
Abstract
Inflammasomes are multi-protein platforms that are organized in the cytosol to cope with pathogens and cellular stress. The pattern recognition receptors NLRP1, NLRP3, NLRC4, AIM2 and Pyrin all assemble canonical platforms for caspase-1 activation, while caspase-11-dependent inflammasomes respond to intracellular Gram-negative pathogens. Inflammasomes are chiefly known for their roles in maturation and secretion of the inflammatory cytokines interleukin-(IL)1β and IL18, but they can also induce regulated cell death. Activation of caspases 1 and 11 in myeloid cells can trigger pyroptosis, a lytic and inflammatory cell death mode. Pyroptosis has been implicated in secretion of IL1β, IL18 and intracellular alarmins. Akin to these factors, it may have beneficial roles in controlling pathogen replication, but become detrimental in the context of chronic autoinflammatory diseases. Inflammasomes are increasingly implicated in induction of additional regulated cell death modes such as pyronecrosis and apoptosis. In this review, we overview recent advances in inflammasome-associated cell death research, illustrating the polyvalent roles of these macromolecular platforms in regulated cell death signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Canonical and non-canonical inflammasome platforms
Inflammasomes are cytosolic multiprotein complexes that are assembled when the cell encounters pathogens, toxins, crystals and a diversity of other agents that may harm the host [1, 2]. These potentially cytotoxic agents or the ensuing cellular stress are monitored by a set of intracellular pattern recognition receptors (PRRs) that auto-oligomerize and recruit additional inflammasome components. Similar to the death-inducing signaling complexes that activate the apoptotic initiator caspases 8 and 10 [3], and the caspase-9-activating apoptosome [4], oligomerization of inactive procaspase-1 zymogens in inflammasomes results in the proximity-induced autoactivation of the protease [1]. The caspase recruitment domain (CARD)- and pyrin domain (PYD)-containing adaptor protein Apoptosis-associated speck-like protein containing a CARD (ASC) bridges the recruitment of procaspase-1, although inflammasomes of CARD-containing PRRs may also recruit the inflammatory protease directly [5–7]. Once active, caspase-1 matures and releases the pleiotropic inflammatory cytokines interleukin(IL)-1β and IL18, thereby contributing importantly to inflammatory and immune responses [8]. Caspase-1 also induces pyroptosis, a lytic regulated cell death mode of myeloid cells that is emerging as a critical host defense mechanism against microbial pathogens [9]. On the other hand, excessive and unwarranted pyroptosis induction is linked to autoinflammatory disease [10–13]. Also, activation of murine caspase-11 and its human orthologs caspases 4 and 5 in the non-canonical inflammasome pathway induces pyroptosis [14–16]. Furthermore, inflammasome-induced cell death responses are increasingly recognized to extend well beyond caspase-1- and -11-induced pyroptosis, and encompass apoptosis and pyronecrosis as discussed in the following paragraphs.
The canonical inflammasomes
By definition, the canonical inflammasomes activate caspase-1. Most canonical inflammasomes are assembled by PRRs that belong to the nucleotide-binding oligomerization domain-like receptor (NLR) family [17], but Pyrin [2] and the HIN200 protein AIM2 [18, 19] also engage well-defined inflammasomes. The roles and signaling mechanisms of the different inflammasomes have been reviewed extensively [1, 20–22], and they will only be briefly discussed here in the context of their roles in regulated cell death signaling.
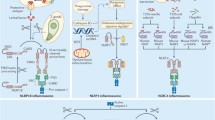
Akin to other NLRs, NLRP3 possesses a central nucleotide-binding NAIP, CIITA, HET-E and TP1 (NACHT) domain and a number of C-terminal leucine-rich repeat (LRR) domains that are thought to regulate NLR activation. NLRP3 is a representative member of the NLRP subfamily, which are characterized by a PYD domain in the amino-terminus. The NLRP3 inflammasome is activated in response to a broad variety of pathogen- and danger-related activators that includes Gram-negative bacteria, Staphylococcus aureus, RNA viruses, pore-forming toxins, ionophores, crystals, etc. [23] (Fig. 1). As it is unlikely for NLRP3 to physically ligate such a diverse set of activators, it is commonly thought that these stimuli may converge on production of a secondary messenger that is monitored by NLRP3. K+ efflux, Ca2+ signaling, mitochondria-derived reactive oxygen species (ROS), cytosolic release of mitochondrial DNA and cardiolipin, release of lysosomal cathepsins and microtubule-assisted relocation of the NLRP3 inflammasome to mitochondria–endoplasmic reticulum (ER) foci all are mechanisms that may possibly contribute to NLRP3 activation [24–30], but further research is required to fully understand NLRP3 activation mechanisms.

Schematic representation of the inflammasome pathways resulting in pyroptosis. Pyroptosis as a regulated lytic cell death mode of myeloid cells is induced in response to a wide variety of endogenous, environmental, and pathogen-derived triggers downstream of inflammasome-associated activation of caspases 1 or 11 in mice, or that of the orthologous caspases 1, 4, and 5 in humans. While the PYD-based NLRP3, AIM2, and Pyrin platforms require the bipartite adaptor protein ASC to recruit and activate caspase-1, the CARD-based NLRP1b and NLRC4 inflammasomes trigger caspase-1-dependent pyroptosis independently of ASC. Both caspases 1 and 11 engage pyroptosis directly by cleaving its substrate gasdermin D. The mechanism by which gasdermin D triggers pyroptosis is currently unknown
The human NLRP1 inflammasome has been implicated in Vitiligo-associated autoimmune disease [31], autoimmune Addison’s disease [32], type I diabetes [33, 34], and autoinflammation [35]. Importantly, humans possess only one NLRP1 gene, whereas the mouse genome contains NLRP1a, NLRP1b, and NLRP1c genes. A number of studies have shown that NLRP1a and NLRP1b assemble inflammasomes that non-redundantly activate caspase-1 in hematopoietic progenitor cells infected with lymphocytic choriomeningitis virus (LCMV) [36], and in macrophages intoxicated with Bacillus anthracis lethal toxin (LeTx), respectively [37]. LeTx consists of two subunits: the protective antigen (PA) subunit attaches to plasma membrane-bound host receptors and assists in cytosolic translocation of the zinc metalloproteinase subunit lethal factor (LF). In the cytosol, LF protease activity triggers assembly of the NLRP1b inflammasome through mechanisms that are not entirely clear yet [38, 39] (Fig. 1).
NLRC4 is another NLR protein that assembles a canonical inflammasome [40]. Unlike NLRP3, NLRC4 contains an amino-terminal CARD. The NLRC4 inflammasome plays an important role in host defense against Salmonella enterica serovar Typhimurium (S. Typhimurium), Legionella pneumophila, Pseudomonas aeruginosa, Shigella flexneri, and Burkholderia thailandensis [23, 41, 42]. Members of the NAIP subfamily—which are characterized by amino-terminal baculovirus IAP repeat (BIR) motifs—bind flagellin or components of bacterial type III secretion systems (T3SS) of pathogenic bacteria that gain access to the cytosol (Fig. 1) [43–45]. NAIP5-independent phosphorylation of NLRC4 is also required for engagement of flagellin-induced inflammasome signaling [46, 47]. Importantly, while rodents encode multiple NAIP genes, humans express different isoforms from a single NAIP gene that detect flagellin and T3SS components, respectively [48].
Also the HIN200 family member Absent in Melanoma 2 (AIM2) assembles a well-characterized inflammasome. Through its dsDNA-binding HIN200 domain, AIM2 detects the presence of viral and bacterial pathogens in the cytosolic compartment [49–51]. AIM2 mediates host defense against cytomegalovirus and influenza virus, but also restricts Francisella tularensis. Guanylate-binding proteins (GBPs) appear dispensable for AIM2 detection of transfected DNA and cytomegalovirus infection, but they are required for responding to AIM2-activating bacterial pathogens [52, 53]. This is explained by the notion that these type I interferon (IFN)-induced GTPases mediate lysis of bacteria-encapsulating vacuoles or the bacterial cell wall, thereby exposing F. tularensis DNA to AIM2 detection.
Pyrin is the latest addition to the list of pattern recognition receptors (PRRs) that engages a canonical inflammasome. Pyrin is mutated in familial Mediterranean Fever patients, and the protein was recently shown to respond to bacterial toxins that post-translationally modify members of the Rho GTPase family [2, 54]. Pathogens such as Clostridium difficile, Clostridium botulinum, Vibrio parahaemolyticus, Histophilus somni, and Burkholderia cenocepacia express such toxins and induce Pyrin-dependent caspase-1 activation (Fig. 1). In light of the reported association of Pyrin with the cell cytoskeleton [55], this suggests that Pyrin may guard disrupted Rho signaling indirectly, possibly by monitoring remodeling of the cytoskeleton downstream of toxin-induced inhibition of the GTPase [2].
The non-canonical inflammasome
Akin to the role of caspase-1 in canonical inflammasomes, caspase-11 acts as the effector protease of the non-canonical inflammasome [14]. Notably, this inflammasome pathway has emerged only recently with the unexpected observation that widely used caspase-1 knockout mice were also deficient in caspase-11 expression [14]. The non-canonical inflammasome pathway targets Gram-negative bacteria that gain access to the cytosolic compartment (Fig. 1). Direct binding of cytosolic LPS was shown to promote caspase-11-mediated pyroptosis as a host defense response that is thought to be important for disposal of macrophages that are infected with Escherichia coli, Citrobacter rodentium, Vibrio cholerae and other Gram-negative pathogens [14, 56]. Akin to their role in lysis of F. tularensis-containing vacuoles to license AIM2 inflammasome activation, GBP GTPases act upstream of caspase-11 for inducing pyroptosis of infected macrophages [57, 58]. Caspase-11 cannot mature IL1β and IL18 directly [59], but it nevertheless promotes secretion of bioactive IL1β and IL18 indirectly through engagement of the NLRP3 inflammasome [14].
Mechanisms of inflammasome-induced pyroptosis
Caspases are well known for their chief roles in apoptosis signaling [60]. Unlike their apoptotic counterparts, the inflammatory caspase subset—i.e., human caspases 1, 4 and 5, and murine caspases 1 and 11—triggers pyroptosis. Maturation of the apoptotic executioner caspases 3 and 7, internucleosomal DNA fragmentation and cleavage of Poly (ADP-ribose) polymerase-1 (PARP1) are considered hallmarks of apoptosis. However, these events may not be apoptosis-selective biomarkers as Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-activity is also detected in S. Typhimurium-infected macrophages [61–63], and occurs downstream of caspase-1 [64] (Fig. 2). Similarly, inflammasome activation promotes caspase-1-dependent maturation of caspase-7, and cleavage of PARP1 [64–66] (Fig. 2). Unlike caspase-1, however, deletion of neither caspase-7 nor PARP1 halted pyroptosis by the Nlrp3 and Nlrc4 inflammasomes [64, 65], suggesting that caspase-7 and PARP1 may contribute to other inflammasome-linked events. In this regard, caspase-7 activation by the Nlrc4 inflammasome was shown to promote endolysosomal destruction of Legionella pneumophila in infected macrophages [67], and caspase-7-mediated PARP1 cleavage enhanced transcription of NF-κB-dependent target genes [66]. Despite these overlapping molecular characteristics, apoptotic and pyroptotic cells differ markedly in other aspects. Apoptosis features cell body shrinkage (pyknosis) and formation of apoptotic body formation, whereas pyroptotic cells undergo cytoplasmic swelling, consequently leading to opposing immunological outcomes. While apoptosis is an immunologically silent regulated cell death mode in which the plasma membrane integrity of the dying cell is not compromised before cells are phagocytosed, pyroptosis represents a lytic regulated cell death mode that is characterized by early membrane rupture and extracellular release of the intracellular contents. Because these hallmarks do not distinguish pyroptosis from other lytic regulated cell death modes such as necroptosis [68], pyroptosis is better defined as ‘a lytic regulated cell death mode that relies on the enzymatic activity of inflammatory caspases’. The term ‘pyroptosis’ combines the Greek roots ‘pyros’ and ‘ptosis’—which, respectively, stand for ‘fire’ and ‘falling’—to highlight the inflammatory nature of this cell death mode [69]. Although the term pyroptosis was coined only in 2001, reports describing caspase-1-mediated regulated cell death of S. Typhimurium- and S. flexneri-infected macrophages date back to as early as 1996, even though at the time these events were designated as apoptotic or necrotic cell death [70–74]. In contrast, caspase-11 only recently emerged as a caspase that shares with caspase-1 the ability to induce pyroptosis in Gram-negative-infected macrophages [14].

Scheme of the morphological and biochemical features of pyroptosis and apoptosis. These forms of cell death represent two very distinct forms of regulated cell death in terms of their final outcome, but share some important characteristics related to their signaling events (highlighted in green boxes)
Though still incompletely understood, understanding of the molecular mechanisms of pyroptosis has gained significant traction lately. The increasingly detailed description of the mechanisms driving caspase-1 and -11 activation in the canonical and non-canonical inflammasome pathways have clarified how pyroptosis is induced by microbial pathogen-associated molecular patterns (PAMPs) and cellular danger signals [23]. Moreover, the recent discovery that gasdermin D cleavage is critical for both caspase-1- and -11-induced pyroptosis revealed a shared pyroptosis execution mechanism in the canonical and non-canonical inflammasome arms [75, 76]. Little is currently known about the physiological roles of gasdermin D and other gasdermin family members, but cleavage of human gasdermin D after Asp275 (corresponding to Asp276 in mouse gasdermin D) by caspase-1 and the caspase-11 orthologues caspases 4 and 5 releases an amino-terminal fragment that suffices to induce pyroptosis when overexpressed (Fig. 2). Gasdermin D is present in humans, mouse, rat and genomes of other mammals, but absent in birds, insects, amphibians and fish. Notably, the amino-terminal domains of other gasdermin proteins also induce cell death when ectopically expressed, but gasdermin D appears the sole family member in which the regulatory carboxy-terminal domain is physically detached from the amino-terminal region by caspase-1/11 cleavage [75, 76]. It would, therefore, be interesting to examine whether the cell death-inducing properties of other gasdermins may be exposed by alternative post-translation modifications in the flexible linker region, and whether this contributes to pyroptosis and/or other lytic cell death modes in macrophages and other cell types.
Remarkably, while hundreds of caspase cleavage events coordinately orchestrate apoptotic cell death [77–79], caspase-11-mediated pyroptosis appears to rely solely on gasdermin D cleavage because gasdermin D-deficient macrophages are fully protected from caspase-11-induced pyroptosis [75, 76]. Because gasdermin D deletion conferred only a temporal protection from caspase-1-induced pyroptosis [76], it is likely that cleavage of additional substrates contributes to caspase-1-mediated pyroptosis. This also suggests that cleavage events associated with pyroptosis via the canonical and non-canonical inflammasomes may overlap significantly because caspase-11-mediated gasdermin D cleavage indirectly engages caspase-1 downstream of the Nlrp3 inflammasome [75, 76].
A central outstanding question concerns the mechanism(s) by which caspase-1/11 cleavage of gasdermin D engages pyroptosis. Given the early plasma membrane rupture in pyroptotic cells, the amino-terminal gasdermin D domain may oligomerize to form a membrane pore akin to how MLKL may act during necroptosis [80, 81]. Alternatively, the pyroptotic gasdermin D domain may induce cell lysis indirectly by damaging intracellular organelles that result in disruption of mitochondrial respiration and ATP synthesis. In this regard, activation of the NLRP3 and AIM2 inflammasomes was shown to be associated with caspase-1-mediated mitochondrial damage that was accompanied by cleavage of the pro-apoptotic Bcl-2 family member Bid and cytosolic release of mitochondrial cytochrome c [82] (Fig. 2). However, transgenic expression of Bcl-2 and deletion of apoptosis-associated mitochondrial outer membrane permeabilization inducers Bid, Bok, Bax, and Bak do not alter the course of pyroptosis induced by the NLRP3 or AIM2 inflammasomes [82, 83], suggesting that mitochondrial destabilization in pyroptotic cells may occur through other mechanisms.
Inflammasome-induced gasdermin D cleavage may also induce pyroptosis by modulating ion fluxes from intracellular stores and/or plasma membrane-bound ion channels, which could explain osmosis and the plasma membrane rupture of pyroptotic cells. In this regard, pyroptosis was suggested to be accompanied by caspase-1-induced formation of transmembrane pores of approximately 1.1–2.4 nm prior to cell lysis [61] (Fig. 2). Unlike caspase-1, caspase-11 was proposed to cleave and degrade the plasma membrane-bound cationic channel subunit transient receptor potential channel 1 (TRPC1) to stimulate unconventional IL1β secretion [84], but how this integrates with the essential role of caspase-11-mediated gasdermin D cleavage for IL1β secretion in the context of non-canonical inflammasome activation requires further investigation (Fig. 2). Without doubt, coming years will provide important progress in understanding the mechanisms driving pyroptosis downstream of caspases 1 and 11, and this is likely to reveal interesting similarities and differences by which the canonical and non-canonical inflammasomes coordinate pyroptotic cell death.
Pyroptosis in inflammation and anti-microbial host defense
Inflammasome activation provides protection against bacterial, viral, fungal, and protozoan pathogens, and pyroptosis induction is thought to contribute importantly to anti-microbial host defense [9, 14, 85–88]. It is hypothesized that it does so by eliminating intracellular replication niches and by externalizing intracellular pathogens for immune recognition and clearance. In addition, recent reports suggest that pyroptosis may also represent a mechanism for the passive extracellular release of bioactive Interleukin (IL)-1β and IL18. Unlike conventional cytokines, IL1β and the related cytokine IL18 are secreted independently of the endoplasmic reticulum (ER)-Golgi secretory pathway, but instead are synthesized as cytosolic precursors that await their proteolytic cleavage by caspase-1 [88]. IL1β promotes fever and infiltration of inflammatory cells indirectly through inflammatory mediators such as PGE2, NOS and adhesion molecules. In addition, it modulates T and B cell responses by inducing Th2 and Th17 polarization of naïve CD4 T cells [8]. IL18 also regulates T cell maturation by polarizing the response towards Th1 or Th2 patterns in conjunction with IL12 [8]. By monitoring caspase-1 activity using an engineered fluorescence resonance energy transfer (FRET) sensor in parallel with extracellular IL1β at the single-cell level, secretion of IL1β was shown to correlate fully with pyroptosis induction by the NLRP3, NLRC4, and AIM2 inflammasomes [89]. Moreover, although a role for gasdermin D in active secretion of these cytokines cannot be ruled out, the observation that its deletion delayed pyroptosis induction by the canonical inflammasomes along with extracellular release of mature IL1β and IL18 suggests a causal link between these inflammasome outcomes [75, 76].
In addition to IL1β and IL18, pyroptosis has been associated with externalization of intracellular danger-associated molecular patterns (DAMPs). In particular, release of IL1α and high mobility group box 1 (HMGB1) have been linked to pyroptosis induction by the canonical and non-canonical inflammasomes [14, 90]. IL1α is a cytokine highly related to IL1β, but with the contrasting difference that it does not need to be cleaved to be functional [8]. Therefore, release of bioactive IL1α likely is a common feature of lytic cell death modes, and may account for some of their inflammatory properties. Also, release of HMGB1 has been documented not only during pyroptosis [14, 90], but also in the context of necroptosis [91, 92]. Intracellular HMGB1 regulates chromosome architecture in the nucleus, and although its intracellular roles confound its analysis as an extracellular DAMP in conditionally targeted mice [93–95], studies using antibody-based HMGB1 neutralization have implicated it in a variety of inflammatory disease models. For example, lethality in the caspase-11-dependent LPS-induced endotoxemia model [14, 96] was more effectively prevented by neutralization of HMGB1 than by the combined deletion of IL1β and IL18 [14, 90, 97]. It is thought that extracellular HMGB1 may act as a chemokine that engages receptor for advanced glycation endproducts (RAGE) and possibly as a Toll-like receptor (TLR) ligand in conjunction with PAMPs [98].
In addition to these DAMPs, pyroptotic cells have been suggested to release ASC specks to increase local inflammatory responses or to amplify inflammasome signaling when these aggregates are phagocytosed by neighboring cells [99, 100]. In the extracellular space, ASC specks continue to promote caspase-1 proteolysis of pro-IL1β, and phagocytosed ASC specks serve as a platform for nucleating inflammasome activation in bystander cells [99, 100]. Thus, by promoting release of ASC specks and DAMPs such as IL1α, IL1β, IL18, and HMGB1, pyroptosis is increasingly regarded as a major effector mechanism by which inflammasomes contribute to inflammatory and host defense responses.
Inflammasome-induced apoptosis and pyronecrosis
Although inflammasomes have primarily been linked with induction of pyroptosis, mounting evidence suggests that they can elicit additional cell death modes, namely apoptosis and pyronecrosis. Similar to pyroptosis, pyronecrosis is a lytic cell death mode that relies on inflammasome adaptors, namely NLRP3 and ASC, but unlike pyroptosis it proceeds independently of caspase-1 activity [101, 102]. Instead, this inflammatory cell death mode possibly relies on the lysosomal cathepsins to induce cell lysis and HMGB1 release in the context of Shigella flexneri- and Neisseria gonorrhoeae-infected THP1 cells and human peripheral blood mononuclear cell (PBMC)-derived monocytes [101, 102].
Current understanding of pyronecrosis is incomplete, but our knowledge on how inflammasomes engage apoptosis is gradually increasing. The first description of apoptosis induction by inflammasomes emerged from studies with ectopically expressed NLRC4 and ASC in HEK293T cells, which lack caspase-1. In this system, NLRC4 and ASC formed a complex that recruited endogenous caspase-8 and induced apoptosis [103]. ASC was proposed to engage in direct heterotypic CARD/DED interactions with caspase-8 for apoptosis induction [103–105], although the possibility of a still unidentified component mediating their interaction cannot be ruled out. Caspase-8 is intimately connected with inflammasome responses, as in addition to its role in mediating inflammasome-associated apoptosis, it mediates transcriptional upregulation of proIL1β in response to TLR4 engagement, thus also serving as a checkpoint for efficient inflammasome-induced cytokine responses [106]. Moreover, caspase-8 is recruited to NLRP3 and NLRC4-engaged ASC specks in the context of pyroptosis signaling [106–108]. Additionally, it was shown to promote IL1β maturation and secretion from macrophages independently of inflammasomes under conditions of ER stress, fungal infection, death receptor engagement and chemotherapy treatment [109–112].
Apoptosis induction with endogenous inflammasome components was more recently demonstrated in caspase-1 and -11-deficient S. Typhimurium-infected mouse macrophages [113]. Using pharmacological caspase-1 inhibition, caspase-1 protease activity was suggested to actively suppress apoptosis in S. Typhimurium-infected macrophages, although the mechanism involved remains unknown [113]. Apoptosis induction in the absence of caspase-1 activity was relayed by the inflammasome adaptors NLRC4 and NLRP3 in this context [113]. In a similar manner, caspase-1/11-deficient macrophages that have been exposed to canonical NLRP3 and AIM2 stimuli also responded with delayed induction of apoptosis [104, 105]. Apoptosis was accompanied by caspase-8 recruitment to ASC specks, but caspase-8 could only be fully activated in the absence of caspase-1 [104, 105]. It is interesting to note in this respect that ASC was originally cloned as an aggresome-forming protein in retinoic acid- and etoposide-treated apoptotic human promyelocytic leukemia HL-60 cells [114], and its expression is suppressed in close to half of primary human breast cancers, suggesting that it may act as a tumor suppressor that induces apoptosis [115].
However, formation of ASC specks is not confined to apoptotic cells, but also observed in the context of inflammasome-induced pyroptosis. ASC deletion prevents both pyroptosis and apoptosis induction in the context of the NLRP3 and AIM2 inflammasomes, but it is difficult to establish whether ASC-dependent apoptosis induction emerges from ASC specks or the ASC-containing inflammasome platforms because ASC is critical for bridging the interaction between NLRP3 and AIM2 with caspase-1 in their respective inflammasomes [23]. Indeed, the formation of the 1–2 µm-sized ASC aggregates is considered a hallmark of inflammasome engagement, and their prion-like physicochemical properties are well documented [116, 117]. ASC specks are also formed in the context of the NLRC4 and NLRP1b inflammasomes, but as both NLRC4 and NLRP1b have a CARD domain, these sensors can directly engage caspase-1 in the absence of ASC utilizing homotypic CARD interactions [5–7]. Consequently, pyroptosis induction by the NLRC4 and NLRP1b inflammasomes is unhampered in the absence of ASC, whereas ASC is essential for pyroptosis in the context of the PYD-based NLRP3 and AIM2 inflammasomes [5–7]. The observations described above suggest that inflammasome-associated pyroptosis and apoptosis induction may generate profoundly different systemic outcomes, and modulation of these cell death responses may offer novel approaches for treating inflammasome-associated diseases. In conclusion, significant progress was made in recent years in characterizing inflammasome-associated apoptosis, but more work is needed to examine when and how they contribute to inflammasome signaling in vivo.
Inflammasome-induced cell death in infection and autoinflammation
While understanding the relevance of inflammasome-mediated apoptosis in vivo is still in its infancy, a clearer picture on how pyroptosis induction by caspases 1 and 11 contributes to host defense against microbial pathogens, and detrimental inflammation in autoinflammatory diseases is emerging. Pyroptosis has been particularly linked to in vivo protection against infection with Bacillus anthracis spores, B. thailandensis, B. pseudomallei, S. Typhimurium, Legionella pneumophila, and F. tularensis [86, 118, 119]. In S. Typhimurium infection, the combined absence of IL1β and IL18 failed to fully recapitulate the more severe phenotype of caspase-1/11-deficient mice in agreement with the notion that pyroptosis not only promotes secretion of IL1β and IL18, but also exposes pathogens to extracellular immune recognition [86, 119].
However, generalized pyroptosis may also become detrimental to the host. For instance, extensive caspase-1-driven pyroptosis was identified as a major cause of immunodepletion in HIV patients that targets CD4 T cells that have been unproductively infected with the virus [120, 121]. Also caspase-11-mediated pyroptosis in the absence of caspase-1-dependent cytokine production was suggested to be disadvantageous to the host in terms of efficiently clearing S. Typhimurium in vivo [122]. Excessive caspase-11-associated pyroptosis may also be pathogenic during LPS-induced endotoxemia because caspase-11 knockout mice are highly resistant to LPS-induced lethality, while animals lacking IL1β and IL18 remain largely sensitive [14, 96]. Nevertheless, detailed analysis of the in vivo roles of pyroptosis was hampered by the absence of specific biomarkers, but the recent identification of gasdermin D cleavage as a pyroptosis-selective event offers a potentially suitable biomarker for monitoring pyroptosis in vivo.
Nevertheless, inflammasome-induced cell death is suspected to contribute to inflammatory pathology in inflammasomopathies, which are hereditary periodic fever syndromes caused by gain-of-function mutations in genes coding for inflammasome components [11, 123]. CAPS (Cryopyrin-associated periodic syndromes) is frequently caused by mutations in and around the central NACHT domain of the inflammasome adaptor NLRP3 [124]. Patients diagnosed with these diseases can be distributed across a spectrum of severity of their clinical outcomes, in which FCAS (Familial cold autoinflammatory syndrome) patients present the mildest form, MWS (Muckle–Wells syndrome) correlates with an intermediate phenotype, while NOMID (Neonatal onset multisystem inflammatory disease) is very severe. CAPS patients exhibit the symptoms of general inflammation, suffering with rash, fever, headache and fatigue that can be triggered by cold exposure, stress or, in its most serious form, even be present in a chronic manner. Severe presentations of CAPS can progress to hearing impairment or even neurological sequelae due to aseptic meningitis [124]. Some NLRP3 SNPs that are associated with CAPS have been shown to render NLRP3 constitutively active, which explains the high levels of inflammation experienced by the patients [123]. CAPS patients highly benefit from IL1 inhibitors that are already prescribed in the clinic [124]. However, in mouse models of CAPS, combined blockade of IL1R/IL18R signaling provided less protection from postnatal lethality than caspase-1 deletion, reinforcing the notion that pyroptosis-related DAMPs may contribute to pathology in this autoinflammatory model [10].
Inflammasome-associated cell death may also be an important driver of pathology in recently described autoinflammatory diseases that are caused by activating mutations in NLRC4 [11–13]. In all studied cases, recurrent fever began early in life but the other symptoms were variable, including rash and intestinal-commitment. High levels of IL18 in the serum of patients, together with other inflammatory markers, confirmed the correlation of NLRC4-activating mutations with disease onset. In accordance with the clinical presentation, these diseases were termed NLCR4-MAS, SCAN4, and NLRC4-FCAS, respectively [11–13]. IL1β neutralization improved a subset of symptoms in NLCR4-MAS patients and in a mouse model of NLRC4-FCAS, but could not rescue the high levels of circulating IL18. The latter suggests that excessive NLRC4-induced cell death linked with IL18 secretion and macrophage activation syndrome may be an important cause of pathology in NLRC4-associated autoinflammation [11–13]. These findings undoubtedly warrant a thorough investigation of inflammasome-induced cell death responses and its roles in CAPS and NLRC4-associated autoinflammation.
Concluding remarks: inflammasomes as polyvalent cell death controllers
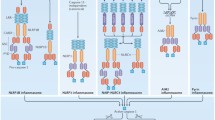
We illustrated throughout this review that inflammasomes may regulate several cell death modes and inflammatory mechanisms (Fig. 3). That a signaling platform would control a variety of downstream cell death pathways is not unprecedented. The death receptor family member tumor necrosis factor (TNF)-receptor 1 (TNF-R1) can engage at least three different complexes, termed complex I, II and IIb, with each complex promoting a distinct cellular response. Complex I induces NF-κB- and AP-1-dependent transcription of pro-inflammatory cytokines and the apoptosis inhibitor cFLIP, therefore constituting a pro-survival signal. When complex I-dependent responses are impaired, assembly of complex II activates caspase-8 for induction of apoptosis. When caspase-8 activation fails, complex IIb leads to induction of necroptosis through RIP kinases 1 and 3. Therefore, signaling through TNF-R1 is highly regulated, thereby skewing cellular responses to TNF stimulation depending on the cellular context [92]. Similarly, inflammasome responses appear to be regulated by an exquisite range of regulatory mechanisms. Although induction of pyroptosis downstream of caspase-1 may be an all–or–none response [89], inflammasome activation itself is tightly controlled at both the transcriptional and post-translational levels. A prime example of transcriptional control is presented by the NF-κB-dependent induction of NLRP3, proIL1β and caspase-11 levels to license inflammasome assembly, pyroptosis, and the release of mature IL1β [23]. Inflammasome activation is also regulated directly through post-translational modifications, as illustrated by the necessity for NLRP1b autocleavage [125] and NLRC4 phosphorylation [46, 47] for inflammasome activation. Primate-specific CARD-only and Pyrin-only proteins represent yet another level of inflammasome regulation [126]. Overall, tight regulation of inflammasome activation may serve to ensure that cells respond adequately to intracellular pathogen invasion while minimizing as much as possible the collateral damage induced by excessive cell death and inflammatory responses.

Representation of inflammasomes as cell death switches. While engagement of NLRP3, AIM2 and NLRC4 leads to pyroptosis in the presence of caspase-1, lack of this inflammatory caspase deviates the response towards caspase-8-dependent apoptosis. Importantly, NLRC4 is able to activate caspase-1 in the absence of the adaptor ASC, but the apoptotic phenotype is highly dependent on the adaptor protein. Considering the downstream effects of pyroptosis and apoptosis, inflammasome engagement could potentially play contrasting roles in immune response depending on the expression level of caspase-1
References
Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161. doi:10.1146/annurev-cellbio-101011-155745
Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, Wang F, Shao F (2014) Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513(7517):237–241. doi:10.1038/nature13449
Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J 16(10):2794–2804. doi:10.1093/emboj/16.10.2794
Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci USA 98(25):14250–14255. doi:10.1073/pnas.231465798
Broz P, von Moltke J, Jones JW, Vance RE, Monack DM (2010) Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8(6):471–483. doi:10.1016/j.chom.2010.11.007
Guey B, Bodnar M, Manie SN, Tardivel A, Petrilli V (2014) Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci USA 111(48):17254–17259. doi:10.1073/pnas.1415756111
Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M (2014) Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nature communications 5:3209. doi:10.1038/ncomms4209
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550. doi:10.1146/annurev.immunol.021908.132612
Jorgensen I, Miao EA (2015) Pyroptotic cell death defends against intracellular pathogens. Immunol Rev 265(1):130–142. doi:10.1111/imr.12287
Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM (2013) Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Investig 123(11):4695–4705. doi:10.1172/JCI71543
Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJ, Sanchez GA, Kim H, Chapelle D, Plass N, Huang Y, Villarino AV, Biancotto A, Fleisher TA, Duncan JA, O’Shea JJ, Benseler S, Grom A, Deng Z, Laxer RM, Goldbach-Mansky R (2014) An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 46(10):1140–1146. doi:10.1038/ng.3089
Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K (2014) An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med 211(12):2385–2396. doi:10.1084/jem.20141091
Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, Overton J, Meffre E, Khokha MK, Huttner AJ, West B, Podoltsev NA, Boggon TJ, Kazmierczak BI, Lifton RP (2014) Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 46(10):1135–1139. doi:10.1038/ng.3066
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479(7371):117–121. doi:10.1038/nature10558
Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, Tanzer MC, Monteleone M, Robertson AA, Cooper MA, Alvarez-Diaz S, Herold MJ, Bedoui S, Schroder K, Masters SL (2015) NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol 45(10):2918–2926. doi:10.1002/eji.201545655
Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V (2015) Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 45(10):2911–2917. doi:10.1002/eji.201545523
Kanneganti TD, Lamkanfi M, Nunez G (2007) Intracellular NOD-like receptors in host defense and disease. Immunity 27(4):549–559. doi:10.1016/j.immuni.2007.10.002
Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G (2009) An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 10(3):266–272. doi:10.1038/ni.1702
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458(7237):509–513. doi:10.1038/nature07710
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13(6):397–411. doi:10.1038/nri3452
Horvath GL, Schrum JE, De Nardo CM, Latz E (2011) Intracellular sensing of microbes and danger signals by the inflammasomes. Immunol Rev 243(1):119–135. doi:10.1111/j.1600-065X.2011.01050.x
Saavedra PH, Demon D, Van Gorp H, Lamkanfi M (2015) Protective and detrimental roles of inflammasomes in disease. Semin Immunopathol. doi:10.1007/s00281-015-0485-5
Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157(5):1013–1022. doi:10.1016/j.cell.2014.04.007
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9(8):847–856. doi:10.1038/ni.1631
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36(3):401–414. doi:10.1016/j.immuni.2012.01.009
Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD (2013) Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191(10):5230–5238. doi:10.4049/jimmunol.1301490
Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39(2):311–323. doi:10.1016/j.immuni.2013.08.001
Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14(5):454–460. doi:10.1038/ni.2550
Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
Horng T (2014) Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol 35(6):253–261. doi:10.1016/j.it.2014.02.007
Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR, Spritz RA (2007) NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med 356(12):1216–1225. doi:10.1056/NEJMoa061592
Jin Y, Birlea SA, Fain PR, Spritz RA (2007) Genetic variations in NALP1 are associated with generalized vitiligo in a Romanian population. J Invest Dermatol 127(11):2558–2562. doi:10.1038/sj.jid.5700953
Magitta NF, Boe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, Undlien DE, Joner G, Njolstad PR, Kvien TK, Forre O, Knappskog PM, Husebye ES (2009) A coding polymorphism in NALP1 confers risk for autoimmune Addison’s disease and type 1 diabetes. Genes Immun 10(2):120–124. doi:10.1038/gene.2008.85
Motta VN, Markle JG, Gulban O, Mortin-Toth S, Liao KC, Mogridge J, Steward CA, Danska JS (2015) Identification of the inflammasome Nlrp1b as the candidate gene conferring diabetes risk at the Idd4.1 locus in the nonobese diabetic mouse. J Immunol 194(12):5663–5673. doi:10.4049/jimmunol.1400913
Grandemange S, Sanchez E, Louis-Plence P, Rittore C, Reed J, Touitou I, Genevieve D (2015) NLRP1 mutations cause autoinflammatory diseases in human. Pediatr Rheumatol 13(Suppl 1):O22
Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, McArthur K, Baldwin TM, Chevrier S, Nowell CJ, Cengia LH, Henley KJ, Collinge JE, Kastner DL, Feigenbaum L, Hilton DJ, Alexander WS, Kile BT, Croker BA (2012) NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37(6):1009–1023. doi:10.1016/j.immuni.2012.08.027
Boyden ED, Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38(2):240–244. doi:10.1038/ng1724
Chavarria-Smith J, Vance RE (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9(6):e1003452. doi:10.1371/journal.ppat.1003452
Wickliffe KE, Leppla SH, Moayeri M (2008) Killing of macrophages by anthrax lethal toxin: involvement of the N-end rule pathway. Cell Microbiol 10(6):1352–1362. doi:10.1111/j.1462-5822.2008.01131.x
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430(6996):213–218. doi:10.1038/nature02664
Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Nunez G (2006) Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol 7(6):576–582. doi:10.1038/ni1346
Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107(7):3076–3080. doi:10.1073/pnas.0913087107
Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, Monack DM, Tsolis RM, Vance RE (2008) Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 9(10):1171–1178. doi:10.1038/ni.1646
Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477(7366):596–600. doi:10.1038/nature10510
Yang J, Zhao Y, Shi J, Shao F (2013) Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA 110(35):14408–14413. doi:10.1073/pnas.1306376110
Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Komuves L, Cupp JE, Arnott D, Monack D, Dixit VM (2012) Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490(7421):539–542. doi:10.1038/nature11429
Matusiak M, Van Opdenbosch N, Vande Walle L, Sirard JC, Kanneganti TD, Lamkanfi M (2015) Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc Natl Acad Sci USA 112(5):1541–1546. doi:10.1073/pnas.1417945112
Kortmann J, Brubaker SW, Monack DM (2015) Cutting edge: inflammasome activation in primary human macrophages is dependent on flagellin. J Immunol 195(3):815–819. doi:10.4049/jimmunol.1403100
Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11(5):385–393. doi:10.1038/ni.1859
Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM (2010) Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci USA 107(21):9771–9776. doi:10.1073/pnas.1003738107
Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11(5):395–402. doi:10.1038/ni.1864
Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, Dussurgey S, Dick MS, Kistner A, Rigard M, Degrandi D, Pfeffer K, Yamamoto M, Henry T, Broz P (2015) Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol 16(5):476–484. doi:10.1038/ni.3119
Man SM, Karki R, Malireddi RK, Neale G, Vogel P, Yamamoto M, Lamkanfi M, Kanneganti TD (2015) The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16(5):467–475. doi:10.1038/ni.3118
Gavrilin MA, Abdelaziz DH, Mostafa M, Abdulrahman BA, Grandhi J, Akhter A, Abu Khweek A, Aubert DF, Valvano MA, Wewers MD, Amer AO (2012) Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J Immunol 188(7):3469–3477. doi:10.4049/jimmunol.1102272
Mansfield E, Chae JJ, Komarow HD, Brotz TM, Frucht DM, Aksentijevich I, Kastner DL (2001) The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood 98(3):851–859
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514(7521):187–192. doi:10.1038/nature13683
Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, Coers J (2014) Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci USA 111(16):6046–6051. doi:10.1073/pnas.1321700111
Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, Yamamoto M, Broz P (2014) Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509(7500):366–370. doi:10.1038/nature13157
Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, Greenberg AH, Yuan J (1996) Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem 271(34):20580–20587
Ashkenazi A, Salvesen G (2014) Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol 30:337–356. doi:10.1146/annurev-cellbio-100913-013226
Fink SL, Cookson BT (2006) Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8(11):1812–1825. doi:10.1111/j.1462-5822.2006.00751.x
Monack DM, Raupach B, Hromockyj AE, Falkow S (1996) Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci USA 93(18):9833–9838
Chen LM, Kaniga K, Galan JE (1996) Salmonella spp. are cytotoxic for cultured macrophages. Mol Microbiol 21(5):1101–1115
Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, Nunez G (2008) Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7(12):2350–2363. doi:10.1074/mcp.M800132-MCP200
Malireddi RK, Ippagunta S, Lamkanfi M, Kanneganti TD (2010) Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J Immunol 185(6):3127–3130. doi:10.4049/jimmunol.1001512
Erener S, Petrilli V, Kassner I, Minotti R, Castillo R, Santoro R, Hassa PO, Tschopp J, Hottiger MO (2012) Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-kappaB target genes. Mol Cell 46(2):200–211. doi:10.1016/j.molcel.2012.02.016
Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, Day C, Sarkar A, Newland C, Butchar J, Marsh CB, Wewers MD, Tridandapani S, Kanneganti TD, Amer AO (2009) Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog 5(4):e1000361. doi:10.1371/journal.ppat.1000361
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147. doi:10.1038/nrm3737
Cookson BT, Brennan MA (2001) Pro-inflammatory programmed cell death. Trends Microbiol 9(3):113–114
Chen Y, Smith MR, Thirumalai K, Zychlinsky A (1996) A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J 15(15):3853–3860
Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A (1999) The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA 96(5):2396–2401
Brennan MA, Cookson BT (2000) Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol 38(1):31–40
Jesenberger V, Procyk KJ, Yuan J, Reipert S, Baccarini M (2000) Salmonella-induced caspase-2 activation in macrophages: a novel mechanism in pathogen-mediated apoptosis. J Exp Med 192(7):1035–1046
Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, Flavell RA, Yuan J, Sansonetti PJ, Zychlinsky A (1998) Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem 273(49):32895–32900
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. doi:10.1038/nature15514
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature. doi:10.1038/nature15541
Fischer U, Janicke RU, Schulze-Osthoff K (2003) Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 10(1):76–100. doi:10.1038/sj.cdd.4401160
Van Damme P, Martens L, Van Damme J, Hugelier K, Staes A, Vandekerckhove J, Gevaert K (2005) Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat Methods 2(10):771–777. doi:10.1038/nmeth792
Dix MM, Simon GM, Cravatt BF (2008) Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 134(4):679–691. doi:10.1016/j.cell.2008.06.038
Su L, Quade B, Wang H, Sun L, Wang X, Rizo J (2014) A plug release mechanism for membrane permeation by MLKL. Structure 22(10):1489–1500. doi:10.1016/j.str.2014.07.014
Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell reports 7(4):971–981. doi:10.1016/j.celrep.2014.04.026
Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T (2014) Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA 111(43):15514–15519. doi:10.1073/pnas.1414859111
Allam R, Lawlor KE, Yu EC, Mildenhall AL, Moujalled DM, Lewis RS, Ke F, Mason KD, White MJ, Stacey KJ, Strasser A, O’Reilly LA, Alexander W, Kile BT, Vaux DL, Vince JE (2014) Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep 15(9):982–990. doi:10.15252/embr.201438463
Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, Dietrich A, Lipinski MM, Henry T, Clapham DE, Yuan J (2014) Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep 6(6):1122–1128. doi:10.1016/j.celrep.2014.02.015
Lamkanfi M, Dixit VM (2010) Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8(1):44–54. doi:10.1016/j.chom.2010.06.007
Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA (2013) Caspase-11 protects against bacteria that escape the vacuole. Science 339(6122):975–978. doi:10.1126/science.1230751
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341(6151):1246–1249. doi:10.1126/science.1240248
Lamkanfi M (2011) Emerging inflammasome effector mechanisms. Nat Rev Immunol 11(3):213–220. doi:10.1038/nri2936
Liu T, Yamaguchi Y, Shirasaki Y, Shikada K, Yamagishi M, Hoshino K, Kaisho T, Takemoto K, Suzuki T, Kuranaga E, Ohara O, Miura M (2014) Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep 8(4):974–982. doi:10.1016/j.celrep.2014.07.012
Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM (2010) Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185(7):4385–4392. doi:10.4049/jimmunol.1000803
Kaczmarek A, Vandenabeele P, Krysko DV (2013) Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38(2):209–223. doi:10.1016/j.immuni.2013.02.003
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517(7534):311–320. doi:10.1038/nature14191
Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, Bansal P, Billiar TR, Tsung A, Wang Q, Bartlett DL, Whitcomb DC, Chang EB, Zhu X, Wang H, Lu B, Tracey KJ, Cao L, Fan XG, Lotze MT, Zeh HJ 3rd, Tang D (2014) Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 146(4):1097–1107. doi:10.1053/j.gastro.2013.12.015
Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR, Ding Q, Loughran P, Zhu X, Beer-Stolz D, Chang EB, Billiar T, Tsung A (2014) Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology 59(5):1984–1997. doi:10.1002/hep.26976
Yanai H, Matsuda A, An J, Koshiba R, Nishio J, Negishi H, Ikushima H, Onoe T, Ohdan H, Yoshida N, Taniguchi T (2013) Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci USA 110(51):20699–20704. doi:10.1073/pnas.1320808110
Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J (1998) Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92(4):501–509
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285(5425):248–251
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ (2010) HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 28:367–388. doi:10.1146/annurev.immunol.021908.132603
Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, Barbera-Cremades M, Yague J, Ruiz-Ortiz E, Anton J, Bujan S, Couillin I, Brough D, Arostegui JI, Pelegrin P (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15(8):738–748. doi:10.1038/ni.2919
Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, Mangan MS, Zimmer S, Monks BG, Fricke M, Schmidt RE, Espevik T, Jones B, Jarnicki AG, Hansbro PM, Busto P, Marshak-Rothstein A, Hornemann S, Aguzzi A, Kastenmuller W, Latz E (2014) The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15(8):727–737. doi:10.1038/ni.2913
Willingham SB, Bergstralh DT, O’Connor W, Morrison AC, Taxman DJ, Duncan JA, Barnoy S, Venkatesan MM, Flavell RA, Deshmukh M, Hoffman HM, Ting JP (2007) Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2(3):147–159. doi:10.1016/j.chom.2007.07.009
Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF, Ting JP (2009) Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol 182(10):6460–6469. doi:10.4049/jimmunol.0802696
Masumoto J, Dowds TA, Schaner P, Chen FF, Ogura Y, Li M, Zhu L, Katsuyama T, Sagara J, Taniguchi S, Gumucio DL, Nunez G, Inohara N (2003) ASC is an activating adaptor for NF-kappa B and caspase-8-dependent apoptosis. Biochem Biophys Res Commun 303(1):69–73
Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ (2013) AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20(9):1149–1160. doi:10.1038/cdd.2013.37
Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, Henry T (2012) AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ 19(10):1709–1721. doi:10.1038/cdd.2012.51
Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD (2014) FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192(4):1835–1846. doi:10.4049/jimmunol.1302839
Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE (2013) Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol 191(10):5239–5246. doi:10.4049/jimmunol.1301581
Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, Bryant CE (2014) Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci USA 111(20):7403–7408. doi:10.1073/pnas.1402911111
Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TB (2012) Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol 13(3):246–254. doi:10.1038/ni.2222
Antonopoulos C, El Sanadi C, Kaiser WJ, Mocarski ES, Dubyak GR (2013) Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1beta via caspase-8 in dendritic cells. J Immunol 191(9):4789–4803. doi:10.4049/jimmunol.1300645
Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, Mocarski ES, Subramanian D, Green DR, Silverman N, Fitzgerald KA, Marshak-Rothstein A, Latz E (2012) Cutting edge: fAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol 189(12):5508–5512. doi:10.4049/jimmunol.1202121
Shenderov K, Riteau N, Yip R, Mayer-Barber KD, Oland S, Hieny S, Fitzgerald P, Oberst A, Dillon CP, Green DR, Cerundolo V, Sher A (2014) Cutting edge: endoplasmic reticulum stress licenses macrophages to produce mature IL-1beta in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J Immunol 192(5):2029–2033. doi:10.4049/jimmunol.1302549
Puri AW, Broz P, Shen A, Monack DM, Bogyo M (2012) Caspase-1 activity is required to bypass macrophage apoptosis upon Salmonella infection. Nat Chem Biol 8(9):745–747. doi:10.1038/nchembio.1023
Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J (1999) ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem 274(48):33835–33838
Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM (2000) TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res 60(22):6236–6242
Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, Chen ZJ (2014) Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156(6):1207–1222. doi:10.1016/j.cell.2014.01.063
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156(6):1193–1206. doi:10.1016/j.cell.2014.02.008
Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, Kuida K, Mariathasan S, Dixit VM, Flavell RA, Dietrich WF, Roy CR (2006) The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol 7(3):318–325. doi:10.1038/ni1305
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11(12):1136–1142. doi:10.1038/ni.1960
Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC (2014) Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505(7484):509–514. doi:10.1038/nature12940
Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC (2014) IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343(6169):428–432. doi:10.1126/science.1243640
Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490(7419):288–291. doi:10.1038/nature11419
Hoffman HM (2007) Hereditary immunologic disorders caused by pyrin and cryopyrin. Curr Allergy Asthma Rep 7(5):323–330
Kuemmerle-Deschner JB (2015) CAPS–pathogenesis, presentation and treatment of an autoinflammatory disease. Semin Immunopathol 37(4):377–385. doi:10.1007/s00281-015-0491-7
Frew BC, Joag VR, Mogridge J (2012) Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog 8(4):e1002659. doi:10.1371/journal.ppat.1002659
Matusiak M, Van Opdenbosch N, Lamkanfi M (2015) CARD- and pyrin-only proteins regulating inflammasome activation and immunity. Immunol Rev 265(1):217–230. doi:10.1111/imr.12282
Acknowledgments
We apologize to colleagues whose work is not cited due to space constraints. N.V.O. is a postdoctoral fellow with the Research Foundation Flanders. Work in ML’s laboratory is supported by Grants from VIB, Ghent University (BOF 01N02313, BOF 01J11113, BOF14/GOA/013), the Fund for Scientific Research-Flanders (Grants G030212 N and G011315 N), and the European Research Council (Grant 281600).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
de Vasconcelos, N.M., Van Opdenbosch, N. & Lamkanfi, M. Inflammasomes as polyvalent cell death platforms. Cell. Mol. Life Sci. 73, 2335–2347 (2016). https://doi.org/10.1007/s00018-016-2204-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2204-3