Sulforaphane suppresses TNF-α-induced activation of p38 MAP kinase, but not JNK, in HAECs
p38 MAP kinase is involved in TNF-α-induced VCAM-1 and MCP-1 expression in endothelial cells [22, 23]. To investigate whether sulforaphane is capable of suppressing TNF-α-induced activation of p38 MAP kinase, HAECs were pretreated with sulforaphane (4 μM) for 1 h and exposed to TNF-α (100 U/ml) for 10 and 20 min. TNF-α-treatment resulted in a marked increase in phosphorylated p38 MAP kinase levels. Treatment with sulforaphane inhibited TNF-α-induced phosphorylation of p38 MAP kinase (Fig. 7). In contrast, treatment with sulforaphane had no effect on TNF-α-induced phosphorylation of JNK in endothelial cells. These data suggest that sulforaphane may inhibit TNF-α-induced MCP-1 and VCAM-1 expression through inhibition of p38 MAP kinase activation.

Sulforaphane suppresses TNF-α-induced activation of p38 MAP kinase. HAEC were pretreated with sulforaphane (4 μM) for 1 h and exposed to TNF-α (100 U/ml) for 10 or 20 min. The activities of p38 MAP kinase and JNK were determined by assaying for the level of the total or phosphorylated form of p38 MAP kinase and JNK by Western blot analysis a and by Elisa b. Values represent mean ± SD, n = 4. *P < 0.05 compared with TNF-α treated cells
Discussion
In the present study, we investigated the effects of sulforaphane on TNF-α-induced inflammatory gene expression in endothelial cells. Our data demonstrated that sulforaphane was able to suppress TNF-α-induced MCP-1 and VCAM-1 expression, as well as monocyte adhesion to endothelial cells. Sulforaphane also inhibited TNF-α-induced activation of p38 MAP kinase, without affecting TNF-α-induced activation of NF-κB or JNK. Although treatment with sulforaphane-induced increases in ARE-driven promoter activity, sulforaphane’s inhibitory effects of VCAM-1 and MCP-1 were not dependent on Nrf2. These data suggest that sulforaphane may suppress inflammatory gene expression through inhibition of the activation of p38 MAP kinase.
Several studies have demonstrated the anti-inflammatory effects of sulforaphane. In cultured Raw 264.7 macrophages, pretreatment with sulforaphane suppressed LPS-induced iNOS as well as Cox-2 gene expression [5]. Similarly, sulforaphane pre-treatment inhibited production of IL-8, GM-CSF, and IL-1β from primary human bronchial epithelial cells upon stimulation with diesel extract [19]. Wu et al. reported that feeding spontaneous hypertension rats with sulforaphane increased GSH reductase and GSH peroxidase activities, decreased oxidative stress and decreased infiltration of activated macrophages in cardiovascular system including inner intimal layers of the aorta, carotid artery and endocardium of the heart [24]. Our results provide further support to the notion that sulforaphane has anti-inflammatory effects and may be beneficial for the prevention and treatment of inflammatory diseases such as atherosclerosis.
The present study showed that sulforaphane has no effects on TNF-α-induced activation of NF-κB. These results are at variance with earlier reports. Heiss et al. reported that sulforaphane at 10 and 20 μM concentrations inhibited LPS-induced DNA binding of NF-κB, without interfering with IκB-α degradation and nuclear translocation of NF-κB in the murine macrophage cell line Raw 264.7. The authors further demonstrated that sulforaphane may directly interfere with NF-κB DNA binding by modifying the critical thiol moiety of the NF-κB subunits [5]. In human prostate cancer PC-3 C4 cells, Xu et al. reported that treatment with 20 and 30 μM sulforaphane inhibited UVC-induced NF-κB activation and expression of NF-κB-regulated genes. In this study, sulforaphane also inhibited UVC-induced activation of IKKβ and IKKα and degradation of IκB-α. [8]. However, our results demonstrated that sulforaphane at 4 μM concentration suppressed TNF-α-induced inflammatory gene expression without affecting IκB-α degradation, NF-κB nuclear translocation and NF-κB transcriptional activity. The difference may be due to concentration used in the studies. These studies were conducted with relatively high concentrations (10–30 μM) of sulforaphane in comparison with our study that uses 4 μM. In our study, endothelial cells are much more sensitive to sulforaphane than other cell types; 30 μM of sulforaphane was toxic to endothelial cells. Furthermore, cell type differences may also contribute to the discrepancies in the observations.
Recent studies have demonstrated that the Nrf2/ARE pathway is involved in immune and inflammatory processes. Nrf2-deficient mice had exacerbated inflammatory responses in hyperoxia and bleomycin-induced lung injury [10, 11], and enhanced bronchial inflammation and susceptibility to cigarette smoke-induced emphysema [13]. We reported earlier that adenovirus-mediated over-expression of Nrf2 suppressed the expression of VCAM-1 and MCP-1 in endothelial cells [14]. Sulforaphane is one of the most potent activators of the Nrf2/ARE pathway and can exert indirect antioxidant effects [1, 2, 25]. However, our data showed that although expression of dominant negative Nrf2 inhibited sulforaphane-induced ARE-driven transcriptional activity, it had no effect on sulforaphane-mediated inhibition of VCAM-1 and MCP-1. Because the total time of exposure to sulforaphane in our experiments was no more than 5 h, it is unlikely that the inhibition of VCAM-1 and MCP-1 observed in our studies is a result of the induction of endogenous antioxidant and/or anti-inflammatory proteins (i.e., HO-1) by Nrf2 subsequent to activation by sulforaphane. Therefore, these data suggest that sulforaphane has anti-inflammatory effects independent of genes activated via the Nrf2/ARE pathway. It is possible that prolonged treatment with sulforaphane will have two components of anti-inflammatory activity: one that is mediated via more acute actions of sulforaphane (i.e., effects described here on p38 activation) and one that may require longer exposure to sulforaphane and which is dependent on the subsequent activation of Nrf2-dependent genes (i.e., HO-1).
In addition to the well-known role of the NF-κB pathway in regulating inflammatory gene expression, the p38 MAP kinase pathway plays an important role in regulating the expression of many inflammatory genes such as MCP-1 [23], VCAM-1 [26], and TNF-β [27]. Using specific p38 MAP kinase inhibitors or a dominant-negative mutant of MKK6 (the upstream kinase activator of p38), it has been reported that p38 MAP kinase is required for the activation of VCAM-1 and MCP-1 expression in response to IL-β and TNF-α [23, 26, 28]. Hwang et al. reported that sulforaphane inhibits H2O2-mediated activation of Erk1/2 and p38 MAP kinase and protects H2O2-induced inhibition of gap junctional intercellular communication in rat liver epithelial cells [29]. ATF2 is a downstream transcription factor activated by p38 MAP kinase and forms heterodimers with members of the AP-1 family [30, 31]. ATF2 knockout mice show decreased LPS-induced inflammation including reduced expression of E-selectin, P-selectin, VCAM-1, IL-6 and KC [32]. Sulforaphane may inhibit activation of ATF2 through inhibition of p38 MAP kinase and suppress the expression of VCAM-1 and MCP-1. The present study demonstrated that sulforaphane is able to suppress TNF-α-mediated activation of the p38 MAP kinase, which suggests that sulforaphane’s anti-inflammatory effects may be mediated through the inhibition of p38 MAP kinase. Additional studies are needed to address how the inhibition of p38 activity by sulforaphane may mediate the selective nature of the inhibition of various inflammatory response genes (i.e. VCAM-1 vs ICAM-1) and to what degree other signaling and transcriptional networks may be involved.
In summary, treatment with sulforaphane suppressed TNF-α-induced MCP-1 and VCAM-1 expression and monocyte adhesion to endothelial cells. The mechanisms of sulforaphane’s action may be mediated by inhibition of p38 MAP kinase. These data provide further support for anti-inflammatory role of sulforaphane and suggest that sulforaphane may be useful as a therapeutic agent for the treatment of inflammatory diseases.
Abbreviations
- ARE:
-
Antioxidant response element
- HAEC:
-
Human aortic endothelial cells
- HMEC:
-
Human microvascular endothelial cells
- ICAM-1:
-
Intercellular adhesion molecule-1
- MCP-1:
-
Monocyte chemoattractant protein-1
- Nrf2:
-
NF-E2-related factor-2
- TNF-α:
-
Tumor necrosis factor-α
- VCAM-1:
-
Vascular cell adhesion molecule-1
- SFN:
-
Sulforaphane
References
Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc Natl Acad Sci USA. 1994;91:3147–50.
Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol. 1999;37:973–9.
Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22.
Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci USA. 1999;96:12731–6.
Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276:32008–15.
Thejass P, Kuttan G. Modulation of cell-mediated immune response in B16F–10 melanoma-induced metastatic tumor-bearing C57BL/6 mice by sulforaphane. Immunopharmacol Immunotoxicol. 2007;29:173–86.
Woo KJ, Kwon TK. Sulforaphane suppresses lipopolysaccharide-induced cyclooxygenase-2 (COX-2) expression through the modulation of multiple targets in COX-2 gene promoter. Int Immunopharmacol. 2007;7:1776–83.
Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24:4486–95.
Zhu M, Zhang Y, Cooper S, Sikorski E, Rohwer J, Bowden GT. Phase II enzyme inducer, sulforaphane, inhibits UVB-induced AP-1 activation in human keratinocytes by a novel mechanism. Mol Carcinogen. 2004;41:179–86.
Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, et al. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–82.
Cho HY, Reddy SP, Yamamoto M, Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004;18:1258–60.
Braun S, Hanselmann C, Gassmann MG, auf dem Keller U, Born-Berclaz C, Chan K, et al. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol Cell Biol. 2002;22:5492–505.
Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–59.
Chen XL, Dodd G, Thomas S, Wasserman MA, Kunsch C. Activation of the Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Phyisol Heart Circ Physiol. 2005;290:H1862–70.
Chen XL, Tummala PE, Olliff L, Medford RM. E-selectin gene expression in vascular smooth muscle cells: evidence for a tissue specific repressor protein. Circ Res. 1997;80:305–11.
Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, et al. Laminar flow induction of antioxidant response element-mediated genes in endothelial cells: a novel anti-inflamatory mechanism. J Biol Chem. 2003;278:703–11.
Kunsch C, Luchoomun J, Chen XL, Dodd GL, Karu KS, Meng CQ, et al. AGIX-4207, a novel antioxidant and anti-inflammatory compound: cellular and biochemical characterization of antioxidant activity and inhibition of redox-sensitive inflammatory gene expression. J Pharm Exp Ther. 2005;313:492–501.
Zhang X, Lu L, Dixon C, Wilmer W, Song H, Chen X, et al. Stress protein activation by the cyclopentenone prostaglandin 15-deoxy-delta12,14-prostaglandin J2 in human mesangial cells. Kidney Int. 2004;65:798–810.
Ritz S, Wan J, Diaz-Sanchez D. Sulforaphane-stimulated phase II enzyme induction inhibits cytokine production by airway epithelial cells stimulated with diesel extract 2006. Am J Physiol Lung Cell Mol Physiol. 2007;292(1):L33–9.
McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, et al. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–307.
Morimitsu Y, Nakagawa Y, Hayashi K, Fujii H, Kumagai T, Nakamura Y, et al. A sulforaphane analogue that potently activates the Nrf2-dependent detoxification pathway. J Biol Chem. 2002;277:3456–63.
Goebeler M, Kilian K, Gillitzer R, Kunz M, Yoshimura T, Brocker EB, et al. The MKK6/p38 stress kinase cascade is critical for tumor necrosis factor-alpha-induced expression of monocyte-chemoattractant protein-1 in endothelial cells. Blood. 1999;93:857–65.
Rovin BH, Wilmer WA, Danne M, Dickerson JA, Dixon CL, Lu L. The mitogen-activated protein kinase p38 is necesssary for interleukin 1beta-induced monocyte chemoattractant protein 1 expression by human mesangial cells. Cytokine. 1999;11:118–26.
Wu L, Noyan Ashraf MH, Facci M, Wang R, Paterson PG, Ferrie A, et al. Dietary approach to attenuate oxidative stress, hypertension, and inflammation in the cardiovascular system. Proc Natl Acad Sci USA. 2004;101:7094–9.
Gao X, Dinkova-Kostova AT, Talalay P. Powerful and prolonged protection of human retinal pigment epithelial cells, keratinocytes, and mouse leukemia cells against oxidative damage: the indirect antioxidant effects of sulforaphane. Proc Natl Acad Sci USA. 2001;98:15221–6.
Pietersma A, Tilly BC, Gaestel M, de Jong N, Lee JC, Koster JF, et al. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem Biophys Res Commun. 1997;230:44–8.
Xu W, Yan M, Lu L, Sun L, Theze J, Zheng Z, et al. The p38 MAPK pathway is involved in the IL-2 induction of TNF-beta gene via the EBS element. Biochem Biophys Res Commun. 2001;289:979–86.
Ju JW, Kim SJ, Jun CD, Chun JS. p38 kinase and c-Jun N-terminal kinase oppositely regulates tumor necrosis factor alpha-induced vascular cell adhesion molecule-1 expression and cell adhesion in chondrosarcoma cells. IUBMB Life. 2002;54:293–9.
Hwang JW, Park JS, Jo EH, Kim SJ, Yoon BS, Kim SH, et al. Chinese cabbage extracts and sulforaphane can protect H2O2-induced inhibition of gap junctional intercellular communication through the inactivation of ERK1/2 and p38 MAP kinases. J Agric Food Chem. 2005;53:8205–10.
Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–55.
Falvo JV, Parekh BS, Lin CH, Fraenkel E, Maniatis T. Assembly of a functional beta interferon enhanceosome is dependent on ATF-2-c-jun heterodimer orientation. Mol Cell Biol. 2000;20:4814–25.
Reimold AM, Kim J, Finberg R, Glimcher LH. Decreased immediate inflammatory gene induction in activating transcription factor-2 mutant mice. Int Immunol. 2001;13:241–8.
Acknowledgments
The authors wish to thank Ms Suzanne Thomas for her expert technical assistance and Dr. Dominick Sinibaldi and Dr. Danny Piper for their critical reading.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: M. J. Parnham.
Rights and permissions
About this article
Cite this article
Chen, XL., Dodd, G. & Kunsch, C. Sulforaphane inhibits TNF-α-induced activation of p38 MAP kinase and VCAM-1 and MCP-1 expression in endothelial cells. Inflamm. Res. 58, 513–521 (2009). https://doi.org/10.1007/s00011-009-0017-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-009-0017-7