
Abstract
Reverse transcription quantitative-polymerase chain reaction (RT-qPCR) has become a routine method in many laboratories. Normalization of data from experimental conditions is critical for data processing and is usually achieved by the use of a single reference gene. Nevertheless, as pointed by the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines, several reference genes should be used for reliable normalization. Ageing is a physiological process that results in a decline of many expressed genes. Reliable normalization of RT-qPCR data becomes crucial when studying ageing. Here, we propose a RT-qPCR study from four mouse brain regions (cortex, hippocampus, striatum and cerebellum) at different ages (from 8 weeks to 22 months) in which we studied the expression of nine commonly used reference genes. With the use of two different algorithms, we found that all brain structures need at least two genes for a good normalization step. We propose specific pairs of gene for efficient data normalization in the four brain regions studied. These results underline the importance of reliable reference genes for specific brain regions in ageing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reverse transcription quantitative-polymerase chain reaction (RT-qPCR) is widely used in gene expression analysis because of its simple, reproducible and high-throughput features. It gives a rapid means of understanding gene variation in various organisms/organs by measuring the expression of target genes compared to reference genes across different samples. On top of that, RT-qPCR has become a low-cost method allowing a generalization of its use in laboratories. However, to analyse RT-qPCR data, several factors such as sample amount, RNA integrity or complementary DNA (cDNA) quality must be controlled because they affect the quantitative measurement of gene expression [1, 2]. Thus, in order to obtain a robust analysis of gene expression, reference genes are used as an internal control to normalize and monitor the variations of expression between samples and reactions [3, 2, 4]. The expression of these reference genes should not vary in the tissues or cells under investigation or in response to experimental treatment [3]. As shown by two surveys realized in 2013, there are still many bias in RT-qPCR studies: the stability of reference genes is not systematically checked, and normalization is generally achieved by the use of a single reference gene [2], which is in discordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines published by Bustin and collaborators to avoid misinterpretations of RT-qPCR results [5].
Ageing is a physiological process associated with deficits in learning and memory tasks, explained by deficits in key brain functions, like motor coordination [6] and sensory and cognitive functions [7]. Ageing is also a risk factor for many neurological diseases, including cerebral ischaemia [8], Alzheimer’s disease [9], Parkinson’s disease [10] and others. To study gene expression during ageing in neurodegenerative pathologies or in normal brain, RT-qPCR has been widely used. It has been suggested that RT-qPCR data should be normalized to at least two reference genes whose expression has been demonstrated to be stable in the conditions studied [3, 11]. The most frequently used genes for normalization are glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and beta-actin (ActinB). However, they can display variable expressions in many paradigms. For example, ActinB has been shown to vary in several studies, like in ischaemic/hypoxic conditions [12, 13], and GAPDH is not stable in a model of apoptosis induced by age on neurones [14]. Also, their sequences overlap with highly homologous segmental duplications sequences [15].
Based on that, Sieber and collaborators published a useful study in 2010 that demonstrated the need to check the stability of normalization genes in ageing and ischaemia for brain studies [16]. However, the authors did not discriminate different structures/areas but rather used the whole brain. It is well known that neurodegenerative diseases/models affect specific brain regions, and gene expression also varies in different structures. By using the geNorm and the Normfinder methods, we studied the stability of common reference genes, such as beta-actin (ActinB), polymerase (RNA) II (DNA directed) polypeptide A (Polr2a), succinate dehydrogenase complex, subunit A (Sdha), protoporphyrinogen oxidase (Ppox), hypoxanthine guanine phosphoribosyl transferase (Hprt), ribosomal protein L13a (Rpl13a), hydroxymethylbilane synthase (Hmbs), peptidylprolyl isomerase B, or cyclophilin B,(Ppib) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in four mouse brain structures (cortex, hippocampus, striatum and cerebellum) during ageing (Figs. 1, 2, 3 and 4). We found that the stability of each gene depends on the studied structure and that reference genes are specific for each structure in ageing. We thus propose, for each of these brain structures, a set of reference genes usable for ageing studies in mice, and we verified our reference genes by measuring the expression of the age-related gene tissue plasminogen activator (tPA) in the hippocampus during ageing.

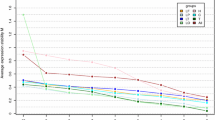
Raw data of RT-qPCRs performed on the cortex of mice aged from 8 weeks to 22 months old. Starting quantity was directly obtained from the Cq value using the Bio-Rad CFX Manager software for Ppib (a), Hmbs (b), Rpl13a (c), ActinB (d), Hprt (e), Ppox (f), Sdha (g), Polr2a (h) and GAPDH (i). n = 5 independent animals in each group

Raw data of RT-qPCRs performed on the hippocampi of mice aged from 8 weeks to 22 months old. Starting quantity was directly obtained from the Cq value using the Bio-Rad CFX Manager software for Ppib (a), Hmbs (b), Rpl13a (c), ActinB (d), Hprt (e), Ppox (f), Sdha (g), Polr2a (h) and GAPDH (i). n = 5 independent animals in each group

Raw data of RT-qPCRs performed on the striatum of mice aged from 8 weeks to 22 months old. Starting quantity was directly obtained from the Cq value using the Bio-Rad CFX Manager software for Ppib (a), Hmbs (b), Rpl13a (c), ActinB (d), Hprt (e), Ppox (F), Sdha (G), Polr2a (H) and GAPDH (i). n = 5 independent animals in each group

Raw data of RT-qPCRs performed on the cerebellum of mice aged from 8 weeks to 22 months old. Starting quantity was directly obtained from the Cq value using the Bio-Rad CFX Manager software for Ppib (a), Hmbs (b), Rpl13a (c), ActinB (d), Hprt (e), Ppox (f), Sdha (g), Polr2a (h) and GAPDH (i). n = 5 independent animals in each group
Results
RT-qPCR Efficiency
Quantification cycle (Cq) was correlated to the starting concentration of the dilution series (from 1:10 to 1:10,000; five orders of magnitude). The coefficient of correlation (R 2) ranged from 0.977 to 0.998 in the cortex, 0.971 to 0.986 in the hippocampus, 0.977 to 0.993 in the striatum and 0.975 to 0.994 in the cerebellum. The efficiency was then calculated as described in the “Material and Methods” and ranged from 82.45 to 126.625 % as shown in the Table 1. Non-normalized Cq values in our experiments were all within the range of the dilution series.
Expression of Reference Genes in the Brain
We measured the raw expression (here called starting quantity) of our nine genes in four cerebral structures: the cortex, hippocampus, striatum and cerebellum. Among the tested genes, we observed variations during ageing in the different brain structures. For example, in the cortex, hmbs, Actinb, Ppox, Polr2a and GAPDH do not vary between 8 weeks, 7 months, 17 months and 22 months in contrast to Ppib, Rpl13a, Hprt and Sdha (Table 2). In the hippocampus, only Ppib, Hprt and Polr2a show no statistical differences of expression during ageing (Table 2). In the striatum, Ppib, Rpl13a, Actinb, Ppox, Sdha, Polr2a and GAPDH do not show statistical differences of expression during ageing, whereas in the cerebellum, only Ppib, Rpl13a and GAPDH show no statistical differences (Table 2). An intragroup analysis (Mann and Whitney test) was also performed on the raw data expression (Supplementary table 2).
These results clearly demonstrate variations in many commonly used reference genes, but also differences between brain structures during normal ageing. It also demonstrates that one reference gene is usually not enough, and normalization to multiple reference genes needs to be done for each cerebral structure during ageing. To address this, we used two different methods: by using the qBase+™ software that contains the geNorm algorithm and by using the Normfinder algorithm.
Determination of the Correct Reference Genes in Cerebral Structures
Cortex
In the cerebral cortex, multiple analyses of the nine reference genes revealed a geNorm V value under the 0.15 threshold for the use of two genes including Actb, Polr2a, Sdha, Ppox, Hprt, Rpl13a, Hmbs, Ppib and GAPDH (Fig. 5a). The geNorm M value [17] showed that the two best genes to use as reference genes in the cerebral cortex during ageing are Rpl13a and GAPDH (Fig. 5b): between 8 weeks and 22 months, these two genes display the same profile of expression in all our studied conditions.

Determination of the best reference genes in the cortex during ageing using the geNorm M algorithm. a Two reference genes are enough in the cortex to normalize RT-qPCR data as the pairwise variation V value for V 2/3 (n/n + 1) is under 0.15. b Best genes to study ageing in the cortex are Rpl13a and GAPDH according to the geNorm M value under 0.5 for these genes. c The use of the Normfinder algorithm also ranked Rpl13a and GAPDH as the best reference genes for normalizing RT-qPCR data in the cortex during ageing
To ensure the choice of the reference genes in the cortex, we also performed a Normfinder analysis that calculates a stability value after the estimation of an intra- and intergroup variation (see “Material and Methods” [1]). The ranking of the reference genes (Fig. 5c) showed that the best genes for normalizing RT-qPCR data in the cortex during ageing are Rpl13a and GAPDH.
Hippocampus
The same method was applied in the hippocampus, aged from 8 weeks to 22 months old. Again, among the nine selected genes, two paired genes were enough to normalize RT-qPCR data in the hippocampus during ageing, as shown by the geNorm V value (Fig. 6a). According to the geNorm M value, Rpl13a and Hprt is the best pair of reference genes to normalize RT-qPCR data in the hippocampus during ageing (Fig. 6b).

Determination of the best reference genes in the hippocampus during ageing using the geNorm M algorithm: a Two reference genes are enough in the hippocampus to normalize RT-qPCR data as the pairwise variation V value for V 2/3 (n/n + 1) is under 0.15. b Best genes to study ageing in the hippocampus are Rpl13a and Hprt according to the geNorm M value under 0.5 for these genes. c The use of the Normfinder algorithm ranked Rpl13a and Sdha as the best reference genes for normalizing RT-qPCR data in the hippocampus during ageing
The Normfinder analysis gave Rpl13a and Sdha as the most stable genes (Fig. 6c). There was a slight difference in the choice of genes between geNorm and Normfinder. However, both analyses ranked Rpl13a as the most stable gene in the hippocampus during ageing.
Striatum
In the striatum of C57BL6/J mice aged from 8 weeks to 22 months old, analysis of the studied reference genes revealed that two genes of the list are enough to normalize RT-qPCR data (Fig. 7a). The geNorm M value showed that Rpl13a and Ppib are the best candidates for the normalization of RT-qPCR data in the striatum during ageing (Fig. 7b), and the Normfinder analysis (Fig. 7c) gave the same result.

Determination of the best reference genes in the striatum during ageing using the geNorm M algorithm. a Two reference genes are enough in the striatum to normalize RT-qPCR data as the pairwise variation V value for V 2/3 (n/n + 1) is under 0.15. b The best genes to study ageing in the striatum are Rpl13a and Ppib according to the geNorm M value under 0.5 for these genes. c The use of the Normfinder algorithm also ranked Rpl13a and Ppib as the best reference genes for normalizing RT-qPCR data in the cortex during ageing
Cerebellum
Regarding the cerebellum in ageing, the analysis of the geNorm V value showed that two reference genes are also enough to normalize RT-qPCR (Fig. 8a). According to the geNorm M value, these genes are GAPDH and pPIB (Fig. 8b). The Normfinder analysis gave a slightly different result, as the two best genes were GAPDH and Hprt (Fig. 8c).

Determination of the best reference genes in the cerebellum during ageing using the geNorm M algorithm: a Two reference genes are enough in the cerebellum to normalize RT-qPCR data as the pairwise variation V value for V 2/3 (n/n + 1) is under 0.15. b The best genes to study ageing in the cerebellum are GAPDH and Ppib according to the geNorm M value under 0.5 for these genes. c The use of the Normfinder algorithm ranked GAPDH and Hprt as the best reference genes for normalizing RT-qPCR data in the cortex during ageing
Evaluation of Reference Genes in RT-qPCR Experiments
To test our hypothesis, we decided to check the expression of a gene during ageing. As it was previously demonstrated that tissue plasminogen activator (tPA) was impacted during ageing [18–20], we decided to study its expression in the hippocampus of mice from 8 weeks to 22 months old.
By using Rpl13a and Hprt, the two recommended reference genes of the geNorm analysis, we observed that the expression of tPA was decreased during ageing (Fig. 9a, p = 0.0079; Kendall’s correlation test). However, when using Hmbs and ActinB, commonly considered as “standard” reference genes, and also under the threshold of 0.5 on the geNorm M value, no changes in the expression of tPA were observed during ageing (Fig. 9b, p = 0.7399; Kendall’s correlation test). These results highlight the importance of efficient normalization for reliable results in gene expression studies in ageing.

Expression of tPA measured by RT-qPCR according to the geNorm-selected reference genes (a) or to random reference genes (b). The correlation between the expression and the age is calculated with the Kendall correlation coefficient
Discussion
RT-qPCR is a useful and sensitive technique that gives a large amount of data within a short amount of time and is able to detect small differences of expression. However, because of technical variations (storage condition, RNA isolation and purification, amount of starting material, enzymatic efficiency…), normalization is an obligatory step [11, 21, 4]. Since RT-qPCR has become a routine technique for molecular biology laboratories, numbers of reference genes have been used, with glyceraldehyde-3-phosphate dehydrogenase (Gapdh) or beta-actin (ActinB) as gold standards. However, in many cases, these genes have been demonstrated to be unstable, like ActinB during cerebral ischaemia [12], or in many other conditions (reviewed in [22]).
In the present study, we show that (i) during ageing, in different cerebral structures, there are variations of expression for nine commonly used reference genes (Actinb, Polr2a, Sdha, Ppox, Hprt, Rpl13a, Hmbs, Ppib and GAPDH), (ii) at least two genes are necessary for a good normalization process in brain structures during ageing and (iii) each cerebral structure studied here (cortex, hippocampus, striatum and cerebellum) requires specific paired-reference genes (Table 3).
It has already been suggested by Sieber and colleagues in 2010 that in physiological brain during ageing, RT-qPCR needs to be normalized to more than one reference gene [16]. They performed the study on whole brains during physiological ageing (from 8 weeks to 24 months) but also in animals subjected to a model of stroke. It appeared that ischaemia leads to the modification of the reference genes to use for normalization of RT-qPCR data in ageing (Hprt and Hmbs vs ActinB and Hmbs).
Despite the importance of reference genes, especially for ageing, many studies use only one reference gene and do not check its stability in experimental conditions as recommended by the MIQE [5]. Here, we show the importance of using at least two reference genes to normalize RT-qPCR data, but also the importance of checking their stability of expression. It is confirmed by studying the expression of tPA during ageing in the hippocampus. By using the geNorm-recommended reference genes, we observed a decrease in tPA expression during ageing, as previously reported [18–20]. However, when we used two other genes commonly considered as “standard” reference genes, we did not see any difference in tPA expression with age.
It is interesting to note that for the hippocampus and the cerebellum, there is a small difference between the two proposed reference genes. Indeed, for the hippocampus, for example, the geNorm analysis recommends Rpl13a and Hprt, while the Normfinder analysis recommends Rpl13a and Sdha. This is probably due to the difference between the two algorithms: geNorm gives a stability value on the principle that the expression ratio of two ideal internal control genes is identical in all samples [3]; whereas Normfinder combines the intra- and the intergroup variation into a stability value [1]. However, when there is a difference between the two methods, the reference genes are always close to the most stable genes, reflecting their ability to be a good reference gene.
Our data also reveal that each structure needs its own reference paired genes. This is very important as many neurological diseases are structure specific. In stroke, depending of the model used, the majority of the infarct lesion is in the cortex and in the striatum [23]; various models of Parkinson’s disease affect the substantia nigra [24]; in models of Alzheimer’s disease, the first amyloid deposits are structure specific depending of the model used [25]. Moreover, a cofactor of all of these diseases is ageing, and it has been previously shown that ageing plays important roles in stroke [18] for example.
This is particularly important for pathological studies in ageing, as the pathology by itself will change the reference genes needed for a good normalization process [16]; however, the same attention should be used for cerebral structure selection, instead of working on whole brain, as we demonstrated here.
Our study highlights the importance of the brain structure studied, by showing that each structure needs its own specific reference genes. It also shows that a stability expression control is primary, as demonstrated by the expression of tPA within the hippocampus. It comes in addition with the MIQE guidelines [5] and other publications which appeal for “good practices” in RT-qPCR studies [2, 1, 3].
Age is a common factor to many neurological disorders, and RT-qPCR made from brain tissue in ageing should be well designed to avoid misinterpretations of the data. Sieber and colleagues were the first ones to underline this specific point in neurological disorders [16]. However, they performed their study on the whole brain. Many neurological studies are more focused on a specific cerebral area (hippocampus, cortex, striatum…) depending on the neurological disease studied. Here, we demonstrate that each cerebral structure needs its own reference genes validation, and we propose the best couple of genes for the cortex, the hippocampus, the striatum and the cerebellum for ageing studies in the C57BL6/J mice. However, for pathological studies in ageing, the stability expression of reference genes needs to be checked.
Material and Methods
Aged Animals
Our study was performed on male C57BL/6 mice of distinct age and approved by local (French Legislation act no. 87–848, Ministère de l’Agriculture et de la Forêt) and European (directives from the European Communities Council, November 24, 1986, 86/609/EEC) authorities. C57BL/6 mice have a lifespan of ~26 months [26]. Four groups were designed: 8 weeks old, 7 months old, 17 months old and 22 months old, with five mice per group. Animals were housed in a temperature-controlled room on a 12-h-light/12-h-dark cycle, with food and water given ad libitum.
Brain Sample Preparation
Mice were anaesthetized with 5 % isoflurane in a 70/30 % mixture of NO2/O2 and perfused with cold heparinized NaCl 0.9 % to wash out blood cells. Brains were excised and precisely dissected under a binocular microscope to isolate the cerebral cortex, hippocampus, cerebellum and striatum. Tissue sections were maintained at −80 °C until messenger RNA (mRNA) extraction.
Total RNA Extraction and Quantification
Total RNA was isolated from brain structures with TRI reagent (Sigma; Lyon, France) according to the manufacturer. Briefly, tissues were dissociated in TRI reagent, and RNA was isolated by the addition of chloroform. Total RNA was washed by ethanol and treated with TURBO DNase (Ambion; Saint Aubin, France) to avoid DNA contamination. Total RNA was quantified by spectrophotometry (NanoDrop Technologies; Wilmington, USA).
Reverse Transcription
We performed the reverse transcription from 1 μg of total RNA and the iScript kit (iScript Select cDNA Synthesis Kit, Bio-Rad; Marnes-la-Coquette, France) in a total volume of 20 μL with the following cycle conditions: 42 °C (90 min); 85 °C (5 min). The cDNA products were then stored at −20 °C until their use.
Quantitative PCR
RT-qPCR were performed from 1 μL of 1:20 diluted cDNA in 15 μL total of a 1× solution of iQ SYBR Green Supermix (Bio-Rad; Marnes-la-Coquette, France) containing 200 nM of each primer. Based on mRNA coding sequences (www.ensembl.org), mouse-specific primers were designed by using the Primer3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) [27] (sequences are in Table 1). Assays were run in triplicate on the CFX96 real-time system c1000 thermal cycler (Bio-Rad; Marnes-la-Coquette, France), with the following cycle conditions: 95 °C (3 min); [95 °C (2 s), 60 °C (20 s)] × 39; 70 °C (30 s).
Data Analysis
Each plate contains the whole samples (triplicate of the 5 mice per group) for one gene and one cerebral structure (8 genes, 4 structures: 32 plates of RT-qPCR), and Cq values were obtained from the Bio-Rad CFX Manager software (Bio-Rad; Marnes-la-Coquette, France). Relative gene expression values (starting quantity) were calculated with the Bio-Rad CFX Manager software and corrected with each efficiency (E) inferred from 5-fold standard dilution series, as described by Pfaffl [28].
Kruskal-Wallis test was performed on the starting quantities to check the stability of each gene in the studied structures.
To investigate the best normalization process to reference genes, we used the geNorm algorithm associated to qBase+™ software and the Normfinder algorithm. geNorm allows the calculation of stability (M value) and the ranking of selected reference genes, on the basis of non-normalized expression levels [3, 17]. It also suggests the number of internal controls to use in the selected experimental conditions by giving the V value. It is stated that the benefit of using an extra (n + 1) reference gene is limited as soon as the V n/n + 1 value drops below the 0.15 threshold. Calculations are detailed as follows [3]:
The qBase+ software calculates relative quantities (RQs) for each gene in each sample by comparing the Cq of a given sample with the average Cq across all samples for that gene, taking into account differences in PCR amplification efficiencies (E) (Eq. 1).
Then, the RQ is normalized by dividing it by the geometric mean Cq of a set of selected reference genes, which results in the normalized relative quantity (NRQ). The reference genes are measured in all samples of the experiment. In theory (if there was no variability), each reference gene should have the same RQ values in all samples. In reality, factors are responsible for variations in the expression levels of the reference genes (amount of cDNA, RNA integrity, enzyme efficiency, …). However, this variation is a direct measure of the noise between the samples and can be used to calculate normalization factors (NF = factor to be multiplied to the observed RQ values so that the expression levels of the housekeeping genes are equalized across samples) for each sample. For NRF, a variation of 2- to 4-fold is generally accepted and based on the geometric mean as shown in Eq. 2.
These normalization factors are then used to adjust the RQ values of the genes accordingly so that the variability is eliminated. Here, we used nine housekeeping genes in our study, and NF varies from 0.398+/−0.031 to 0.946+/−0.103 in our four groups.
Normfinder estimates first variations within the group and then variations between different groups. Having estimated both the intra- and intergroup variation, the algorithm combines the two into a stability value, which adds the two sources of variation and thus represents a practical measure of the systematic error that will be introduced when using the investigated genes [1].
References
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64(15):5245–5250. doi:10.1158/0008-5472.CAN-04-0496
Bustin SA, Benes V, Garson J, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley G, Wittwer CT, Schjerling P, Day PJ, Abreu M, Aguado B, Beaulieu JF, Beckers A, Bogaert S, Browne JA, Carrasco-Ramiro F, Ceelen L, Ciborowski K, Cornillie P, Coulon S, Cuypers A, De Brouwer S, De Ceuninck L, De Craene J, De Naeyer H, De Spiegelaere W, Deckers K, Dheedene A, Durinck K, Ferreira-Teixeira M, Fieuw A, Gallup JM, Gonzalo-Flores S, Goossens K, Heindryckx F, Herring E, Hoenicka H, Icardi L, Jaggi R, Javad F, Karampelias M, Kibenge F, Kibenge M, Kumps C, Lambertz I, Lammens T, Markey A, Messiaen P, Mets E, Morais S, Mudarra-Rubio A, Nakiwala J, Nelis H, Olsvik PA, Perez-Novo C, Plusquin M, Remans T, Rihani A, Rodrigues-Santos P, Rondou P, Sanders R, Schmidt-Bleek K, Skovgaard K, Smeets K, Tabera L, Toegel S, Van Acker T, Van den Broeck W, Van der Meulen J, Van Gele M, Van Peer G, Van Poucke M, Van Roy N, Vergult S, Wauman J, Tshuikina-Wiklander M, Willems E, Zaccara S, Zeka F, Vandesompele J (2013) The need for transparency and good practices in the qPCR literature. Nat Methods 10(11):1063–1067. doi:10.1038/nmeth.2697
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome biology 3 (7):RESEARCH0034
Huggett J, Dheda K, Bustin S, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6(4):279–284. doi:10.1038/sj.gene.6364190
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622. doi:10.1373/clinchem.2008.112797
Seidler RD, Bernard JA, Burutolu TB, Fling BW, Gordon MT, Gwin JT, Kwak Y, Lipps DB (2010) Motor control and aging: links to age-related brain structural, functional, and biochemical effects. Neurosci Biobehav Rev 34(5):721–733. doi:10.1016/j.neubiorev.2009.10.005
Baltes PB, Lindenberger U (1997) Emergence of a powerful connection between sensory and cognitive functions across the adult life span: a new window to the study of cognitive aging? Psychol Aging 12(1):12–21
Ganesh SK, Arnett DK, Assimes TL, Basson CT, Chakravarti A, Ellinor PT, Engler MB, Goldmuntz E, Herrington DM, Hershberger RE, Hong Y, Johnson JA, Kittner SJ, McDermott DA, Meschia JF, Mestroni L, O'Donnell CJ, Psaty BM, Vasan RS, Ruel M, Shen WK, Terzic A, Waldman SA (2013) Genetics and genomics for the prevention and treatment of cardiovascular disease: update: a scientific statement from the American Heart Association. Circulation 128(25):2813–2851. doi:10.1161/01.cir.0000437913.98912.1d
Reitz C, Mayeux R (2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol 88(4):640–651. doi:10.1016/j.bcp.2013.12.024
Hindle JV (2010) Ageing, neurodegeneration and Parkinson's disease. Age Ageing 39(2):156–161. doi:10.1093/ageing/afp223
Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29(1):23–39
Harrison DC, Medhurst AD, Bond BC, Campbell CA, Davis RP, Philpott KL (2000) The use of quantitative RT-PCR to measure mRNA expression in a rat model of focal ischemia—caspase-3 as a case study. Brain Res Mol Brain Res 75(1):143–149
Foldager CB, Munir S, Ulrik-Vinther M, Soballe K, Bunger C, Lind M (2009) Validation of suitable house keeping genes for hypoxia-cultured human chondrocytes. BMC Mol Biol 10:94. doi:10.1186/1471-2199-10-94
Ishitani R, Sunaga K, Hirano A, Saunders P, Katsube N, Chuang DM (1996) Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. J Neurochem 66(3):928–935
Ghani M, Sato C, Rogaeva E (2013) Segmental duplications in genome-wide significant loci and housekeeping genes; warning for GAPDH and ACTB. Neurobiol Aging 34(6):1710 e1711–1714. doi:10.1016/j.neurobiolaging.2012.11.006
Sieber MW, Guenther M, Kohl M, Witte OW, Claus RA, Frahm C (2010) Inter-age variability of bona fide unvaried transcripts normalization of quantitative PCR data in ischemic stroke. Neurobiol Aging 31(4):654–664. doi:10.1016/j.neurobiolaging.2008.05.023
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8(2):R19. doi:10.1186/gb-2007-8-2-r19
Roussel BD, Macrez R, Jullienne A, Agin V, Maubert E, Dauphinot L, Potier MC, Plawinski L, Castel H, Hommet Y, Munuera J, Montaner J, Yepes M, Ali C, Vivien D (2009) Age and albumin D site-binding protein control tissue plasminogen activator levels: neurotoxic impact. Brain 132(Pt 8):2219–2230. doi:10.1093/brain/awp162
Cacquevel M, Launay S, Castel H, Benchenane K, Cheenne S, Buee L, Moons L, Delacourte A, Carmeliet P, Vivien D (2007) Ageing and amyloid-beta peptide deposition contribute to an impaired brain tissue plasminogen activator activity by different mechanisms. Neurobiol Dis 27(2):164–173. doi:10.1016/j.nbd.2007.04.004
Obiang P, Maubert E, Bardou I, Nicole O, Launay S, Bezin L, Vivien D, Agin V (2011) Enriched housing reverses age-associated impairment of cognitive functions and tPA-dependent maturation of BDNF. Neurobiol Learn Mem 96(2):121–129. doi:10.1016/j.nlm.2011.03.004
Bustin SA, Nolan T (2004) Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech: JBT 15(3):155–166
Suzuki T, Higgins PJ, Crawford DR (2000) Control selection for RNA quantitation. BioTechn 29(2):332–337
Orset C, Macrez R, Young AR, Panthou D, Angles-Cano E, Maubert E, Agin V, Vivien D (2007) Mouse model of in situ thromboembolic stroke and reperfusion. Stroke 38(10):2771–2778. doi:10.1161/STROKEAHA.107.487520
Duty S, Jenner P (2011) Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 164(4):1357–1391. doi:10.1111/j.1476-5381.2011.01426.x
Seeman P, Seeman N (2011) Alzheimer's disease: beta-amyloid plaque formation in human brain. Synapse 65(12):1289–1297. doi:10.1002/syn.20957
Forster MJ, Morris P, Sohal RS (2003) Genotype and age influence the effect of caloric intake on mortality in mice. FASEB J : Off Publ Fed Am Soc Exp Biol 17(6):690–692. doi:10.1096/fj.02-0533fje
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Acknowledgments
We would like to thank the Basse-Normandie region for funding Bruckert G and Dr Roussel BD and its credits for the laboratory.
Author information
Authors and Affiliations
Corresponding author
Additional information
Neurodegenerative diseases represent a real challenge for our societies as their frequencies increase with ageing of the population. These diseases are often associated to gene regulation disorders that make their study useful for the comprehension of the disease. Currently, quantitative PCR is the most efficient tool for this kind of studies because it allows a fast and accurate measurement of gene expression levels. However, the reliability of RT-qPCR is influenced by the selection of reference genes, whose expression must be stable in the experimental conditions studied. However, many studies use “common” reference genes such as GAPDH or actin-beta, which have been reported to be unstable in certain conditions. Moreover, Sieber and collaborators have demonstrated the importance of using more than one reference gene to strengthen the exactness of measurements when studying ageing or brain insults such as ischaemia. However, the authors focused on the whole brain instead of studying several structures separately.
By using different algorithms, we identified specific combinations of housekeeping genes in the cortex, the hippocampus, the striatum and the cerebellum, allowing a reliable measurement of gene expression during a longitudinal study in RT-qPCR.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary table 1
An intragroup analysis was performed using the Mann and Whitney non-parametric test (Grap Prism software) on the raw data for each genes studied in the cortex, hippocampus, striatum and cerebellum. *: p < 0.05; **: p < 0.01; ***: p < 0.001. (DOCX 23 kb)
Rights and permissions
About this article

Cite this article
Bruckert, G., Vivien, D., Docagne, F. et al. Normalization of Reverse Transcription Quantitative PCR Data During Ageing in Distinct Cerebral Structures. Mol Neurobiol 53, 1540–1550 (2016). https://doi.org/10.1007/s12035-015-9114-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9114-5