
Abstract
Caspase cysteine proteases are factors widely recognized for their role in the induction of apoptotic cell death. Caspases induce apoptosis during the inflammatory response to pathogen infection; in addition, caspases such as caspase-1 and caspase-11 are known to be involved in the production of inflammatory cytokines in response to bacterial infections. Caspase-1 is activated in the inflammasome, an intracellular protein complex that is formed by the recognition of intracellular ligands or cellular stresses by sensor molecules such as NOD-like receptors. Under certain conditions, caspase-11 is required for the activation of the caspase-1 inflammasome, referred to as the non-canonical inflammasome. In addition to these caspases, accumulating evidence indicates that caspase-8 also contributes to the production of inflammatory cytokines. In contrast to caspase-1, caspase-8 is activated by receptors located on the plasma membrane including dectin-1, TLR-3/4, and Fas. Recently, Fas-mediated caspase-8 activation and inflammatory cytokine production have been shown to play a significant role in the regulation of bacterial infections. This review highlights the functional roles and activation mechanisms of caspase-1/-11 in innate immune responses against bacterial infection. In addition, we discuss the novel aspects of caspase-8 function in comparison with caspase-1/-11 during innate inflammatory responses.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Caspases are cysteinyl aspartate-specific proteases that play a pivotal role in the induction of apoptotic cell death (McIlwain et al. 2013). The first caspase identified in mammalian cells was the interleukin (IL)-1β-converting enzyme (ICE, now known as caspase-1). ICE cleaves pro-IL-1β to yield active cytokine; however, it does so in the absence of apoptosis induction (Black et al. 1989a; Kostura et al. 1989). Based on multiple reports revealing the functional roles and activation mechanisms of caspases, caspases are now divided into three functional groups: apoptosis induction (caspase-2, -3, -6, -7, -8, -9, and -10) (Guo et al. 2002; Kischkel et al. 2001; Parrish et al. 2013), inflammatory responses (caspase-1, -4, -5, and -12) (Bian et al. 2011; Black et al. 1989a; Kostura et al. 1989; Lamkanfi et al. 2004; Sollberger et al. 2012), and differentiation (caspase-14) (Denecker et al. 2008) (Fig. 1). In addition to the major contributions to apoptosis induction, multiple lines of evidence indicate the significant importance of caspases in the inflammatory responses against microbial infection.

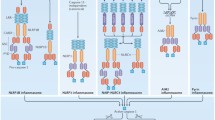
Classical caspase classification. Based on the classical classification, caspases are divided into three major groups: apoptosis (initiator or effector), inflammation, and differentiation. Increasing evidence indicates that the apoptosis initiator caspase-8 also plays a significant role in induction of inflammation. Asterisk mice lack a homolog of human caspase-10. Double asterisk murine caspase-11 is orthologous to human caspase-4/-5. DED death effector domain, CARD caspase recruitment domain
The innate immune system is the first line of defense against pathogenic infections (Kawai and Akira 2011). The recognition and phagocytosis of invasive pathogens, and the ensuing production of inflammatory cytokines by phagocytes, such as macrophages (Mϕ) and dendritic cells (DC), are critical steps in the activation of innate immunity. Several lines of evidence indicate that pathogen recognition is accomplished by germline-encoded pattern recognition receptors (PRRs) including Toll-like receptors (TLRs) and C-type lectin receptors that are expressed at the plasma membrane or on endosomes of Mϕ and DC (Ahmad-Nejad et al. 2002; Geijtenbeek and Gringhuis 2009; Kawai and Akira 2011; Matsumoto et al. 2003). PRRs recognize pathogenic ligands, collectively known as pathogen-associated molecular patterns (PAMPs), or self-derived damage-associated molecular patterns (DAMPs) that arise in response to cellular stress (Newton and Dixit 2012). Because PRRs are located at the plasma membrane or on endosomes, they recognize extracellular or endocytosed ligands. Ligand-activated TLRs recruit the adaptor molecule MyD88 or Toll/IL-1 receptor (TIR)-domain-containing adaptor-inducing interferon (IFN)-β (TRIF) to their cytoplasmic TIR domain, leading to the activation of transcription factors such as nuclear factor (NF)-κB and IFN regulatory factor. This activation leads to the gene expression of inflammatory cytokines including tumor necrosis factor (TNF)-α, IL-12, IL-6 or Type I IFNs that are directly secreted without undergoing post-translational modifications (Kawai and Akira 2011).
In addition to PRRs at the cell surface or on endosome, intracellular sensors of PAMPs/DAMPs such as those belonging to the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) or the pyrin and hematopoietic IFN-inducible nuclear proteins (HIN)-200 (PYHIN) families, have been shown to play a significant role in innate immunity (Schattgen and Fitzgerald 2011; Tsuchiya and Hara 2014). In contrast to TLRs, NLRs recognize pathogenic or stress-associated molecules inside the cell and their activation leads to the formation of a cytoplasmic protein complex including the caspase-1 inflammasome. Through auto-processing, caspase-1 is activated in this complex and subsequently cleaves the pro-forms of the inflammatory cytokines IL-1β/IL-18 to yield their active forms (Broz et al. 2010; Thornberry et al. 1992). The activated inflammasome also induces pyroptosis, a type of cell death that contributes to the removal of intracellular pathogens (Miao et al. 2010). The cytosolic components associated with NLRs and caspase-1 have been well characterized and are referred to as the canonical caspase-1 inflammasome or simply, the canonical inflammasome. Multiple reports have underscored the importance of the canonical inflammasome in regulation of the cellular response to invasive bacteria and the pathogenesis of microbial infection (Koizumi et al. 2012; Tsuchiya and Hara 2014). In addition, the murine ortholog of human caspase-4/-5, caspase-11, is required for caspase-1 inflammasome activation under certain conditions (Wang et al. 1998). The caspase-11-dependent inflammasome is referred to as the non-canonical inflammasome (Kayagaki et al. 2011). Furthermore, in response to pathogen infection, caspase-8 is involved in the processing of pro-IL-1β/IL-18 to yield active cytokines in an inflammasome-independent manner (Latz et al. 2013). In contrast to caspase-1, caspase-8 is activated by dectin-1, TLRs, and Fas located on the plasma membrane (Gringhuis et al. 2012; Lemmers et al. 2007; Strasser et al. 2009). Recently, Fas-activated caspase-8 has been shown to contribute to the production of inflammatory cytokines and the host defense towards pathogenic bacterial infections (Uchiyama et al. 2013). Based on this evidence, it is possible to conclude that in addition to inflammatory caspases, such as caspase-1/-11, caspases-8 plays an important role in the induction of innate inflammatory response in bacterial infections, independent of their role in apoptosis induction. In this review, we summarize the functional roles and induction mechanisms of caspase-1/-11 in innate inflammatory responses against bacterial infection. In addition, we discuss the role of other caspases, such as caspase-8, in comparison with caspase-1/-11 in the induction of the innate immune responses and regulation of infectious bacteria.
Caspase-1
IL-1β/IL-18 Processing and Pyroptosis Induction
In 1989, caspase-1 was first identified as “monocyte specific pre-interleukin 1β convertase” called as ICE, in cell lysates of human monocytes (Black et al. 1989b; Kostura et al. 1989). The cDNA encoding ICE (caspase-1) was cloned in 1992 (Cerretti et al. 1992; Thornberry et al. 1992). Although the caspase-1 ortholog of Caenorhabditis elegans, Ced-3, contributes to programmed cell death, mammalian caspase-1 is dispensable for the induction of apoptosis. Caspase-1 deficient mice do not exhibit a spontaneous apoptosis-related phenotype (Kuida et al. 1995; Li et al. 1995). Based on the reports revealing the functional roles of caspase-1, it is now generally accepted that caspase-1 plays a pivotal role as a component of the inflammasome producing mature IL-1β/IL-18. Activated caspase-1 also induces “pyroptosis”, a form of cell death distinguishable from apoptosis by the absence of certain features such as nuclear condensation (Miao et al. 2010; Tsuchiya and Hara 2014).
The core components of each canonical caspase-1 inflammasome include intracellular pattern recognition receptors such as NLRs [e.g., NLR family pyrin domain containing (NLRP) 1/3 and NLR family caspase recruitment domain (CARD) containing (NLRC) 4] and PYHIN family members [e.g., absent in melanoma 2 (AIM2)]. NLRs contain NODs that are required for oligomerization and leucine-rich repeats that likely function as sensors of PAMPs or endogenous DAMPs ligands (Tsuchiya and Hara 2014). However, there is no direct evidence thus far to indicate a direct molecular interaction between these ligands and NLRs. AIM2 contains a HIN-200 domain that acts as a sensor for double-stranded DNA (Roberts et al. 2009). Activated NLRs oligomerize through their NOD and, in the case of NLRPs that lack the CARD, recruit pro-caspase-1 through the adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC) (Fig. 2) (Schattgen and Fitzgerald 2011; Tsuchiya and Hara 2014). Once recruited to the inflammasome, pro-caspase-1 undergoes autoproteolytic cleavage to yield activated caspase-1 that, in turn, cleaves pro-IL-1β/IL-18. The cleaved, biologically active inflammatory cytokines are subsequently released from cells into the extracellular matrix through an unknown mechanism. Caspase-1-induced pyroptosis contributes to the regulation of bacterial infection by removing infectious microbes inside the cell; however, the molecular mechanisms underlying this process are still unknown.

Molecular mechanisms underlying canonical caspase-1 inflammasome activation. Nucleotide oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) responds to intracellular stress through the detection of reactive oxygen species (ROS), fluctuations in cationic concentration, and lysosome or phagolysosome destabilization. Through its NOD, activated NLRP3 oligomerizes and recruits the adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC) by interacting with its pyrin domain (PYD). In addition to a PYD, ASC contains a CARD that recruits pro-caspase-1. Caspase-1 is activated autoproteolytically and either cleaves the pro-form of cytokines including interleukin (IL)-1β and IL-18, or induces pyroptosis
NLRP3 is a well-studied canonical caspase-1 inflammasome component that can respond to microbial pathogen infections and endogenous noxious molecules that may result from cellular stress. Accumulating evidence indicates that NLRP3 inflammasomes play a significant role in the recognition of and response to bacterial infection. Although there is no direct evidence for the interaction of bacterial ligands with NLRP3, a variety of pathogenic bacteria including Staphylococcus aureus (Craven et al. 2009; Holzinger et al. 2012; Kebaier et al. 2012; Maher et al. 2013; McGilligan et al. 2013; Muñoz-Planillo et al. 2009), Streptococcus pneumoniae (Hoegen et al. 2011; McNeela et al. 2010; Witzenrath et al. 2011) and Listeria monocytogenes (Meixenberger et al. 2010) have been shown to activate the NLRP3 inflammasome. Three main triggers have been proposed to activate the NLRP3 inflammasome: intracellular reactive oxygen species (ROS)/oxidized mitochondrial DNA from damaged mitochondria, destabilization of the lysosome/phagolysosome, and changes in cellular cation concentration (Fig. 2). However, recent reports indicate that NLRP3 agonists have a common ability to render the cell membrane permeable to K+ and Na+, and a reduction in intracellular K+ concentration is sufficient for NLRP3 inflammasome activation (Muñoz-Planillo et al. 2013). In accordance with this, NLRP3 inflammasome activation is induced by changes in the concentration and flux of K+ caused by bacterial cytotoxins that form pores on the target cell membrane (McNeela et al. 2010; Zheng et al. 2011). For example, pore-forming toxins such as α-hemolysin or pneumolysin from S. aureus (Kebaier et al. 2012) or S. pneumoniae (McNeela et al. 2010), respectively, can activate the NLRP3 inflammasome through efflux of K+. In addition, extracellular ATP, released from cells stimulated with surface protein of the periodontopathogen Treponema denticola, induced K+ efflux through a cell membrane ATP receptor, P2X7, eventually leading to NLRP3 inflammasome activation (Jun et al. 2012). These reports indicate that K+ efflux in cells infected with bacteria plays a significant role in the activation of NLRP3 inflammasome, signifying danger towards the host.
In addition to the activation mechanisms triggered by cellular stresses, caspase-1-NLRP3 inflammasomes have been shown to be regulated by unique mechanisms (Tsuchiya and Hara 2014). Type I IFNs suppress the activation of the NLRP3 inflammasome and mature IL-1β production in a STAT1/3-dependent manner (Guarda et al. 2011). In addition, nitrogen oxide (NO) negatively regulated NLRP3 inflammasome activation through S-nitrosylation (Hernandez-Cuellar et al. 2012). In actual bacterial infections, NO suppresses the activation of the NLRP3 inflammasome and the production of mature IL-1β in M. tuberculosis infection. NO-mediated inhibition of NLRP3 inflammasome activation resulted in the suppression of persistent neutrophil recruitment and progressive tissue damage in M. tuberculosis infection (Mishra et al. 2013). In contrast, phosphorylation by downstream kinases including Syk and c-jun N-terminal kinase has recently been shown to regulate ASC (Hara et al. 2013), and phosphorylated ASC-induced NLRP3 inflammasome activation. These results indicate the importance of NLRP3/ASC modification in response to bacterial infection.
In addition to the knowledge gained from using mouse models, human caspase-1 has also been shown to be involved in the induction of inflammatory responses. Human caspase-1 induced mature IL-1β production (Chae et al. 2006), and the caspase-1 inflammasome contributes to inflammatory disorders such as familial Mediterranean fever (Chae et al. 2006), Crohn’s disease (Villani et al. 2009) and Muckle–Wells autoinflammatory disorder (Agostini et al. 2004).
Membrane Stabilization
In addition to the processing of pro-IL-1β/IL-18 and induction of pyroptosis, caspase-1 has been shown to support the survival of cells in response to membrane damage caused by pore-forming toxins derived from invasive bacteria (Gurcel et al. 2006). In response to stimulation with aerolysin from Aeromonas hydrophila, caspase-1 is activated by K+ efflux-mediated NLRP3 and NLRC4 inflammasome formation. Activated caspase-1 facilitates the activation of the transcription factor sterol regulatory element-binding protein 2 (SREBP-2) located in the endoplasmic reticulum (ER) membrane yielding SRE that functions as transcription factor. Activated SRE regulates the expression of genes related to cholesterol and fatty acid biosynthesis, eventually resulting in increased cell survival, possibly by facilitating membrane repair. These results suggest that active caspase-1 contributes to the maintenance of cell membranes; however, the precise molecular mechanisms that lead to caspase-1-mediated SREBP activation, and the survival and resistance of cells towards bacterial infections are unclear.
Phagosomal Acidification
In the innate response towards microbial infection, acidification of phagosomes containing the infectious microbe is a crucial process. Recently, it was shown that caspase-1 is involved in the regulation of the phagosome acidification in response to Gram-positive bacterial infections (Sokolovska et al. 2013). In response to infections such as those caused by S. aureus, caspase-1 is activated, associated with in NLRP3 inflammasomes, and becomes localized within bacteria-containing phagosomes. Caspase-1 acts on the components of NADPH oxidase 2 (NOX2) complex, and cause the pH of vacuole to decrease by controlling the activity of NOX2. These data indicate that the primary role of caspase-1 in infected cells is to induce the innate defense responses against engulfed bacteria through the regulation of phagosome acidification.
Caspase-11
The Non-Canonical Inflammasome
The murine ortholog of human caspase-4/-5, caspase-11, was first identified in the mouse as an essential factor for caspase-1 activation in response to endotoxic shock (Wang et al. 1998). Recent evidence has revealed that strain 129 mice such as 129S6/SvEvTac, lack functional caspase-11, because they express a mis-spliced and truncated form of the protein (Kayagaki et al. 2011). Furthermore, caspase-1-deficient mice generated from strain 129 lack functional caspase-11 even after extensive backcrossing with C57BL/6 or BALB/c mice because the caspase-1 and -11 gene loci are in too close proximity to be segregated by recombination. An examination of C57BL/6 strain mice lacking functional caspase-11 resulting from a deletion in exon 5, revealed that lipopolysaccharide (LPS)/cholera toxin B (CTB) or Citrobacter rodentium infection-induced caspase-1 inflammasome activation required caspase-11 (Fig. 3) (Kayagaki et al. 2011). This caspase-11-dependent inflammasome is referred to as the non-canonical inflammasome. In contrast, caspase-11 is sufficient to induce pyroptosis and the production of IL-1α/high mobility group protein box 1 in response to infections by Gram-negative bacteria including E. coli or Vibrio cholerae (Fig. 3). Furthermore, caspase-11, rather than caspase-1, is required for LPS-induced lethality in mice, which serves as a model for endotoxic shock (Kayagaki et al. 2011). These results demonstrate the importance of caspase-11 in the immune response towards Gram-negative bacterial infections through the activation of non-canonical inflammasomes (Table 1).

The non-canonical caspase-11 inflammasome. Expression of caspase-11 requires Type I interferons (IFNs) signaling, and is auto-activated or activated by unidentified plasma membrane or cytoplasmic receptors. Activated caspase-11 stimulates the production of IL-1α/high mobility group protein box 1 (HMGB1) in a caspase-1-independent manner. However, NLRP3-mediated canonical caspase-1 inflammasomes, induced by some types of bacterial infections, acts in conjunction with caspase-11 through an unknown mechanism
The mechanisms responsible for caspase-11 activation have been progressively elucidated. Type I IFNs play an essential role in the production of precursor caspase-11 (Fig. 3). Caspase-11 protein is either auto-activated or activated by unknown receptors that are induced by endogenous Type I IFNs (Broz et al. 2012; Rathinam et al. 2012). In addition, LPS from E. coli O111:B4 is found to bind CTB and the LPS-CTB conjugate is engulfed by bone marrow-derived Mϕ. In a TLR4-independent manner, endocytosed LPS activates caspase-11 and stimulates the production of IL-1β/IL-18 (Fig. 3) (Kayagaki et al. 2013). In addition, another group has shown that cytosolic LPS activates caspase-11, resulting in endotoxic shock in mice (Hagar et al. 2013). Furthermore, bacterial secretion systems to translocate effector proteins such as flagellin into host cells are involved in the activation of caspase-11. Caspase-11 is involved in the pyroptosis induction in LPS-primed macrophages via the type IV secretion system (T4SS) in Legionella pneumophila infection (Case et al. 2013). In addition to the T4SS in L. pneumophila, the type III secretion system (T3SS) in Yersinia pseudotuberculosis is involved in caspase-11-dependent IL-1α production and cell death induction (Casson et al. 2013). These results suggest that intracellular LPS and effector molecules injected via T3SS/T4SS are recognized by a cytoplasmic receptor that is yet to be identified, and activates the non-canonical inflammasome while inducing endotoxic shock.
One of the human orthologs of murine caspase-11, caspase-4 is required for the UVB-induced mature IL-1β secretion from skin-derived keratinocytes (Sollberger et al. 2012). In the process of cytokine production, caspase-4 interacts with caspase-1, and this interaction is required for the activation of caspase-1 (Sollberger et al. 2012). Caspase-5 has also been shown to be required for caspase-1 activation using human retinal pigment epithelial cells stimulated with LPS/ATP (Bian et al. 2011). These results indicate the possibility that caspase-4/-5 contributes to the activation of the inflammasome, as with murine caspase-11.
Phagosome-Lysosome Fusion
A novel function of caspase-11 was identified in regulation of pathogenic bacterial infection in macrophages (Akhter et al. 2012). In Legionella pneumophila-infected caspase-11-deficient cells, the level of bacterial degradation inside macrophages was minimal compared to that in normal cells. Caspase-11 was shown to be required for the fusion of L. pneumophila-containing phagosomes with lysosomes, and the caspase-11-mediated phagosome–lysosome fusion was only induced when cells were infected with a virulent strain of L. pneumophila. In normal cells, L. pneumophila-containing phagosomes are surrounded by polymerized F-actin. In contrast, sparse amount of F-actin was found around the phagosomes in caspase-11-deficient cells infected with L. pneumophila. Caspase-11 is known to interact with actin interacting protein 1 to promote cofilin-mediated actin depolymerization (Li et al. 2007). In addition, caspase-11 has been shown to interact with actin and modulates the phosphorylation state of cofilin in response to L. pneumophila infection, leading to the maturation of phagosomes.
Caspase-8
Caspase-8 is a well-described protease that induces apoptosis following stimulation with Fas or TNF-α signaling (Strasser et al. 2009). Here, we provide a brief summary of caspase-8 activation with an example of the Fas ligand (FasL)-Fas signaling pathway. Ligation of Fas by FasL causes an interaction between the intracellular domain of Fas and an adaptor molecule called “Fas-associated protein with death domain (FADD)” via their death domains (DD). FADD recruits caspase-8 by the interaction of death effector domains, which are present in both caspase-8 and FADD (Fig. 4). The dimerization of caspase-8 is induced in the protein complex known as the “death-inducing signaling complex (DISC)”, containing the DD of Fas, caspase-8, FADD and modulator protein c-FLIP, leading to the autoproteolytic processing and activation of caspase-8. The activated caspase-8 leaves the DISC, and then interacts with the substrates for induction of apoptosis. In certain human cells, Fas signaling causes the recruitment of caspase-10 to DISC for the induction of apoptosis.

Molecular mechanism of caspase-8 activation. Activation of Fas by FasL causes the recruitment of “Fas-associated protein with death domain (FADD)” via their death domains (DD) inside the cytosolic space. Caspase-8 then interacts with FADD via death effector domains (DED), leading to the dimerization and autoproteolytic processing of caspase-8. Activated caspase-8 then cleaves substrates related to apoptosis induction
Multiple reports indicate that in addition to its function as an apoptosis inducer, caspase-8 is also as a regulator of innate inflammatory responses in microbial infections (Table 1).
Caspase-8 Activation and IL-1β Processing Through Dectin-1 and TLR-3/4
Dectin-1, a member of the C-type lectin receptor family, is expressed on the cell surface of phagocytes, acting as a sensor of glycans in fungi (e.g., Candida albicans) or mycobacteria (Hardison and Brown 2012; Rothfuchs et al. 2007; Yadav and Schorey 2006). Activated dectin-1 induces the expression of various genes through NF-κB, leading to the production of cytokines that initiate the T helper 1 (Th1) or Th17 responses (Carvalho et al. 2012; Gow et al. 2007). In addition, dectin-1 contributes to IL-1β production against fungal or mycobacterial infection. In response to infection with C. albicans or mycobacteria, dectin-1 induces gene expression of Il1β via the CARD9-Bcl-10-MALT1 mediated signaling pathway (Fig. 5). Dectin-1-mediated signaling also stimulates the formation of the caspase-8-MALT1-ASC complex, and then the activated caspase-8 produces mature IL-1β (Fig. 5) (Gringhuis et al. 2012). The dectin-1 pathway is also known for its ability to activate the canonical caspase-1 inflammasome (Hise et al. 2009). Thus, dectin-1 ligands activate multiple pathways that converge to induce IL-1β release following fungal and mycobacterial infection.

Caspase-8 activation by dectin-1 and Toll-like receptor (TLR)-3/4. Dectin-1 and TLR-3/4 induce expression of cytokine genes in response to pathogen infection. These receptors also activate caspase-8 to produce active IL-1β/IL-18
TLRs are PRRs that recognize PAMPs/DAMPs and induce the expression of genes encoding inflammatory cytokines including TNF-α, IL-12, or IL-6. In addition, TLR-3/4 signaling that involves ligands such as polyinosinic-polycytidylic acid (poly(I:C)) or LPS induces IL-1β production. TLR-3/4-TRIF-dependent caspase-8 activation results in the production of mature IL-1β (Fig. 5) (Maelfait et al. 2008). TLR signaling also contributes to the formation of a protein complex called the “ripoptosome”, which contains caspase-8, FADD, receptor-interacting protein (RIP)1 and c-FLIP. This protein complex and the RIP3-ROS signaling pathway are required for the activation of caspase-8, leading to the production of mature IL-1β (Fig. 5) (Vince et al. 2012). Endogenous apoptosis inhibitors such as “inhibitor of apoptosis” inhibit ripoptosome formation (Fig. 5). In addition, ER stress induces the production of IL-1β in cells stimulated with LPS through TLR-4-TRIF-caspase-8 axis (Shenderov et al. 2014). These results raise the possibility that TLR-activated caspase-8 plays a significant role in the inflammatory response towards bacterial infection. However, these experiments were carried out in in vitro using purified ligands. Therefore, the role of TLR-mediated caspase-8 activation in the innate immune response towards bacterial infection in vivo is unclear.
Caspase-8 Activation and IL-1β/IL-18 Processing Through Fas
Fas (CD95 or Apo-1), a member of the TNF receptor family, is important for the induction of apoptotic cell death. In the context of the host defense system, it has been shown that Fas signaling contributes to the apoptotic elimination of virus-infected or tumor cells (Strasser et al. 2009). In addition, Fas signaling contributes to the production of active IL-1β/IL-18; injection of FasL-expressing cells into the peritoneal cavity of mice induces the production of active IL-1β and the accumulation of neutrophils (Miwa et al. 1998). FasL-Fas signaling also contributes to the production of active IL-18 in a mice model of graft-versus-host disease (Tsutsui et al. 1999). In caspase-8/RIP3-deficient cells, Fas-activated caspase-8 has been shown to contribute to the processing of pro-IL-1β/IL-18 to yield active cytokines (Bossaller et al. 2012). These reports suggest that Fas-caspase-8 signaling play a significant role in the innate immune system in response to pathogenic microbe infection. Apart from the recognition of PAMPs/DAMPs by dectin-1 or TLR-3/4, and the stimulation of the Fas receptor by FasL on host cells, little is known about this mechanism.
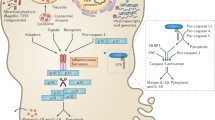
Recently, insight into this mechanism was attained using a mouse infection model involving pathogenic L. monocytogenes (Fig. 6) (Uchiyama et al. 2013). In response to L. monocytogenes infection, a lower level of mature IL-1β/IL-18 is produced from Fas −/− Mϕ than wild type cells, regulating L. monocytogenes in vivo. Natural killer (NK) cells induced FasL expression in response to L. monocytogenes infection and the FasL on NK cells stimulated macrophages, leading to the caspase-8 activation and cleavage of cytokine pro-forms to yield active ones.

Schematic representation of Fas-mediated caspase-8 activation and cytokine production. Fas ligand (FasL), expressed on natural killer (NK) cells, induces caspase-8 activation through an intracellular ROS-dependent mechanism that potentially contributes to the production of mature IL-1β/IL-18 and ASC. Type I IFNs negatively regulate the production of active IL-1β through an unknown mechanism
Caspase-1, -9, and -11 do not contribute to Fas-mediated cytokine production; cells deficient in these genes produced level of mature IL-1β/IL-18 that are comparable to wild type cells. In addition, NLRP3, NLRC4, and P2X7R, which are required for the caspase-1 inflammasome, are dispensable. However, the adaptor molecule of caspase-1 inflammasome ASC is partially required for IL-1β/IL-18 production in response to FasL stimulation. ASC is known to contribute to the activation of caspase-1; however, in the absence of ASC, caspase-8 is activated to a level comparable to wild type cells in response to FasL stimulation. These results suggest that ASC contributes to the production of mature cytokines through an unknown mechanism that does not involve caspase-8 activation.
The production of IL-1β/IL-18 is regulated by Type I IFNs (Guarda et al. 2011; Rathinam et al. 2012). In addition, L. monocytogenes infections induce the expression of Type I IFNs including IFN-β (Reimer et al. 2007; Reutterer et al. 2008; Yamamoto et al. 2012). Fas-mediated cytokine production is also regulated by Type I IFNs. IL-18 production is abolished in L. monocytogenes-infected Ifnαr1 −/− cells. The IFN consensus sequence binding protein, which is activated by Type I IFNs, regulates the promoter region of Il18. In accordance with this, Ifnαr1 −/− cells failed to upregulate Il18 expression in response to L. monocytogenes infection, leading to the low-level production of active IL-18 (Fig. 6).
NF-κB Regulation
Double-stranded RNA-stimulated NF-κB activation and inflammatory cytokine gene expression regulated by retinoic acid-inducible gene-I and melanoma differentiation-associated gene-5, requires caspase-8/-10 (Buskiewicz et al. 2012; Takahashi et al. 2006). TLR-4 signaling also requires caspase-8 for NF-κB activation, because caspase-8-deficient B cells show a delayed nuclear translocation of NF-κB (Lemmers et al. 2007). In addition, multiple reports indicate that caspase-8 activation is required for the proliferation and activation of T cells induced by antibodies against CD3 and recombinant IL-2 stimulation (Maelfait and Beyaert 2008). In this context, activated caspase-8 was also shown to play a role in the activation of NF-κB (Maksimow et al. 2006). The differentiation of monocytes into macrophages also requires caspase-8 activity. The deletion of the caspase-8 gene in myeloid cells results in the inhibition of differentiation (Maelfait and Beyaert 2008). Stimulation with macrophage colony-stimulating factor activates the caspase-8-mediated cleavage of RIP1. Processed RIP1 is involved in the inhibition of sustained NF-κB activation, favoring proper macrophage differentiation (Rébé et al. 2007). These results suggest that caspase-8 plays a significant role in the regulation of NF-κB in the innate immune response towards bacterial infection; however, the functional role in the regulation of the infectious bacteria is not clear.
Human caspase-8 also contributes to NF-κB activation, leading to IL-8 production in response to muramyl dipeptide stimulation (Hasegawa et al. 2005). Individuals containing a mutation in the caspase-8 gene show immunodeficiency characterized by recurrent sinopulmonary and herpes simplex virus infections (Chun et al. 2002). These results indicate the possibility that human caspase-8 also contributes to the inflammatory responses against microbial infection.
Inhibition of Inflammatory Responses
In addition to the induction of inflammation, caspase-8 is shown to contribute to the inhibition of inflammatory cytokine production. Caspase-8-deficient epidermal cells show an increased inflammatory response towards transfected DNA, leading to chronic skin inflammation (Kovalenko et al. 2009).
Regulation of the Inflammasome
In addition to its role in cytokine production, caspase-8 also contributes to the regulation of inflammasome activation. Caspase-8 deficiency augments the NLRP3 inflammasome activation and IL-1β production in response to LPS stimulation (Kang et al. 2013). In a basal condition, RIP3 contributes to the activation of the NLRP3 inflammasome, which is inhibited by caspase-8 under certain conditions (Kang et al. 2013). In contrast, deletion of caspase-8 or FADD in a RIP3-deficient background eliminates the caspase-1 activation and the production of mature IL-1β in response to LPS/ATP stimulation or C. rodentium infection, indicating positive regulation of inflammasome activation by caspase-8 (Gurung et al. 2014). Caspase-8 was shown to be involved in the expression of inflammasome-related and pro-IL-1β genes and activation of the NLRP3 inflammasome (Gurung et al. 2014). These results indicate that caspase-8 plays important roles in the positive and negative regulation of inflammatory responses mediated by the NLRP3 inflammasome.
Other Caspases
Caspase-12-deficient mice were shown to have greater resistance in a murine model of sepsis (Saleh et al. 2006). Interestingly, the proteolytic activity of caspase-12 is not required for this phenomenon. In addition, it has been shown that full-length caspase-12 (caspase-12L) associates with caspase-1, inhibiting caspase-1 activity and leading to the inhibition of IL-1β, IL-18, and IFN-γ production (Saleh et al. 2006). Caspase-12 also associates with NOD2 via RIP2, leading to the inhibition of NF-κB activation. This results in the downregulation of antimicrobial responses (LeBlanc et al. 2008).
Caspase-9 is well-known effector involved in apoptosis induction through the mitochondrial intrinsic pathway. Caspase-9 was shown to inhibit necrotic cell death in macrophages infected with M. tuberculosis, leading to maintain the place of growth for bacterium in vitro (Uchiyama et al. 2007). However, it is not clear whether caspase-9 contributes to the regulation of M. tuberculosis in vivo.
Caspase-7 was identified as a substrate of caspase-1 (Lamkanfi et al. 2008). Later, it was shown that L. pneumophila infection activates caspase-1 through NLRC4-inflammasome formation, leading to the cleavage of caspase-7. Mice and macrophages lacking caspase-7 allow substantial L. pneumophila replication. It has been shown that caspase-7 is involved in the maturation of L. pneumophila-containing phagosomes and induces cell death in infected macrophages (Akhter et al. 2009). In addition, caspase-7 contributes to the activation of NF-κB through the cleavage of PARP1 in response to LPS stimulation in macrophages (Erener et al. 2012); however, it is not clear whether this mechanism is involved in the inflammatory response towards genuine bacterial infection.
Concluding Remarks
Caspases play a well-characterized role in apoptosis induction during the innate inflammatory response towards bacterial infection. In this review, we have summarized the novel functions and activation mechanisms of caspases including those that are involved in not only the activation, but also the inhibition of inflammation. Furthermore, in an in vivo context, caspases are activated in numerous cell type including Mϕ, DC, T, and B cells. Additional investigation into the functional roles of caspases will be required to further our understanding of the mechanism by which caspases control the innate defense response towards bacterial infections in vivo.
Abbreviations
- AIM2:
-
Absent in melanoma 2
- ASC:
-
Apoptosis-associated speck-like protein containing a CARD
- CARD:
-
Caspase recruitment domain
- CTB:
-
Cholera toxin B
- DAMPs:
-
Damage-associated molecular patterns
- DC:
-
Dendritic cells
- DD:
-
Death domain
- DISC:
-
Death-inducing signaling complex
- ER:
-
Endoplasmic reticulum
- FADD:
-
Fas-associated protein with death domain
- FasL:
-
Fas ligand
- HIN:
-
Hematopoietic IFN-inducible nuclear proteins
- ICE:
-
Interleukin-1β-converting enzyme
- IFN:
-
Interferon
- LPS:
-
Lipopolysaccharide
- Mϕ:
-
Macrophage
- NLRC4:
-
NLR family CARD domain containing 4
- NLRP3:
-
NLR family pyrin domain containing 3
- NLRs:
-
NOD-like receptors
- NO:
-
Nitrogen oxide
- NOD:
-
Nucleotide oligomerization domain
- NOX2:
-
NADPH oxidase 2
- PAMPs:
-
Pathogen-associated molecular patterns
- PRRs:
-
Pattern recognition receptors
- RIP:
-
Receptor-interacting protein
- ROS:
-
Reactive oxygen species
- SREBP-2:
-
Sterol regulatory element-binding protein 2
- T3SS:
-
Type three secretion system
- T4SS:
-
Type four secretion system
- Th1/17:
-
T helper 1/17
- TIR:
-
Toll/interleukin-1 receptor
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor α
- TRIF:
-
Toll/IL-1 receptor-domain-containing adaptor-inducing IFN-β
References
Agostini L, Martinon F, Burns K et al (2004) NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-wells autoinflammatory disorder. Immunity 20:319–325
Ahmad-Nejad P, Häcker H, Rutz M et al (2002) Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur J Immunol 32:1958–1968
Akhter A, Gavrilin MA, Frantz L et al (2009) Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog 5:e1000361
Akhter A, Caution K, Abu Khweek A et al (2012) Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37:35–47
Bian ZM, Elner SG, Khanna H et al (2011) Expression and functional roles of caspase-5 in inflammatory responses of human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 52:8646–8656
Black RA, Kronheim SR, Sleath PR (1989a) Activation of interleukin-1 beta by a co-induced protease. FEBS Lett 247:386–390
Black RA, Kronheim SR, Merriam JE et al (1989b) A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J Biol Chem 264:5323–5326
Bossaller L, Chiang PI, Schmidt-Lauber C et al (2012) Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol 189:5508–5512
Broz P, von Moltke J, Jones JW et al (2010) Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microb 8:471–483
Broz P, Ruby T, Belhocine K et al (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490:288–291
Buskiewicz IA, Koenig A, Huber SA et al (2012) Caspase-8 and FLIP regulate RIG-I/MDA5-induced innate immune host responses to picornaviruses. Future Virol 7:1221–1236
Carvalho A, Giovannini G, De Luca A et al (2012) Dectin-1 isoforms contribute to distinct Th1/Th17 cell activation in mucosal candidiasis. Cell Mol Immunol 9:276–286
Case CL, Kohler LJ, Lima JB et al (2013) Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci USA 110:1851–1856
Casson CN, Copenhaver AM, Zwack EE et al (2013) Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 9:e1003400
Cerretti DP, Kozlosky CJ, Mosley B et al (1992) Molecular cloning of the interleukin-1 beta converting enzyme. Science 256:97–100
Chae JJ, Wood G, Masters SL et al (2006) The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci USA 103:9982–9987
Chun HJ, Zheng L, Ahmad M et al (2002) Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419:395–399
Craven RR, Gao X, Allen IC et al (2009) Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One 4:e7446
Denecker G, Ovaere P, Vandenabeele P et al (2008) Caspase-14 reveals its secrets. J Cell Biol 180:451–458
Erener S, Pétrilli V, Kassner I et al (2012) Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-κB target genes. Mol Cell 46:200–211
Geijtenbeek TB, Gringhuis SI (2009) Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9:465–479
Gow NA, Netea MG, Munro CA et al (2007) Immune recognition of Candida albicans beta-glucan by dectin-1. J Infect Dis 196:1565–1571
Gringhuis SI, Kaptein TM, Wevers BA et al (2012) Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol 13:246–254
Guarda G, Braun M, Staehli F et al (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223
Guo Y, Srinivasula SM, Druilhe A et al (2002) Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem 277:13430–13437
Gurcel L, Abrami L, Girardin S et al (2006) Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126:1135–1145
Gurung P, Anand PK, Malireddi RK et al (2014) FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192:1835–1846
Hagar JA, Powell DA, Aachoui Y et al (2013) Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341:1250–1253
Hara H, Tsuchiya K, Kawamura I et al (2013) Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol 14:1247–1255
Hardison SE, Brown GD (2012) C-type lectin receptors orchestrate antifungal immunity. Nat Immunol 13:817–822
Hasegawa M, Imamura R, Kinoshita T et al (2005) ASC-mediated NF-kappaB activation leading to interleukin-8 production requires caspase-8 and is inhibited by CLARP. J Biol Chem 280:15122–15130
Hernandez-Cuellar E, Tsuchiya K, Hara H et al (2012) Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol 189:5113–5117
Hise AG, Tomalka J, Ganesan S et al (2009) An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microb 5:487–497
Hoegen T, Tremel N, Klein M et al (2011) The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol 187:5440–5451
Holzinger D, Gieldon L, Mysore V et al (2012) Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol 92:1069–1081
Jun HK, Lee SH, Lee HR et al (2012) Integrin α5β1 activates the NLRP3 inflammasome by direct interaction with a bacterial surface protein. Immunity 36:755–768
Kang TB, Yang SH, Toth B et al (2013) Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38:27–40
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650
Kayagaki N, Warming S, Lamkanfi M et al (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121
Kayagaki N, Wong MT, Stowe IB et al (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341:1246–1249
Kebaier C, Chamberland RR, Allen IC et al (2012) Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis 205:807–817
Kischkel FC, Lawrence DA, Tinel A et al (2001) Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem 276:46639–46646
Koizumi Y, Toma C, Higa N et al (2012) Inflammasome activation via intracellular NLRs triggered by bacterial infection. Cell Microbiol 14:149–154
Kostura MJ, Tocci MJ, Limjuco G et al (1989) Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci USA 86:5227–5231
Kovalenko A, Kim JC, Kang TB et al (2009) Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med 206:2161–2177
Kuida K, Lippke JA, Ku G et al (1995) Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267:2000–2003
Lamkanfi M, Kalai M, Vandenabeele P (2004) Caspase-12: an overview. Cell Death Differ 11:365–368
Lamkanfi M, Kanneganti TD, Van Damme P et al (2008) Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7:2350–2363
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411
LeBlanc PM, Yeretssian G, Rutherford N et al (2008) Caspase-12 modulates NOD signaling and regulates antimicrobial peptide production and mucosal immunity. Cell Host Microb 3:146–157
Lemmers B, Salmena L, Bidère N et al (2007) Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J Biol Chem 282:7416–7423
Li P, Allen H, Banerjee S et al (1995) Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80:401–411
Li J, Brieher WM, Scimone ML et al (2007) Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat Cell Biol 9:276–286
Maelfait J, Beyaert R (2008) Non-apoptotic functions of caspase-8. Biochem Pharmacol 76:1365–1373
Maelfait J, Vercammen E, Janssens S et al (2008) Stimulation of Toll-like receptor 3 and 4 induces interleukin-1β maturation by caspase-8. J Exp Med 205:1967–1973
Maher BM, Mulcahy ME, Murphy AG et al (2013) Nlrp-3-driven interleukin 17 production by γδT cells controls infection outcomes during Staphylococcus aureus surgical site infection. Infect Immun 81:4478–4489
Maksimow M, Söderström TS, Jalkanen S et al (2006) Fas costimulation of naive CD4 T cells is controlled by NF-kappaB signaling and caspase activity. J Leukoc Biol 79:369–377
Matsumoto M, Funami K, Tanabe M et al (2003) Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol 171:3154–3162
McGilligan VE, Gregory-Ksander MS, Li D et al (2013) Staphylococcus aureus activates the NLRP3 inflammasome in human and rat conjunctival goblet cells. PLoS One 8:e74010
McIlwain DR, Berger T, Mak TW (2013) Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5:a008656
McNeela EA, Burke A, Neill DR et al (2010) Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog 6:e1001191
Meixenberger K, Pache F, Eitel J et al (2010) Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol 184:922–930
Miao EA, Leaf IA, Treuting PM et al (2010) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11:1136–1142
Mishra BB, Rathinam VA, Martens GW et al (2013) Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol 14:52–60
Miwa K, Asano M, Horai R et al (1998) Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat Med 4:1287–1292
Muñoz-Planillo R, Franchi L, Miller LS et al (2009) A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol 183:3942–3948
Muñoz-Planillo R, Kuffa P, Martínez-Colón G et al (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153
Newton K, Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4(3)
Parrish AB, Freel CD, Kornbluth S et al (2013) Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol 5(6)
Rathinam VA, Vanaja SK, Waggoner L et al (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150:606–619
Rébé C, Cathelin S, Launay S et al (2007) Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood 109:1442–1450
Reimer T, Schweizer M, Jungi TW (2007) Type I IFN induction in response to Listeria monocytogenes in human macrophages: evidence for a differential activation of IFN regulatory factor 3 (IRF3). J Immunol 179:1166–1177
Reutterer B, Stockinger S, Pilz A et al (2008) Type I IFN are host modulators of strain-specific Listeria monocytogenes virulence. Cell Microbiol 10:1116–1129
Roberts TL, Idris A, Dunn JA et al (2009) HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323:1057–1060
Rothfuchs AG, Bafica A, Feng CG et al (2007) Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol 179:3463–3471
Saleh M, Mathison JC, Wolinski MK et al (2006) Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440:1064–1068
Schattgen SA, Fitzgerald KA (2011) The PYHIN protein family as mediators of host defenses. Immunol Rev 243:109–118
Shenderov K, Riteau N, Yip R et al (2014) Cutting edge: endoplasmic reticulum stress licenses macrophages to produce mature IL-1β in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J Immunol 192:2029–2033
Sokolovska A, Becker CE, Ip WK et al (2013) Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol 14:543–553
Sollberger G, Strittmatter GE, Kistowska M et al (2012) Caspase-4 is required for activation of inflammasomes. J Immunol 188:1992–2000
Strasser A, Jost PJ, Nagata S (2009) The many roles of FAS receptor signaling in the immune system. Immunity 30:180–192
Takahashi K, Kawai T, Kumar H et al (2006) Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J Immunol 176:4520–4524
Thornberry NA, Bull HG, Calaycay JR et al (1992) A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356:768–774
Tsuchiya K, Hara H (2014) The inflammasome and its regulation. Crit Rev Immunol 34:41–80
Tsutsui H, Kayagaki N, Kuida K et al (1999) Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 11:359–367
Uchiyama R, Kawamura I, Fujimura T et al (2007) Involvement of caspase-9 in the inhibition of necrosis of RAW 264 cells infected with Mycobacterium tuberculosis. Infect Immun 75:2894–2902
Uchiyama R, Yonehara S, Tsutsui H (2013) Fas-mediated inflammatory response in Listeria monocytogenes infection. J Immunol 190:4245–4254
Villani AC, Lemire M, Fortin G et al (2009) Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet 41:71–76
Vince JE, Wong WW, Gentle I et al (2012) Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36:215–227
Wang S, Miura M, Jung YK et al (1998) Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92:501–509
Witzenrath M, Pache F, Lorenz D et al (2011) The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol 187:434–440
Yadav M, Schorey JS (2006) The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108:3168–3175
Yamamoto T, Hara H, Tsuchiya K et al (2012) Listeria monocytogenes strain-specific impairment of the TetR regulator underlies the drastic increase in cyclic di-AMP secretion and beta interferon-inducing ability. Infect Immun 80:2323–2332
Zheng Y, Lilo S, Brodsky IE et al (2011) A Yersinia effector with enhanced inhibitory activity on the NF-κB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog 7:e1002026
Acknowledgments
The authors thank Prof. Shin Yonehara (Kyoto University) and Prof. Kenji Nakanishi (Hyogo College of Medicine) for helpful discussions. This work was supported by the Japan Society for the Promotion of Science KAKENHI Grant 20790334 and by a Grant-in-Aid for Researchers, Hyogo College of Medicine, 2010.
Conflict of interest
The authors have no financial conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Uchiyama, R., Tsutsui, H. Caspases as the Key Effectors of Inflammatory Responses Against Bacterial Infection. Arch. Immunol. Ther. Exp. 63, 1–13 (2015). https://doi.org/10.1007/s00005-014-0301-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-014-0301-2