
DNA viruses, as well as their host cells, require a DNA-dependent DNA polymerase to faithfully replicate their genomes. Viruses with small DNA genomes, such as papillomaviruses and polyomaviruses, have a limited coding capacity and utilize mainly the host replication machinery for their genome amplification. In contrast, large DNA viruses encode a specific polymerase equipped with a proofreading 3ʹ–5ʹ-exonuclease activity and other replication proteins that assure the replication of their genomic information. As a critical component of the viral replication machinery, viral DNA polymerases are the specific target of a number of antiviral drugs currently used to inhibit viral replication. Most antiviral drugs approved by the US Food and Drug Administration (FDA) inhibit viral genome replication, nearly all of these inhibit a DNA polymerase and most of these drugs are nucleoside analogs. FDA-approved inhibitors of viral polymerases target certain human herpesviruses, the retrovirus HIV (human immunodeficiency virus) and the hepadnavirus HBV (hepatitis B virus). This chapter focuses on the description of viral DNA polymerase inhibitors, whether currently approved or candidate drugs, that are particularly active against herpesviruses.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
DNA viruses, as well as their host cells, require a DNA-dependent DNA polymerase to faithfully replicate their genomes. Viruses with small DNA genomes, such as papillomaviruses and polyomaviruses, have a limited coding capacity and utilize mainly the host replication machinery for their genome amplification. In contrast, large DNA viruses encode a specific polymerase equipped with a proofreading 3ʹ–5ʹ-exonuclease activity and other replication proteins that assure the replication of their genomic information. As a critical component of the viral replication machinery, viral DNA polymerases are the specific target of a number of antiviral drugs currently used to inhibit viral replication. Most antiviral drugs approved by the US Food and Drug Administration (FDA) inhibit viral genome replication, nearly all of these inhibit a DNA polymerase and most of these drugs are nucleoside analogs. FDA-approved inhibitors of viral polymerases target certain human herpesviruses, the retrovirus HIV (human immunodeficiency virus) and the hepadnavirus HBV (hepatitis B virus). This chapter focuses on the description of viral DNA polymerase inhibitors, whether currently approved or candidate drugs, that are particularly active against herpesviruses.
Four types of polymerase inhibitors are recognized: substrate analogs (nucleoside and nucleotide analogs), product analogs (pyrophosphate analogs), allosteric inhibitors (non-nucleoside inhibitors), and inhibitors that intercalate or directly interact with nucleic acids.
The development of antiviral drugs against large DNA viruses (i.e., herpesviruses and poxviruses) was initially focused on viral DNA polymerase inhibitors and, as a consequence, most of the antiviral drugs currently on the market target viral DNA polymerases (Table 22.1). However, advances in the molecular biology of DNA virus replication allowed the identification of novel targets for drug development (Kleymann 2005; Biron 2006). Progress in different areas of research, such as gene expression, protein purification, proteomics, bioinformatics, and efficient high-throughput screening, has facilitated the characterization and functional assays of these new targets (DeFilippis et al. 2003; Coen & Schaffer 2003).
Currently Approved DNA Pol Inhibitors
Nucleoside Analogs
Acyclic Nucleoside Analogues
Nucleoside analogs represent the most productive source of antiviral agents. These agents need to be phosphorylated to their active form, the triphosphate form, to be able to target the viral DNA polymerase. The active forms inhibit polymerases by competing with natural dNTP substrates and/or incorporation into the growing DNA chain, where they can often terminate DNA elongation. Nucleoside analogs, acting as competitive inhibitors or substrates for viral polymerases, afford a reduction in viral DNA synthesis in infected cells. The selectivity of a nucleoside analog as inhibitor of viral replication depends on two parameters: (i) the efficiency by which viral enzymes phosphorylate the drug compared to cellular enzymes and (ii) the potency and efficiency by which viral genome replication is inhibited in comparison with cellular functions.
The first nucleoside analogs were synthesized as part of anti-metabolite cancer research programs in the late 1970s. Retrospectively, it can be stated that antiviral chemotherapy came of age with the discovery of acyclovir (ACV) in 1977 (Elion et al. 1977). This compound is a potent and selective inhibitor of herpes simplex virus (HSV) and varicella-zoster virus (VZV) replication and has demonstrated an excellent safety profile in clinical practice. Notably, in 1988 Dr. G. Elion received the Nobel Prize award for her work on the mechanism of action of nucleoside analogs including acyclovir.
Prior to ACV, nucleoside analogs such as vidarabine (adenine arabinoside, Ara-A), idoxuridine (IDU), and trifluridine (TFT) were associated with variable antiviral activity in humans and significant toxicity when systemically administered. The use of IDU and TFT was limited to topical therapy of herpetic keratitis (Kaufman et al. 1962; Kaufman 1963; Kaufman & Heidelberger 1964; Kaufman 1962). Neither IDU nor TFT can be used systemically because they are too toxic, especially to bone marrow. Vidarabine can be administered systemically, and it was the first antiviral drug used to treat herpetic encephalitis (Whitley et al. 1977). The therapeutic window for vidarabine is very narrow since this drug is phosphorylated by cellular adenosine kinase, resulting in significant side effects (mostly megaloblastic anemia) in patients. Shortly after the introduction of ACV as a highly specific anti-HSV agent, vidarabine was replaced by ACV in the treatment of herpetic encephalitis (Whitley et al. 1986; Whitley et al. 1981; Skoldenberg et al. 1984).
ACV represents the first generation of effective antiherpesvirus drugs. Despite its safety profile and potency against HSV and VZV, this compound had two major limitations: modest activity against other herpesviruses and poor oral bioavailability (only 15–30%). Three different approaches have been followed to obtain better oral bioavailability, intracellular pharmacokinetics, and antiviral activity: (i) synthesis of oral prodrugs of ACV, (ii) improvement nucleoside structure, and (iii) development of entirely novel structures. Soon after the discovery of ACV, other purine analogs, including penciclovir (PCV) and ganciclovir (GCV) and pyrimidine analogs, such as BVDU (brivudin) were described.
Acyclovir and Valacyclovir
ACV [9-(2-hydroxyethoxymethyl)guanine, Zovirax®] (Fig. 22.1), a structural analog of the natural 2ʹ-deoxyguanosine, can be considered the first truly specific antiviral agent (Elion et al. 1977; Schaeffer et al. 1978) with potent activity against herpes simplex type 1 (HSV-1) and type 2 (HSV-2), VZV, and Epstein–Barr virus (EBV) and modest activity against human cytomegalovirus (HCMV). It consists of a guanine base attached to an acyclic sugar-like molecule. Shortly after its discovery, ACV became the drug of choice for the treatment of HSV and VZV infections, particularly primary and recurrent genital herpes, and mucocutaneous HSV lesions and VZV infections in immunosuppressed patients (Table 22.1). Nowadays, ACV and its prodrug, the l-valyl ester valacyclovir, have become the gold standard for prophylaxis and treatment of diseases caused by HSV and VZV, and both compounds have shown benefit in the management of HCMV diseases in transplant recipients.


Structural formulae of viral DNA polymerase inhibitors that have been marketed.
The mechanism of action of ACV is shown in Fig. 22.2. The critical determinant of the selective activity of ACV against HSV and VZV is its preferential phosphorylation by the virus-encoded thymidine kinase (TK) (Fyfe et al. 1978; Keller et al. 1981). This enzyme converts ACV to ACV monophosphate (ACV-MP) which is then phosphorylated by the cellular GMP kinase to ACV diphosphate (ACV-DP) and further to ACV triphosphate (ACV-TP) by nucleoside diphosphate kinase or other cellular enzymes (Miller & Miller 1980; Miller & Miller 1982). ACV-TP, the active form of ACV, is more inhibitory to HSV DNA polymerase than to cellular DNA polymerases (Furman et al. 1979; St Clair et al. 1980) and this inhibition is competitive with respect to the natural substrate dGTP. High concentrations of dGTP can reverse the antiviral activity of ACV. In addition, ACV-TP can also serve as a substrate of the DNA polymerase reaction and hence be incorporated into DNA at its 3ʹ-terminus. As 3ʹ-terminal ACV-MP residues cannot be excised by the DNA polymerase-associated 3ʹ–5ʹexonuclease (Derse et al. 1981), they prevent further chain elongation and thus act as DNA chain terminators because ACV does not contain the 3ʹ-hydroxyl group required for DNA elongation. This explains the occurrence of short DNA fragments in HSV-infected cells exposed to ACV (McGuirt et al. 1984). It has also been demonstrated that following incorporation of ACV-TP, the viral polymerase becomes trapped on the ACV-terminated DNA chain when the next deoxynucleoside triphosphate binds (Martin et al. 1994; Ilsley et al. 1995; Reardon 1989). ACV has been described as a “suicide” inhibitor, because it inactivates the HSV DNA polymerase as well as being a substrate for it (Furman et al. 1984).

Intracellular metabolism and mechanism of action of nucleoside analogs. Acyclic nucleoside analogs such as ACV, PCV, and GCV need to be selectively phosphorylated intracellularly in three steps, to the triphosphate (TP) active forms. The first phosphorylation step to the monophosphate forms (MP) is carried out by the HSV- or VZV-encoded thymidine kinase (TK), the HCMV UL97 open reading frame that encodes for a protein kinase or its HHV-6 homolog UL69. Therefore, the first phosphorylation is limited to virus-infected cells. Further phosphorylation to the diphosphate (DP) and triphosphate (TP) forms is carried out by cellular enzymes [i.e., dGMP kinase and nucleoside 5ʹ-diphosphate (NDP) kinase] The triphosphate forms inhibit viral DNA polymerases acting as competitive inhibitors of dGTP binding and/or as alternative substrates if incorporated into the growing DNA chain. In the case of ACV, the incorporation of ACV-TP into the viral DNA leads to termination of chain elongation and trapping of the viral polymerase on the terminated DNA chain when the next deoxynucleoside triphosphate binds. For the pyrimidine nucleoside analog BVDU, following uptake by the (virus-infected) cells the compound is phosphorylated by the HSV-1- or VZV-encoded TK to the 5ʹ-monophosphate (BVDU-MP) and 5ʹ-diphosphate (BVDU-DP), and further to the 5ʹ-triphosphate (BVDU-TP) by cellular kinases, i.e., NDP kinase. BVDU-TP can act as competitive inhibitor/alternative substrate of the viral DNA polymerase and as a substrate it can be incorporated internally (via nucleotide linkages) into the (growing) DNA chain
As ACV has limited oral bioavailability and limited solubility in water (∼0.2%, 25°C) relatively large doses and frequent administration were thus required to maintain plasma levels high enough to achieve viral inhibition. Much research effort was directed to improve the solubility and oral bioavailability of ACV, and several water-soluble esters of ACV were investigated. The valine ester of ACV, valacyclovir (VACV, Valtrex®, Zelitrex®) (Fig. 22.1), proved to be a safe and efficacious drug (Perry & Faulds 1996; Darby 1994). The absolute oral bioavailability of ACV following oral administration of VACV is 54% (Weller et al. 1993). The increased oral bioavailability of VACV is due to a carrier-mediated intestinal absorption, via the human intestinal peptide transporter hPEPT1 (Guo et al. 1999), followed by the rapid conversion to ACV by ester hydrolysis in the small intestine (Perry & Faulds 1996; De Clercq & Field 2006). Following oral administration, VACV is rapidly metabolized to yield ACV and the essential amino acid l-valine (Perry & Faulds 1996).
Several clinical studies have demonstrated that VACV has a safety profile comparable to ACV in patients with genital herpes, herpes labialis, and herpes zoster (Warren et al. 2004; Corey et al. 2004; DeJesus et al. 2003; Corey et al. 2004; Beutner 1995; Beutner et al. 1995; Gupta et al. 2004). VACV appears a more attractive option in the treatment of HSV and VZV infections due to a less frequent dosing regimen, which may contribute to increased patient adherence to therapy. Although VACV is not potent enough for the treatment of established HCMV disease, it has been approved in several countries for prophylaxis of HCMV infection in transplant recipients (Biron 2006). The safety and efficacy of VACV for prevention of HCMV have been documented in several studies (Hodson et al. 2005; Lowance et al. 1999).
Penciclovir and Famciclovir
Penciclovir [PCV, 9-(4-hydroxy-3-hydroxymethyl-but-1-yl)guanine Denavir®, Vectavir®] (Fig. 22.1) is also a 2ʹ-deoxyguanosine analog resembling ACV in chemical structure, mechanism of action, and spectrum of antiviral activity (Perry & Wagstaff 1995). Like ACV, PCV depends on the HSV- and VZV-encoded TK for activation to the monophosphate form (Fig. 22.2). Cellular enzymes are responsible for further phosphorylation to the triphosphate form. PCV-TP inhibits the viral DNA polymerase through competition with 2ʹ-deoxyguanosine triphosphate and is incorporated in the viral DNA. Unlike ACV-TP, PCV-TP is not an obligate chain terminator as PCV has two hydroxyl groups on the acyclic chain and can be incorporated into the growing DNA chain. Intracellular concentrations of PCV-TP are at least 30-fold higher than those of ACV-TP (Vere Hodge et al. 1989). However, HSV and VZV DNA polymerases have higher affinity for ACV-TP than for PCV-TP. As a result, the relative activities ACV and PCV against HSV and VZV in cell culture are similar. PCV-TP is more stable within infected cells than ACV-TP and, therefore, it has longer antiviral action (Boyd et al. 1987).
Like ACV, PCV is very poorly absorbed when given orally. Famciclovir (FCV) (Fig. 22.1), the diacetylester of 6-deoxypenciclovir, was developed as an oral prodrug. After oral administration, FCV is rapidly and extensively absorbed and efficiently converted to PCV in two steps: (i) removal of the two acetyl groups (the first acetyl side chain of FCV is cleaved by esterases found in the intestinal wall, and the second is removed on first pass through the liver), and (ii) oxidation at the six position catalyzed by aldehyde oxidase that account for the conversion of 6-deoxypenciclovir to PCV (Rashidi et al. 1997; Perry & Wagstaff 1995). The total oral bioavailability of PCV from FCV is 77%. Data collected from several clinical studies demonstrated that FCV is well tolerated in patients and is effective against HSV-1 and HSV-2 for both therapy and long-term suppression of recurrences and is also efficacious in the treatment of herpes zoster (Table 22.1) (Simpson & Lyseng-Williamson 2006; Mertz et al. 1997; Sacks et al. 2005; Wald et al. 2006).
Ganciclovir and Valganciclovir
ACV has poor activity against HCMV due to the much lower accumulation of phosphorylated ACV in HCMV-infected cells than in HSV- or VZV-infected cells (Furman et al. 1981). The lack of activity of ACV against HCMV prompted the development of other acyclic guanosine analogs, including ganciclovir [GCV, 9-(1,3-dihydroxy-2-propoxymethyl)guanine, DHPG, Cymevene®, Cytovene®] (Fig. 22.1), which is more potent against HCMV than ACV. GCV was the first antiviral agent approved for the treatment of infections caused by HCMV. It has become the treatment of choice for the management of HCMV diseases and remains the first-line treatment of HCMV disease in transplant recipients (Razonable & Emery 2004; Biron 2006).
GCV is an acyclic guanosine analog of 2ʹ-deoxyguanosine and unlike ACV, but similar to PCV, has the equivalent of a 3ʹ-hydroxyl group in the acyclic chain (Fig. 22.1). Like ACV and PCV, GCV is converted to GCV-TP (the active form against HCMV) in a multi-step process involving viral and cellular enzymes (Fig. 22.2). Because HCMV does not encode for a TK, in HCMV-infected cells the initial phosphorylation of GCV is catalyzed by a protein kinase encoded by the HCMV UL97 open reading frame (Sullivan et al. 1992; Littler et al. 1992). Cellular kinases further convert GCV-MP to GCV-TP, which is both a competitive inhibitor and a substrate for the viral DNA polymerase. GCV-TP is a better inhibitor for the HCMV DNA polymerase than for cellular DNA polymerases (Martin et al. 1994; Biron et al. 1985), and it is a better substrate for HCMV DNA polymerase than for cellular DNA polymerases (Reid et al. 1988). GCV-TP is not an obligate chain terminator; however, after incorporating GCV-MP, HCMV DNA polymerase stalls after incorporating one additional nucleotide (Reid et al. 1988). The preferential phosphorylation of GCV in HCMV-infected cells and the higher inhibition of viral DNA polymerization than cellular DNA synthesis account for the selectivity of GCV against HCMV. However, it appears that the selectivity of GCV at each of these steps is lower than the selectivity of ACV against HSV or VZV. This correlates with a higher toxicity of GCV in the clinic. The side effects of GCV are mostly hematologic abnormalities (neutropenia, anemia and thrombocytopenia) and probable long-term reproductive toxicity. The HCMV-encoded protein kinase UL97 also catalyses the initial phosphorylation step of ACV (Talarico et al. 1999); however, ACV is a less efficient substrate than GCV, which explains in part the lower activity of ACV compared to GCV against HCMV. Another difference between these two compounds is the four- to five-fold shorter half-life of ACV-TP compared to GCV-TP in infected cells, resulting in lower intracellular levels of the active ACV-TP.
GCV can be given intravenously (IV), orally (Cytovene®), or as an ocular implant (Vitraset®, Chiron) for the treatment of HCMV retinitis (Table 22.1). The low bioavailability of GCV (∼6%) prompted the development of the prodrug, valganciclovir (VGCV, Valcyte®), the l-valyl ester of GCV (Fig. 22.1). Following oral administration, it is metabolized to the active form (GCV) in the intestinal wall and liver. VGCV exhibits oral bioavailability of about 60%. VGCV has now replaced oral GCV, and there is still a debate whether prophylaxis therapy or preemptive therapy should be used for HCMV infections in asymptomatic high-risk transplant recipients (Biron 2006).
Human herpesvirus 6 (HHV-6) encodes UL69 phosphotransferase, a homolog of HCMV UL97, which has been shown to be able to phosphorylate GCV (Michel & Mertens 2004; Ansari & Emery 1999). This compound shows reasonable activity against HHV-6 in vitro, although 20-fold lower levels of GCV-MP are attained by pUL69 phosphotransferase than by its HCMV homolog pUL97 (De Bolle et al. 2002).
Pyrimidine Nucleoside Analogs
Brivudin
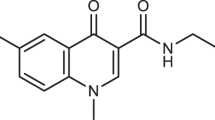
In the late 1970 s, brivudin [BVDU, (E)-5-(2-bromovinyl)-2ʹ-deoxyuridine, bromovinyldeoxyuridine Zostex®, Brivirac®, Zerpex®] (Fig. 22.1) was described as a highly selective antiviral agent (De Clercq et al. 1979; De Clercq 2004), which proved specifically active against HSV-1 and VZV (De Clercq 2004). Several congeners of BVDU have been synthesized, including BVaraU (sorivudine), the arabinofuranosyl counterpart of BVDU. The 5-2-(E)-bromovinyl group, with the bromine in the trans configuration, is crucial for the antiviral selectivity of all BVDU derivatives.
BVDU and BVaraU are among the most potent inhibitors of VZV. The selective activity of BVDU against HSV-1 and VZV is dependent on specific phosphorylation by HSV-1 or VZV TK which converts BVDU subsequently to its monophosphate (BVDU-MP) and diphosphate (BVDU-DP) (Fig. 22.2). The latter is then converted to the triphosphate BVDU-TP by a nucleoside diphosphate (NDP) kinase or other cellular kinase, whereupon BVDU-TP enters in competition with the natural substrate dTTP for the viral DNA polymerase. It can inhibit the incorporation of dTTP into viral DNA, or, as an alternate substrate, it can be itself incorporated, thus leading to the formation of structurally and functionally disabled viral DNA (De Clercq 2004). The conversion of BVDU-MP to BVDU-DP does not occur in HSV-2-infected cells due to the lack of thymidylate kinase activity of the HSV-2 TK (Fyfe 1982). This explains the poor activity of BVDU against HSV-2. Thus, the predominant determinant in the antiviral activity of BVDU, as well as of the different BVDU derivatives, is the virus-encoded TK and its associated thymidylate (dTMP) kinase activity. As seen in HSV-2, in some clinical isolates and in in vitro-selected BVDU-resistant mutants, thymidylate kinase activity can be regulated independently from TK activity (Docherty et al. 1991; Andrei et al. 2005a).
Following oral administration of BVDU, approximately 90% is absorbed and about 70% of the oral dose is rapidly transformed to bromovinyl–uracil (BVU) during first passage through the liver (Wutzler 1997). Clinical studies have confirmed that BVDU is effective in treatment of herpes zoster, both in short-term (formation of new lesions) and long-term effects (prevention of post-herpetic neuralgia), BVDU being more efficient and/or convenient than the other anti-VZV drugs acyclovir, valacyclovir, and famciclovir (De Clercq 2004). BVDU has been marketed in several European countries for the treatment of herpes zoster (shingles, zona) (Table 22.1).
There is one limitation for the use of BVDU: it should not be given to patients under 5-fluorouracil therapy since BVU, the degradation product of BVDU, is a potent inhibitor of dihydropyrimidine dehydrogenase (DHP), the enzyme responsible for the first step in the catabolic pathway of pyrimidines. DHP is also needed for the degradation of 5-fluorouracil. Therefore, concomitant administration of this drug together with BVDU results in increased exposure to 5-fluorouracil since BVU protects 5-fluorouracil against breakdown by DHP and significantly increases the half-life of BVU (Desgranges et al. 1986). Like BVDU, sorivudine is metabolized to bromovinyl–uracil and, therefore, its administration with 5-fluorouracil is contraindicated. Sorivudine was licensed in Japan in 1993 for the treatment of herpes zoster, but the product was withdrawn following several deaths related to co-administration with 5-fluorouracil (Okuda et al. 1997; Okuda et al. 1998)
Nucleotide Analogs
Acyclic Nucleoside Phosphonates (ANPs)
The discovery of acyclic nucleoside phosphonates (ANPs) represented a breakthrough in the treatment of DNA virus and retrovirus infections (De Clercq & Holý 2005). According to their activity spectrum, the first generation of ANPs can be classified in two categories: (i) the “HPMP” (i.e., 3-hydroxy-2-phosphonylmethoxypropyl) derivatives, represented by HPMPC (cidofovir, CDV) (Fig. 22.1), which displays activity against a broad variety of DNA viruses, and (ii) the “PME” (i.e., 2-phosphonylmethoxyethyl) and “PMP” (i.e., 2-phosphonylmethoxypropyl) derivatives, represented by PMEA (adefovir) and PMPA (tenofovir), respectively. These three representative compounds have been licensed for treatment of HCMV retinitis in AIDS patients (cidofovir, Vistide®), chronic hepatitis B virus (HBV) infections (adefovir dipivoxil, Hepsera®), and HIV infections (tenofovir disoproxil fumarate, TDF, Viread®). CDV can also be used “off-label” in treatment of herpesvirus (other than HCMV) infections, as well as polyoma-, papilloma-, adeno-, and poxvirus infections (Safrin et al. 1997; Snoeck & De Clercq 2002; De Clercq 2003). TDF is also available in a fixed-dose combination form with emtricitabine (Truvada®), with emtricitabine, and efavirenz (Atripla®) for the treatment of AIDS and has recently been approved for treatment of HBV infections as well.
In natural nucleotides (or nucleoside phosphates), the phosphate group is attached through an ester bound (–P–O–C–) to the nucleoside, as, for example, when a phosphate group has been linked to ACV (ACV-MP) during the first step of phosphorylation carried out by HSV- or VZV-encoded TK. In ANPs, the phosphate group, in the form of a phosphonate group, has already been attached to the (acyclic) nucleoside analog, thus resulting in formation of a phosphonomethyl ether (–P–C–O–), which unlike the phosphate ester linkage should resis attack by esterases.
The first ANP, identified in 1986, was (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine (HPMPA), emerging as a broad-spectrum anti-DNA agent (De Clercq et al. 1986). This compound can be considered a hybrid between acyclic nucleoside analogs, such as (S)-9-(2,3-dihydroxypropyl)adenine (DHPA), which was previously described (De Clercq et al. 1978) as an acyclic nucleoside analog with broad-spectrum antiviral activity and a phosphonate analog such as phosphonoformic acic (PFA) or phosphonoacetic acid (PAA). PMEA was developed in parallel with HPMPA, whereas CDV was derived from HPMPA by simply substituting a pyrimidine (cytosine) for the purine (adenine) moiety. Further modifications of the acyclic side chain of HPMPA and PMEA led to PMPA and PMPDAP. The transition of HPMPA to PMEA allowed the activity spectrum to be extended to retroviruses (while maintaining activity against herpes- and hepadnaviruses); further modification to PMPA restricted the activity spectrum to retro- and hepadnaviruses.
ANPs can be taken up by cells and need only two phosphorylation steps to be converted to their active phosphoryl derivatives (Fig. 22.3) (Ho et al. 1992). These two phosphorylation steps are carried out by cellular enzymes. In this way, ANPs are independent of the first phosphorylation step which in the nucleoside analogs is catalyzed by the HSV- or VZV-encoded TK or the HCMV-encoded protein kinase UL97. Therefore, ANPs are active against TK-deficient HSV and VZV mutants. The diphosphoryl derivatives of the ANPs (i.e., CDVpp and PMEApp) interact with the viral DNA polymerase as either competitive inhibitors [with respect to the natural substrates (i.e., dCTP, dATP)] or alternate substrates (thus leading to incorporation of ANP into DNA). For PMEA, this incorporation inevitably leads to DNA chain termination, but CDV and HPMPA contain a hydroxyl function in the acyclic side chain that would allow further chain elongation. Incorporation of CDVpp into viral DNA slows down elongation and results in chain termination when two consecutive CDVpp residues are incorporated as has been demonstrated for HCMV (Xiong et al. 1997; Xiong et al. 1996). The diphosphorylated forms of ANPs inhibit viral DNA polymerases more potently than the cellular DNA polymerases.

Intracellular metabolism and mechanism of action of CDV (cidofovir) and PMEA (adefovir) against DNA viruses. Once inside the cells, ANPs need to be activated by cellular enzymes, which are different for the pyrimidine (i.e., CDV) and for the purine (i.e., PMEA) series. Pyrimidine nucleoside monophosphate (PNMP) kinase catalyses conversion of CDV to CDV-monophosphoryl (CDVp), which is then further phosphorylated to the active form, CDV-diphosphoryl (CDVpp) by nucleoside 5ʹ-diphosphate (NDP) kinase. CDVp-choline is considered to serve as an intracellular reservoir for the mono- and diphosphoryl derivatives of CDV. Two different pathways have been suggested for the phosphorylation of PMEA and related purines. PMEA can be converted directly to PMEA-diphosphoryl (PMEApp) by 5ʹ-phosphoribosyl-1-pyrophosphate (PRPP) synthetase. Alternatively, AMP kinase may be involved in the two consecutive steps of phosphorylation of PMEA to PMEAp and PMEApp. The diphosphoryl derivatives of the ANPs (i.e., CDVpp and PMEApp) interact with the viral DNA polymerase as either competitive inhibitors [with respect to natural substrates (i.e., dCTP, dATP)] or alternative substrates (thus leading to incorporation of ANPs into DNA). For PMEA, this incorporation inevitably leads to DNA chain termination, but CDV has a hydroxyl function in the acyclic side chain that might allow further chain elongation. Chain termination occurs when two consecutive CDVpp are incorporated into the growing DNA chain.
The metabolites of the ANPs (i.e., CDV) show an unusually long intracellular half-life; this may account for the long-lasting antiviral activity of the compounds. This prolonged antiviral action may be attributed to the formation of the CDVp-choline adduct, which could serve as an intracellular reservoir for the mono- and diphosphoryl derivatives of CDV.
CDV administered by the intravenous route has been approved for the treatment of HCMV retinitis in AIDS patients, but has also been used successfully in the treatment of HSV-1, HSV-2, and VZV infections (i.e., those that are resistant to first-line therapy), EBV, human herpesvirus 6 (HHV-6), HHV-7, and HHV-8 infections, polyomavirus infections [i.e., progressive multifocal leukoencephalopathy (PML) due to JC virus and hemorrhagic cystitis due to BK virus], papillomavirus infections (i.e., disseminated respiratory papillomatosis), adenovirus infections, and poxvirus infections (Table 22.1). When given topically, CDV has also proven beneficial in the treatment of mucocutaneous HSV-1 and HSV-2 infections, HPV-associated papillomatosous lesions [i.e., anogenital warts, plantar warts, recurrent laryngeal papillomatosis, cervical intraepithelial neoplasia (CIN) grade III], and poxvirus infections [i.e., molluscum contagiosum and orf virus] (Safrin et al. 1997; Snoeck & De Clercq 2002; De Clercq 2003). CDV present two major disadvantages that have restrained its use: (i) low oral bioavailability (<5%) requiring IV administration, usually once a week (or every other week), and (ii) dose-dependent nephrotoxicity that can be limited by pre-hydration and co-administration of probenecid.
Pyrophosphate Analogs
Foscarnet
Foscarnet (PFA, Foscavir®, FOS) is the trisodium salt of phosphonoformic acid (Fig. 22.1), a pyrophosphate analogue, which is a product of polymerization of nucleic acids. PFA, which does not require phosphorylation by viral or cellular kinases, inhibits directly the activity of the viral DNA polymerase. PFA acts as a product analog by binding to the pyrophosphate binding site and blocking the release of pyrophosphate from the terminal nucleoside triphosphate when added onto the growing DNA chain (Ostrander & Cheng 1980; Eriksson et al. 1982; Crumpacker 1992).
PFA can be considered as a second-line therapy and its use is reserved to patients that have failed ACV or GCV therapy due to viral resistance or that cannot be treated with GCV due to side effects of the drug (Table 22.1). Because PFA does not require a phosphorylation step to inhibit the viral DNA polymerase, it remains active against ACVr HSV and VZV strains due to mutations in the viral TK or against GCVr HCMV strains harboring mutations in the UL97 gene. Indeed, several reports have documented the efficacy of PFA in management of infections caused by these types of drug-resistant viruses (Morfin & Thouvenot 2003; Gilbert et al. 2002; Baldanti & Gerna 2003).
Mechanisms of Resistance
Since the introduction of ACV, now 25 years ago, for treatment of herpesvirus infections (Bacon et al. 2003), several studies have characterized drug-resistant herpesvirus mutants isolated either in vitro or in vivo. Different mechanisms exist by which HSV can acquire resistance to ACV. Three of these mechanisms involve the viral TK: (i) alteration of viral TK (TKaltered) resulting in less efficient phosphorylation of the drug, (ii) deletion of the viral TK gene (TKnegative, TK−), and (iii) reduction of the expression level of TK (TKpartial) (Coen 1991; Hill et al. 1991; Harris et al. 2003; Chibo et al. 2004). Alternatively, alterations at the level of the viral DNA polymerase gene have also been observed (Collins et al. 1989; Sacks et al. 1989; Hwang et al. 1992; Gaudreau et al. 1998). Although viruses of all four phenotypes have been isolated from patients, the predominant drug-resistant phenotype recovered in vivo (similar to the in vitro situation) exhibited TK deficiency (TKnegative or TKpartial, albeit rather low activity), as shown by several investigators (Gaudreau et al. 1998; Pottage, Jr. & Kessler 1995; Sacks et al. 1989; Coen et al. 1982).
Surveys among immunocompetent individuals have shown that ACV-resistant (ACVr) HSV is extremely rare in this group of patients, the prevalence varying between 0 and 0.6% (Englund et al. 1990; Nugier et al. 1992; Christophers et al. 1998; Danve-Szatanek et al. 2004). In contrast to usually self-limiting infections in healthy individuals, in immunocompromised individuals (i.e., patients with HIV infection and recipients of solid organ or bone marrow transplants) HSV infection can be severe and persistent. In these cases, prolonged antiviral therapy is required for management of the infection, resulting in emergence of drug-resistant mutants in approximately 4–7% of immunocompromised patients (Christophers et al. 1998; Chakrabarti et al. 2000; Chen et al. 2000). The probability of developing unresponsive lesions appears to be related to degree of immunosuppression.
The TK gene is not essential for virus replication in cell culture, although in vivo it is involved in HSV virulence, pathogenicity, and reactivation from latency (Coen et al. 1989; Efstathiou et al. 1989; Jacobson et al. 1993). Nevertheless, about 95% of clinical HSV isolates resistant to ACV contain mutations in the viral TK and not in the viral DNA polymerase (Pottage, Jr. & Kessler 1995; Christophers et al. 1998). The lower frequency of DNA polymerase mutations could be due to fewer mutations that can result in a drug-resistant phenotype relative to TK, where there are several mutations that can confer resistance. Mutations in the TK gene that are associated with ACV resistance are mostly due to the addition or deletion of nucleotides in long homopolymer runs of G’s and C’s resulting in frame shift mutations and consequently in a truncated enzyme (Hwang & Chen 1995; Sasadeusz et al. 1997; Morfin et al. 2000; Sarisky et al. 2001). In fact, two studies have demonstrated that about 50% of the clinical ACV-resistant (ACVr) HSV strains contain such type of mutations and the other half harbor single nucleotide substitutions in conserved and/or non-conserved regions of the TK gene (Gilbert et al. 2002). Mutations identified in PCVr mutant herpesviruses isolated in vitro were generally not found within homopolymeric G and C nucleotide stretches (Sarisky et al. 2001). In a subsequent study (Suzutani et al. 2003), it was found that mutations in the TK genes from ACVr HSV-1 mutants consisted of 50% single nucleotide substitutions and 50% frameshift mutations, while the corresponding figures for the PCVr mutants were 4 and 96%, respectively. Recently, it was described that mutations in the TK genes of mutant viruses produced under single-round high-dose selection with BVDU consisted of 41.7% frameshift mutations within homopolymer repeats of Gs and Cs and single nucleotide substitutions (58.3 %) (Andrei et al. 2005a). The A168T change, which proved to be associated with an altered TK phenotype, appeared to be the most common substitution.
HSV-1 DNA polymerase, encoded by the UL30 gene, has 1,235 amino acid residues. It exhibits all the enzymatic functions of a polymerase and belongs to the pol α family, which includes human polymerase α and DNA polymerases from animals and other viruses. HSV-1 DNA polymerase exhibits in addition to a deoxyribonucleotide polymerizing (catalytic) function, 5ʹ→3ʹexonuclease/RNase H function and 3ʹ→5ʹ exonuclease editing activity (Knopf 1979; Boehmer & Lehman 1997). The COOH terminus of the polymerase interacts with an accessory factor, UL42 that serves to increase processivity of the polymerase (Digard et al. 1993; Zuccola et al. 2000). The overall architecture of HSV-1 DNA polymerase closely resembles that of other polymerases belonging to the polymerase α family despite being at least 300 amino acids longer and exhibiting low sequence similarity (range 16–50%). The crystal structure of the HSV-1 DNA polymerase has recently been reported (Liu et al. 2006) showing that the 3ʹ–5ʹ-exonuclease domain and the polymerase palm, fingers, and thumb domains of HSV polymerase can be individually superimposed with the equivalent domain structures from other polymerase α structures. HSV-1 polymerase is comprised of six structural domains: a pre-NH2 domain, an NH2 domain terminal, a 3ʹ–5ʹ-exonuclease domain, and polymerase palm, fingers, and thumb domains. Based on sequence conservation in the pol α polymerase family, the exonuclease domain contains conserved regions exo I, exo II (region IV), and exo III (δ- region C). Regions III and VI are located in the fingers subdomain, regions I, II, and VII belong to the palm subdomain, and the thumb subdomain contains conserved region V (Fig. 22.4). These regions appear to flank the catalytic site in the palm subdomain and may play a role in positioning the template and primer strands.

Organization of HSV-1 DNA polymerase and amino acid substitutions leading to drug resistance. Locations of functional sites on HSV-1 DNA polymerase and linear N terminus to C terminus configuration of the polypeptide are shown. Solid boxes indicate regions numbered I to VII based on conservation among the DNA polymerases and region A, a region showing slight conservation. Locations of point mutations that result in altered drug sensitivity are shown in the lower part of the figure.
Most of the work with regard to mutations in the HSV DNA polymerase has been performed with ACV and PFA, the majority of the mutations being mapped to regions I, II, and III and δ-region C, indicating that these regions are important for the binding of dNTP and pyrophosphate. Although PFA inhibits the viral DNA polymerase by a different mechanism than that of nucleoside analogs, mutations in the HSV DNA polymerase that confer resistance to ACV also confer resistance to PFA. Mutations associated with resistance to HPMP derivatives such as CDV and HPMPA have been linked to changes in non-conserved regions of HSV DNA polymerase. Interestingly, these mutations did not confer resistance to PME derivatives such as PMEA (Andrei et al. 2000). These findings suggest that these two subclasses of ANPs differ in their mode of interaction with the viral DNA polymerase. Moreover, mutants resistant to HPMP derivatives remained sensitive to PFA and ACV, while different degrees of cross-resistance between PME derivatives, PFA, and ACV were noted (Andrei et al. 2007a; Andrei et al. 2007b; Bestman-Smith & Boivin 2002). Most DNA polymerase mutations conferring resistance to PFA have been associated with cross-resistance to PME derivatives, but some HSV polymerase mutations have been linked to resistance to PFA and sensitivity to PME derivatives (Andrei et al. 2007a). These data support possible use of CDV and not PMEA in treatment of PFAr HSV infections. Indeed, CDV has proven efficacious in treatment of ACVr and/or PFAr HSV-associated diseases (Snoeck et al. 1994a; LoPresti et al. 1998; Safrin et al. 1997).
Pathogenicity of drug-resistant HSV mutants has been mostly studied in mouse models (Coen 1994). One assay of pathogenesis in mice entails infection of the central nervous system via intracerebral inoculation (i.c.), which leads to encephalitis and death (neurovirulence). In a second assay of pathogenesis, virus is inoculated at a peripheral site, where it replicates and, following axonal transport, reaches the trigeminal ganglia (secondary site of replication). There, HSV establishes and maintains a latent infection. The assay that can be considered as most sensitive to drug-resistance mutation is ability of virus to kill mice after i.c. inoculation (Coen 1994). In this assay, mutants described as TKnegative and TKpartial have been shown to be significantly less virulent than wild-type viruses (Field & Wildy 1978; Field & Darby 1980; Darby et al. 1981; Tenser et al. 1983; Chrisp et al. 1989; Suzutani et al. 1995; Andrei et al. 2005a; Andrei et al. 2007a). However, Pelosi and colleagues have reported a TK mutant with a large deletion mutation which was only slightly impaired in neurovirulence and one TKpartial mutant which was fully neurovirulent following i.c. inoculation (Pelosi et al. 1998a). Degrees of attenuation of TKaltered mutants and some DNA polymerase mutants may vary substantially (Pelosi et al. 1998a; Pelosi et al. 1998b; Darby et al. 1981; Darby et al. 1984; Field & Darby 1980; Field & Coen 1986). Among TKaltered and DNA polymerase mutants the most pathogenic drug-resistant mutants can be found, although it appears that drug-resistant mutants arising under pressure of the HPMP derivatives have the lowest levels of neurovirulence (Andrei et al. 2007a) Although it is generally accepted that TKdeficient viruses are unable to be reactivated from latency (Coen et al. 1989; Efstathiou et al. 1989; Jacobson et al. 1993), some reports have emerged that describe individual TKdeficient isolates (in particular, mutants harboring mutations within the 7G homopolymer repeat of the HSV-1 TK gene) that can be recovered, albeit inefficiently, from latently infected animals and can cause recurrent infections in patients (Hwang et al. 1994; Sasadeusz & Sacks 1996; Horsburgh et al. 1998; Morfin et al. 2000; Harris et al. 2003). Three different mechanisms can be exploited by HSV-1 to compensate for a mutation that would otherwise inactivate TK and prevent reactivation from latency: (i) ribosomal frameshifting during which low levels of TK are expressed (Hwang et al. 1994; Griffiths et al. 2003), (ii) replication errors due to reduced fidelity of the DNA polymerase when replicating homopolymeric runs that create subpopulations of wild-type virus (Sasadeusz & Sacks 1996; Griffiths & Coen 2003; Grey et al. 2003), and (iii) alleles in loci other than TK that have potential to complement TK function and influence the ability of HSV to replicate in the nervous system and to reactivate from latency (Horsburgh et al. 1998). An important factor that influences drug-resistance and pathogenicity is heterogeneity of the viral population. Mixtures of different drug-resistant mutants or mixtures of drug-resistant virus and wild-type virus can complement for both drug resistance and pathogenicity. This can be seen in the case of TK frameshift mutations in homopolymeric sequences, the most common drug-resistant mutations, which can easily revert resulting in mixed populations that are reactivated from latency (Sasadeusz & Sacks 1996; Griffiths & Coen 2003; Grey et al. 2003).
Similarly to HSV, ACV treatment for VZV infections does not generate ACVr viruses in immunocompetent hosts. However, in immunocompromised hosts, VZV infection tends to be severe and prolonged and ACVr mutants have been isolated after long-term treatment with ACV (Pahwa et al. 1988; Snoeck et al. 1994b; Talarico et al. 1993; Boivin et al. 1994). Resistance to ACV in VZV appears as a consequence of mutations either in TK or DNA polymerase genes. These are the most frequent TK mutants isolated both in cell culture and in the clinic (Gilbert et al. 2002). Mutations conferring resistance to nucleoside analogs have been found all along the VZV TK gene, although specific regions including ATP- and nucleoside-binding sites are recognized as mutagenic hot spots as well as amino acid 231 (Boivin et al. 1994; Morfin et al. 1999; Talarico et al. 1993). Resistances to PFA associated with mutations in the VZV DNA polymerase gene have also been described in immunocompromised patients (Visse et al. 1998; Visse et al. 1999). Amino acid substitutions in VZV DNA polymerase described in ACVr and PFAr mutants corresponded to changes described in the HSV DNA polymerase, although some mutants exhibited a discrepancy in their sensitivity to ACV or aphidicolin in comparison with corresponding HSV-1 mutants (Kamiyama et al. 2001; Visse et al. 1998; Visse et al. 1999). These findings indicate that identical or similar amino acid substitutions may create different conformations in HSV and VZV DNA polymerase that account for a discrepancy in drug susceptibility of the ACVr mutants (Kamiyama et al. 2001). In contrast, VZV mutants selected in vitro under pressure with PME derivatives harbored mutations in viral DNA not corresponding to those described in HSV. Interestingly, it appears that ACV and PCV select in vitro for different drug-resistant VZV genotypes: ACV selects for TK mutants, while PCV selects for DNA polymerase mutants (Andrei et al. 2004). Furthermore, alterations in the viral DNA polymerase that confer resistance to PCV were shown to be also responsible for cross-resistance to PFA (Andrei et al. 2004). Several reports have indicated that PCV remains active against some HSV-1 and VZV TK and DNA polymerase mutants that are resistant to ACV (Boyd et al. 1987; Pelosi et al. 1998a; Hasegawa et al. 1995; Andrei et al. 2004). These findings indicate that interactions between HSV or VZV TK and PCV or ACV, and likewise between viral DNA polymerases and triphosphates of PCV or ACV, are distinct and may account for the differences observed between ACVr and PCVr VZV strains. Furthermore, emergence frequency of resistant VZV mutants proved to be significantly higher following ACV exposure than following PCV exposure (Ida et al. 1999)
HCMV resistance to GCV may arise from mutations in either the UL97 (phosphotransferase) or UL54 (DNA polymerase) genes (Baldanti & Gerna 2003; Gilbert et al. 2002; Chou 1999; Chou & Meichsner 2000). In contrast to ACV, the selection of GCVr HCMV mutants requires a longer time in cell culture than for HSV. This is probably due to higher fidelity of HCMV DNA polymerase compared to HSV DNA polymerase (Sullivan & Coen 1991). Also different from HSV and VZV TK gene mutations, HCMV UL97 mutations have limited distribution in the gene: changes clustered in codons 460–520 (proposed ATP-binding site) or codons 590–607 (function in substrate recognition) (Wolf et al. 1995; Chou et al. 1995; Gilbert et al. 1998; Abraham et al. 1999). This is probably due to the fact that HCMV UL97 is very important for viral replication, presenting various UL97 mutants a fitness loss compared to wild-type strains. The HCMV pUL97 has been characterized as a protein which is autophosphorylated and is capable of phosphorylating GCV. The role of pUL97 protein kinase in HCMV replication and pathogenesis is still under investigation. Wolf and colleagues reported that UL97 kinase has an impact on at least two distinct phases of viral replication: DNA synthesis as well as capsid assembly and nuclear egress, indicating that protein phosphorylation mediated by this kinase increases efficiency of these two phases of virus replication (Wolf et al. 2001). The fact that HCMV UL97 protein kinase mutations associated with antiviral resistance are localized at specific codons simplifies identification of mutations by targeted PCR sequencing or restriction length polymorphism analysis (Scott et al. 2004; Chou 1999; Lurain et al. 2001). As PFA and CDV are independent of pUL97 protein kinase for their antiviral action, these two drugs are recommended for treatment of HCMV infections resistant to GCV due to alterations in UL97. It is worth mentioning that valganciclovir does not appear to select for an increased number of GCVr strains (Boivin et al. 2004).
A number of different mutations in the HCMV UL54 gene have been associated with resistance to GCV (Jabs et al. 1998; Smith et al. 1997). However, mutations in the UL54 gene are less common than in the UL97 gene. Most mutations conferring resistance to GCV cluster in specific regions of the HCMV DNA polymerase that are conserved among α-like DNA polymerases and show simultaneous cross-resistance to CDV (Baldanti & Gerna 2003; Gilbert et al. 2002; Chou 1999). Most of the GCVr/CDVr UL54 mutants retained sensitivity to PFA, while mutations in domains II, III, and IV of HCMV DNA polymerase responsible for PFA resistance do not show cross-resistance to GCV or CDV (Cihlar et al. 1998a; Cihlar et al. 1998b; Baldanti et al. 1996). However, multi-drug-resistant HCMV strains have been recently isolated in immunocompromised patients emphasizing the necessity of developing new treatment options. Furthermore, previously unrecognized mutations in the HCMV DNA polymerase gene continue to be isolated from patients receiving antiviral therapy (Weinberg et al. 2003; Scott et al. 2004). CDVr strains isolated in vitro were shown to be cross-resistant to GCV. Because CDV is not the first-line therapy for HCMV infections due to its renal toxicity, selection of CDVr strains is an extremely rare event and it appears to be mostly driven by long-term administration of GCV (Smith et al. 1997).
Similar to HCMV, resistance to GCV in HHV-6 has been mapped in pUL69 and DNA polymerase (encoded by the U38 gene). GCVr HHV-6 have been detected in clinical specimens and generated in the laboratory (Manichanh et al. 2001). Amino acid changes in the HHV-6 UL69 protein kinase, homologous to those in the HCMV UL97 phosphotransferase, were shown to cause resistance to GCV (Safronetz et al. 2003). Mutations in the UL38 gene associated with resistance to GCV and CDV were shown to be different from those conferring resistance to PFA (Manichanh et al. 2001; Bonnafous et al. 2007).
Amino acid substitutions in vaccinia virus DNA polymerase (encoded by the E9L gene) which are linked to CDV resistance have been recently reported (Andrei et al. 2006). These mutations are located within the 3ʹ–5ʹ exonuclease (A314T) and polymerase (A684V) catalytic domain. By marker transfer experiments it could be demonstrated that either mutation alone could confer a drug-resistant phenotype although degree of resistance was significantly lower than in virus encoding both mutations. A314T recombinant virus was shown to be associated with hypersensitivity to the pyrophosphate analogue phosphonoacetic acid (PAA). A684V appeared to increase resistance to PAA. Presence of both mutations resulted in no change in susceptibility to PAA. All CDVr viruses exhibited reduced virulence in mice, demonstrating that these E9L mutations are inextricably linked to reduced fitness in vivo. Interestingly, it was observed that treatment for 5 days with CDV still protected mice against a lethal intranasal challenge with drug-resistant virus bearing both mutations. Sequence analysis of adenovirus DNA polymerase of in vitro-selected CDVr strains allowed identification of amino acid changes associated with drug resistance (Kinchington et al. 2002). These substitutions are located in conserved regions of adenovirus DNA polymerase predicted to be involved in nucleotide binding.
Different reports based on crystallographic studies have provided details on how mutations in different viral DNA polymerases domains affect binding and catalysis (Liu et al. 2006; Zuccola et al. 2000; Loregian et al. 2004; Appleton et al. 2006; Huang et al. 1999; Shi et al. 2006; Tchesnokov et al. 2006).
Candidate Viral DNA Polymerase Inhibitors
In Table 22.2 are summarized the spectrum of antiviral activity and mechanism of action of nucleoside analogs, nucleotide analogs, and non-nucleoside analogs that target viral DNA polymerases which have been developed or are under development.


Structural formulae of candidate viral DNA polymerase inhibitors that are candidates for further development.
Nucleoside Analogs
Lobucavir
Lobucavir (LBV) is a cyclobutyl analog of guanine (Fig. 22.5) that has activity against most herpesviruses and also against HIV and HBV. LBV is a potent inhibitor of HCMV DNA polymerase in vitro. However, this nucleoside analogue is phosphorylated intracellularly to its triphosphate form both in infected and in uninfected cells, being the phosphorylated metabolite levels in HCMV-infected cells being only two- to three-fold higher compared to uninfected cells (Tenney et al. 1997). Due to lack of selective metabolism of LBV in virus-infected cells, the compound can be used as a substrate by host cell polymerases thus increasing toxicity and safety risks. Although promising results were obtained in early clinical trials against HCMV and HBV, development of LBV was halted due to safety concerns. Toxicologic studies in rodents showed an increase in number of different cancers with long-term administration of the drug.
H2G
H2G, (R)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine (omaciclovir), is an acyclic nucleoside analog (Fig. 22.5) that has shown potent activity against different herpesviruses, especially against VZV (Abele et al. 1988). This compound has a mode of action similar to that of ACV, but with less selectivity as a substrate for TK. Also, resistance to H2G has been mapped in TK (Ng et al. 2001). In contrast to ACV, H2G is not an obligate chain terminator, although incorporation of the triphosphate form (H2G-TP) results in limited chain elongation. Another difference with ACV-TP is the longer intracellular half-life of H2G-TP. A prodrug of H2G, MIV-606, the l-valine ester of H2G, with higher oral bioavailability has been synthesized and phase I/II clinical trails have been initiated with this compound.
A-5021
In a search for novel nucleoside analogs with antiherpesvirus activity, (1'S,2'R)-9-[[1',2'-bis(hydroxymethyl)cycloprop-1'-yl]methyl]-guanine (A-5021) (Fig. 22.5) emerged as a potent inhibitor of HSV-1, HSV-2, VZV, and HCMV (Iwayama et al. 1998). In vitro antiviral activity of this guanosine analog was higher than that of ACV or PCV against HSV-1 and VZV; however, activity against HSV-2 was comparable to that of the gold standards ACV and PCV (Iwayama et al. 1998). A-5021 also proved active against EBV and HHV-6 but not HHV-8 (De Clercq et al. 2001). The mechanism of action of this compound was shown to be similar to that of ACV and PCV (Ono et al. 1998). A-5021 is monophosphorylated by viral TK and to the di- and triphosphate forms by cellular kinases. A-5021-TP appeared to accumulate more than ACV-TP but less than PCV-TP in MRC-5 cells infected with HSV-1 or VZV, whereas HSV-2-infected MRC-5 cells had comparable levels of A-5021 and ACV triphosphates. The intracellular half-life of A-5021-TP was shown to be considerably longer than that of ACV-TP and shorter than that of PCV-TP. A-5021-TP is a competitive inhibitor of herpes DNA polymerases with respect to dGTP and it can be incorporated into DNA instead of dGTP and terminate elongation, although it permits limited chain extension. Thus, the strong antiviral activity of A-5021 appears to depend on a more rapid and stable accumulation of its triphosphate in infected cells; however, a disadvantage for the compound is its cross-resistance with ACV and PCV. In vivo efficacy of A-5021 against HSV-1 has been demonstrated in different animal models, its activity being superior to that of ACV in all HSV-1 models of infection (Iwayama et al. 1999). However, no advantage over PCV was seen against a model of systemic HSV-2 infection. The compound has entered clinical development, albeit transiently, and it remains to be demonstrated whether the stronger potency and prolonged antiviral activity compared to ACV observed in vitro and in animal models will be observed in the clinical studies.
S2242
The acyclic purine nucleoside analog, 2-amino-7-[(1,3-dihydroxy-2-propoxy) methyl]purine (S2242) (Fig. 22.5), the only known antivirally active acyclic nucleoside analogue with the side chain substituted at the N7 position of the purine ring, has been reported to possess potent activity against several herpesviruses including HSV, VZV, HCMV, EBV, HHV-6, HHV-7, and HHV-8 and also against poxviruses (Neyts & De Clercq 1997; Meerbach et al. 1998; Neyts et al. 1994; Zhang et al. 1999; De Clercq et al. 2001). Of special interest is the potent activity of the compound against HCMV and against TKdeficient mutants of HSV and VZV. Indeed, it was demonstrated that S2242 is not phosphorylated by either HSV TK or HCMV-encoded UL-97 kinase (Zimmermann et al. 1997). S2242 was found to be a substrate for deoxycytidine (dCK) kinase and for deoxyguanosine (dGK) kinase. S2242 was shown to be phosphorylated in a time- and concentration-dependent manner to its monophosphate, diphosphate, and triphosphate (Neyts et al. 1998). This nucleoside analog was not preferentially phosphorylated in HSV-1-, VZV-, or HHV-6-infected cells. In HCMV-infected human embryonic lung cells, a 5- to 25-fold increase in S2242 metabolite formation was observed compared with noninfected cells, suggesting that an HCMV-encoded or -induced enzyme causes specific phosphorylation of S2242.
S2242 proved to be more effective than ACV in different mouse models of HSV-1 infection and it was also far more effective than GCV in preventing or delaying murine cytomegalovirus-induced mortality in immunocompetent and severe combined immune deficiency (SCID) mice (Neyts et al. 1995; Neyts & De Clercq 2001). In addition, S2242 and its orally active diacetate ester prodrug (HOE961) were reported to be potent inhibitors of vaccinia virus and cowpox virus replication in cell culture and in infected mice (Smee et al. 2002). However, development of the compound was stopped due to safety concerns.
BCNAs
In 1999, some unusual bicyclic nucleoside analogues (BCNAs) with significant and selective anti-VZV activity were reported (McGuigan et al. 1999). The early drug leads had a long alkyl side chain on the bicyclic base part, with optimum length of ca. C8–C10. In vitro potencies of these compounds against VZV were ca. 300-fold more potent than the clinically established anti-herpetic agent ACV. Replacement of the alkyl chain by a p-alkylphenyl unit led to a significant boost in potency against VZV (McGuigan & Balzarini 2006). The most potent analogue was the p-pentylphenyl BCNA analogue Cf1743 (Fig. 22.5), which exhibited activity against a broad range of VZV clinical isolates at subnanomolar concentrations. After extensive studies of structure–activity relationship and pharmacology of the BCNAs, Cf1743 was identified as a potential clinical candidate. Pharmocokinetic studies indicated the need to improve its bioavailability. The HCl salt of the 5’-valyl ester of Cf1743 (i.e., FV-100 designed in analogy with valaciclovir with respect to acyclovir) emerged as the most promising prodrug (McGuigan et al. 2007). Phase 1 studies have been planned. Unlike ACV, which is a broad-spectrum anti-herpetic agent, the BCNAs are highly specific for VZV with no significant activity against any other virus, including other members of the herpesvirus family and the closely related simian varicella virus (SVV).
Although the precise mechanism of action of these compounds remains to be elucidated, it is clear that for their antiviral activity they depend on phosphorylation by the VZV-encoded thymidine kinase, since Cf1743 completely lost its anti-VZV efficacy against both laboratory and clinical virus isolates with a deficient TK (Andrei et al. 2005b). Furthermore, mutant viruses emerging under selective pressure with the BCNAs present amino acid changes in the viral TK. The BCNAs owe at least part of their antiviral selectivity to a specific activation/phosphorylation by the VZV-encoded TK and associated thymidylate kinase (dTMP-K) activity, while not being recognized by the closely related HSV-1-encoded TK/dTMP-K enzyme (Sienaert et al. 2002). Strikingly, there was no close correlation between affinity for VZV TK and antiviral activity, pointing to a different structure–activity relationship for the eventual antiviral target for the BCNAs. It should be noted that the closely related SVV TK is also able to recognize the BCNAs as a substrate, but SVV replication is not affected by the BCNAs (Sienaert et al. 2004). Notably, this class of compounds is not recognized by cellular kinases that participate in the anabolism of other pyrimidine analogs. Among the cellular kinases that do not recognize the BCNAs as substrate is the nucleoside diphosphate (NDK) kinase, which converts BVDU-DP to the active triphosphate form (Sienaert et al. 2003). Consequently, no 5ʹ-triphosphate of BCNAs could be detected in VZV-infected cells. Further studies are highly warranted to decipher the mode of action of the BCNAs and to determine the effects of the BCNAs anabolites on the VZV DNA polymerase.
BCNAs are not susceptible to degradation by human or bacterial thymidine phosphorylase, and thus are not cleaved to their free (inactive) base (Balzarini et al. 2002). Also, the latter is not inhibitory to dihydropyrimidine dehydrogenase, an enzyme involved in the degradation of thymine, uracil, and the anticancer agent 5-fluorouracil. This means that BCNAs do not interfere with degradation of 5-fluorouracil and, in contrast to BVDU, they could eventually be used in therapy of VZV infections in patients under 5-fluorouracil treatment. It is evident that BCNAs represent highly promising anti-VZV compounds that are not susceptible to breakdown by nucleoside/nucleobase catabolic enzymes and are not expected to interfere with cellular catabolic processes such as those involved in 5-fluorouracil catabolism.
Nucleotide Analogs
Cidofovir Esters
The clinical use of cidofovir is limited by its poor oral bioavailability and renal toxicity. To overcome these restrictions, Hostetler’s group has synthesized alkoxyalkyl esters of CDV and its cyclic form, i.e., cCDV (Beadle et al. 2002). In these prodrugs, a fatty acid been linked to the parent molecule to facilitate drug absorption in the gastrointestinal tract. These alkoxyalkyl esters of CDV and cCDV were much more active in vitro than the parent compounds against several herpesviruses, including HSV, VZV, CMV, EBV, HHV-6, and HHV-8 and poxviruses. A 2.5- to 4-log increase in antiviral activity against HCMV replication in vitro was observed. In addition, these derivatives showed improved uptake and absorption and had oral bioavailabilities in mice of 88–97%, compared to less than 5% for CDV. Studies with radiolabeled compound confirmed increased cell penetration (10 to 20-fold) and higher intracellular levels (100-fold) of CDV-PP (the active form of the compound) than those measured following treatment of the cells with CDV (Aldern et al. 2003). In vivo, oral administration of hexadecyloxypropyl-CDV (HDP-CDV) (Fig. 22.5) proved as effective as parental CDV in treatment of herpes- and poxvirus infection in several mouse models (Kern et al. 2004; Bidanset et al. 2004; Wan et al. 2005; Keith et al. 2004; Keith et al. 2004; Quenelle et al. 2004). Importantly, diminished accumulation of the drug in the kidney was reported according to studies evaluating tissue distribution of radiolabeled HDP-CDV and other alkoxyalkyl esters of CDV in mice (Ciesla et al. 2003; Kern et al. 2004).
HDP-CDV (CMX001) in an oral formulation is presently under development by Chimerix. A Phase I clinical study to evaluate the safety and pharmacokinetics of orally administered CMX001 in healthy volunteers has been announced (Painter & Hostetler, 2004). CMX001 will be developed for potential use in treatment of smallpox or vaccine-related side effects. This drug could also provide a safer therapy for ACV- and GCV-resistant herpesviruses in the immunocompromised host.
In addition to herpes- and poxviruses, increased activity of alkoxyalkyl esters of CDV compared to parent compound CDV was also shown against adenovirus, polyomavirus, and papillomavirus (Hartline et al. 2005; Hostetler et al., 2006). A similar prodrug strategy was applied to other ANPs, such as HPMPA, the enhancement of antiviral potency being similar to that of CDV (Ruiz et al. 2007; Lebeau et al. 2006; Choo et al. 2007; Dal Pozzo et al. 2007; Ruiz et al. 2006; Beadle et al. 2006).
New Generations of Acyclic Nucleoside Phosphonates
Following the success of the first ANPs, two new generations of ANPs have been recently described. The “second generation” ANPs include the “open ring” or “O-linked” ANP analogues or 6-[2-phosphonomethoxyalkoxy]-2,4-diaminopyrimidines (DAPys), which showed substantial potential for the treatment of a broad range of DNA virus and retrovirus infections (Balzarini et al. 2004; Hockova et al. 2003; Hockova et al. 2004; De Clercq et al. 2005). HPMPO-DAPy (Fig. 22.5) offers an activity similar to that of CDV except for HCMV that is poorly inhibited, while PMEO-DAPy (Fig. 22.5) displays an analogous activity spectrum as that of PMEA. The “third generation” of ANPs encompasses HPMP derivatives with a 5-azacytosine moiety such as HPMP-5-azaC (Fig. 22.3) and its cyclic form (i.e., cHPMP-5-azaC) (Krecmerova et al. 2007a). These compounds were at least as potent as CDV against several DNA viruses, including HCMV. Furthermore, CDV and HPMP-5-azaC proved equally potent in pathogenic models of HSV and poxvirus infections in mice. Among several prodrugs of cHPMP-5-azaC synthesized, the hexadecyloxyethyl ester proved to be about 250-fold more active than the parent compound (Krecmerova et al. 2007b). Further studies are needed to determine the clinical potential of these compounds.
Non-nucleoside Analogs
The use of an in vitro HCMV DNA polymerase assay in high-throughput screening allowed identification of a novel class of non-nucleoside herpesvirus polymerase inhibitors, the naphthalene–carboxamides, PNU-26370 being the lead compound of this series of non-nucleoside DNA polymerase inhibitors (Vaillancourt et al. 2000; Tucker et al. 2000). Structure–activity relationship (SAR) studies demonstrated that a quinoline ring could be substituted for naphthalene, leading to the discovery of the 4-oxo-dihydroquinoline-3-carboxamides (4-oxo-DHQ), represented by PNU-181128, PNU-181465, and PNU-183792 (Fig. 22.5), that demonstrated inhibition of HCMV, HSV, and VZV polymerases (Brideau et al. 2002; Oien et al. 2002). High specificity for viral DNA polymerases compared to human alpha (α), gamma (γ), delta (δ) polymerases was observed. PNU-183792 displays broad spectrum of activity in cell culture against different herpesviruses, including HSV-1, HSV-2, VZV, SVV, HCMV, murine and rat cytomegaloviruses, EBV, and HHV-8 (Kaposi-associated herpesvirus), exceptions being HHV-6 and HHV-7 (Wathen 2002). PNU-183792 was inactive against unrelated DNA or RNA viruses indicating specificity for herpesviruses. A strong correlation between inhibition of the viral DNA polymerases and antiviral activity for this class of compounds supports inhibition of the viral DNA polymerase as the mechanism of antiviral activity. The 4-oxo-DHQs were found to be competitive inhibitors of nucleoside binding; however, no cross-resistance could be detected with GCV-resistant HCMV or ACV-resistant HSV mutants. In vitro antiviral activity of the 4-oxo-DHQs was comparable or superior to existing antiherpesvirus drugs and drug resistance to these compounds correlated with point mutations in conserved domain III of the HCMV DNA polymerase (V823A + V824L) (Thomsen et al. 2003). V823 is conserved in the DNA polymerases of human herpesviruses, except for HHV-6 and HHV-7 that contain an alanine at this position. Mutations associated with resistance to 4-oxo-DHQs did not confer resistance to existing anti-herpesvirus nucleoside analogs. Based on the crystal structure of the HSV DNA polymerase, Liu et al. proposed a HSV DNA polymerase model, suggesting that the 4-oxo-DHQs bind at the polymerase active site interacting non-covalently with the DNA duplex–DNA polymerase complex and not with enzyme or DNA duplex alone (Liu et al. 2006). PNU-183792 is orally bioavailable, and activity was demonstrated in a model of lethal murine cytomegalovirus (MCMV) infection (Brideau et al. 2002). So far, these non-nucleoside DNA polymerase inhibitors have not been evaluated in any clinical trials.
Further SAR studies led to the discovery of 4-oxo-4,7-dihydrothienopyridines (DHTPs) (Schnute et al. 2005; Schnute et al. 2007) and 7-oxo-4,7-dihydrothieno [3,2-b]pyridine-6-carboxamides (Larsen et al. 2007). Some of these compounds demonstrated broad-spectrum inhibition of the herpesvirus polymerases HCMV, HSV-1, EBV, and VZV with high specificity compared to human DNA polymerases. DHTPs, in contrast to the kinetics determined for the 4-oxo-DHQs, proved to be competitive inhibitors of dTTP incorporation into primer template by HCMV DNA polymerase (Schnute et al. 2005).
Inhibitors of Protein–Protein Interactions
Protein–protein interactions among proteins implicated in herpesvirus DNA replication are essential for viral genome replication and are considered as attractive potential drug targets (Coen & Schaffer 2003). Similar to other herpesviruses, HCMV DNA polymerase contains a catalytic subunit (UL54) and an accessory protein (UL44) that is thought to increase the processivity of the enzyme. Loregian and collaborators (Loregian et al. 2003) have identified peptides from the C terminus of UL54 which could efficiently disrupt the physical interaction between UL54 and UL44 and specifically inhibit the stimulation of UL54 by UL44. These findings provide the basis for developing new classes of anti-HCMV inhibitors that act by disrupting the UL54/UL44 interaction. Indeed, small molecules that disrupt the in vitro interaction between the HSV DNA polymerase (UL30) and the accessory protein (UL42) and exhibit anti-HSV activity have been identified (Coen & Schaffer 2003). Recently, mutations that decrease DNA binding of the processivity factor of HSV DNA polymerase were shown to reduce viral yield, to alter kinetics of viral replication, and to decrease fidelity of DNA replication (Jiang et al. 2007b; Jiang et al. 2007a).
Small Interfering RNAs
RNA interference (RNAi) is a natural mechanism of post-transcriptional gene silencing, widely conserved in multicellular organisms. This pathway is thought to be an ancient mechanism for protecting the host and its genome against viruses and transposable genetic elements (Hannon 2002). The molecular mediators of RNAi are double-stranded RNAs of 21–23 nucleotides in length that induce the sequence-specific degradation of homologous RNAs. RNAi has been used as a means to manipulate gene expression experimentally and to probe gene function. It has also been proposed that this biological response might be exploited therapeutically as an antiviral defense mechanism. The siRNA approaches have been shown to be effective against a variety of viruses in cell culture (Leonard & Schaffer 2006; Silva et al. 2002; Dykxhoorn & Lieberman 2006). Recently, it has been described that synthetic siRNA against essential gene products of HCMV such as UL54 (DNA polymerase) and UL97 (protein kinase) can trigger RNAi in infected cells leading to effective inhibition of viral replication (Wiebusch et al. 2004; Shin et al. 2006). These results demonstrated the effectiveness of siRNAs against experimental HCMV infection and open new possibilities for antiviral strategies.
References
Abele, G., B. Eriksson, J. Harmenberg & B. Wahren, 1988. Inhibition of varicella-zoster virus-induced DNA polymerase by a new guanosine analog, 9-[4-hydroxy-2-(hydroxymethyl) butyl]guanine triphosphate, Antimicrob. Agents Chemother. 32: 1137–1142.
Abraham, B., S. Lastere, J. Reynes, F. Bibollet-Ruche, N. Vidal & M. Segondy, 1999. Ganciclovir resistance and UL97 gene mutations in cytomegalovirus blood isolates from patients with AIDS treated with ganciclovir, J. Clin. Virol. 13: 141–148.
Aldern, K. A., S. L. Ciesla, K. L. Winegarden & K. Y. Hostetler, 2003. Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism, Mol. Pharmacol. 63: 678–681.
Andrei, G., J. Balzarini, P. Fiten, E. De Clercq, G. Opdenakker & R. Snoeck, 2005a. Characterization of herpes simplex virus type 1 thymidine kinase mutants selected under a single round of high-dose brivudin, J. Virol. 79: 5863–5869.
Andrei, G., E. De Clercq & R. Snoeck, 2004. In vitro selection of drug-resistant varicella-zoster virus (VZV) mutants (OKA strain): differences between acyclovir and penciclovir ? Antiviral Res. 61: 181–187.
Andrei, G., P. Fiten, M. Froeyen, E. De Clercq, G. Opdenakker & R. Snoeck, 2007a. DNA polymerase mutations in drug-resistant herpes simplex virus mutants determine in vivo neurovirulence and drug-enzyme interactions, Antiviral. Ther. 12: 719–732.
Andrei, G., P. Fiten, P. Goubau, H. van Landuyt, B. Gordts, D. Selleslag, E. De Clercq, G. Opdenakker & R. Snoeck, 2007b. Dual infection with polyomavirus BK and acyclovir-resistant herpes simplex virus successfully treated with cidofovir in a bone marrow transplant recipient, Transpl. Infect. Dis. 9: 126–131.
Andrei, G., D. B. Gammon, P. Fiten, E. De Clercq, G. Opdenakker, R. Snoeck & D. H. Evans, 2006. Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice, J. Virol. 80: 9391–9401.
Andrei, G., R. Sienaert, C. McGuigan, E. De Clercq, J. Balzarini & R. Snoeck, 2005b. Susceptibilities of several clinical varicella-zoster virus (VZV) isolates and drug-resistant VZV strains to bicyclic furano pyrimidine nucleosides, Antimicrob. Agents Chemother. 49: 1081–1086.
Andrei, G., R. Snoeck, E. De Clercq, R. Esnouf, P. Fiten & G. Opdenakker, 2000. Resistance of herpes simplex virus type 1 against different phosphonylmethoxyalkyl derivatives of purines and pyrimidines due to specific mutations in the viral DNA polymerase gene, J. Gen. Virol. 81: 639–648.
Ansari, A.& V. C. Emery, 1999. The U69 gene of human herpesvirus 6 encodes a protein kinase which can confer ganciclovir sensitivity to baculoviruses, J. Virol. 73: 3284–3291.
Appleton, B. A., J. Brooks, A. Loregian, D. J. Filman, D. M. Coen & J. M. Hogle, 2006. Crystal structure of the cytomegalovirus DNA polymerase subunit UL44 in complex with the C terminus from the catalytic subunit. Differences in structure and function relative to unliganded UL44, J. Biol. Chem. 281: 5224–5232.
Bacon, T. H., M. J. Levin, J. J. Leary, R. T. Sarisky & D. Sutton, 2003. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy, Clin. Microbiol. Rev. 16: 114–128.
Baldanti, F. & G. Gerna, 2003. Human cytomegalovirus resistance to antiviral drugs: diagnosis, monitoring and clinical impact, J. Antimicrob. Chemother. 52: 324–330.
Baldanti, F., M. R. Underwood, S. C. Stanat, K. K. Biron, S. Chou, A. Sarasini, E. Silini & G. Gerna, 1996. Single amino acid changes in the DNA polymerase confer foscarnet resistance and slow-growth phenotype, while mutations in the UL97-encoded phosphotransferase confer ganciclovir resistance in three double-resistant human cytomegalovirus strains recovered from patients with AIDS, J. Virol. 70: 1390–1395.
Balzarini, J., C. Pannecouque, L. Naesens, G. Andrei, R. Snoeck, E. De Clercq, D. Hockova & A. Holy, 2004. 6-[2-phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines: a new class of acyclic pyrimidine nucleoside phosphonates with antiviral activity, Nucleosides Nucleotides Nucleic Acids 23: 1321–1327.
Balzarini, J., R. Sienaert, S. Liekens, A. Van Kuilenburg, A. Carangio, R. Esnouf, E. De Clercq & C. McGuigan, 2002. Lack of susceptibility of bicyclic nucleoside analogs, highly potent inhibitors of varicella-zoster virus, to the catabolic action of thymidine phosphorylase and dihydropyrimidine dehydrogenase, Mol. Pharmacol. 61: 1140–1145.
Beadle, J. R., C. Hartline, K. A. Aldern, N. Rodriguez, E. Harden, E. R. Kern & K. Y. Hostetler, 2002. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro, Antimicrob. Agents Chemother. 46: 2381–2386.
Beadle, J. R., W. B. Wan, S. L. Ciesla, K. A. Keith, C. Hartline, E. R. Kern & K. Y. Hostetler, 2006. Synthesis and antiviral evaluation of alkoxyalkyl derivatives of 9-(S)-(3-hydroxy-2-phosphonomethoxypropyl)adenine against cytomegalovirus and orthopoxviruses, J. Med. Chem. 49: 2010–2015.
Bestman-Smith, J.& G. Boivin, 2002. Herpes simplex virus isolates with reduced adefovir susceptibility selected in vivo by foscarnet therapy, J. Med. Virol. 67: 88–91.
Beutner, K. R., 1995. Valacyclovir: a review of its antiviral activity, pharmacokinetic properties, and clinical efficacy, Antiviral Res. 28: 281–290.
Beutner, K. R., D. J. Friedman, C. Forszpaniak, P. L. Andersen & M. J. Wood, 1995. Valacyclovir compared with acyclovir for improved therapy for herpes zoster in immunocompetent adults, Antimicrob. Agents Chemother. 39: 1546–1553.
Bidanset, D. J., J. R. Beadle, W. B. Wan, K. Y. Hostetler & E. R. Kern, 2004. Oral activity of ether lipid ester prodrugs of cidofovir against experimental human cytomegalovirus infection, J. Infect. Dis. 190: 499–503.
Biron, K. K., 2006. Antiviral drugs for cytomegalovirus diseases, Antiviral Res. 71: 154–163.
Biron, K. K., S. C. Stanat, J. B. Sorrell, J. A. Fyfe, P. M. Keller, C. U. Lambe & D. J. Nelson, 1985. Metabolic activation of the nucleoside analog 9-[(2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine in human diploid fibroblasts infected with human cytomegalovirus, Proc. Natl. Acad. Sci. U. S. A 82: 2473–2477.
Boehmer, P. E. & I. R. Lehman, 1997. Herpes simplex virus DNA replication, Annu. Rev. Biochem. 66: 347–384.
Boivin, G., C. K. Edelman, L. Pedneault, C. L. Talarico, K. K. Biron & H. H. Balfour, Jr., 1994. Phenotypic and genotypic characterization of acyclovir-resistant varicella-zoster viruses isolated from persons with AIDS, J. Infect. Dis. 170: 68–75.
Boivin, G., N. Goyette, C. Gilbert, N. Roberts, K. Macey, C. Paya, M. D. Pescovitz, A. Humar, E. Dominguez, K. Washburn, E. Blumberg, B. Alexander, R. Freeman, N. Heaton & E. Covington, 2004. Absence of cytomegalovirus-resistance mutations after valganciclovir prophylaxis, in a prospective multicenter study of solid-organ transplant recipients, J. Infect. Dis. 189: 1615–1618.
Bonnafous, P., L. Naesens, S. Petrella, A. Gautheret-Dejean, D. Boutolleau, W. Sougakoff & H. Agut, 2007. Different mutations in the HHV-6 DNA polymerase gene accounting for resistance to foscarnet, Antivir. Ther. 12: 877–888.
Boyd, M. R., T. H. Bacon, D. Sutton & M. Cole, 1987. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL 39123) in cell culture, Antimicrob. Agents Chemother. 31: 1238–1242.
Brideau, R. J., M. L. Knechtel, A. Huang, V. A. Vaillancourt, E. E. Vera, N. L. Oien, T. A. Hopkins, J. L. Wieber, K. F. Wilkinson, B. D. Rush, F. J. Schwende & M. W. Wathen, 2002. Broad-spectrum antiviral activity of PNU-183792, a 4-oxo-dihydroquinoline, against human and animal herpesviruses, Antiviral Res. 54: 19–28.
Chakrabarti, S., D. Pillay, D. Ratcliffe, P. A. Cane, K. E. Collingham & D. W. Milligan, 2000. Resistance to antiviral drugs in herpes simplex virus infections among allogeneic stem cell transplant recipients: risk factors and prognostic significance, J. Infect. Dis. 181: 2055–2058.
Chen, Y., C. Scieux, V. Garrait, G. Socie, V. Rocha, J. M. Molina, D. Thouvenot, F. Morfin, L. Hocqueloux, L. Garderet, H. Esperou, F. Selimi, A. Devergie, G. Leleu, M. Aymard, F. Morinet, E. Gluckman & P. Ribaud, 2000. Resistant herpes simplex virus type 1 infection: an emerging concern after allogeneic stem cell transplantation, Clin. Infect. Dis. 31: 927–935.
Chibo, D., J. Druce, J. Sasadeusz & C. Birch, 2004. Molecular analysis of clinical isolates of acyclovir resistant herpes simplex virus, Antiviral Res. 61: 83–91.
Choo, H., J. R. Beadle, Y. Chong, J. Trahan & K. Y. Hostetler, 2007. Synthesis of the 5-phosphono-pent-2-en-1-yl nucleosides: a new class of antiviral acyclic nucleoside phosphonates, Bioorg. Med. Chem. 15: 1771–1779.
Chou, S., 1999. Antiviral drug resistance in human cytomegalovirus, Transpl. Infect. Dis. 1: 105–114.
Chou, S., S. Guentzel, K. R. Michels, R. C. Miner & W. L. Drew, 1995. Frequency of UL97 phosphotransferase mutations related to ganciclovir resistance in clinical cytomegalovirus isolates, J. Infect. Dis. 172: 239–242.
Chou, S.& C. L. Meichsner, 2000. A nine-codon deletion mutation in the cytomegalovirus UL97 phosphotransferase gene confers resistance to ganciclovir, Antimicrob. Agents Chemother. 44: 183–185.
Chrisp, C. E., J. C. Sunstrum, D. R. Averill, Jr., M. Levine & J. C. Glorioso, 1989. Characterization of encephalitis in adult mice induced by intracerebral inoculation of herpes simplex virus type 1 (KOS) and comparison with mutants showing decreased virulence, Lab Invest 60: 822–830.
Christophers, J., J. Clayton, J. Craske, R. Ward, P. Collins, M. Trowbridge & G. Darby, 1998. Survey of resistance of herpes simplex virus to acyclovir in northwest England, Antimicrob. Agents Chemother. 42: 868–872.
Ciesla, S. L., J. Trahan, W. B. Wan, J. R. Beadle, K. A. Aldern, G. R. Painter & K. Y. Hostetler, 2003. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney, Antiviral Res. 59: 163–171.
Cihlar, T., M. D. Fuller & J. M. Cherrington, 1998a. Characterization of drug resistance-associated mutations in the human cytomegalovirus DNA polymerase gene by using recombinant mutant viruses generated from overlapping DNA fragments, J. Virol. 72: 5927–5936.
Cihlar, T., M. D. Fuller, A. S. Mulato & J. M. Cherrington, 1998b. A point mutation in the human cytomegalovirus DNA polymerase gene selected in vitro by cidofovir confers a slow replication phenotype in cell culture, Virology 248: 382–393.
Coen, D. M., 1991. The implications of resistance to antiviral agents for herpesvirus drug targets and drug therapy, Antiviral Res. 15: 287–300.
Coen, D. M., 1994. Acyclovir-resistant, pathogenic herpesviruses, Trends Microbiol. 2: 481–485.
Coen, D. M., M. Kosz-Vnenchak, J. G. Jacobson, D. A. Leib, C. L. Bogard, P. A. Schaffer, K. L. Tyler & D. M. Knipe, 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate, Proc. Natl. Acad. Sci. U. S. A 86: 4736–4740.
Coen, D. M.& P. A. Schaffer, 2003. Antiherpesvirus drugs: a promising spectrum of new drugs and drug targets, Nat. Rev. Drug Discov. 2: 278–288.
Coen, D. M., P. A. Schaffer, P. A. Furman, P. M. Keller & M. H. St Clair, 1982. Biochemical and genetic analysis of acyclovir-resistant mutants of herpes simplex virus type 1, Am. J. Med. 73: 351–360.
Collins, P., B. A. Larder, N. M. Oliver, S. Kemp, I. W. Smith & G. Darby, 1989. Characterization of a DNA polymerase mutant of herpes simplex virus from a severely immunocompromised patient receiving acyclovir, J. Gen. Virol. 70 (Pt 2): 375–382.
Corey, L., A. Wald, R. Patel, S. L. Sacks, S. K. Tyring, T. Warren, J. M. Douglas, Jr., J. Paavonen, R. A. Morrow, K. R. Beutner, L. S. Stratchounsky, G. Mertz, O. N. Keene, H. A. Watson, D. Tait & M. Vargas-Cortes, 2004. Once-daily valacyclovir to reduce the risk of transmission of genital herpes, N. Engl. J. Med. 350: 11–20.
Crumpacker, C. S., 1992. Mechanism of action of foscarnet against viral polymerases, Am. J. Med. 92: 3S-7S.
Dal Pozzo, F., G. Andrei, I. Lebeau, J. R. Beadle, K. Y. Hostetler, E. De Clercq & R. Snoeck, 2007. In vitro evaluation of the anti-orf virus activity of alkoxyalkyl esters of CDV, cCDV and (S)-HPMPA, Antiviral Res. 75: 52–57.
Danve-Szatanek, C., M. Aymard, D. Thouvenot, F. Morfin, G. Agius, I. Bertin, S. Billaudel, B. Chanzy, M. Coste-Burel, L. Finkielsztejn, H. Fleury, T. Hadou, C. Henquell, H. Lafeuille, M. E. Lafon, A. Le Faou, M. C. Legrand, L. Maille, C. Mengelle, P. Morand, F. Morinet, E. Nicand, S. Omar, B. Picard, B. Pozzetto, J. Puel, D. Raoult, C. Scieux, M. Segondy, J. M. Seigneurin, R. Teyssou & C. Zandotti, 2004. Surveillance network for herpes simplex virus resistance to antiviral drugs: 3-year follow-up, J. Clin. Microbiol. 42: 242–249.
Darby, G., 1994. Acyclovir–and beyond, J. Int. Med. Res. 22 Suppl 1: 33A–42A.
Darby, G., M. J. Churcher & B. A. Larder, 1984. Cooperative effects between two acyclovir resistance loci in herpes simplex virus, J. Virol. 50: 838–846.
Darby, G., H. J. Field & S. A. Salisbury, 1981. Altered substrate specificity of herpes simplex virus thymidine kinase confers acyclovir-resistance, Nature 289: 81–83.
De Bolle, L., D. Michel, T. Mertens, C. Manichanh, H. Agut, E. De Clercq & L. Naesens, 2002. Role of the human herpesvirus 6 u69-encoded kinase in the phosphorylation of ganciclovir, Mol. Pharmacol. 62: 714–721.
De Clercq, E., 2003. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections, Clin. Microbiol. Rev. 16: 569–596.
De Clercq, E., 2004. Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster, Biochem. Pharmacol. 68: 2301–2315.
De Clercq, E., J. Descamps, P. De Somer & A. Holý, 1978. (S)-9-(2,3-Dihydroxypropyl)adenine: an aliphatic nucleoside analog with broad spectrum antiviral activity. Science 200: 563–565.
De Clercq, E., G. Andrei, J. Balzarini, P. Leyssen, L. Naesens, J. Neyts, C. Pannecouque, R. Snoeck, C. Ying, D. Hockova & A. Holý, 2005. Antiviral potential of a new generation of acyclic nucleoside phosphonates, the 6-[2-(phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines, Nucleosides Nucleotides Nucleic Acids 24: 331–341.
De Clercq, E., J. Descamps, P. de Somer, P. J. Barr, A. S. Jones & R. T. Walker, 1979. (E)-5-(2-Bromovinyl)-2'-deoxyuridine: a potent and selective anti-herpes agent, Proc. Natl. Acad. Sci. U. S. A 76: 2947–2951.
De Clercq, E.& H. J. Field, 2006. Antiviral prodrugs - the development of successful prodrug strategies for antiviral chemotherapy, Br. J. Pharmacol. 147: 1–11.
De Clercq, E.& A. Holý, 2005. Acyclic nucleoside phosphonates: a key class of antiviral drugs, Nat. Rev. Drug Discov. 4: 928–940.
De Clercq, E., A. Holy, I. Rosenberg, T. Sakuma, J. Balzarini & P. C. Maudgal, 1986. A novel selective broad-spectrum anti-DNA virus agent, Nature 323: 464–467.
De Clercq, E., L. Naesens, L. De Bolle, D. Schols, Y. Zhang & J. Neyts, 2001. Antiviral agents active against human herpesviruses HHV-6, HHV-7 and HHV-8, Rev. Med. Virol. 11: 381–395.
DeFilippis, V., C. Raggo, A. Moses & K. Fruh, 2003. Functional genomics in virology and antiviral drug discovery, Trends Biotechnol. 21: 452–457.
DeJesus, E., A. Wald, T. Warren, T. W. Schacker, S. Trottier, M. Shahmanesh, J. L. Hill & C. A. Brennan, 2003. Valacyclovir for the suppression of recurrent genital herpes in human immunodeficiency virus-infected subjects, J. Infect. Dis. 188: 1009–1016.
Derse, D., Y. C. Cheng, P. A. Furman, M. H. St Clair & G. B. Elion, 1981. Inhibition of purified human and herpes simplex virus-induced DNA polymerases by 9-(2-hydroxyethoxymethyl)guanine triphosphate. Effects on primer-template function, J. Biol. Chem. 256: 11447–11451.
Desgranges, C., G. Razaka, E. De Clercq, P. Herdewijn, J. Balzarini, F. Drouillet & H. Bricaud, 1986. Effect of (E)-5-(2-bromovinyl)uracil on the catabolism and antitumor activity of 5-fluorouracil in rats and leukemic mice, Cancer Res. 46: 1094–1101.
Digard, P., W. R. Bebrin, K. Weisshart & D. M. Coen, 1993. The extreme C terminus of herpes simplex virus DNA polymerase is crucial for functional interaction with processivity factor UL42 and for viral replication, J. Virol. 67: 398–406.
Docherty, J. J., A. T. Dobson, J. J. Trimble & B. A. Jennings, 1991. Herpes simplex virus type 1 that exhibits herpes simplex virus type 2 sensitivity to (E)-5-(2-bromovinyl)-2'-deoxyuridine, Intervirology 32: 308–315.
Dykxhoorn, D. M.& J. Lieberman, 2006. Silencing viral infection, PLoS. Med. 3: e242.
Efstathiou, S., S. Kemp, G. Darby & A. C. Minson, 1989. The role of herpes simplex virus type 1 thymidine kinase in pathogenesis, J. Gen. Virol. 70 (Pt 4): 869–879.
Elion, G. B., P. A. Furman, J. A. Fyfe, P. de Miranda, L. Beauchamp & H. J. Schaeffer, 1977. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine, Proc. Natl. Acad. Sci. U. S. A 74: 5716–5720.
Englund, J. A., M. E. Zimmerman, E. M. Swierkosz, J. L. Goodman, D. R. Scholl & H. H. Balfour, Jr., 1990. Herpes simplex virus resistant to acyclovir. A study in a tertiary care center, Ann. Intern. Med. 112: 416–422.
Eriksson, B., B. Oberg & B. Wahren, 1982. Pyrophosphate analogues as inhibitors of DNA polymerases of cytomegalovirus, herpes simplex virus and cellular origin, Biochim. Biophys. Acta 696: 115–123.
Field, H. J.& D. M. Coen, 1986. Pathogenicity of herpes simplex virus mutants containing drug resistance mutations in the viral DNA polymerase gene, J. Virol. 60: 286–289.
Field, H. J. & G. Darby, 1980. Pathogenicity in mice of strains of herpes simplex virus which are resistant to acyclovir in vitro and in vivo, Antimicrob. Agents Chemother. 17:209–216.
Field, H. J.& P. Wildy, 1978. The pathogenicity of thymidine kinase-deficient mutants of herpes simplex virus in mice, J. Hyg. (Lond) 81: 267–277.
Furman, P. A., P. de Miranda, M. H. St Clair & G. B. Elion, 1981. Metabolism of acyclovir in virus-infected and uninfected cells, Antimicrob. Agents Chemother. 20: 518–524.
Furman, P. A., M. H. St Clair, J. A. Fyfe, J. L. Rideout, P. M. Keller & G. B. Elion, 1979. Inhibition of herpes simplex virus-induced DNA polymerase activity and viral DNA replication by 9-(2-hydroxyethoxymethyl) guanine and its triphosphate, J. Virol. 32: 72–77.
Furman, P. A., M. H. St Clair & T. Spector, 1984. Acyclovir triphosphate is a suicide inactivator of the herpes simplex virus DNA polymerase, J. Biol. Chem. 259: 9575–9579.
Fyfe, J. A., 1982. Differential phosphorylation of (E)-5-(2-bromovinyl)-2'-deoxyuridine mono- phosphate by thymidylate kinases from herpes simplex viruses types 1 and 2 and varicella zoster virus, Mol. Pharmacol. 21: 432–437.
Fyfe, J. A., P. M. Keller, P. A. Furman, R. L. Miller & G. B. Elion, 1978. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine, J. Biol. Chem. 253: 8721–8727.
Gaudreau, A., E. Hill, H. H. Balfour, Jr., A. Erice & G. Boivin, 1998. Phenotypic and genotypic characterization of acyclovir-resistant herpes simplex viruses from immunocompromised patients, J. Infect. Dis. 178: 297–303.
Gilbert, C., J. Bestman-Smith & G. Boivin, 2002. Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms, Drug Resist. Updat. 5: 88–114.
Gilbert, C., J. Handfield, E. Toma, R. Lalonde, M. G. Bergeron & G. Boivin, 1998. Emergence and prevalence of cytomegalovirus UL97 mutations associated with ganciclovir resistance in AIDS patients, AIDS 12: 125–129.
Grey, F., M. Sowa, P. Collins, R. J. Fenton, W. Harris, W. Snowden, S. Efstathiou & G. Darby, 2003. Characterization of a neurovirulent acyclovir-resistant variant of herpes simplex virus, J. Gen. Virol. 84: 1403–1410.
Griffiths, A., S. H. Chen, B. C. Horsburgh & D. M. Coen, 2003. Translational compensation of a frameshift mutation affecting herpes simplex virus thymidine kinase is sufficient to permit reactivation from latency, J. Virol. 77: 4703–4709.
Griffiths, A.& D. M. Coen, 2003. High-frequency phenotypic reversion and pathogenicity of an acyclovir-resistant herpes simplex virus mutant, J. Virol. 77: 2282–2286.
Guo, A., P. Hu, P. V. Balimane, F. H. Leibach & P. J. Sinko, 1999. Interactions of a nonpeptidic drug, valacyclovir, with the human intestinal peptide transporter (hPEPT1) expressed in a mammalian cell line, J. Pharmacol. Exp. Ther. 289: 448–454.
Gupta, R., A. Wald, E. Krantz, S. Selke, T. Warren, M. Vargas-Cortes, G. Miller & L. Corey, 2004. Valacyclovir and acyclovir for suppression of shedding of herpes simplex virus in the genital tract, J. Infect. Dis. 190: 1374–1381.
Hannon, G. J., 2002. RNA interference, Nature 418: 244–251.
Harris, W., P. Collins, R. J. Fenton, W. Snowden, M. Sowa & G. Darby, 2003. Phenotypic and genotypic characterization of clinical isolates of herpes simplex virus resistant to aciclovir, J. Gen. Virol. 84: 1393–1401.
Hartline, C. B., K. M. Gustin, W. B. Wan, S. L. Ciesla, J. R. Beadle, K. Y. Hostetler & E. R. Kern, 2005. Ether lipid-ester prodrugs of acyclic nucleoside phosphonates: activity against adenovirus replication in vitro, J. Infect. Dis. 191: 396–399.
Hasegawa, T., M. Kurokawa, T. A. Yukawa, M. Horii & K. Shiraki, 1995. Inhibitory action of acyclovir (ACV) and penciclovir (PCV) on plaque formation and partial cross-resistance of ACV-resistant varicella-zoster virus to PCV, Antiviral Res. 27: 271–279.
Hill, E. L., G. A. Hunter & M. N. Ellis, 1991. In vitro and in vivo characterization of herpes simplex virus clinical isolates recovered from patients infected with human immunodeficiency virus, Antimicrob. Agents Chemother. 35: 2322–2328.
Ho, H. T., K. L. Woods, J. J. Bronson, H. De Boeck, J. C. Martin & M. J. Hitchcock, 1992. Intracellular metabolism of the antiherpes agent (S)-1-[3-hydroxy-2-(phosphonylmethoxy) propyl]cytosine, Mol. Pharmacol. 41: 197–202.
Hockova, D., A. Holý, M. Masojidkova, G. Andrei, R. Snoeck, E. De Clercq & J. Balzarini, 2003. 5-Substituted-2,4-diamino-6-[2-(phosphonomethoxy)ethoxy]pyrimidines-acycli c nucleoside phosphonate analogues with antiviral activity, J. Med. Chem. 46: 5064–5073.
Hockova, D., A. Holý, M. Masojidkova, G. Andrei, R. Snoeck, E. De Clercq & J. Balzarini, 2004. Synthesis and antiviral activity of 2,4-diamino-5-cyano-6-[2-(phosphonomethoxy) ethoxy]pyrimidine and related compounds, Bioorg. Med. Chem. 12: 3197–3202.
Hodson, E. M., C. A. Jones, A. C. Webster, G. F. Strippoli, P. G. Barclay, K. Kable, D. Vimalachandra & J. C. Craig, 2005. Antiviral medications to prevent cytomegalovirus disease and early death in recipients of solid-organ transplants: a systematic review of randomised controlled trials, Lancet 365: 2105–2115.
Horsburgh, B. C., S. H. Chen, A. Hu, G. B. Mulamba, W. H. Burns & D. M. Coen, 1998. Recurrent acyclovir-resistant herpes simplex in an immunocompromised patient: can strain differences compensate for loss of thymidine kinase in pathogenesis?, J. Infect. Dis. 178: 618–625.
Hostetler, K. Y., S. Rought, K. A. Aldern, J. Trahan, J. R. Beadle & J. Corbeil, 2006. Enhanced antiproliferative effects of alkoxyalkyl esters of cidofovir in human cervical cancer cells in vitro, Mol. Cancer Ther. 5: 156–159.
Huang, L., K. K. Ishii, H. Zuccola, A. M. Gehring, C. B. Hwang, J. Hogle & D. M. Coen, 1999. The enzymological basis for resistance of herpesvirus DNA polymerase mutants to acyclovir: relationship to the structure of alpha-like DNA polymerases, Proc. Natl. Acad. Sci. U. S. A 96: 447–452.
Hwang, C. B.& H. J. Chen, 1995. An altered spectrum of herpes simplex virus mutations mediated by an antimutator DNA polymerase, Gene 152: 191–193.
Hwang, C. B., B. Horsburgh, E. Pelosi, S. Roberts, P. Digard & D. M. Coen, 1994. A net +1 frameshift permits synthesis of thymidine kinase from a drug-resistant herpes simplex virus mutant, Proc. Natl. Acad. Sci. U. S. A 91: 5461–5465.
Hwang, C. B., K. L. Ruffner & D. M. Coen, 1992. A point mutation within a distinct conserved region of the herpes simplex virus DNA polymerase gene confers drug resistance, J. Virol. 66: 1774–1776.
Ida, M., S. Kageyama, H. Sato, T. Kamiyama, J. Yamamura, M. Kurokawa, M. Morohashi & K. Shiraki, 1999. Emergence of resistance to acyclovir and penciclovir in varicella-zoster virus and genetic analysis of acyclovir-resistant variants, Antiviral Res. 40: 155–166.
Ilsley, D. D., S. H. Lee, W. H. Miller & R. D. Kuchta, 1995. Acyclic guanosine analogs inhibit DNA polymerases alpha, delta, and epsilon with very different potencies and have unique mechanisms of action, Biochemistry 34: 2504–2510.
Iwayama, S., Y. Ohmura, K. Suzuki, N. Ono, H. Nakazawa, M. Aoki, I. Tanabe, T. Sekiyama, T. Tsuji, M. Okunishi, K. Yamanishi & Y. Nishiyama, 1999. Evaluation of anti-herpesvirus activity of (1'S,2'R)-9-[[1',2'-bis(hydroxymethyl)cycloprop-1'-yl]methyl]- guanine (A-5021) in mice, Antiviral Res. 42: 139–148.
Iwayama, S., N. Ono, Y. Ohmura, K. Suzuki, M. Aoki, H. Nakazawa, M. Oikawa, T. Kato, M. Okunishi, Y. Nishiyama & K. Yamanishi, 1998. Antiherpesvirus activities of (1'S,2'R)-9-[[1',2'-bis(hydroxymethyl)cycloprop-1'-yl]methyl]guanine (A-5021) in cell culture, Antimicrob. Agents Chemother. 42: 1666–1670.
Jabs, D. A., C. Enger, M. Forman & J. P. Dunn, 1998. Incidence of foscarnet resistance and cidofovir resistance in patients treated for cytomegalovirus retinitis. The cytomegalovirus retinitis and viral resistance study group, Antimicrob. Agents Chemother. 42: 2240–2244.
Jacobson, J. G., K. L. Ruffner, M. Kosz-Vnenchak, C. B. Hwang, K. K. Wobbe, D. M. Knipe & D. M. Coen, 1993. Herpes simplex virus thymidine kinase and specific stages of latency in murine trigeminal ganglia, J. Virol. 67: 6903–6908.
Jiang, C., Y. T. Hwang, J. C. Randell, D. M. Coen & C. B. Hwang, 2007a. Mutations that decrease DNA binding of the processivity factor of the herpes simplex virus DNA polymerase reduce viral yield, alter the kinetics of viral DNA replication, and decrease the fidelity of DNA replication, J. Virol. 81: 3495–3502.
Jiang, C., Y. T. Hwang, G. Wang, J. C. Randell, D. M. Coen & C. B. Hwang, 2007b. Herpes simplex virus mutants with multiple substitutions affecting DNA binding of UL42 are impaired for viral replication and DNA synthesis, J. Virol. 81: 12077–12079.
Kamiyama, T., M. Kurokawa & K. Shiraki, 2001. Characterization of the DNA polymerase gene of varicella-zoster viruses resistant to acyclovir, J. Gen. Virol. 82: 2761–2765.
Kaufman, H., E. L. Martola & C. Dohlman, 1962. Use of 5-iodo-2'-deoxyuridine (IDU) in treatment of herpes simplex keratitis, Arch. Ophthalmol. 68: 235–239.
Kaufman, H. E., 1962. Clinical cure of herpes simplex keratitis by 5-iodo-2-deoxyuridine, Proc. Soc. Exp. Biol. Med. 109: 251–252.
Kaufman, H. E., 1963. Chemotherapy of Herpes Keratitis, Invest Ophthalmol. 2: 504–518.
Kaufman, H. E.& C. Heidelberger, 1964. Therapeutic Antiviral Action of 5-trifluoromethyl-2'-deoxyuridine in herpes simplex keratitis, Science 145: 585–586.
Keith, K. A., W. B. Wan, S. L. Ciesla, J. R. Beadle, K. Y. Hostetler & E. R. Kern, 2004. Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro, Antimicrob. Agents Chemother. 48: 1869–1871.
Keller, P. M., J. A. Fyfe, L. Beauchamp, C. M. Lubbers, P. A. Furman, H. J. Schaeffer & G. B. Elion, 1981. Enzymatic phosphorylation of acyclic nucleoside analogs and correlations with antiherpetic activities, Biochem. Pharmacol. 30: 3071–3077.
Kern, E. R., D. J. Collins, W. B. Wan, J. R. Beadle, K. Y. Hostetler & D. C. Quenelle, 2004. Oral treatment of murine cytomegalovirus infections with ether lipid esters of cidofovir, Antimicrob. Agents Chemother. 48: 3516–3522.
Kinchington, P. R., T. Araullo-Cruz, J. P. Vergnes, K. Yates & Y. J. Gordon, 2002. Sequence changes in the human adenovirus type 5 DNA polymerase associated with resistance to the broad spectrum antiviral cidofovir, Antiviral Res. 56: 73–84.
Kleymann, G., 2005. Agents and strategies in development for improved management of herpes simplex virus infection and disease, Expert. Opin. Investig. Drugs 14: 135–161.
Knopf, K. W., 1979. Properties of herpes simplex virus DNA polymerase and characterization of its associated exonuclease activity, Eur. J. Biochem. 98: 231–244.
Krecmerova, M., A. Holý, A. Piskala, M. Masojidkova, G. Andrei, L. Naesens, J. Neyts, J. Balzarini, E. De Clercq & R. Snoeck, 2007a. Antiviral activity of triazine analogues of 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (cidofovir) and related compounds, J. Med. Chem. 50: 1069–1077.
Krecmerova, M., A. Holý, R. Pohl, M. Masojidkova, G. Andrei, L. Naesens, J. Neyts, J. Balzarini, E. De Clercq & R. Snoeck, 2007b. Ester Prodrugs of Cyclic 1-(S)- [3-Hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine: Synthesis and antiviral activity, J. Med. Chem. 50(23): 5765–5772.
Larsen, S. D., Z. Zhang, B. A. DiPaolo, P. R. Manninen, D. C. Rohrer, M. J. Hageman, T. A. Hopkins, M. L. Knechtel, N. L. Oien, B. D. Rush, F. J. Schwende, K. J. Stefanski, J. L. Wieber, K. F. Wilkinson, K. M. Zamora, M. W. Wathen & R. J. Brideau, 2007. 7-Oxo-4,7-dihydrothieno[3,2-b]pyridine-6-carboxamides: synthesis and biological activity of a new class of highly potent inhibitors of human cytomegalovirus DNA polymerase, Bioorg. Med. Chem. Lett. 17:3840–3844.
Lebeau, I., G. Andrei, F. Dal Pozzo, J. R. Beadle, K. Y. Hostetler, E. De Clercq, O. J. van den Oord & R. Snoeck, 2006. Activities of alkoxyalkyl esters of cidofovir (CDV), cyclic CDV, and (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine against orthopoxviruses in cell monolayers and in organotypic cultures, Antimicrob. Agents Chemother. 50: 2525–2529.
Leonard, J. N.& D. V. Schaffer, 2006. Antiviral RNAi therapy: emerging approaches for hitting a moving target, Gene Ther. 13: 532–540.
Littler, E., A. D. Stuart & M. S. Chee, 1992. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir, Nature 358: 160–162.
Liu, S., J. D. Knafels, J. S. Chang, G. A. Waszak, E. T. Baldwin, M. R. Deibel, Jr., D. R. Thomsen, F. L. Homa, P. A. Wells, M. C. Tory, R. A. Poorman, H. Gao, X. Qiu & A. P. Seddon, 2006. Crystal structure of the herpes simplex virus 1 DNA polymerase, J. Biol. Chem. 281: 18193–18200.
LoPresti, A. E., J. F. Levine, G. B. Munk, C. Y. Tai & D. B. Mendel, 1998. Successful treatment of an acyclovir- and foscarnet-resistant herpes simplex virus type 1 lesion with intravenous cidofovir, Clin. Infect. Dis. 26: 512–513.
Loregian, A., B. A. Appleton, J. M. Hogle & D. M. Coen, 2004. Specific residues in the connector loop of the human cytomegalovirus DNA polymerase accessory protein UL44 are crucial for interaction with the UL54 catalytic subunit, J. Virol. 78: 9084–9092.
Loregian, A., R. Rigatti, M. Murphy, E. Schievano, G. Palu & H. S. Marsden, 2003. Inhibition of human cytomegalovirus DNA polymerase by C-terminal peptides from the UL54 subunit, J. Virol. 77: 8336–8344.
Lowance, D., H. H. Neumayer, C. M. Legendre, J. P. Squifflet, J. Kovarik, P. J. Brennan, D. Norman, R. Mendez, M. R. Keating, G. L. Coggon, A. Crisp & I. C. Lee, 1999. Valacyclovir for the prevention of cytomegalovirus disease after renal transplantation. International Valacyclovir Cytomegalovirus Prophylaxis Transplantation Study Group, N. Engl. J. Med. 340: 1462–1470.
Lurain, N. S., A. Weinberg, C. S. Crumpacker & S. Chou, 2001. Sequencing of cytomegalovirus UL97 gene for genotypic antiviral resistance testing, 1, Antimicrob. Agents Chemother. 45: 2775–2780.
Manichanh, C., C. Olivier-Aubron, J. P. Lagarde, J. T. Aubin, P. Bossi, A. Gautheret-Dejean, J. M. Huraux & H. Agut, 2001. Selection of the same mutation in the U69 protein kinase gene of human herpesvirus-6 after prolonged exposure to ganciclovir in vitro and in vivo, J. Gen. Virol. 82: 2767–2776.
Martin, J. L., C. E. Brown, N. Matthews-Davis & J. E. Reardon, 1994. Effects of antiviral nucleoside analogs on human DNA polymerases and mitochondrial DNA synthesis, Antimicrob. Agents Chemother. 38: 2743–2749.
McGuigan, C. & J. Balzarini, 2006. Aryl furano pyrimidines: the most potent and selective anti-VZV agents reported to date, Antiviral Res. 71: 149–153.
McGuigan, C., R. N. Pathirana, M. Migliore, R. Adak, G. Luoni, A. T. Jones, A. Diez-Torrubia, M. J. Camarasa, S. Velazquez, G. Henson, E. Verbeken, R. Sienaert, L. Naesens, R. Snoeck, G. Andrei & J. Balzarini, 2007. Preclinical development of bicyclic nucleoside analogues as potent and selective inhibitors of varicella zoster virus, J. Antimicrob. Chemother. 60(6): 1316–1330.
McGuigan, C., C. J. Yarnold, G. Jones, S. Velazquez, H. Barucki, A. Brancale, G. Andrei, R. Snoeck, E. De Clercq & J. Balzarini, 1999. Potent and selective inhibition of varicella-zoster virus (VZV) by nucleoside analogues with an unusual bicyclic base, J. Med. Chem. 42: 4479–4484.
McGuirt, P. V., J. E. Shaw, G. B. Elion & P. A. Furman, 1984. Identification of small DNA fragments synthesized in herpes simplex virus-infected cells in the presence of acyclovir, Antimicrob. Agents Chemother. 25: 507–509.
Meerbach, A., A. Holý, P. Wutzler, E. De Clercq & J. Neyts, 1998. Inhibitory effects of novel nucleoside and nucleotide analogues on Epstein-Barr virus replication, Antivir. Chem. Chemother. 9: 275–282.
Mertz, G. J., M. O. Loveless, M. J. Levin, S. J. Kraus, S. L. Fowler, D. Goade & S. K. Tyring, 1997. Oral famciclovir for suppression of recurrent genital herpes simplex virus infection in women. A multicenter, double-blind, placebo-controlled trial. Collaborative Famciclovir Genital Herpes Research Group, Arch. Intern. Med. 157: 343–349.
Michel, D. & T. Mertens, 2004. The UL97 protein kinase of human cytomegalovirus and homologues in other herpesviruses: impact on virus and host, Biochim. Biophys. Acta 1697: 169–180.
Miller, W. H. & R. L. Miller, 1980. Phosphorylation of acyclovir (acycloguanosine) monophosphate by GMP kinase, J. Biol. Chem. 255: 7204–7207.
Miller, W. H. & R. L. Miller, 1982. Phosphorylation of acyclovir diphosphate by cellular enzymes, Biochem. Pharmacol. 31: 3879–3884.
Morfin, F. & D. Thouvenot, 2003. Herpes simplex virus resistance to antiviral drugs, J. Clin. Virol. 26: 29–37.
Morfin, F., D. Thouvenot, M. Aymard & G. Souillet, 2000. Reactivation of acyclovir-resistant thymidine kinase-deficient herpes simplex virus harbouring single base insertion within a 7 Gs homopolymer repeat of the thymidine kinase gene, J. Med. Virol. 62: 247–250.
Morfin, F., D. Thouvenot, M. Turenne-Tessier, B. Lina, M. Aymard & T. Ooka, 1999. Phenotypic and genetic characterization of thymidine kinase from clinical strains of varicella-zoster virus resistant to acyclovir, Antimicrob. Agents Chemother. 43: 2412–2416.
Neyts, J., G. Andrei, R. Snoeck, G. Jahne, I. Winkler, M. Helsberg, J. Balzarini & E. De Clercq, 1994. The N-7-substituted acyclic nucleoside analog 2-amino-7-[(1,3-dihydroxy-2-propoxy)methyl]purine is a potent and selective inhibitor of herpesvirus replication, Antimicrob. Agents Chemother. 38: 2710–2716.
Neyts, J., J. Balzarini, G. Andrei, Z. Chaoyong, R. Snoeck, A. Zimmermann, T. Mertens, A. Karlsson & E. De Clercq, 1998. Intracellular metabolism of the N7-substituted acyclic nucleoside analog 2-amino-7-(1,3-dihydroxy-2-propoxymethyl)purine, a potent inhibitor of herpesvirus replication, Mol. Pharmacol. 53: 157–165.
Neyts, J. & E. De Clercq, 1997. Antiviral drug susceptibility of human herpesvirus 8, Antimicrob. Agents Chemother. 41: 2754–2756.
Neyts, J. & E. De Clercq, 2001. Efficacy of 2-amino-7-(1,3-dihydroxy-2-propoxymethyl)purine for treatment of vaccinia virus (orthopoxvirus) infections in mice, Antimicrob. Agents Chemother. 45: 84–87.
Neyts, J., G. Jahne, G. Andrei, R. Snoeck, I. Winkler & E. De Clercq, 1995. In vivo antiherpesvirus activity of N-7-substituted acyclic nucleoside analog 2-amino-7-[(1,3-dihydroxy-2-propoxy)methyl]purine, Antimicrob. Agents Chemother. 39: 56–60.
Ng, T. I., Y. Shi, H. J. Huffaker, W. Kati, Y. Liu, C. M. Chen, Z. Lin, C. Maring, W. E. Kohlbrenner & A. Molla, 2001. Selection and characterization of varicella-zoster virus variants resistant to (R)-9-[4-hydroxy-2-(hydroxymethy)butyl]guanine, Antimicrob. Agents Chemother. 45: 1629–1636.
Nugier, F., J. N. Colin, M. Aymard & M. Langlois, 1992. Occurrence and characterization of acyclovir-resistant herpes simplex virus isolates: report on a two-year sensitivity screening survey, J. Med. Virol. 36: 1–12.
Oien, N. L., R. J. Brideau, T. A. Hopkins, J. L. Wieber, M. L. Knechtel, J. A. Shelly, R. A. Anstadt, P. A. Wells, R. A. Poorman, A. Huang, V. A. Vaillancourt, T. L. Clayton, J. A. Tucker & M. W. Wathen, 2002. Broad-spectrum antiherpes activities of 4-hydroxyquinoline carboxamides, a novel class of herpesvirus polymerase inhibitors, Antimicrob. Agents Chemother. 46: 724–730.
Okuda, H., T. Nishiyama, K. Ogura, S. Nagayama, K. Ikeda, S. Yamaguchi, Y. Nakamura, K. Kawaguchi, T. Watabe & Y. Ogura, 1997. Lethal drug interactions of sorivudine, a new antiviral drug, with oral 5-fluorouracil prodrugs, Drug Metab. Dispos. 25: 270–273.
Okuda, H., K. Ogura, A. Kato, H. Takubo & T. Watabe, 1998. A possible mechanism of eighteen patient deaths caused by interactions of sorivudine, a new antiviral drug, with oral 5-fluorouracil prodrugs, J. Pharmacol. Exp. Ther. 287: 791–799.
Ono, N., S. Iwayama, K. Suzuki, T. Sekiyama, H. Nakazawa, T. Tsuji, M. Okunishi, T. Daikoku & Y. Nishiyama, 1998. Mode of action of (1'S,2'R)-9-[[1',2'-bis(hydroxymethyl) cycloprop-1'-yl]methyl]guanine (A-5021) against herpes simplex virus type 1 and type 2 and varicella-zoster virus, Antimicrob. Agents Chemother. 42: 2095–2102.
Ostrander, M.& Y. C. Cheng, 1980. Properties of herpes simplex virus type 1 and type 2 DNA polymerase, Biochim. Biophys. Acta 609: 232–245.
Pahwa, S., K. Biron, W. Lim, P. Swenson, M. H. Kaplan, N. Sadick & R. Pahwa, 1988. Continuous varicella-zoster infection associated with acyclovir resistance in a child with AIDS, JAMA 260: 2879–2882.
Painter, G. R.& K. Y. Hostetler, 2004. Design and development of oral drugs for the prophylaxis and treatment of smallpox infection, Trends Biotechnol. 22: 423–427.
Pelosi, E., G. B. Mulamba & D. M. Coen, 1998a. Penciclovir and pathogenesis phenotypes of drug-resistant Herpes simplex virus mutants, Antiviral Res. 37: 17–28.
Pelosi, E., F. Rozenberg, D. M. Coen & K. L. Tyler, 1998b. A herpes simplex virus DNA polymerase mutation that specifically attenuates neurovirulence in mice, Virology 252: 364–372.
Perry, C. M. & D. Faulds, 1996. Valaciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in herpesvirus infections, Drugs 52: 754–772.
Perry, C. M. & A. J. Wagstaff, 1995. Famciclovir. A review of its pharmacological properties and therapeutic efficacy in herpesvirus infections, Drugs 50: 396–415.
Pottage, J. C., Jr. & H. A. Kessler, 1995. Herpes simplex virus resistance to acyclovir: clinical relevance, Infect. Agents Dis. 4: 115–124.
Quenelle, D. C., D. J. Collins, W. B. Wan, J. R. Beadle, K. Y. Hostetler & E. R. Kern, 2004. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir, Antimicrob. Agents Chemother. 48: 404–412.
Rashidi, M. R., J. A. Smith, S. E. Clarke & C. Beedham, 1997. In vitro oxidation of famciclovir and 6-deoxypenciclovir by aldehyde oxidase from human, guinea pig, rabbit, and rat liver, Drug Metab Dispos. 25: 805–813.
Razonable, R. R. & V. C. Emery, 2004. Management of CMV infection and disease in transplant patients. 27–29 February 2004, Herpes. 11: 77–86.
Reardon, J. E., 1989. Herpes simplex virus type 1 and human DNA polymerase interactions with 2'-deoxyguanosine 5'-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition, J. Biol. Chem. 264: 19039–19044.
Reid, R., E. C. Mar, E. S. Huang & M. D. Topal, 1988. Insertion and extension of acyclic, dideoxy, and ara nucleotides by herpesviridae, human alpha and human beta polymerases. A unique inhibition mechanism for 9-(1,3-dihydroxy-2-propoxymethyl)guanine triphosphate, J. Biol. Chem. 263: 3898–3904.
Ruiz, J. C., K. A. Aldern, J. R. Beadle, C. B. Hartline, E. R. Kern & K. Y. Hostetler, 2006. Synthesis and antiviral evaluation of alkoxyalkyl esters of phosphonopropoxymethyl-guanine and phosphonopropoxymethyl-diaminopurine, Antivir. Chem. Chemother. 17: 89–95.
Ruiz, J. C., J. R. Beadle, K. A. Aldern, K. A. Keith, C. B. Hartline, E. R. Kern & K. Y. Hostetler, 2007. Synthesis and antiviral evaluation of alkoxyalkyl-phosphate conjugates of cidofovir and adefovir, Antiviral Res. 75: 87–90.
Sacks, S. L., F. Y. Aoki, A. Y. Martel, S. D. Shafran & M. Lassonde, 2005. Clinic-initiated, twice-daily oral famciclovir for treatment of recurrent genital herpes: a randomized, double-blind, controlled trial, Clin. Infect. Dis. 41: 1097–1104.
Sacks, S. L., R. J. Wanklin, D. E. Reece, K. A. Hicks, K. L. Tyler & D. M. Coen, 1989. Progressive esophagitis from acyclovir-resistant herpes simplex. Clinical roles for DNA polymerase mutants and viral heterogeneity? Ann. Intern. Med. 111: 893–899.
Safrin, S., J. Cherrington & H. S. Jaffe, 1997. Clinical uses of cidofovir, Rev. Med. Virol. 7: 145–156.
Safronetz, D., M. Petric, R. Tellier, B. Parvez & G. A. Tipples, 2003. Mapping ganciclovir resistance in the human herpesvirus-6 U69 protein kinase, J. Med. Virol. 71: 434–439.
Sarisky, R. T., M. R. Quail, P. E. Clark, T. T. Nguyen, W. S. Halsey, R. J. Wittrock, B. J. O'Leary, M. M. Van Horn, G. M. Sathe, S. Van Horn, M. D. Kelly, T. H. Bacon & J. J. Leary, 2001. Characterization of herpes simplex viruses selected in culture for resistance to penciclovir or acyclovir, J. Virol. 75: 1761–1769.
Sasadeusz, J. J. & S. L. Sacks, 1996. Spontaneous reactivation of thymidine kinase-deficient, acyclovir-resistant type-2 herpes simplex virus: masked heterogeneity or reversion?, J. Infect. Dis. 174: 476–482.
Sasadeusz, J. J., F. Tufaro, S. Safrin, K. Schubert, M. M. Hubinette, P. K. Cheung & S. L. Sacks, 1997. Homopolymer mutational hot spots mediate herpes simplex virus resistance to acyclovir, J. Virol. 71: 3872–3878.
Schaeffer, H. J., L. Beauchamp, P. de Miranda, G. B. Elion, D. J. Bauer & P. Collins, 1978. 9-(2-hydroxyethoxymethyl) guanine activity against viruses of the herpes group, Nature 272: 583–585.
Schnute, M. E., D. J. Anderson, R. J. Brideau, F. L. Ciske, S. A. Collier, M. M. Cudahy, M. Eggen, M. J. Genin, T. A. Hopkins, T. M. Judge, E. J. Kim, M. L. Knechtel, S. K. Nair, J. A. Nieman, N. L. Oien, A. Scott, S. P. Tanis, V. A. Vaillancourt, M. W. Wathen & J. L. Wieber, 2007. 2-Aryl-2-hydroxyethylamine substituted 4-oxo-4,7-dihydrothieno[2,3-b]pyridines as broad-spectrum inhibitors of human herpesvirus polymerases, Bioorg. Med. Chem. Lett. 17: 3349–3353.
Schnute, M. E., M. M. Cudahy, R. J. Brideau, F. L. Homa, T. A. Hopkins, M. L. Knechtel, N. L. Oien, T. W. Pitts, R. A. Poorman, M. W. Wathen & J. L. Wieber, 2005. 4-Oxo-4,7-dihydrothieno[2,3-b]pyridines as non-nucleoside inhibitors of human cytomegalovirus and related herpesvirus polymerases, J. Med. Chem. 48: 5794–5804.
Scott, G. M., M. A. Isaacs, F. Zeng, A. M. Kesson & W. D. Rawlinson, 2004. Cytomegalovirus antiviral resistance associated with treatment induced UL97 (protein kinase) and UL54 (DNA polymerase) mutations, J. Med. Virol. 74: 85–93.
Shi, R., A. Azzi, C. Gilbert, G. Boivin & S. X. Lin, 2006. Three-dimensional modeling of cytomegalovirus DNA polymerase and preliminary analysis of drug resistance, Proteins 64: 301–307.
Shin, M. C., S. K. Hong, J. S. Yoon, S. S. Park, S. G. Lee, D. G. Lee, W. S. Min, W. S. Shin & S. Y. Paik, 2006. Inhibition of UL54 and UL97 genes of human cytomegalovirus by RNA interference, Acta Virol. 50: 263–268.
Sienaert, R., G. Andrei, R. Snoeck, E. De Clercq, C. McGuigan & J. Balzarini, 2004. Inactivity of the bicyclic pyrimidine nucleoside analogues against simian varicella virus (SVV) does not correlate with their substrate activity for SVV-encoded thymidine kinase, Biochem. Biophys. Res. Commun. 315: 877–883.
Sienaert, R., L. Naesens, A. Brancale, A. Carangio, G. Andrei, R. Snoeck, A. Van Kuilenburg, E. De Clercq, C. McGuigan & J. Balzarini, 2003. Metabolic and pharmacological characteristics of the bicyclic nucleoside analogues (BCNAs) as highly selective inhibitors of varicella-zoster virus (VZV), Nucleosides Nucleotides Nucleic Acids 22: 995–997.
Sienaert, R., L. Naesens, A. Brancale, E. De Clercq, C. McGuigan & J. Balzarini, 2002. Specific recognition of the bicyclic pyrimidine nucleoside analogs, a new class of highly potent and selective inhibitors of varicella-zoster virus (VZV), by the VZV-encoded thymidine kinase, Mol. Pharmacol. 61: 249–254.
Silva, J. M., S. M. Hammond & G. J. Hannon, 2002. RNA interference: a promising approach to antiviral therapy?, Trends Mol. Med. 8: 505–508.
Simpson, D. & K. A. Lyseng-Williamson, 2006. Famciclovir: a review of its use in herpes zoster and genital and orolabial herpes, Drugs 66: 2397–2416.
Skoldenberg, B., M. Forsgren, K. Alestig, T. Bergstrom, L. Burman, E. Dahlqvist, A. Forkman, A. Fryden, K. Lovgren & K. Norlin, 1984. Acyclovir versus vidarabine in herpes simplex encephalitis. Randomised multicentre study in consecutive Swedish patients, Lancet 2: 707–711.
Smee, D. F., K. W. Bailey & R. W. Sidwell, 2002. Treatment of lethal cowpox virus respiratory infections in mice with 2-amino-7-[(1,3-dihydroxy-2-propoxy)methyl]purine and its orally active diacetate ester prodrug, Antiviral Res. 54: 113–120.
Smith, I. L., J. M. Cherrington, R. E. Jiles, M. D. Fuller, W. R. Freeman & S. A. Spector, 1997. High-level resistance of cytomegalovirus to ganciclovir is associated with alterations in both the UL97 and DNA polymerase genes, J. Infect. Dis. 176: 69–77.
Snoeck, R., G. Andrei, M. Gerard, A. Silverman, A. Hedderman, J. Balzarini, C. Sadzot-Delvaux, G. Tricot, N. Clumeck & E. De Clercq, 1994a. Successful treatment of progressive mucocutaneous infection due to acyclovir- and foscarnet-resistant herpes simplex virus with (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC), Clin. Infect. Dis. 18: 570–578.
Snoeck, R. & E. De Clercq, 2002. Role of cidofovir in the treatment of DNA virus infections, other than CMV infections, in immunocompromised patients, Curr. Opin. Investig. Drugs 3: 1561–1566.
Snoeck, R., M. Gerard, C. Sadzot-Delvaux, G. Andrei, J. Balzarini, D. Reymen, N. Ahadi, J. M. De Bruyn, J. Piette & B. Rentier, 1994b. Meningoradiculoneuritis due to acyclovir-resistant varicella zoster virus in an acquired immune deficiency syndrome patient, J. Med. Virol. 42: 338–347.
St Clair, M. H., P. A. Furman, C. M. Lubbers & G. B. Elion, 1980. Inhibition of cellular alpha and virally induced deoxyribonucleic acid polymerases by the triphosphate of acyclovir, Antimicrob. Agents Chemother. 18: 741–745.
Sullivan, V. & D. M. Coen, 1991. Isolation of foscarnet-resistant human cytomegalovirus patterns of resistance and sensitivity to other antiviral drugs, J. Infect. Dis. 164: 781–784.
Sullivan, V., C. L. Talarico, S. C. Stanat, M. Davis, D. M. Coen & K. K. Biron, 1992. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells, Nature 359: 85.
Suzutani, T., K. Ishioka, E. De Clercq, K. Ishibashi, H. Kaneko, T. Kira, K. Hashimoto, M. Ogasawara, K. Ohtani, N. Wakamiya & M. Saijo, 2003. Differential mutation patterns in thymidine kinase and DNA polymerase genes of herpes simplex virus type 1 clones passaged in the presence of acyclovir or penciclovir, Antimicrob. Agents Chemother. 47: 1707–1713.
Suzutani, T., S. Koyano, M. Takada, I. Yoshida & M. Azuma, 1995. Analysis of the relationship between cellular thymidine kinase activity and virulence of thymidine kinase-negative herpes simplex virus types 1 and 2, Microbiol. Immunol. 39: 787–794.
Talarico, C. L., T. C. Burnette, W. H. Miller, S. L. Smith, M. G. Davis, S. C. Stanat, T. I. Ng, Z. He, D. M. Coen, B. Roizman & K. K. Biron, 1999. Acyclovir is phosphorylated by the human cytomegalovirus UL97 protein, Antimicrob. Agents Chemother. 43: 1941–1946.
Talarico, C. L., W. C. Phelps & K. K. Biron, 1993. Analysis of the thymidine kinase genes from acyclovir-resistant mutants of varicella-zoster virus isolated from patients with AIDS, J. Virol. 67: 1024–1033.
Tchesnokov, E. P., C. Gilbert, G. Boivin & M. Gotte, 2006. Role of helix P of the human cytomegalovirus DNA polymerase in resistance and hypersusceptibility to the antiviral drug foscarnet, J. Virol. 80: 1440–1450.
Tenney, D. J., G. Yamanaka, S. M. Voss, C. W. Cianci, A. V. Tuomari, A. K. Sheaffer, M. Alam & R. J. Colonno, 1997. Lobucavir is phosphorylated in human cytomegalovirus-infected and -uninfected cells and inhibits the viral DNA polymerase, Antimicrob. Agents Chemother. 41: 2680–2685.
Tenser, R. B., J. C. Jones, S. J. Ressel & F. A. Fralish, 1983. Thymidine plaque autoradiography of thymidine kinase-positive and thymidine kinase-negative herpesviruses, J. Clin. Microbiol. 17: 122–127.
Thomsen, D. R., N. L. Oien, T. A. Hopkins, M. L. Knechtel, R. J. Brideau, M. W. Wathen & F. L. Homa, 2003. Amino acid changes within conserved region III of the herpes simplex virus and human cytomegalovirus DNA polymerases confer resistance to 4-oxo-dihydroquinolines, a novel class of herpesvirus antiviral agents, J. Virol. 77: 1868–1876.
Tucker, J. A., T. L. Clayton, C. G. Chidester, M. W. Schulz, L. E. Harrington, S. J. Conrad, Y. Yagi, N. L. Oien, D. Yurek & M. S. Kuo, 2000. Structure-activity relationships of acyloxyamidine cytomegalovirus DNA polymerase inhibitors, Bioorg. Med. Chem. 8: 601–615.
Vaillancourt, V. A., M. M. Cudahy, S. A. Staley, R. J. Brideau, S. J. Conrad, M. L. Knechtel, N. L. Oien, J. L. Wieber, Y. Yagi & M. W. Wathen, 2000. Naphthalene carboxamides as inhibitors of human cytomegalovirus DNA polymerase, Bioorg. Med. Chem. Lett. 10: 2079–2081.
Vere Hodge, R. A., D. Sutton, M. R. Boyd, M. R. Harnden & R. L. Jarvest, 1989. Selection of an oral prodrug (BRL 42810; famciclovir) for the antiherpesvirus agent BRL 39123 [9-(4-hydroxy-3-hydroxymethylbut-l-yl)guanine; penciclovir], Antimicrob. Agents Chemother. 33: 1765–1773.
Visse, B., B. Dumont, J. M. Huraux & A. M. Fillet, 1998. Single amino acid change in DNA polymerase is associated with foscarnet resistance in a varicella-zoster virus strain recovered from a patient with AIDS, J. Infect. Dis. 178 Suppl 1: S55–S57.
Visse, B., J. M. Huraux & A. M. Fillet, 1999. Point mutations in the varicella-zoster virus DNA polymerase gene confers resistance to foscarnet and slow growth phenotype, J. Med. Virol. 59: 84–90.
Wald, A., S. Selke, T. Warren, F. Y. Aoki, S. Sacks, F. Diaz-Mitoma & L. Corey, 2006. Comparative efficacy of famciclovir and valacyclovir for suppression of recurrent genital herpes and viral shedding, Sex Transm. Dis. 33: 529–533.
Wan, W. B., J. R. Beadle, C. Hartline, E. R. Kern, S. L. Ciesla, N. Valiaeva & K. Y. Hostetler, 2005. Comparison of the antiviral activities of alkoxyalkyl and alkyl esters of cidofovir against human and murine cytomegalovirus replication in vitro, Antimicrob. Agents Chemother. 49: 656–662.
Warren, T., J. Harris & C. A. Brennan, 2004. Efficacy and safety of valacyclovir for the suppression and episodic treatment of herpes simplex virus in patients with HIV, Clin. Infect. Dis. 39 Suppl 5: S258-S266.
Wathen, M. W., 2002. Non-nucleoside inhibitors of herpesviruses, Rev. Med. Virol. 12: 167–178.
Weinberg, A., D. A. Jabs, S. Chou, B. K. Martin, N. S. Lurain, M. S. Forman & C. Crumpacker, 2003. Mutations conferring foscarnet resistance in a cohort of patients with acquired immunodeficiency syndrome and cytomegalovirus retinitis, J. Infect. Dis. 187: 777–784.
Weller, S., M. R. Blum, M. Doucette, T. Burnette, D. M. Cederberg, P. de Miranda & M. L. Smiley, 1993. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers, Clin. Pharmacol. Ther. 54: 595–605.
Whitley, R. J., C. A. Alford, M. S. Hirsch, R. T. Schooley, J. P. Luby, F. Y. Aoki, D. Hanley, A. J. Nahmias & S. J. Soong, 1986. Vidarabine versus acyclovir therapy in herpes simplex encephalitis, N. Engl. J. Med. 314: 144–149.
Whitley, R. J., S. J. Soong, R. Dolin, G. J. Galasso, L. T. Ch'ien & C. A. Alford, 1977. Adenine arabinoside therapy of biopsy-proved herpes simplex encephalitis. National Institute of Allergy and Infectious Diseases collaborative antiviral study, N. Engl. J. Med. 297: 289–294.
Whitley, R. J., S. J. Soong, M. S. Hirsch, A. W. Karchmer, R. Dolin, G. Galasso, J. K. Dunnick & C. A. Alford, 1981. Herpes simplex encephalitis: vidarabine therapy and diagnostic problems, N. Engl. J. Med. 304: 313–318.
Wiebusch, L., M. Truss & C. Hagemeier, 2004. Inhibition of human cytomegalovirus replication by small interfering RNAs, J. Gen. Virol. 85: 179–184.
Wolf, D. G., C. T. Courcelle, M. N. Prichard & E. S. Mocarski, 2001. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation, Proc. Natl. Acad. Sci. U. S. A 98: 1895–1900.
Wolf, D. G., D. J. Lee & S. A. Spector, 1995. Detection of human cytomegalovirus mutations associated with ganciclovir resistance in cerebrospinal fluid of AIDS patients with central nervous system disease, Antimicrob. Agents Chemother. 39: 2552–2554.
Wutzler, P., 1997. Antiviral therapy of herpes simplex and varicella-zoster virus infections, Intervirology 40: 343–356.
Xiong, X., J. L. Smith & M. S. Chen, 1997. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation, Antimicrob. Agents Chemother. 41: 594–599.
Xiong, X., J. L. Smith, C. Kim, E. S. Huang & M. S. Chen, 1996. Kinetic analysis of the interaction of cidofovir diphosphate with human cytomegalovirus DNA polymerase, Biochem. Pharmacol. 51: 1563–1567.
Zhang, Y., D. Schols & E. De Clercq, 1999. Selective activity of various antiviral compounds against HHV-7 infection, Antiviral Res. 43: 23–35.
Zimmermann, A., D. Michel, I. Pavic, W. Hampl, A. Luske, J. Neyts, E. De Clercq & T. Mertens, 1997. Phosphorylation of aciclovir, ganciclovir, penciclovir and S2242 by the cytomegalovirus UL97 protein: a quantitative analysis using recombinant vaccinia viruses, Antiviral Res. 36: 35–42.
Zuccola, H. J., D. J. Filman, D. M. Coen & J. M. Hogle, 2000. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase, Mol. Cell 5: 267–278.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Andrei, G., De Clercq, E., Snoeck, R. (2009). Viral DNA Polymerase Inhibitors. In: Raney, K., Gotte, M., Cameron, C. (eds) Viral Genome Replication. Springer, Boston, MA. https://doi.org/10.1007/b135974_22
Download citation
DOI: https://doi.org/10.1007/b135974_22
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-0-387-89425-6
Online ISBN: 978-0-387-89456-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)