
Abstract
Glioblastoma (GBM) is the most common and lethal primary brain tumor. Over the past few years tremendous genomic and proteomic characterization along with robust animal models of GBM have provided invaluable data that show that “GBM”, although histologically indistinguishable from one another, are comprised of molecularly heterogenous diseases. In addition, robust pre-clinical models and a better understanding of the core pathways disrupted in GBM are providing a renewed optimism for novel strategies targeting these devastating tumors. Here, we summarize a brief history of the disease, our current molecular knowledge, lessons from animal models and emerging concepts of angiogenesis, invasion, and metabolism in GBM that may lend themselves to therapeutic targeting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Historical Overview of Gliomas
Gliomas are the most common form of primary brain tumors with glioblastoma (GBM) being the most malignant form of glioma (Kleihues and Cavanee 2000). Before the advent of molecular biology and large-scale genomic studies, which most recently have allowed for detailed sub-classification of primary brain tumors, research into primary brain tumors consisted of two main eras, the classical period (~1860–1920) and the “histological period” (~1920–1940) (Scherer 1940). Gliomas were originally known as “medullary sarcomas” by the English medical community and “fungus medullare” by the German medical community and this era of research in gliomas is largely known as the “macroscopic era” (Scherer 1940). GBM was first identified in 1863 by Dr. Rudolf Virchow as a tumor with glial cell origins using macroscopic and microscopic techniques which ultimately gave rise to the modern day classification of brain tumors (Globus and Strauss 1925; Virchow 1863). In addition to coining the term glioma, he ushered in the “classical period” of glioma research, which combined macroscopic observations with microscopic observations. In the 1920s, neurosurgeon, Dr. Walter Dandy took a radical step by removing the entire hemisphere of two comatose patients suffering from GBM. Despite this intervention, these patients ultimately succumbed to the disease, providing the first evidence of how truly invasive GBM is (Dandy 1928). GBM was originally known as spongioblastoma multiforme, but in 1926 neurosurgeon/neuropathologist Dr. Percival Bailey and neurosurgeon Dr. Harvey Cushing renamed the tumor as GBM (Bailey and Cushing 1926). Bailey and Cushing were instrumental in leading the “histological era” of glioma research. Bailey and Cushing noted that many patients after incomplete removal of a cerebral glioma survived longer than expected compared with others (Bailey and Cushing 1926). This fact combined with their observation that not all gliomas have similar microscopic features led them to make a thorough study of the structure and clinical history of over 400 verified gliomas. In their work they were able to classify over 13 glioma types and observed varying degrees of prognosis associated with different gliomas (Bailey and Cushing 1926). Bailey and Cushing’s careful characterization laid a foundation on which the field of neurooncology and neurosurgery would grow. These studies from past eras coupled with tremendous advances in clinical, pathological, molecular, and genetic research today reveal a detailed identity of GBM.
Epidemiological Features of GBM
The incidence of all of primary brain and central nervous system (CNS) tumors is estimated at 18.71 per 100,000 individuals per year (CBTRUS 2010). Primary brain tumors are one of the top ten causes of cancer related deaths accounting for ~2.3 % of cancer-related deaths in Europe and North America (Canadian Cancer Society’s Steering Committee 2010). The most common primary brain tumor is the glioma, accounting for 32 % of CNS tumors and 80 % of malignant CNS tumors (Fig. 1). Gliomas are classified according to their presumed cell of origin and extent of brain infiltration (circumscribed or diffusely infiltrating) and include astrocytomas, oligodendrogliomas, and ependymomas. The most common type of glioma is astrocytoma, which account for about 76 % of gliomas (Fig. 1). GBMs are the most malignant glioma and common astrocytoma accounting for ~54 % of all astrocytic tumors (CBTRUS 2010). GBMs are more common among men than woman (male:female; ratio: 1.58:1) and twice as common in Caucasians versus African-Americans (CBTRUS 2010). The median age of patients at the time of diagnosis is approximately 64 years. No underlying cause has been identified for the majority of malignant gliomas. Although there has been some concern about an increased risk of gliomas in association with the use of cellular telephones, the largest studies have not demonstrated this assertion (Deltour et al. 2009; Rickman et al. 2001). Approximately 5 % of patients with malignant gliomas have a family history of gliomas. Some of these familial cases are associated with rare genetic syndromes, including neurofibromatosis types 1 and 2, and Li-Fraumeni syndrome (Farrell and Plotkin 2007).

Primary brain tumor distributions. Percent distribution of all a primary brain and CNS tumors by histology, b primary brain gliomas in the United States from 2004 to 2006
Pathological Features of GBM
Astrocytomas are heterogeneous in nature and may show diffuse infiltration of adjacent and distant brain structures (Kleihues and Cavanee 2000). These neoplasms are histologically graded based on the criteria set by the World Health Organization into four prognostic grades (Fig. 2). Pilocytic astrocytomas, which are benign and relatively circumscribed, are classified as grade I tumors. Low-grade diffuse astrocytomas (LGA; grade II) are histologically characterized by moderate cellularity, mild nuclear atypia, and rare or absent mitotic figures and are moderately proliferative and invasive. Grade III astrocytomas, or anaplastic astrocytomas (AA), are characterized histologically by increased cellularity, nuclear atypia, and mitotic activity. These tumors are more proliferative and infiltrative compared with grade II gliomas. GBMs, classified as grade IV gliomas, are histologically similar to AA but in addition are characterized by the presence of necrosis and glomeruloid microvascular proliferation. GBMs are significantly more proliferative, invasive, and angiogenic compared with grade II and III astrocytomas (Kleihues and Cavanee 2000). Malignant gliomas typically contain both neoplastic and stromal tissues, which contribute to their histologic heterogeneity and survival heterogeneity.

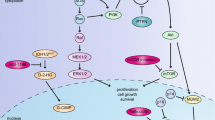
Genetic and molecular pathogenesis of GBM. Genetic and chromosomal alterations involved in primary and secondary GBMs are shown. a Primary GBMs present de novo, whereas secondary GBMs arise and progress from a lower grade astrocytoma. Both primary and secondary GBMs contain similar aberrations, although certain aberrations are more prevalent in one subtype over the other (bold). b Recent integrated genomic analysis has defined primary and secondary GBMs into a further four distinct subtypes characterized by specific molecular alterations. OLIG2 oligodendrocyte transcription factor 2, PTEN phosphatase and tensin homolog, LOH loss of heterozygosity, Amp amplification, INK4A inhibitor kinase 4A, ARF alternate reading frame, MDM2 mouse double minute 2, RB retinoblastoma, PDGFR platelet derived growth factor receptor, PI3K phosphatidylinositol 3-kinase, VEGF vascular endothelial growth factor, NES nestin, NF1 neurofibromin 1, MET met proto-oncogene (hepatocyte growth factor receptor), CHI3L1 chitinase 3-like 1, CD44 cd44 antigen, MERTK c-mer proto-oncogene tyrosine kinase, TNF tumor necrosis factor, NFKB nuclear factor kappa-B, IDH1 isocitrate dehydrogenase 1, PIK3R1 phosphatidylinositol 3-kinase, regulatory subunit 1
Molecular Genetics of Malignant Gliomas
Traditionally, GBMs have been classified into primary or secondary subtypes on the basis of clinical presentation and features although both subtypes are indistinguishable at a morphological level. Approximately, 95 % of GBMs present as de novo primary tumors and typically occur in older patients (Ohgaki et al. 2004). Secondary GBMs arise and progress from lower grade astrocytomas, are quite rare, and tend to occur in patients below the age of 45. It is now known that both subtypes constitute distinct diseases with specific genetic differences documented between primary and secondary GBMs (Fig. 2). Primary GBMs are characterized by epidermal growth factor receptor (EGFR) gene amplification and mutation; loss of heterozygosity (LOH) of chromosome 10q containing phosphatase and tensin homolog (PTEN); overexpression of mouse double minute 2 (MDM2); and deletion of p16. The hallmarks of secondary GBMs include mutations of TP53 and retinoblastoma (RB); overexpression of platelet-derived growth factor A, and platelet-derived growth factor receptor alpha (PDGFA/PDGFRα); and LOH of 19q (Furnari et al. 2007; Wen and Kesari 2008). Recent studies from The Cancer Genome Atlas (TCGA) Research Network have reinforced our existing knowledge of the molecular genetics of GBMs and provided additional insight. TCGA is a multi-institute effort to accelerate understanding of the molecular basis of GBMs and other cancers through the use of genome analysis technologies. Ongoing gene copy number, gene sequencing, epigenetic methylation, gene expression profile, and micro RNA level characterization of a large cohort of GBM specimens have provided preliminary results that are reshaping our understanding of this disease.
Initial findings confirmed that three core pathways are deregulated in most, if not all GBMs: receptor tyrosine kinase signaling, and the TP53 and RB tumor suppressor pathways (Cancer Genome Atlas Research Network 2008). In fact, 74 % of GBMs analyzed harbored alterations in all three pathways. This study, along with other integrated genomic analysis studies, has characterized the importance of mutations in NF1, and ERBB2 (Cancer Genome Atlas Research Network 2008). Interestingly, mutations in isocitrate dehydrogenase 1 (IDH1) were observed in about 70 % of grade II/III astrocytomas and secondary GBMs that developed from these lower-grade lesions suggesting a potentially more specific marker of secondary GBMs (Yan et al. 2009). The most frequent mutation of IDH1 is a point mutation of R132H, which is a gain of function mutation that allows production of 2 hydroxyglutarate and contributes to a CpG island methylator phenotype (Dang et al. 2010; Noushmehr et al. 2010; Xu et al. 2011). Three differentially expressed IDH isoforms exist in humans, IDH1 being cytosolic, whereas IDH2 and IDH3 are mitochondrial. In addition to IDH1 mutations, IDH2 mutations at R172 have also been identified in low-grade gliomas and GBM. Only 3–7 % of primary GBM have been reported to have the IDH1 mutation compared with 60–80 % in secondary GBM.
Molecular Subtypes of GBM
Our latest insight into the molecular pathogenesis of GBMs comes from very recent TCGA data. Integrated genomic analysis described a gene expression-based molecular classification of GBMs into classical, mesenchymal, proneural, and neural subtypes characterized by aberrations and gene expression of EGFR, NF1, PDGFRα, and ERBB2, respectively. The researchers of this study also found that response to aggressive chemotherapy and radiation differed by subtype (Verhaak et al. 2010). Using unsupervised hierarchical clustering, Verhaak et al. (2010), identified four molecular subtypes of GBM using an 840 gene signature which was validated in separate data sets. The four subtypes identified are proneural, neural, mesenchymal, and classical. Each subtype was enriched for different mutation, genomic, and transcript alterations. The proneural subgroup was enriched for mutations in IDH1/2, mutation in TP53, and amplifications of PDGFRA, CDK6, CDK4, and MET. Additionally this group contained the highest percentage of young patients, likely due to the enrichment of IDH1 mutations which is associated with younger age. The classical sub-type is characterized by EGFR amplification and loss of PTEN. The classical subtype also harbors the mutant EGFR variant III or EGFRvIII mutation, which is a constitutively active and has in frame deletion of exons 2–7. The messenchymal subclass is associated with poor overall survival and contains NF1 mutations and loss of TP53 and CDKN2A. Last, the neural subtype has elevated levels of neural markers such as NEFL but has no unique distinguishing alterations from other classes, although elevated rates of ERBB2 mutation were observed. Figure 2 summarizes the four subclasses of GBM.
The Cell of Origin in GBM and the Cancer Stem Cell Hypothesis
Here, we define the cell of origin as the original cell or group of cells that acquired neoplastic lesions to induce transformation or cancer. The putative cell of origin of GBM is weighted around three possible theories: first, mature glia will dedifferentiate to acquire unregulated “stem-cell” like properties through acquisition of mutations or epigenetic lesions. Second, restricted neural progenitors, which have limited self-renewal potential need to acquire mutations which lead to the gain of unregulated “stem-cell” like properties, and third, adult neural stem cells (NSCs) which normally have tight regulation over their proliferative and differentiation potential acquire mutations which render them tumourigenic (Dirks 2001, 2008; Stiles and Rowitch 2008) (Fig. 3). This concept in GBM where a subset of cells within the tumor that has stem-cell like properties, limitless expansion potential and can drive tumor formation, has been designated the cancer stem cell (CSC) hypothesis. The stem cell compartment of tumors has been termed CSCs, tumor initiating cell (TIC), or tumor-propagating cell (Hadjipanayis and Van Meir 2009). The definition of brain TICs (BTICs) does not encompass the cell of origin, which refers to the original cell or group of cells from which the tumor arose. TICs have been isolated from several cancers including brain tumors such as GBM and medulloblastoma (Galli et al. 2004; Singh et al. 2004; Taylor et al. 2005). BTICs are able to propagate in an undifferentiated manner and are able to recapitulate the tumor when injected in low numbers compared with non-purified tumor cells (Clarke et al. 2006; Galli et al. 2004; Singh et al. 2004; Taylor et al. 2005). BTICs candidates express markers including CD133, Nestin, SOX2, and can behave similar to normal NSCs and are isolated using several means. For more detailed review on CSC please refer to the following reviews highlighting emerging concepts and controversies (Dietrich et al. 2010; Lefranc et al. 2009; Munoz and Guha 2011; Park and Rich 2009; Sanchez-Martin 2008).

Putative cells of origin in GBM. Normal CNS differentiations and CNS tumor formation. Neural stem cells (NSC) give rise to neural and glial progenitors that then differentiate into the major cell types of the CNS neurons, oligodendrocytes and astrocytes. BTIC have been theorized to come from terminally differentiated cells (1) which acquire genetic mutations which endow them with a proliferative advantage a slow accumulation of critical mutations result in its transformation. Normal NSCs give rise to progenitors with limited proliferative and self-replicative capacity. BTIC have been theorized to form as a result of NSC (2) or neural progenitor transformation GBM can also arise through differentiation of mature glial or neuronal lineages (3). Curved arrow represents ability to self renew
Genetically Engineered Mouse Models (GEMs)
The Need for GEMs in Glioblastoma Tumor Research
Recent novel therapies like immuno- and gene therapy have shown some promise in existing pre-clinical models, but have failed to demonstrate therapeutic benefit in patients. The reason(s) for such failures include our incomplete understanding of the molecular pathogenesis of these tumors and also the testing of novel biological therapies in less than ideal pre-clinical models. Transgenic mouse models offer an opportunity to develop and utilize an easily replenished, reproducible, manipulated spontaneous, and more accurate pre-clinical model of human cancers, which we can use to add to our molecular knowledge and test promising therapies. The recent completion of the sequencing of the human and mouse genomes have revealed a high homology between the two genomes; thus, mouse models that recapitulate human GBM may be an invaluable tool.
GEM Models of Glioma
Molecular progression of gliomas, like many tumors, involves the accumulation of genetic and epigenetic alterations that result in the loss of tumor suppressor gene function (PTEN, TP53, INK4A/p16, ARF/p19, Rb) or activation of oncogenic pathways (p21-Ras, PI3-kinase, EGFR, CDK4, MDM2) (Cancer Genome Atlas Research Network 2008; Furnari et al. 2007; Parsons et al. 2008). Here, we briefly highlight some seminal observations and results using mouse models of known GBM modifier genes. The importance of the p53 pathway signaling pathway early in gliomagenesis is also demonstrated by the small incidence of gliomas in mice null for p19Arf, a protein involved in regulating the stability of p53 through its negative regulation of MDM2 (King et al. 2002). Alternatively, overexpression of relevant oncogenic receptors or downstream signaling pathways has also been employed in developing mouse glioma models. These have been either alone or in combination with mice harboring specific knockouts of relevant cell cycle regulatory proteins. For example, the S100 glial precursor promoter regulated v-ERBB (an activated member of the EGFR family) transgenic mice develop oligodendrogliomas, which are potentiated in terms of shorter latency and increased malignancy when undertaken in mice deficient for both p16 and p19 (Ink4a/ARF null mice) (Weiss et al. 2003). Since wild-type EGFR and mutant EGFRvIII are the most common gain of function alterations in malignant human astrocytomas, our group has recently created mice expressing these proteins under regulation of the Glial fibrillary acidic protein (GFAP) promoter (Ding et al. 2003). Mice with GFAP regulated expression of EGFR and EGFRvIII; both did not result in gliomas, suggesting that like in human gliomas, these are not initiation factors. However, when these mice were mated with a mouse that is prone to develop gliomas, such as the GFAP regulated activated p21-Ras mouse described below, the EGFRvIII but not the EGFR mice potentiated glioma formation. In addition, expression of EGFRvIII also altered the glioma sub-type, with the double transgenic mice (activated p21-Ras + EGFRvIII) developing oligodendrogliomas and mixed oligo-astrocytomas tumors, compared with the mainly astrocytic lineage tumors in the single transgenic activated p21-Ras mouse.
There are several examples of aberrant expression of relevant downstream signaling pathways in mouse glioma modeling. These include astrocytomas of varying grades resulting from GFAP regulated expression of v-src (Weissenberger et al. 1997). Our laboratory has utilized our initial observation of aberrant activation of the p21-Ras signaling pathway in astrocytomas to develop glioma models using ES transgenesis (Ding et al. 2001, 2003). GFAP regulated oncogenic/activated p21-Ras led to multi-focal astrocytomas of varying grades, in a p21-Ras activity dose-dependent manner. Mice with extremely high levels of p21-Ras activity in the brain died as chimeras with GBM like tumors. Moderate levels of p21-Ras activation led to germline transmission, with mice that are born normally but start to develop and die from their multi-focal astrocytomas of varying grades commencing at ~12–14 weeks. Of interest, expression of activated p21-Ras in adult astrocytes leads to senescence, so the developmental expression of p21-Ras in early glial progenitor cells is of importance. Furthermore, expression of activated p21-Ras in the early glial progenitors leads to genetic instability, but is not sufficient alone for transformation, since the astrocytomas occur after a period of development post birth. When the GBM-like tumors are examined in these mice, additional genetic alterations such as those found in human GBMs (over-expression of EGFR, CDK4, MDM2; decreased expression of p16, p19, p53, PTEN) (Holland 2001) are present, which we hypothesize are facilitated by the genetic instability created by activated p21-Ras. As described above, experiments involving mating these p21-Ras mice to other genetically defined mice such as EGFRvIII have already commenced and are continuing, with the potential of these mouse models not only serving as useful pre-clinical reagents, but also to increase our understanding of the in vivo interactions of these molecular alterations in the pathogenesis of gliomas. Table 1 summarizes currently used and relevant glioma mouse models which recapitulate hallmarks of human GBM.
Virally transduced expression of relevant gain of function alterations, in combination with transgenic mouse technology, allows one to model such somatic alterations at a later stage in life, though it does not lead to germline colonies. Although the link between a viral etiology and human gliomas is weak, retroviruses that have been engineered to express relevant gain of function genes have been used to create glioma models in mice and other mammals (Holland et al. 2000; Uhrbom et al. 1998, 2002). This includes members of the Rous sarcoma virus family and simian sarcoma virus, whose transforming properties are due to over expression of the viral oncogene v-sis, the cellular counterpart of which is c-sis or PDGF-B. Over expression of PDGF and activation of PDGF-receptors are well recognized in gliomagenesis (Uhrbom et al. 1998, 2002). Retroviruses carrying v-sis (PDGF-B), when injected into normal mice, have yielded astrocytic tumors, with varying glioma types when injected in Ink4a/ARF null mice (Uhrbom et al. 1998). The frequency of gliomas using this retroviral strategy was between 40 and 80 %, with the most frequent histology being high-grade gliomas that have characteristics similar to GBMs. One of the best examples of coupling retroviruses to express somatically defined gain of function genes in varying cell lineages and genetic backgrounds to model gliomas is the RCAS-tva system, detailed previously. This system develops focal gliomas, the sub-type and grade of which varies with the injected retroviral transduced gene (i.e. PDGF-B, EGFRvIII, activated p21-Ras, activated Akt), the lineage of the cell expressing the tva receptor (GFAP, Nestin) and underlying genetic cell cycle alterations in the mice (null for Ink4a/ARF, p53, etc.). For example, retroviral transduced expression of v-sis or PDGF-B in GFAP-tva mice developed oligodendroglioma or mixed oligo-astrocytoma in 40 % of the mice, with 60 % developing similar gliomas in Nestin-tva mice (Dai et al. 2001). When these experiments were undertaken in Ink4a/ARF null mice, the gliomas formed with a shorter latency and were of higher grade. Another example with the RCAS-tva system are experiments with retroviral transduction of activated p21-Ras and Akt, representing the two most implicated aberrant signaling pathways in gliomas, as previously discussed. Neither of these activated signaling molecules by itself formed gliomas, but when expressed together in more primitive Nestin+’ve precursor glial cells, astrocytomas developed.
Similar results with EGFRvIII have also been obtained by our ES transgenesis model (Ding et al. 2003) and those with the RCAS-tva system (Holland et al. 1998). In both, expression of EGFRvIII by itself is not transforming, as it is a progression and not an initiation factor. In the RCAS-tva system, retroviral transduction of both EGFRvIII and CDK4 in mice null for p53 was required for formation of mixed gliomas. In addition to the use of retroviruses or adeno-cre virus injections and lentiviruses may also be used to initiate glioma formation. Briefly, the injection of adenovirus containing the EGFRvIII mutant into mice harboring activated Ras lead to efficient formation of GBM (Wei et al. 2006). Lentiviruses expressing oncogenes such as H-Ras or AKT were efficiently introduced into mice expressing Cre-recobinase under varying promoters such as GFAP. These constructs contained a locus of cross over (lox)-stop-lox sequence in front of the oncogene. Upon integration of the lentivirus, Cre-excision of the lox-stop-lox codon would allow for activation of the oncogenes. GBM tumors were efficiently formed when lentiviruses harboring activated Ras and AKT were injected into mice expressing GFAP-Cre on a TP53 heterozygous background (Ikawa et al. 2003; Marumoto et al. 2009). The advent of inducible transgenic systems will also allow for temporal and spatial regulation of gene-expression. With relevance to glioma and CNS biology two genetically engineered mice strains express Cre recombinase fused to the estrogen receptor ligand-biding domain. Upon administration of Tamoxifen, the Cre recombinase is active, translocates to the nucleus, and excises DNA flanked by locus of cross over (Flox) sites as detailed earlier. With the generation of GFAP-CreER, Nestin-CreER mice gene activation/inactivation will allow to be regulated not only spatially as dictated by the promoter but also temporally as dictated by administration of tamoxifen (Casper et al. 2007; Chen et al. 2009; Chow et al. 2008). Current studies have allowed for determining that gliomagenic potential of mice is greater at a younger age with excision of GBM-relevant genes such as PTEN, NF1, and TP53 (Alcantara Llaguno et al. 2009). Currently, inducible tetrocycline systems are being generated for regulation of glioma and GBM genes under cell specific manners by our lab and others, which may be of potential promise. Last, utilization of orthotopic xenograft models for gene discovery or as preclinical models is also of use (Carlson et al. 2009; Pandita et al. 2004; Sarkaria et al. 2006; Verhaak et al. 2010). They retain hallmark features of GBM such as invasion and necrosis that cell models lack. In addition, serial passaging tumors in mice better retain genetic hallmarks of GBM such as EGFRvIII which is lost in culture or IDH1 which is also lost in culture (Piaskowski et al. 2011; Schulte et al. 2012). Last, several xenograft models developed by our lab and others have GBM molecular profiles similar to the human molecular GBM subtypes recently identified (Piaskowski et al. 2011).
Current Treatment Paradigms
The standard therapy for newly diagnosed malignant gliomas involves maximal surgical resection, radiotherapy, and chemotherapy, yet this does little to improve life expectancy. The surgical incurability of most GBMs is underscored by the invasive, diffuse, and ill-defined borders of these tumors. Following surgery, intensity-modulated or image-guided radiotherapy is delivered and enhances median survival from 3 to 12 months. However, tumor recurrence occurs from invasive tumor cells that have escaped surgical removal and dodged lethal radiation exposure and inevitably, 90 % of tumors recur at the site of surgery (Berens and Giese 1999). Although the prognosis remains poor, recent changes to standard of care treatment strategies have significantly improved the median survival of patients with GBMs. A clinical study carried out by Stupp and colleagues found that concomitant treatment with radiation and the alkylating agent temozolomide (TMZ) followed by adjuvant TMZ increased median overall survival from 12.1 to 14.6 months compared with that of patients receiving radiation alone (Stupp et al. 2005). The 2-year survival rate increased significantly from 10.4 % in those receiving radiation alone to 26.5 % in those receiving concurrent treatment. Furthermore, a subgroup of patients who responded better to concurrent TMZ treatment was identified; patients who had silencing of the DNA repair enzyme O6-methylguanine DNA methyltransferase (MGMT) through promoter methylation and underwent concurrent treatment as described above, had a median overall survival of 21.7 months compared with 12.7 months in those patients receiving a similar treatment but with no MGMT promoter methylation (Hegi et al. 2005). The two-year survival rate in patients with MGMT promoter methylation and receiving concomitant treatment was 46 % compared with 13.8 % for patients receiving concurrent treatment but lacking MGMT promoter methylation. However, there is still a significant proportion of patients who do not benefit from TMZ and in whom MGMT promoter methylation fails to predict response. Emerging evidence has suggested that the mismatch repair pathway and base excision repair pathway also promote resistance to TMZ. In particular, a recent study identified that the DNA base excision repair enzyme, alkyl purine DNA glycosylase (APNG), mediated resistance to TMZ in vitro and in vivo. APNG directly repairs alkylated bases at N7 guanine and N3 adenine, the later cytotoxic (Elder et al. 1998; Smith and Engelward 2000). Interestingly, patients with elevated levels of APNG also had poorer overall survival compared with pateints that were APNG negative (Agnihotri et al. 2011, 2012).
Novel and Investigational Therapies
The dismal prognosis of patients diagnosed with GBMs is attributed to the highly infiltrative nature of these tumors, as well as the heterogeneous molecular profiles of seemingly histologically similar GBMs. The objective of many investigational therapies is to (1) target cellular pathways or specific molecules implicated in pathogenesis and (2) identify molecular features of the tumor that predict a response, so that patients who are most likely to benefit can be selected for a particular treatment. The number and types of current experimental therapies are too numerous to describe here, but suffice it to say that inhibition of all features of tumor function, survival, proliferation, apoptosis, invasion, and angiogenesis, are under investigation. Table 2 summarizes current clinical trials underway in GBM. Current experimental strategies to increase the effectiveness of targeted molecular therapies include the use of a single agent targeted against several kinases (e.g. ZD6474), combinations of agents that inhibit complementary targets such as EGFR and mTOR (e.g. erlotinib + temsirolimus), and targeted agents combined with radiotherapy and chemotherapy (e.g. Cetuximab + TMZ + radiation) (Furnari et al. 2007).
Targeting Angiogenesis as a Therapeutic Strategy
It is well established that angiogenesis plays an important role in tumor progression, especially in the case of GBMs which due to the rapidity of their progression have a high metabolic demand for efficient oxygen delivery and waste removal (Carmeliet and Jain 2000; Darland 2001; Folkman 1990; Holash et al. 1999; Kargiotis et al. 2006; Klein and Weinhouse 1974; Risau 1991; Risau and Flamme 1995). The end result of uncontrolled vessel formation is a highly torturous and dilated physiological abnormal vascular network that provides an insufficient blood supply creating increased edema, and hypoxic conditions (Voest 2004), in turn, trigger a continuous cycle of dysfunctional vessel formation (Bergers and Benjamin 2003; Carmeliet and Jain 2000; Darland 2001; Wilting et al. 1995). The initiation of vessel formation is reliant on the ‘angiogenic switch’, a balance of multiple pro- and anti-angiogenic factors secreted by both host and tumor cells, favoring the pro-angiogenic factors (Bergers and Benjamin 2003; Darland 2001). Multiple candidate factors that signal tumor cells to initiate the cascade of angiogenic factor signaling have been proposed (Carmeliet and Jain 2000; Cavallaro and Christofori 2000; Darland 2001; Durairaj et al. 2000; Kerbel 2000; Lutsenko et al. 2003). The main contributors are hypoxia, increased physical forces, and the products of oncogenes and mutated tumor suppressor genes (Carmeliet and Jain 2000; Cavallaro and Christofori 2000; Darland 2001; Durairaj et al. 2000; Kerbel 2000; Lutsenko et al. 2003). The molecular mechanisms of tumor angiogenesis recapitulates loosely those of normal physiological vessel development, which occurs via two processes, “vasculogenesis” and “angiogenesis” (Houck et al. 1991; Papetti and Herman 2002; Pardanaud et al. 1987; Patan 2000; Risau 1991, 1997; Risau and Flamme 1995). During angiogenesis new vessels are formed via sprouting and elongation of pre-existing vessels to create a stabilized vascular network (Hillen and Griffioen 2007; Jain and Carmeliet 2001; Risau 1991, 1997; Risau and Flamme 1995; Wilting et al. 1995; Wilting and Christ 1996; Yancopoulos et al. 2000). Vasculogenesis is primarily an embryonic process whereby angioblasts differentiate from the mesoderm into endothelial cells (ECs) (Risau and Flamme 1995), which in turn proliferate to form de novo primitive vascular networks (Risau 1991, 1997; Risau and Flamme 1995). Recent studies suggest circulating bone marrow-derived progenitor cells (BMDPCs) are the adult counterpart of embryonic angioblasts and demonstrate that vasculogenesis occurs in adulthood in response to pathological stimuli such as tumor growth (Asahara et al. 1999; Kioi et al. 2010; Ribatti 2004), whereby BMDPCs differentiate into ECs to produce de novo blood vessels (Aghi and Chiocca 2005; Ahn and Brown 2009; Ribatti 2004; Zhang et al. 2009). Other more indirect mechanisms facilitating access of tumor cells to the blood supply include “vessel co-option” where tumor cells physically migrate toward blood vessels in response to signals within the tumor microenvironment and grow alongside existing vessels. Alternatively, there have been suggestions that tumor cells themselves can form vascular channels to transport blood independently of EC lined vessels, a phenomenon referred to as “vascular mimicry” (El Hallani et al. 2010; Hillen and Griffioen 2007; Ribatti et al. 2003; Ricci-Vitiani et al. 2010; Wang et al. 2010). The challenges of studying the origins of vascularity come in understanding the dynamic contribution of progenitor cells to the tumor vasculature. In our recent work we have taken advantage of two-photon laser microscopy and green fluorescent protein positive bone marrow (GFP + BM) chimeric mouse models to study the true origin of tumor vasculature through repeat intra-vital long-term imaging. Using this strategy, we have been able to study in real-time at a single-cell level the recruitment GFP + BMDPCs (bone marrow derived precursor cells) into the vasculature at the tumor site. We have demonstrated conclusively that BMDPCs have an intimate relationship with the newly forming vascular networks and so vasculogenesis does in fact occur in response to tumor (Fig. 4a, b). However, by highlighting the vasculature with a CD31 antibody we can also demonstrate that this relationship is not endothelial in nature, but instead, the BMDPCs appear to provide a more supportive role in the form of immuno-modulatory peri-vascular cells which in ex vivo tissue stain positively for MAC3, CD11b and IBA1 (Fig. 4c–e) (Burrell et al. 2012). Built around the knowledge that tumor angiogenesis was vital for tumor growth, anti-angiogenic treatment held much promise in arresting tumor growth and progression, maintaining patients in a stable asymptomatic state (Brem 1999; Durairaj et al. 2000; Muehlbauer 2003; Puduvalli and Sawaya 2000; Risau 1991; Sipos et al. 1994). Levels of vascularization observed in all cancers results in vascular networks which are readily accessible and contain altered and up-regulated EC receptors. This would allow for identification of exclusive therapeutic targets of aberrant tumour vasculature. Combinatorial therapy, using anti-angiogenic strategies concomitantly with radiation therapy, however, has become the primary focus, due to the concept of “vascular normalization” following anti-angiogenic therapy where abnormal tumor vasculature is somewhat corrected allowing a transiently more efficient tumor blood flow to the tumor, improving delivery of therapeutics and tumor oxygenation, and ultimately response to radiation (RT) and chemotherapy (Carmeliet and Jain 2011a, b; Jain 2001, 2005; Jain et al. 2007). There are some pre-clinical studies and early clinical trials that have explored the therapeutic benefits of combining anti-angiogenesis with RT, but results have been inconsistent (Abdollahi et al. 2003a, b; Browder et al. 2000; Bruns et al. 2002; Choy and Kim 2003; Geng et al. 2001; Gorski et al. 2003; Jain 2001; Kerbel and Folkman 2002; Kozin et al. 2001, 2007; Schueneman et al. 2003; Willett et al. 2006) and the optimal scheduling of these adjuvant treatments remains unknown. Moreover, individual tumor type plays a significant role in determining the correct combinatorial scheduling and very few studies have focused on brain tumors. Current anti-angiogenic drugs can take many forms including more traditional forms such as monoclonal antibodies and antibody fragments, as well as proteins and other small organic molecules. The drugs are designed to target the many pro-angiogenic factors involved in the initiation of tumor vascularization including PDGF, SDF1, HIF1a, FDF, and EDFR, although many drugs at present target a single pathway, the pro-angiogenic signal vascular endothelial growth factor (VEGF), as it is thought to be the main driver of angiogenesis in tumor pathophysiology. Strategies include interfering with either the VEGF ligand and its receptor VEGFR2 (Batchelor et al. 2010; Vredenburgh et al. 2007) or its downstream signaling cascade. Bevacizumab (avastin) is an anti-VEGF antibody which is now used readily in a clinical setting for the treatment of recurrent GBM. Initial phase II trials of bevacizumab used concomitantly with irinotecan demonstrated a 60 % radiographic response and increased progression-free survival (Vredenburgh et al. 2007). Similarly, two follow-up phase II trials demonstrated a radiographic response and increased progression-free survival (Kreisl et al. 2009), as such phase III trials, including the RTOG-0825 study, are currently under way. The recent approval of bevacizumab as a therapeutic agent in the treatment of recurrent GBMs provides the foundation for future studies in this disease and the use of anti-VEGF angiogenics in the disease treatment paradigm. In the context of GBMs there are other redundant factors which work in conjunction with VEGF to initiate the pro-angiogenic switch including PDGF, FGF, the ANG/TIE system, notch signaling, integrins, ephrins, interleukin 8, and SDF-1a (Bergers and Benjamin 2003; Carmeliet and Jain 2011a, b). As such therapeutics have also been targeted at the inhibition and blockade of these signaling pathways including the multiple tyrosine kinase receptor inhibitors such as cedirinab (AZD2171) and Sunitunib (sutent) both of which are able to pan block all tyrosine kinase receptors and as such sense both VEGF and PDGF signaling. Preliminary clinical trials, however, did not demonstrate as much promise as the VEGF inhibition exclusively and showed a greater effect in the cancer types than GBM suggesting as previously reported tumor stage and microenvironment play a great effect in what signals are initiating angiogenesis in tumor.

Angiogenesis and bone marrow recruitment in GBM. a 2 Photon image demonstrating the recruitment of GFP + BMDPC to the RFP + tumor area. Vasculature is highlighted with a CD31APC tagged antibody magnification ×10. b Zoomed in 2 photon intravital images demonstrate GFP + BMDCs are intimately involved in the vasculature of the tumor but remain surrounding the vasculature and as such do not co-localize with the CD31 ×20 magnification and magnification ×40. c IF staining on ex vivo tissue confirms the relationship the RFP + BMDPCs have with vasculature but in the form of immune cells staining positively for MAC3, CD11b and IBA1 with 488 tagged secondaries. These immune markers suggest a macrophage/microglial cell fate and that the BMDPCs play more of a support role to newly forming vasculature magnification ×20
In addition to the micro-stage selectivity of angiogenic signals it will be important to account for the level of redundancy between angiogenic signals and any compensatory up-regulation in other signals upon inhibition of certain pathways, resulting in decreased drug effect (Batchelor et al. 2007; Du et al. 2008). Remaining concerns include the ability to measure the effect of drugs which have a dramatic effect on existing vasculature affecting the magnetic resonance imaging, the basis for looking at tumor volume in patients (Wen et al. 2010).
The lack of clinical trials means to-date the long-term effects of these drugs are not evident; however, preliminary data suggest that reoccurrences post anti-angiogenic treatment vary dramatically from the original tumor and are harder to treat. In addition, the invasiveness of tumor cells has been shown to increase when under angiogenic therapy possible as a result of oxygen starvation forcing them to home to other existing vessels (Mrugala 2009; Narayana et al. 2012). All of these remaining questions will be essential in optimizing the potential use of anti-angiogenics in therapy of GBMs.
Invasion of Glioblastoma Negatively Impacts Therapy and Survival
One of the hallmark features of glioma is their diffuse infiltrative nature. Both low- and high-grade astrocytomas grow as masses amongst normal brain, yet their borders are poorly defined. This invasive nature of GBM in particular contributes to the surgical incurability. This is in contrast to tumors that have metastasized to the brain, in which defined tumor borders are observed. Tumor cells, single or in groups can disseminate from the primary tumor and travel following migration routes that are defined by architecture of the brain and the related extra cellular matrix (ECM). Common routes of invasion and migration include dispersion along the white matter tracks, the basal lamina of brain blood vessels, or in between the glia limitans and the pia mater (Bellail et al. 2004). The ECM in the brain mainly consists of the polysaccharide hyaluronan and proteoglycan-based matrix, mostly hyaluronic acid-binding secreted chondroitin sulfate proteoglycans of the lectican family. This environment also lacks many ECM elements found in other organs, including most basal lamina matrix (collagens, fibronectin, and type I laminin) and supporting stromal tissue (Bellail et al. 2004). This unique brain parenchyma may explain why many tumors do not metastasize to the brain and also why primary brain tumors such as GBM do not metastasize outside the brain, perhaps due to lack of proper microenvironment. The intrinsic ability of glioma cells to deeply invade within normal brain structures poses serious clinical challenges because these cells are ultimately believed to be responsible for tumor recurrence after maximal surgery, radiation, and chemotherapy. Invasion of GBM likely involves sequential adhesion to the ECM, degradation of the ECM, and altered cell contractility/motility (Giese and Westphal 1996; Nakada et al. 2007). Glioma cells adhere to the ECM using several methods. The immunoglobulin superfamily member CD44 and the hyaluronan-mediated motility receptor (HMMR or RHAMM) are receptors for hyaluronan and are expressed in GBM (Akiyama et al. 2001). CD44 is cleaved by both ADAM proteases (Murai et al. 2004) and matrix metalloproteinase (MMP)-9 (Chetty et al. 2012) that promotes motility. CD44 is repressed by p53 (Godar et al. 2008; Sohr and Engeland 2008), highly suggestive that cellular progression through cell cycle checkpoints and the ability to migrate/invade are intertwined. Integrins also contribute to cell invasion and migration, particularly the avb3 and avb5 heterodimers (Bello et al. 2001). Other factors that contribute to cell adhesion/migration and may be of therapeutic relevance include focal adhesion kinase (Riemenschneider et al. 2005; Rutka et al. 1999). MMP proteins have been implicated in altering the ECM that ultimately leads to both promotion of invasion and migration or alternatively loss of adhesion (Rao 2003). MMP-2 and MMP-9 also drive glioma invasion (Forsyth et al. 1999) and are regulated through several molecular cascades. Both enzymes promoted invasion when up-regulated via a CD95-mediated activation of AKT1 involving recruitment of Src family member Yes and p85 (Kleber et al. 2008). Collectively, a better understanding of invasion and the key mediators may allow for specific inhibitors that may reduce or prevent glioma invasion. Although in its infancy, clinical trials targeting the GBM invasion machinery are underway and may be of benefit. In particular, suberoylanilide hydoxamic acid has shown to have anti-invasion properties in vitro and in vivo through inhibition of MMP2 and MMP9 (An et al. 2010).
The Emerging Role of Tumor Metabolism
Altered glycolytic metabolism is one of most commonly altered biological processes in solid tumors. Cancer cells demonstrate a number of alterations in their metabolism to survive unfavorable and harsh microenvironments, while being able to proliferate and evade cell death (Vander Heiden et al. 2009). A classic metabolic adaptation of tumor cells is a shift to aerobic glycolysis as a main source of ATP rather than oxidative phosphorylation (OXPHOS), irrespective of oxygen availability, a phenomenon referred to as the “Warburg effect” (Warburg 1956). This phenotype may promote a state of apoptosis resistance (Kroemer and Pouyssegur 2008; Plas and Thompson 2002), the generation of biosynthetic precursors for proliferation (Vander Heiden et al. 2009), as well as increased invasive ability (Stern et al. 2002). Genetic and epigenetic alterations in key enzymes resulting in metabolic modification include primary mutations, altered isoform expression profile, and altered regulation/function secondary to oncogenic signaling pathways or the tumor microenvironment (Vander Heiden et al. 2009). An example of alterations in isoform expression profile of metabolic enzymes is exemplified by a switch in splice isoforms from the adult pyruvate kinase M1 (PKM1) to the fetal PKM2, believed to promote aerobic glycolysis and tumor growth in lung cancer cell lines (Christofk et al. 2008). IDH1, a metabolic enzyme, was recently identified in GBM, the most common and lethal of all primary human CNS tumors (Mellinghoff et al. 2005; Stupp et al. 2005). High-throughput genomic screening of GBMs identified point mutation (predominantly R132H) in the IDH1 gene in ~12 % of all GBMs (Parsons et al. 2008) and ~80 % of LGA or secondary GBMs that developed from their malignant progression (Watanabe et al. 2009; Yan et al. 2009). Mutation in IDH1 results in neomorphic (not familiar with this word) activity producing a different metabolite, 2-hydroxyglutaric acid, while wild-type IDH1 normally converts isocitrate to α-ketoglutarate coupled with NADP+/NADPH. The impact of this mutation and of the accumulation of the metabolite 2-hydroxyglutarate on GBM metabolism and glucose utilization and subsequent growth is still under investigation but may contribute to a hypermethylator phenotype in a subset of GBM (Dang et al. 2009; Noushmehr et al. 2010; Zhao et al. 2009). However, more than 90 % of GBMs are primary GBMs and the molecular basis of the Warburg effect in this subset of GBMs is under active investigation. An important role of the glycolytic enzyme HK2 was first demonstrated in hepatomas in which it was shown that the glucokinase isoform expressed in liver is substituted by HK2 (Mathupala et al. 2006). Similarly, several reports including TCGA dataset support up-regulation of HK2 in GBMs (Dong et al. 2005) and our own work with variable levels of HK1 expressed in normal brain. As the first enzyme of the glycolytic pathway, HK controls glucose flux into glycolysis or the PPP. HK2 is a highly regulated form of hexokinase, whose transcript is regulated by HIF1α, glucose, insulin, glucagon, cAMP, and p53, among others (Pedersen et al. 2002). Mitochondrial binding of HK2 to the outer membrane promotes its stability and reduces feedback inhibition from its product G6P (Bustamante and Pedersen 1977). HK2 interacts with voltage-dependent anion channel (VDAC) at the mitochondria and regulates the release of cytochrome c and intrinsic apoptosis, although the exact mechanisms of this association are not well understood (Majewski et al. 2004; Pastorino et al. 2002). Pyruvate dehydrogenase (PDH) is a mitochondrial multi-enzyme complex that catalyzes the oxidative decarboxylation of pyruvate, whose enzymatic activity is regulated by a phosphorylation/dephosphorylation cycle. The mitochondrial matrix protein Pyruvate dehyrogenase kinase (PDK1–4) is an important inhibitor of OXPHOS via its phosphorylation of the E1 α subunit of PDH. HIF1 α transactivates PDK1 resulting in decreased conversion of pyruvate to acetyl-coA and compromising OXPHOS (Kim et al. 2006). Treatment of cancer cells with the small-molecule inhibitor of PDK dichloroacetate (DCA), currently employed for the treatment of congenital lactic acidosis, was found to activate OXPHOS and promote apoptosis in cancer cells (Bonnet et al. 2007). DCA is believed to sensitize cells to apoptosis via two mechanisms: (1) enhanced flux of electrons through the ETC resulting in greater depolarization of the mitochondrial membrane, which is generally hyperpolarized in tumor cells, and enhanced release of apoptotic cytochrome c; (2) return in OXPHOS function generating greater reactive oxygen species, up-regulating voltage-dependent K+ channel leading to an efflux of K+ and activation of caspases (Bonnet et al. 2007). DCA, which crosses the blood–brain barrier, has been administered in a small number of GBM patients with limited toxicity aside from a dose-dependent, reversible peripheral neuropathy (Michelakis et al. 2010). However, large, randomized controlled trials are warranted to ascertain its effectiveness in GBM patients. Furthermore, whether DCA can sensitize GBM cells to TMZ or radiation remains to be determined. The metabolic divergence between GBM and normal cells may provide novel therapeutic strategies to be exploited. Selective targeting of HK2 may have dual effects on cells by inhibiting glycolysis and promoting OXPHOS, thereby impacting proliferation as well as dissociating the anti-apoptotic interaction between HK2 and VDAC. Current agents targeting HK2 (e.g. 3-bromopyruvate, 2-deoxyglucose) have limited clinical potential in GBMs due to non-specificity (e.g. impact on HK1 or other metabolic proteins) and systemic toxicity. Newer agents currently under development are aimed at interfering more selectively the interaction between HK2 and VDAC (e.g. methyl jasmonate) (Goldin et al. 2008). However, to date, no specific inhibitor of HK2 has been developed. It also remains to be investigated whether combined targeting of metabolic enzymes, including inhibitors to HK2, PKM2, and DCA for PDK, will further enhance GBM cell death.
Future Directions and Conclusions
The past decade of research has been invaluable in increasing our understanding of GBM. However, several outstanding questions remain. First, tumor heterogeneity is still poorly characterized in GBM. GBM has both molecular and pathological heterogeneity and future studies will need to delineate the role of microenvironment and the influence it has on tumor regions. Second, based on recent evidence it is becoming apparent that there is no single cell of origin in GBM. GBM can arise from stem cells, progenitor cells or through de-differentiation, and this may already be reflected by the observation that GBM comprises of distinct molecular subtypes. Last, overcoming therapeutic resistance is of great importance as GBM is a highly aggressive and resistant tumor. The fast pace of genomic, epigenetic, and proteomic discoveries in GBM will likely help integrate this molecular and histological data into clinical practice by improving diagnosis, prognosis, response to therapy, and ultimately delivery of more specialized and targeted therapy.
References
Abdollahi A, Lipson KE, Han X et al (2003a) SU5416 and SU6668 attenuate the angiogenic effects of radiation-induced tumor cell growth factor production and amplify the direct anti-endothelial action of radiation in vitro. Cancer Res 63:3755–3763
Abdollahi A, Lipson KE, Sckell A et al (2003b) Combined therapy with direct and indirect angiogenesis inhibition results in enhanced antiangiogenic and antitumor effects. Cancer Res 63:8890–8898
Aghi M, Chiocca EA (2005) Contribution of bone marrow-derived cells to blood vessels in ischemic tissues and tumors. Mol Ther 12:994–1005
Agnihotri S, Wolf A, Munoz DM et al (2011) A GATA4-regulated tumor suppressor network represses formation of malignant human astrocytomas. J Exp Med 208:689–702
Agnihotri S, Gajadhar AS, Ternamian C et al (2012) Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J Clin Invest 122:253–266
Ahn G-O, Brown JM (2009) Role of endothelial progenitors and other bone marrow-derived cells in the development of the tumor vasculature. Angiogenesis 12:159–164
Akiyama Y, Jung S, Salhia B et al (2001) Hyaluronate receptors mediating glioma cell migration and proliferation. J Neurooncol 53:115–127
Alcantara Llaguno S, Chen J, Kwon CH et al (2009) Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15:45–56
An Z, Gluck CB, Choy ML et al (2010) Suberoylanilide hydroxamic acid limits migration and invasion of glioma cells in two and three dimensional culture. Cancer Lett 292:215–227
Asahara T, Masuda H, Takahashi T et al (1999) Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res 85:221–228
Bailey P, Cushing H (1926) A classification of the tumors of the Glioma group on histogenetic basis with correlated study of prognosis. Lipponcott, Philadelphia, p 175
Batchelor TT, Sorensen AG, di Tomaso E et al (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11:83–95
Batchelor TT, Duda DG, Di Tomaso E et al (2010) Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol 28:2817–2823
Bellail AC, Hunter SB, Brat DJ et al (2004) Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol 36:1046–1069
Bello L, Francolini M, Marthyn P et al (2001) Alpha(v)beta3 and alpha(v)beta5 integrin expression in glioma periphery. Neurosurgery 49:380–389 (discussion 390)
Berens ME, Giese A (1999) “…those left behind.” Biology and oncology of invasive glioma cells. Neoplasia 1:208–219
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3:401–410
Bonnet S, Archer SL, Allalunis-Turner J et al (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11:37–51
Brem S (1999) Angiogenesis and cancer control: from concept to therapeutic trial. Cancer Control 6:436–458
Browder T, Butterfield CE, Kraling BM et al (2000) Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 60:1878–1886
Bruns CJ, Shrader M, Harbison MT et al (2002) Effect of the vascular endothelial growth factor receptor-2 antibody DC101 plus gemcitabine on growth, metastasis and angiogenesis of human pancreatic cancer growing orthotopically in nude mice. Int J Cancer 102:101–108
Burrell K, Hill RP, Zadeh G (2012) High-resolution in vivo analysis of normal brain response to cranial irradiation. PLoS One 7:e38366
Bustamante E, Pedersen PL (1977) High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci USA 74:3735–3739
Canadian Cancer Society’s Steering Committee (2010) Canadian cancer statistics 2010. Canadian Cancer Society, Toronto
Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Carlson BL, Grogan PT, Mladek AC et al (2009) Radiosensitizing effects of temozolomide observed in vivo only in a subset of O6-methylguanine-DNA methyltransferase methylated glioblastoma multiforme xenografts. Int J Radiat Oncol Biol Phys 75:212–219
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407:249–257
Carmeliet P, Jain RK (2011a) Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307
Carmeliet P, Jain RK (2011b) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 10:417–427
Casper KB, Jones K, McCarthy KD (2007) Characterization of astrocyte-specific conditional knockouts. Genesis 45:292–299
Cavallaro U, Christofori G (2000) Molecular mechanisms of tumor angiogenesis and tumor progression. J Neurooncol 50:63–70
CBTRUS (2010) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2004–2006. Central Brain Tumor Registry of the United States, Hinsdale http://www.cbtrusorg
Chen J, Kwon CH, Lin L et al (2009) Inducible site-specific recombination in neural stem/progenitor cells. Genesis 47:122–131
Chetty C, Vanamala SK, Gondi CS et al (2012) MMP-9 induces CD44 cleavage and CD44 mediated cell migration in glioblastoma xenograft cells. Cell Signal 24:549–559
Chow LM, Zhang J, Baker SJ (2008) Inducible cre recombinase activity in mouse mature astrocytes and adult neural precursor cells. Transgenic Res 17:919–928
Choy H, Kim DW (2003) Chemotherapy and irradiation interaction. Semin Oncol 30:3–10
Christofk HR, Vander Heiden MG, Harris MH et al (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230–233
Clarke MF, Dick JE, Dirks PB et al (2006) Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res 66:9339–9344
Dai C, Celestino JC, Okada Y et al (2001) PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 15:1913–1925
Dandy WE (1928) Removal of right cerebral hemisphere for certain tumors with hemiplegia: preliminary report. JAMA 90:3
Dang L, White DW, Gross S et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Dang L, White DW, Gross S et al (2010) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 465:966
Darland D (2001) Tumor angiogenesis and microcirculation, 1st edn. Marcel Dekker Inc., New York
Deltour I, Johansen C, Auvinen A et al (2009) Time trends in brain tumor incidence rates in Denmark, Finland, Norway, and Sweden, 1974–2003. J Natl Cancer Inst 101:1721–1724
Dietrich J, Diamond EL, Kesari S (2010) Glioma stem cell signaling: therapeutic opportunities and challenges. Exp Rev Anticancer Ther 10:709–722
Ding H, Roncari L, Shannon P et al (2001) Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res 61:3826–3836
Ding H, Shannon P, Lau N et al (2003) Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res 63:1106–1113
Dirks PB (2001) Glioma migration: clues from the biology of neural progenitor cells and embryonic CNS cell migration. J Neurooncol 53:203–212
Dirks PB (2008) Brain tumor stem cells: bringing order to the chaos of brain cancer. J Clin Oncol 26:2916–2924
Dong S, Nutt CL, Betensky RA et al (2005) Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J Neuropathol Exp Neurol 64:948–955
Du R, Lu KV, Petritsch C et al (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13:206–220
Durairaj A, Mehra A, Singh RP et al (2000) Therapeutic angiogenesis. Cardiol Rev 8:279–287
El Hallani S, Boisselier B, Peglion F et al (2010) A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain 133:973–982
Elder RH, Jansen JG, Weeks RJ et al (1998) Alkylpurine-DNA-N-glycosylase knockout mice show increased susceptibility to induction of mutations by methyl methanesulfonate. Mol Cell Biol 18:5828–5837
Farrell CJ, Plotkin SR (2007) Genetic causes of brain tumors: neurofibromatosis, tuberous sclerosis, von Hippel-Lindau, and other syndromes. Neurol Clin 25:925–946 (viii)
Folkman J (1990) What is the evidence that tumors are angiogenesis-dependent? J Natl Cancer Inst 82:4–6
Forsyth PA, Wong H, Laing TD et al (1999) Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer 79:1828–1835
Furnari FB, Fenton T, Bachoo RM et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710
Galli R, Binda E, Orfanelli U et al (2004) Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 64:7011–7021
Geng L, Donnelly E, McMahon G et al (2001) Inhibition of vascular endothelial growth factor receptor signaling leads to reversal of tumor resistance to radiotherapy. Cancer Res 61:2413–2419
Giese A, Westphal M (1996) Glioma invasion in the central nervous system. Neurosurgery 39:235–250 (discussion 250–252)
Globus J, Strauss I (1925) Spongioblastoma multiforme. Arch Neurol Psychiatry 14:139–151
Godar S, Ince TA, Bell GW et al (2008) Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell 134:62–73
Goldin N, Arzoine L, Heyfets A et al (2008) Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 27:4636–4643
Gorski DH, Mauceri HJ, Salloum RM et al (2003) Prolonged treatment with angiostatin reduces metastatic burden during radiation therapy. Cancer Res 63:308–311
Hadjipanayis CG, Van Meir EG (2009) Brain cancer propagating cells: biology, genetics and targeted therapies. Trends Mol Med 15:519–530
Hegi ME, Diserens AC, Gorlia T et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003
Hillen F, Griffioen AW (2007) Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev 26:489–502
Holash J, Wiegand SJ, Yancopoulos GD (1999) New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoiesis and VEGF. Oncogene 18:5356–5362
Holland EC (2001) Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2:120–129
Holland EC, Hively WP, DePinho RA et al (1998) A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev 12:3675–3685
Holland EC, Celestino J, Dai C et al (2000) Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 25:55–57
Houck KA, Ferrara N, Winer J et al (1991) The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol 5:1806–1814
Ikawa M, Tanaka N, Kao WW et al (2003) Generation of transgenic mice using lentiviral vectors: a novel preclinical assessment of lentiviral vectors for gene therapy. Mol Ther 8:666–673
Jain RK (2001) Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 7:987–989
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307:58–62
Jain RK, Carmeliet PF (2001) Vessels of death or life. Sci Am 285:38–45
Jain RK, Tong RT, Munn LL (2007) Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res 67:2729–2735
Kamijo T, Bodner S, van de Kamp E (1999) Tumor spectrum in ARF-deficient mice. Cancer Res 59:2217–2222
Kargiotis O, Rao JS, Kyritsis AP (2006) Mechanisms of angiogenesis in gliomas. J Neurooncol 78:281–293
Kerbel RS (2000) Tumor angiogenesis: past, present and the near future. Carcinogenesis 21:505–515
Kerbel R, Folkman J (2002) Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2:727–739
Kim JW, Tchernyshyov I, Semenza GL et al (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185
King D, Yang G, Thompson MA et al (2002) Loss of neurofibromatosis-1 and p19(ARF) cooperate to induce a multiple tumor phenotype. Oncogene 21:4978–4982
Kioi M, Vogel H, Schultz G et al (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120:694–705
Kleber S, Sancho-Martinez I, Wiestler B et al (2008) Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 13:235–248
Kleihues P, Cavanee WK (2000) World Health Organization classification of tumors: pathology and genetic: tumors of the nervous system. IARC Press, Lyon
Klein G, Weinhouse S (1974) Tumor angiogenesis. In: Folkman J (ed) Advances in cancer research. Academic Press, New York, pp 43–52
Kozin SV, Boucher Y, Hicklin DJ et al (2001) Vascular endothelial growth factor receptor-2-blocking antibody potentiates radiation-induced long-term control of human tumor xenografts. Cancer Res 61:39–44
Kozin SV, Winkler F, Garkavtsev I et al (2007) Human tumor xenografts recurring after radiotherapy are more sensitive to anti-vascular endothelial growth factor receptor-2 treatment than treatment-naive tumors. Cancer Res 67:5076–5082
Kreisl TN, Kim L, Moore K et al (2009) Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27:740–745
Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13:472–482
Lefranc F, Rynkowski M, DeWitte O et al (2009) Present and potential future adjuvant issues in high-grade astrocytic glioma treatment. Adv Tech Stand Neurosurg 34:3–35
Lutsenko SV, Kiselev SM, Severin SE (2003) Molecular mechanisms of tumor angiogenesis. Biochemistry 68:286–300
Majewski N, Nogueira V, Bhaskar P et al (2004) Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell 16:819–830
Marumoto T, Tashiro A, Friedmann-Morvinski D et al (2009) Development of a novel mouse glioma model using lentiviral vectors. Nat Med 15:110–116
Mathupala SP, Ko YH, Pedersen PL (2006) Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25:4777–4786
Mellinghoff IK, Wang MY, Vivanco I et al (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353:2012–2024
Michelakis ED, Sutendra G, Dromparis P et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31–34
Mrugala MM (2009) Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 72:773
Muehlbauer PM (2003) Anti-angiogenesis in cancer therapy. Semin Oncol Nurs 19:180–192
Munoz DM, Guha A (2011) Mouse models to interrogate the implications of the differentiation status in the ontogeny of gliomas. Oncotarget 2:590–598
Murai T, Miyazaki Y, Nishinakamura H et al (2004) Engagement of CD44 promotes Rac activation and CD44 cleavage during tumor cell migration. J Biol Chem 279:4541–4550
Nakada M, Nakada S, Demuth T et al (2007) Molecular targets of glioma invasion. Cell Mol Life Sci 64:458–478
Narayana A, Kunnakkat SD, Medabalmi P et al (2012) Change in pattern of relapse after antiangiogenic therapy in high-grade glioma. Int J Radiat Oncol Biol Phys 82:77–82
Noushmehr H, Weisenberger DJ, Diefes K et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Ohgaki H, Dessen P, Jourde B et al (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64:6892–6899
Pandita A, Aldape KD, Zadeh G et al (2004) Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosom Cancer 39:29–36
Papetti M, Herman IM (2002) Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol 282:C947–C970
Pardanaud L, Altmann C, Kitos P et al (1987) Vasculogenesis in the early quail blastodisc as studied with a monoclonal antibody recognizing endothelial cells. Development 100:339–349
Park DM, Rich JN (2009) Biology of glioma cancer stem cells. Mol Cells 28:7–12
Parsons DW, Jones S, Zhang X et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Pastorino JG, Shulga N, Hoek JB (2002) Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem 277:7610–7618
Patan S (2000) Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J Neurooncol 50:1–15
Pedersen PL, Mathupala S, Rempel A et al (2002) Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta 1555:14–20
Piaskowski S, Bienkowski M, Stoczynska-Fidelus E et al (2011) Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Br J Cancer 104:968–970
Plas DR, Thompson CB (2002) Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab 13:75–78
Puduvalli VK, Sawaya R (2000) Antiangiogenesis—therapeutic strategies and clinical implications for brain tumors. J Neurooncol 50:189–200
Rao JS (2003) Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 3:489–501
Reilley KM, Loisen DM, Bronson RT et al (2000) Nf1; Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 26:109–113
Ribatti D (2004) The involvement of endothelial progenitor cells in tumor angiogenesis. J Cell Mol Med 8:294–300
Ribatti D, Vacca A, Dammacco F (2003) New non-angiogenesis dependent pathways for tumour growth. Eur J Cancer 39:1835–1841
Ricci-Vitiani L, Pallini R, Biffoni M et al (2010) Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468:824–828
Rickman DS, Bobek MP, Misek DE et al (2001) Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res 61:6885–6891
Riemenschneider MJ, Mueller W, Betensky RA et al (2005) In situ analysis of integrin and growth factor receptor signaling pathways in human glioblastomas suggests overlapping relationships with focal adhesion kinase activation. Am J Pathol 167:1379–1387
Risau W (1991) Embryonic angiogenesis factors. Pharmacol Ther 51:371–376
Risau W (1997) Mechanisms of angiogenesis. Nature 386:671–674
Risau W, Flamme I (1995) Vasculogenesis. Annu Rev Cell Dev Biol 11:73–91
Rutka JT, Muller M, Hubbard SL et al (1999) Astrocytoma adhesion to extracellular matrix: functional significance of integrin and focal adhesion kinase expression. J Neuropathol Exp Neurol 58:198–209
Sanchez-Martin M (2008) Brain tumour stem cells: implications for cancer therapy and regenerative medicine. Curr Stem Cell Res Ther 3:197–207
Sarkaria JN, Carlson BL, Schroeder MA et al (2006) Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res 12:2264–2271
Scherer HJ (1940) A critical review: the pathology of cerebral gliomas. J Neurol Psychiatry 3:147–177
Schueneman AJ, Himmelfarb E, Geng L et al (2003) SU11248 maintenance therapy prevents tumor regrowth after fractionated irradiation of murine tumor models. Cancer Res 63:4009–4016
Schulte A, Gunther HS, Martens T et al (2012) Glioblastoma stem-like cell lines with either maintenance or loss of high-level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res 18:1901–1913
Singh SK, Hawkins C, Clarke ID et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Sipos EP, Tamargo RJ, Weingart JD et al (1994) Inhibition of tumor angiogenesis. Ann N Y Acad Sci 732:263–272
Smith SA, Engelward BP (2000) In vivo repair of methylation damage in Aag 3-methyladenine DNA glycosylase null mouse cells. Nucleic Acids Res 28:3294–3300
Sohr S, Engeland K (2008) RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle 7:3448–3460
Stern R, Shuster S, Neudecker BA et al (2002) Lactate stimulates fibroblast expression of hyaluronan and CD44: the Warburg effect revisited. Exp Cell Res 276:24–31
Stiles CD, Rowitch DH (2008) Glioma stem cells: a midterm exam. Neuron 58:832–846
Stupp R, Mason WP, van den Bent MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Taylor MD, Poppleton H, Fuller C et al (2005) Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8:323–335
Uhrbom L, Hesselager G, Nister M et al (1998) Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res 58:5275–5279
Uhrbom L, Dai C, Celestino JC et al (2002) Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res 62:5551–5558
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Verhaak RG, Hoadley KA, Purdom E et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Virchow R (1863) Die Krankhaften Geschwulste. Hirschwald, Berlin
Voest EE (2004) Angiogenesis: from understanding to targeting. Biochim Biophys Acta 1654:1
Vredenburgh JJ, Desjardins A, Herndon JE et al (2007) Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 13:1253–1259
Wang R, Chadalavada K, Wilshire J et al (2010) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468:829–833
Warburg O (1956) On respiratory impairment in cancer cells. Science 124:269–270
Watanabe T, Nobusawa S, Kleihues P et al (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153
Wei Q, Clarke L, Scheidenhelm DK et al (2006) High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res 66:7429–7437
Weiss WA, Burns MJ, Hackett C et al (2003) Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res 63:1589–1595
Weissenberger J, Steinbach JP, Malin G et al (1997) Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene 14:2005–2013
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507
Wen P, Macdonald D, Reardon D et al (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28:1963–1972
Willett CG, Kozin SV, Duda DG et al (2006) Combined vascular endothelial growth factor-targeted therapy and radiotherapy for rectal cancer: theory and clinical practice. Semin Oncol 33:S35–S40
Wilting J, Christ B (1996) Embryonic angiogenesis: a review. Naturwissenschaften 83:153–164
Wilting J, Brand-Saberi B, Kurz H et al (1995) Development of the embryonic vascular system. Cell Mol Biol Res 41:219–232
Xiao A, Wu H, Pandolfi PP et al (2002) Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell 1:157–168
Xu W, Yang H, Liu Y et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Yan H, Parsons DW, Jin G et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Yancopoulos GD, Davis S, Gale NW et al (2000) Vascular-specific growth factors and blood vessel formation. Nature 407:242–248
Zhang HR, Chen FL, Xu CP et al (2009) Incorporation of endothelial progenitor cells into the neovasculature of malignant glioma xenograft. J Neurooncol 93:165–174
Zhao S, Lin Y, Xu W et al (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324:261–265
Zhu Y, Guignard F, Zhao D et al (2005) Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 8:119–130
Acknowledgments
We would like to dedicate this review in memory of Dr. Abhijit Guha, our mentor whose dedication and love for research and neurosurgery will always inspire us.
Author information
Authors and Affiliations
Corresponding authors
About this article
Cite this article
Agnihotri, S., Burrell, K.E., Wolf, A. et al. Glioblastoma, a Brief Review of History, Molecular Genetics, Animal Models and Novel Therapeutic Strategies. Arch. Immunol. Ther. Exp. 61, 25–41 (2013). https://doi.org/10.1007/s00005-012-0203-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-012-0203-0