
Abstract
Hypoxia that originates from disturbed growth of solid tumors initiates a cascade of intracellular events engaging hypoxia-inducible factors, HIF-1 and HIF-2. Overexpression of HIF has been confirmed in solid tumors and was unfortunately accompanied with chemo- and radioresistance observed in many patients. Multiple cellular pathways resulting in HIF activation could be successfully inhibited by use of different kinds of drugs (e.g. topotecan, heat shock protein 90 and mTOR inhibitors, YC-1, pleurotin or 2-methoxyestradiol), which are being subjected into intensive investigation in clinical trials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
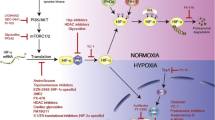
Most solid tumors exhibit hypoxic conditions inside their mass. Tumor hypoxia originates from disturbed growth of tumors and initiates not only nuclear factor (NF)-κB activation (Koong et al. 1994) but also a cascade of intracellular events engaging hypoxia-inducible factors, HIF-1 and the less well-studied HIF-2 (Duechler and Wilczynski 2010; Powis and Kirkpatrick 2004). Overexpression of HIF-1 has been confirmed in many solid tumors including ovarian, bladder, uterus, breast, colon, brain, pancreatic, renal, and prostate (Talks et al. 2000), and has been found to be associated with worse prognosis (Birner et al. 2001). Hypoxia is not the only factor which triggers HIF-1 activity. Some tumors show constitutive HIF-1 over-activity irrespectively to hypoxic conditions due to inactivation of the von Hippel-Lindau protein (pVHL) gene (Krieg et al. 2000), activation of PI3K/AKT signaling pathway, or mutation of PTEN gene (Zundel et al. 2000). Hypoxia might impede cancer therapy as it increases resistance to radiotherapy and chemotherapy through activation of HIF factors and directly alters the function of tumor cells, stimulating them to de-differentiate and to release angiogenic factors to increase blood and oxygen supply (Holmquist et al. 2006). Tumor hypoxia also promotes malignant progression and metastasis formation and contributes to cancer immune suppression. Inhibitors of HIF-1 have been subjected to preclinical and clinical trials to estimate their usefulness in anti-cancer therapy (Fig. 1).

Targeting the HIF pathway. Under normoxic conditions (right part), HIF-α proteins are marked for degradation by hydroxylation by prolyl hydroxylases (PHD) and ubiquitination mediated by von Hippel-Lindau (VHL). Degradation occurs in the proteasome. Under hypoxia (left part), PHD is inactive, and HIF-α protein accumulates and translocates into the nucleus where it interacts with HIF-β to bind to specific DNA sequences. As a consequence, transcription of target genes is initiated
Hypoxia-Inducible Factors
Most human solid tumors contain areas which are not well oxygenated. The adaptation to hypoxic conditions is primarily promoted by two hypoxia-inducible factors (HIF-1 and HIF-2), the master transcription factors controlling the cellular response to low oxygen tension. These transcription factors share pronounced similarities in structure, function and regulation, but are not redundant and serve different functions. HIF-1 and HIF-2 are heterodimeric transcription factors composed from a tightly regulated α-subunit and constitutively expressed β-subunit (aryl hydrocarbon receptor nuclear translocator) (Brahimi-Horn and Pouysségur 2009). The α-subunits are continuously produced, but rapidly degraded under normoxic conditions. During hypoxia they become stabilized and activated. The degradation of an α-subunit is initiated by post-translational prolyl hydroxylation which promotes the interaction of HIF-1/2α protein with pVHL ubiquitin-ligase complex. pVHL then covalently adds an ubiquitin chain which targets the protein for proteasomal degradation. Another oxygen sensing enzyme with a prolyl hydroxylase domain, factor inhibiting HIF, suppresses the interaction of HIF-1 with its cofactor p300/CBP (CREB binding protein) in normoxic conditions (Brahimi-Horn and Pouysségur 2009).
HIF-1α is not only activated in response to oxygen shortage, but is also stabilized under normoxic conditions by transcriptional and post-transcriptional regulation: (1) upon growth factor signaling the transcription rate of HIF-1α gene is increased which involves activation of protein kinase C and NF-κB; (2) activation of the PI3K/Akt/mTOR/p70S6K pathway for instance by reactive oxygen species increases the translational rate of HIF-1α protein (Semenza 2003); (3) tyrosine kinase transactivation upon receptor stimulation promotes HIF-1 complex activation (Pagé et al. 2008). HIF-1α is also stabilized by inflammatory cytokines (Jung et al. 2003, Gerber and Pober 2008, Zhou et al. 2003), by transforming growth factor β (Abdul-Hafez et al. 2009), by lipopolysaccharide (Nishi et al. 2008), and by stem cell factor in various cell types (Pedersen et al. 2008).
In many human cancers, HIF-1 and/or HIF-2 are constitutively active. The PI3-K/Akt pathway is activated in most human cancers and plays a key role in cell proliferation and survival. Alternatively, loss of proteins which are involved in the degradation of HIF-α subunits results in accumulation of the protein. For example, loss of pVHL is a cause of renal cancer. Cancer-related growth factor signals that induce HIF-1α include human epidermal growth factor receptor 2 and epidermal growth factor (EGF) in breast cancer (Milani and Harris 2008), endothelins in ovarian cancer and melanoma (Grimshaw 2007), prostaglandin E2 in prostate cancer cells (Liu et al. 2002), activated Src in pancreatic and prostate carcinomas (Gray et al. 2005), and interleukin (IL)-4 in transformed intestinal cells (Scharte et al. 2006).
Specific Roles for HIF-2 in Cancer
Most tumors investigated so far showed coexpression of both HIF-α subunits (Talks et al. 2000). For a long time, HIF-1 was the major factor investigated in tumor hypoxia; in recent years, however, some specific contributions of HIF-2 were elucidated. HIF-2α becomes stabilized at higher oxygen tensions than HIF-1α in both non-malignant and malignant cells. Therefore, active HIF-2α can contribute to the development of tumor aggressiveness by inducing the program for a hypoxic phenotype also at near-physiological oxygen tensions. In clinical materials, a strong correlation between HIF-2α expression and increased levels of pro-angiogenic factors, such as vascular-endothelial growth factor (VEGF), was found suggesting a direct role of HIF-2α in angiogenesis (Löfstedt et al. 2007). High HIF-2α expression has been linked to poor prognosis in several tumor types, including clear cell renal carcinoma (ccRCC), non-small cell lung cancer, and neuroblastoma (Qing and Simon 2009). Recently, it has been demonstrated in xenograft studies that inhibition of HIF-2 prevents the tumorigenesis of genetically diverse human cancers including glioblastoma, colorectal and lung carcinoma (Franovic et al. 2009). In ccRCC, accumulation of HIF-2α resulted in Hdm2-mediated suppression of p53 (Roberts et al. 2009). In hepatoma cells, mitochondrial dysfunction caused by mitochondrial DNA mutations or deletions increased the angiogenic potential of tumor cells through increased HIF-2α mRNA expression (Cheon et al. 2010). HIF-2 also contributes to dedifferentiation of neuroblastoma cells toward a phenotype of tumor initiating cells (cancer stem cells) (Pietras et al. 2009; Semenza 2010).
The Cross-Talk between NF-κB and HIF-1
Under normoxic conditions, HIF-1α expression can be increased through growth factor signaling. Most growth factor signals activate the PI3K/Akt-pathway and hence NF-κB. Also reactive oxygen species like H2O2 stimulate NF-κB activity (Gloire et al. 2006). NF-κB was shown to bind to an element in the promoter region of the HIF-1α gene and to stimulate its transcription (Belaiba et al. 2007). NF-κB also acts as hypoxia sensitive factor; decreased prolyl hydroxylation of IκB kinase during hypoxia may activate NF-κB (Cummins et al. 2006). Thus, binding to the HIF-1α promotor and up-regulation of transcription also happen as a consequence of hypoxia (Yu et al. 1998). The levels of HIF-1α may be critical for a cells readiness to respond to hypoxia. NF-κB activity seems to determine the degree and speed of HIF-1 activation, and the hypoxic response and the sensitivity of an individual cell to hypoxia might depend on NF-κB activity (Görlach and Bonello 2008).
The communication between NF-κB and HIF-1 is a real cross-talk: NF-κB stimulates the transcription of HIF-1, and HIF-1 was shown to contribute to the activation of NF-κB (Scortegagna et al. 2008). In mice overexpressing HIF-1 in keratinocytes, increased expression of pro-inflammatory NF-κB target genes was demonstrated. Two pathways were elucidated for HIF-1-induced NF-κB activation: IκB turned out to be hyperphosphorylated leading to its degradation and to enhanced p65 nuclear localization, and p65 was phosphorylated on Ser 276 mediated by ERK1/2, a modification necessary for cofactor binding and transcriptional activity. In general, inhibition of NF-κB might influence the activity of HIF-1 and vice versa, and this influence might spread further through the complex network of signaling molecules.
Inhibitors of HIF-1 Activation and HIF-1-Dependent Pathways
Topoisomerase Inhibitors: Topotecan
Topotecan, an analog of campthotecin and inhibitor of topoisomerase I, was demonstrated to decrease HIF-1α, but not HIF-1β, accumulation inside tumor cells in a dose-dependent manner. This mechanism was independent of proteasome degradation and did not affect HIF-1α mRNA accumulation, but was caused by a decrease of HIF-1α translation (Rapisarda et al. 2004a). As a result, inhibition of VEGF-dependent angiogenesis occurs, as shown by in vitro studies performed on glioma and neuroblastoma cell lines (Brown et al. 2006; Puppo et al. 2008). In preclinical studies glioblastoma xenografted animals had significantly reduced angiogenesis and tumor growth upon low-dose topotecan treatment. Thus, anti-cancer activity was accompanied not only by HIF-1 inhibition but also by inhibition of HIF-1 target genes expression (Rapisarda et al. 2004b). Although hypoxia is a major regulator of HIF-1-dependent VEGF over-expression, also insulin-like growth factor (IGF)-I induces VEGF expression in cancer cells through a PI3K-dependent mechanism, which, however, is also mediated by HIF-1. Investigations performed on neuroblastoma cell lines indicated that topotecan was able to inhibit not only hypoxia-induced but also IGF-I-induced VEGF expressions showing multi-directional blocking activity (Beppu et al. 2005). Besides neuroblastoma, topotecan has revealed limited activity as a second line therapy for advanced or recurrent endometrial cancer; however, some patients suffered from severe and potentially fatal toxicities (Miller et al. 2002). Some in vitro studies seem to confirm that higher topotecan efficacy is obtained only in subtoxic or toxic doses (Brown et al. 2006).
Heat Shock Protein 90 Inhibitors
Heat shock protein 90 (Hsp90) functions as a chaperone to control the activity, intracellular localization, and turnover of a plenty of proteins (Maloney and Workman 2002). The client proteins for Hsp90 in cancer cells include anti-apoptotic factors, proteins stimulating cell growth, proliferation, and metastatic potential. The spectrum of proteins being controlled by Hsp90 includes also HIF-1 (Powers and Workman 2006). Inhibition of Hsp90 results in degradation of misfolded proteins via ubiquitination inside the proteasome, resulting in the decreased levels of multiple oncoproteins (Connell et al. 2001). One of the most thoroughly investigated Hsp90 inhibitors are geldanamycin and its analogs. Geldanamycin is a benzoquinone ansamycin antibiotic leading to the degradation of Hsp90 client proteins, which due to its hepatotoxicity has been replaced by less toxic and more stable 17-allylamino-17-desmethoxygeldanamycin analog (17-AAG or tanespimycin) (Supko et al. 1995). Its water-soluble derivative is called 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG). Both analogs have been reported to diminish HIF-1 cellular concentrations and activity independently from tissue oxygenation status and activity of pVHL (Mabjeesh et al. 2002).
The efficacy of Hsp90 inhibition was tested during phase I clinic trials by administration of 17-AAG to heavily pretreated multiple myeloma (MM) patients. The first study showed that 17-AAG was a relatively safe drug giving less peripheral neuropathy events compared to bortezomib or thalidomide. Clinical benefit described as partial/minor response or stabilization of the disease was obtained in 41% of patients (Chanan-Khan et al. 2005). The second study demonstrated that the use of 17-AAG in bortezomib-naive MM patients resulted in 71% clinical response, while among bortezomib-refractory patients the overall response reached 33% (Richardson et al. 2006). A recent phase I dose-escalation study demonstrated that, in the group of patients with refractory/recurrent MM, 17-AAG is a well-tolerated drug. However, only 3% of patients showed minor response, and 52% had stabilization of the disease for 2 months (Richardson et al. 2010).
The Hsp90 protein seems to be a promising target for inhibitory therapy as tumor cells show about ten times greater expression of Hsp90 compared to normal tissues (Gooljarsingh et al. 2006). The efficacy of 17-AAG and 17-DMAG has been demonstrated in preclinical trials. In vitro studies on gastric cancer cell lines indicated that treatment with 17-AAG significantly reduced intracellular signaling through EGF receptor and HIF-1, as well as VEGF secretion. A murine xenograft model of gastric cancer led to the observation that low-dose application of 17-DMAG resulted in reduced tumor growth and vascularization (Lang et al. 2007a). Blocking of Hsp90 in pancreatic cancer cell lines effectively deactivated IGF-I receptor signaling and disrupted IL-6-dependent HIF-1 and signal transducer and activator of transcription 3 (STAT3) activation (Lang et al. 2007b). Geldanamycin analogs are currently being investigated in phase I/II clinical trials in malignant melanoma, prostate, and breast cancer patients (Banerji et al. 2005; Grem et al. 2005).
Soluble Guanyl-Cyclase Stimulator YC-1
Agent YC-1 (3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole) is an activator of soluble guanyl cyclase and acts as inhibitor of platelet aggregation and vasodilator, what is associated with increased cGMP levels inside platelets and the vascular wall (Teng et al. 1997). It is capable of blocking HIF-1 activity in vitro, what could be used for management of tumors. Preclinical studies on SCID mice xenografted with human tumors indicated that YC-1-treated animals had statistically smaller tumors than controls and expressed less HIF-1-dependent downstream products, regardless of the tumor type (Yeo et al. 2003). The YC-1 agent may be a valuable option for supplement therapy for advanced cancer due to its low toxicity and lack of negative influence on patient’s immunological status; however, the possibility of bleeding and hypotension during treatment needs standardization of the dose before application for humans and should be further tested in clinical trials (Yeo et al. 2003).
Thioredoxin Inhibitors: Pleurotin and PX-12
Thioredoxin (Trx)-1 is a redox protein which is subjected to reversible NADPH-catalyzed reduction by flavoprotein Trx-reductases. Binding of Trx-1 to a variety of enzymes in different redox conditions regulates their activity. The enzymes demonstrated to be regulated in this manner include apoptosis signal-regulating kinases, protein kinases C, transcription factor NF-κB and p53 (Hayashi et al. 1993; Saitoh et al. 1998; Ueno et al. 1999). HIF-1α can be stabilized in normoxia by overexpression of thioredoxin (Muniyappa et al. 2009). Many human solid cancers were shown to increase Trx-1 activity, which correlated with increased HIF-1 expression independently of oxygen status inside tumor environment (Welsh et al. 2002), as well as a more vigorous proliferation of tumors and worse survival of the patients (Kakolyris et al. 2001). Several Trx-1 inhibitors were identified, and among them PX-12 (1-methylpropyl 2-imidazolyl disulfide) and pleurotin (a natural fungal antibiotic agent produced by Pleurotia griseus) are the best known ones. The mechanisms of inhibition differ, as PX-12 produces irreversible thioalkylation of Trx-1, while pleurotin blocks thioredoxin reductases (Kunkel et al. 1997). Preclinical studies performed on breast, renal, and colon cancer cell lines indicated that both PX-12 and pleurotin were effective in blocking HIF-1 activity in hypoxic conditions, which resulted in a decrease in VEGF and inducible nitric oxide synthase expression in vitro (Welsh et al. 2003). Inhibitory properties of both agents were independent of the pVHL (Welsh et al. 2003). Antitumor activity of PX12 and pleurotin was also proved in vivo where mice xenografted with human breast cancer tissue demonstrated decreased intra tumor HIF-1 and VEGF expression, and microvessel density (Welsh et al. 2003). The investigations using Trx-1 inhibitors have entered clinical phase I trials.
Anti-Microtubule Agents: 2-Methoxyestradiol and ENMD-1198
Anti-microtubule agents are a group of drugs that cause disruption of microtubule cytoskeleton during the interphase of cell division. Destabilization of the cytoskeleton leads to decrease of HIF-1 levels and activity in hypoxic conditions, inhibition of downstream products such as VEGF and occurs by a translation-dependent mechanism independently from proteasomal HIF-1 degradation (Mabjeesh et al. 2003). 2-Methoxyestradiol (2ME2) and its analog ENMD-1198 belong to this group of drugs (together with vincristine and taxol) (Moser et al. 2008). An additional mechanism by which 2ME2 could down-regulate HIF-1 might be based on induction of superoxide radicals (Salceda and Caro 1997). Because 2ME2 is a metabolite of 17β-estradiol (E2), a reasonable question arises if estrogen receptors α or β (ERα or ERβ) could be molecular targets for 2ME2. It was demonstrated that although 2ME2 had binding affinity for ERs, it was much lower compared to E2, especially in the case of ERβ. However, despite the ability to interact with ERs, 2ME2 did not engage them as antagonists; therefore, the antiproliferative effects of 2ME2 were not mediated through ERs (LaVallee et al. 2002).
The efficacy of 2ME2 was studied both in preclinical and clinical trials, where it showed antiproliferative and antiangiogenic properties (James et al. 2007; Kang et al. 2006). The treatment of head and neck cancer cell lines with 2ME2 combined with paclitaxel resulted in down-regulation of VEGF and tumor xenograft reduction (Ricker et al. 2004). ENMD-1198 was shown in vitro to inhibit the MAPK/Erk, PI3K/Akt, and STAT3 pathways in hepatocellular cancer cultures, thus leading to reduction of tumor growth and vascularization in xenografted mice (Moser et al. 2008). Its effectiveness is being studied in phase I and II trials in patients with solid cancers (prostate and breast cancer) and multiple myeloma refractory to different therapies (Ricker et al. 2004).
mTOR Inhibitors
An alternative way of HIF-1 regulation is based on the function of the mammalian target of rapamycin (mTOR) molecule, which promotes translation of HIF-1 mRNA into active HIF-1 protein (Hudson et al. 2002). Inhibitors of mTOR function act through decrease of tyrosine kinase activity and include drugs such as Temsirolimus (CCI-779), Everolimus (RAD001) and Deforolimus (AP23573) (Chon et al. 2006). The mTOR inhibitors showed their efficacy in preclinical studies, what encouraged to further investigations. Several phase I and II clinical trials in solid tumors, including endometrial, ovarian, breast, lung, and colorectal cancer, have demonstrated modest response to mTOR inhibitors as estimated by partial clinical response or stabilization of the disease (Chan et al. 2005; Hidalgo et al. 2006; Slomovitz et al. 2010).
Future Perspectives
The main problem which should be solved when using NF-κB and HIF-1 modulators and inhibitors is how to avoid unintentional drug action in healthy tissues. The complex activity of the described drugs is from one point of view their strength, as it helps to attack tumor from many directions, but from another perspective their disadvantage, as they could affect many intercellular pathways in normal tissues. This is connected directly with the problem of toxicity of therapeutic doses and under-treatment at safer dosages. In this context, some discriminator between normal and tumor tissues, allowing for “switch on” of NF-κB and HIF-1 inhibitors only inside tumors, would be helpful. To avoid long-term immunosuppression, the desired transcription factor inhibition, especially of NF-κB, should be transient and highly reversible. Of course, there is a variety of potential molecules which could more effectively and selectively modulate NF-κB and HIF-1 functions. None of the described drugs, however, could be recognized as targeting exclusively those transcription factors. One of the alternative solutions is molecular therapy based on use of double-stranded oligodeoxynucleotides which are capable of blocking NF-κB binding to promoter regions of targeted genes, or on use of single-stranded antisense oligonucleotides complementary to mRNA transcribed from the target gene (Milligan et al. 1993). It is also of importance to analyze the effects of drugs or their combinations in a cancer-cell-type specific manner. Genomic technology should help to identify subsets of patients’ best responding to NF-κB and HIF-1 inhibition. Another question is the use of some of the molecules discussed above as prophylactic agents against cancer, for instance as diet supplements. Moreover, randomized clinical trials estimating the combined treatment using NF-κB and HIF-1 inhibitors together with chemotherapeutics applied in advanced and recurrent solid cancer cases are urgently needed. This would allow for better comparison of efficacy with traditional therapy. A similar change of strategy resulted in approval of bortezomib for routine treatment of MM patients. Future research will probably bring new solutions to some of these problems.
Abbreviations
- HIF:
-
Hypoxia-inducible factor
- pVHL:
-
von Hippel-Lindau protein
- CREB:
-
cAMP response element-binding
- NF-κB:
-
Nuclear factor κB
- PI3K:
-
Phosphatidylinositol 3-kinase
- Akt:
-
Serine/threonine protein kinase
- mTOR:
-
Mammalian target of rapamycin
- EGF:
-
Epidermal growth factor
- VEGF:
-
Vascular-endothelial growth factor
- IGF-I:
-
Insulin-like growth factor-I
- 17-AAG:
-
17-Allylamino-17-desmethoxygeldanamycin
- 17-DMAG:
-
17-(Dimethylaminoethylamino)-17-demethoxygeldanamycin
- MM:
-
Multiple myeloma
- Trx-1:
-
Thioredoxin-1
- ccRCC:
-
Clear cell renal carcinoma
- STAT3:
-
Signal transducer and activator of transcription 3
- YC-1:
-
3-(5′-Hydroxymethyl-2′-furyl)-1-benzylindazole
- PX-12:
-
1-Methylpropyl 2-imidazolyl disulfide
- 2ME2:
-
2-Methoxyestradiol
- MAPK:
-
Mitogen-activated protein kinase
References
Abdul-Hafez A, Shu R, Uhal BD (2009) JunD and HIF-1alpha mediate transcriptional activation of angiotensinogen by TGF-beta1 in human lung fibroblasts. FASEB J 23:1655–1662
Banerji U, O’Donnell A, Scurr M et al (2005) Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 23:4152–4161
Belaiba RS, Bonello S, Zahringer C et al (2007) Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell 18:4691–4697
Beppu K, Nakamura K, Linehan WM et al (2005) Topotecan blocks hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression induced by insulin-like growth factor-I in neuroblastoma cells. Cancer Res 65:4775–4781
Birner P, Schindl M, Obermair A et al (2001) Expression of hypoxia-inducible factor-1alpha in epithelial ovarian tumors: its impact on prognosis and on response to chemotherapy. Clin Cancer Res 7:1661–1668
Brahimi-Horn MC, Pouysségur J (2009) HIF at a glance. J Cell Sci 122(Pt 8):1055–1057
Brown LM, Cowen RL, Debray C et al (2006) Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol Pharmacol 69:411–418
Chan S, Scheulen ME, Johnston S et al (2005) Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 23:5314–5322
Chanan-Khan AA, Richardson PG, Alsina M et al (2005) Phase 1 clinical trial of KOS-953 + Bortezomib (BZ) in relapsed refractory multiple myeloma (MM). Blood 106:109a–110a
Cheon H, Moon HE, Lee MS et al (2010) Loss of mitochondrial DNA enhances angiogenic and invasive potential of hepatoma cells. Oncol Rep 23:779–786
Chon HS, Hu W, Kavanagh JJ (2006) Targeted therapies in gynecologic cancers. Curr Cancer Drug Targets 6:333–363
Connell P, Ballinger CA, Jiang J et al (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol 3:93–96
Cummins EP, Berra E, Comerford KM et al (2006) Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 103:18154–18159
Duechler M, Wilczyński JR (2010) Hypoxia inducible factor-1 in cancer immune suppression. Curr Immunol Rev 6:260–271
Franovic A, Holterman CE, Payette J et al (2009) Human cancers converge at the HIF-2alpha oncogenic axis. Proc Natl Acad Sci USA 106:21306–21311
Gerber SA, Pober JS (2008) IFN-alpha induces transcription of hypoxia-inducible factor-1alpha to inhibit proliferation of human endothelial cells. J Immunol 181:1052–1062
Gloire G, Legrand-Poels S, Piette J (2006) NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 72:1493–1505
Gooljarsingh LT, Fernandes C, Yan K et al (2006) A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc Natl Acad Sci USA 103:7625–7630
Görlach A, Bonello S (2008) The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison. Biochem J 412:e17–e19
Gray MJ, Zhang J, Ellis LM et al (2005) HIF-1alpha, Stat3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 24:3110–3120
Grem JL, Morrison G, Guo XD et al (2005) Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol 23:1885–1893
Grimshaw MJ (2007) Endothelins and hypoxia-inducible factor in cancer. Endoc Relat Cancer 14:233–244
Hayashi T, Ueno Y, Okamoto T (1993) Oxidoreductive regulation of nuclear factor kappaB: involvement of a cellular reducing catalyst thioredoxin. J Biol Chem 268:11380–11388
Hidalgo M, Buckner JC, Erlichman C et al (2006) A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res 12:5755–5763
Holmquist L, Lofstedt T, Pahlman S (2006) Effect of hypoxia on the tumor phenotype: the neuroblastoma and breast cancer models. Adv Exp Med Biol 587:179–193
Hudson CC, Liu M, Chiang GG et al (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 22:7004–7014
James J, Murry DJ, Treston AM et al (2007) Phase I safety, pharmacokinetic and pharmacodynamic studies of 2-methoxyestradiol alone or in combination with docetaxel in patients with locally recurrent or metastatic breast cancer. Invest New Drugs 25:41–48
Jung YJ, Isaacs JS, Lee S et al (2003) IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J 17:2115–2117
Kakolyris S, Giatromanolaki A, Koukourakis M et al (2001) Thioredoxin expression is associated with lymph node status and prognosis in early operable non small cell lung cancer. Clin Cancer Res 7:3087–3091
Kang SH, Cho HT, Devi S et al (2006) Antitumor effect of 2-methoxyestradiol in a rat orthotopic brain tumor model. Cancer Res 66:11991–11997
Koong AC, Chen EY, Giaccia AJ (1994) Hypoxia causes the activation of nuclear factor kappaB through the phosphorylation of IkappaB α on tyrosine residues. Cancer Res 54:1425–1430
Krieg M, Haas R, Brauch H et al (2000) Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19:5435–5443
Kunkel MW, Kirkpatrick DL, Johnson JI et al (1997) Cell line-directed screening assay for inhibitors of thioredoxin reductase signaling as potential anti-cancer drugs. Anticancer Drug Des 12:659–670
Lang SA, Klein D, Moser C et al (2007a) Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastric cancer cells and reduces tumor growth and vascularization in vivo. Mol Cancer Ther 6:1123–1132
Lang SA, Moser C, Gaumann A et al (2007b) Targeting heat shock protein 90 in pancreatic cancer impairs insulin-like growth factor-I receptor signaling, disrupts an interleukin-6/signal-transducer and activator of transcription 3/hypoxia-inducible factor-1α autocrine loop, and reduces orthotopictumor growth. Clin Cancer Res 13:6459–6468
LaVallee TM, Zhan XH, Herbstritt CJ et al (2002) 2-Methoxyestradiol inhibits proliferation and induces apoptosis independently of estrogen receptors alpha and beta. Cancer Res 62:3691–3697
Liu XH, Kirschenbaum A, Lu M et al (2002) Prostaglandin E2 induces hypoxia-inducible factor-1alpha stabilization and nuclear localization in a human prostate cancer cell line. J Biol Chem 277:50081–50086
Löfstedt T, Fredlund E, Holmquist-Mengelbier L et al (2007) Hypoxia inducible factor-2alpha in cancer. Cell Cycle 6:919–926
Mabjeesh NJ, Post DE, Willard MT et al (2002) Geldanamycin induces degradation of hypoxia-inducible factor 1alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res 62:2478–2482
Mabjeesh NJ, Escuin D, LaVallee TM et al (2003) 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 3:363–375
Maloney A, Workman P (2002) HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther 2:3–24
Milani M, Harris AL (2008) Targeting tumour hypoxia in breast cancer. Eur J Cancer 44:2766–2773
Miller DS, Blessing JA, Lentz SS et al (2002) A phase II trial of topotecan in patients with advanced, persistent, or recurrent endometrial carcinoma: a gynecologic oncology group study. Gynecol Oncol 87:247–251
Milligan JF, Matteucci MD, Martin JC (1993) Current concepts in antisense drug design. J Med Chem 36:1923–1937
Moser C, Lang SA, Mori A et al (2008) ENMD-1198, a novel tubulin-binding agent reduces HIF-1alpha and STAT3 activity in human hepatocellular carcinoma (HCC) cells, and inhibits growth and vascularization in vivo. BMC Cancer 8:206
Muniyappa H, Song S, Mathews CK et al (2009) Reactive oxygen species-independent oxidation of thioredoxin in hypoxia: inactivation of ribonucleotide reductase and redox-mediated checkpoint control. J Biol Chem 284:17069–17081
Nishi K, Oda T, Takabuchi S (2008) LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal 10:983–995
Pagé EL, Chan DA, Giaccia AJ et al (2008) Hypoxia-inducible factor-1alpha stabilization in non hypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell 19:86–94
Pedersen M, Löfstedt T, Sun J et al (2008) Stem cell factor induces HIF-1alpha at normoxia in hematopoietic cells. Biochem Biophys Res Commun 377:98–103
Pietras A, Hansford LM, Johnsson AS et al (2009) HIF-2alpha maintains an undifferentiated state in neural crest-like human neuroblastoma tumor-initiating cells. Proc Natl Acad Sci USA 106:16805–16810
Powers MV, Workman P (2006) Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer 13(Suppl 1):S125–S135
Powis G, Kirkpatrick L (2004) Hypoxia inducible factor-1alpha as a cancer drug target. Mol Cancer Ther 3:647–654
Puppo M, Battaglia F, Ottaviano C et al (2008) Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha. Mol Cancer Ther 7:1974–1984
Qing G, Simon MC (2009) Hypoxia inducible factor-2alpha: a critical mediator of aggressive tumor phenotypes. Curr Opin Genet Dev 19:60–66
Rapisarda A, Uranchimeg B, Sordet O et al (2004a) Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res 64:1475–1482
Rapisarda A, Zalek J, Hollingshead M et al (2004b) Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res 64:6845–6848
Richardson PG, Chanan-Khan A, Lonial S et al (2006) A multicenter phase 1 clinical trial of tanespimycin (KOS-953) + bortezomib (BZ): encouraging activity and manageable toxicity in heavily pre-treated patients with relapsed refractory multiple myeloma (MM). In: Annual meeting of the American Society of Hematology, Orlando, FL
Richardson PG, Chanan-Khan AA, Alsina M et al (2010) Tanespimycin monotherapy in relapsed multiple myeloma: results of a phase 1 dose-escalation study. Br J Haematol 150:438–445
Ricker JL, Chen Z, Yang XP et al (2004) 2-Methoxy estradiol inhibits hypoxia-inducible factor 1alpha, tumor growth, and angiogenesis and augments paclitaxel efficacy in head and neck squamous cell carcinoma. Clin Cancer Res 10:8665–8673
Roberts AM, Watson IR, Evans AJ et al (2009) Suppression of hypoxia-inducible factor 2alpha restores p53 activity via Hdm2 and reverses chemoresistance of renal carcinoma cells. Cancer Res 69:9056–9064
Saitoh M, Nishitoh H, Fujii M et al (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17:2596–2606
Salceda S, Caro J (1997) Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 272:22642–22647
Scharte M, Jurk K, Kehrel B et al (2006) IL-4 enhances hypoxia induced HIF-1alpha protein levels in human transformed intestinal cells. FEBS Lett 580:6399–6404
Scortegagna M, Cataisson C, Martin RJ et al (2008) HIF-1alpha regulates epithelial inflammation by cell autonomous NFkappaB activation and paracrine stromal remodeling. Blood 111:3343–3354
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Semenza GL (2010) Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29:625–634
Slomovitz BM, Lu KH, Johnston T et al (2010) A phase 2 study of oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer 116:5415–5419
Supko JG, Hickman RL, Grever MR et al (1995) Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol 36:305–315
Talks KL, Turley H, Gatter KC et al (2000) The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 157:411–421
Teng CM, Wu CC, Ko FN et al (1997) YC-1, a nitric oxide independent activator of soluble guanylate cyclase, inhibits platelet-rich thrombosis in mice. Eur J Pharmacol 320:161–166
Ueno M, Masutani H, Arai RJ et al (1999) Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem 274:35809–35815
Welsh SJ, Bellamy WT, Briehl M et al (2002) The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor-1alpha protein expression: Trx-1 over-expression results in increased vascular endothelial growth factor production and enhanced angiogenesis. Cancer Res 62:5089–5095
Welsh SJ, Williams RR, Birmingham A et al (2003) The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther 2:235–243
Yeo EJ, Chun YS, Cho YS et al (2003) YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J Natl Cancer Inst 95:516–525
Yu AY, Frid MG, Shimoda LA et al (1998) Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol 275(4Pt 1):L818–L826
Zhou J, Fandrey J, Schümann J et al (2003) NO and TNF-alpha released from activated macrophages stabilize HIF-1alpha in resting tubular LLC-PK1 cells. Am J Physiol Cell Physiol 284:C439–C446
Zundel W, Schindler C, Haas-Kogan D et al (2000) Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev 14:391–396
Acknowledgments
The work was supported by the grant no. N402 458738 from the Ministry of Science and Higher Education (Poland).
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Wilczynski, J., Duechler, M. & Czyz, M. Targeting NF-κB and HIF-1 Pathways for the Treatment of Cancer: Part II. Arch. Immunol. Ther. Exp. 59, 301–307 (2011). https://doi.org/10.1007/s00005-011-0132-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-011-0132-3