
Abstract
Melatonin and N-acetylserotonin (NAS) are tryptophan metabolites that have potent anti-oxidant, anti-inflammatory and neuroprotective properties in several animal models of neurological injury and disease including multiple sclerosis (MS). The therapeutic effect of NAS has not been reported previously in experimental autoimmune encephalomyelitis (EAE), a commonly used animal model of MS. Using a MOG-peptide induced EAE mouse model we examined the effects of melatonin and NAS on clinical score, inflammatory markers, free radical generation, and sparing of axons, oligodendrocytes and myelin. We found that NAS and melatonin reduced clinical scores when administered prior to or after symptom onset. This effect was more pronounced when melatonin and NAS were administrated prior to symptom onset whereby the appearance of motor symptoms was significantly delayed. Activated microglia and CD4+ T-cells were increased in the white matter of untreated EAE mice, with a return to near control levels after melatonin or NAS treatment. The expression of the NADPH oxidase component p67phox and inducible nitric oxide synthase (iNOS) was increased in the EAE mice as compared with controls, and both drug treated groups had significant reductions in their expression. Melatonin and NAS treatment significantly reduced the loss of mature oligodendrocytes, demyelination and axonal injury. Both compounds also significantly attenuated iNOS induction and reactive oxygen species (ROS) generation in lipopolysaccharide-activated microglia in culture. Our results show for the first time the therapeutic effects of NAS and confirm previous reports on the effectiveness of melatonin in the EAE model of MS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a chronic neuro-inflammatory disease that affects young adults with a prevalence in different populations of 5–200 cases per 100,000 individuals (Milo and Kahana 2010). Cytokines derived from CD4+ T helper (Th) lymphocytes including Th1 and Th17 cells drive the inflammatory process in MS, which is amplified by CNS microglia targeting myelin proteins such as myelin basic protein, myelin oligodendrocyte glycoprotein (MOG) and proteolipid protein. Peroxynitrite, the reaction product of nitric oxide and superoxide, is a powerful oxidant released by reactive microglia (Li et al. 2005) which can be toxic to oligodendrocytes (Li et al. 2011a; Li et al. 2011b). The Th1 cytokines interferon-γ and tumor necrosis factor-α (IFN-γ, TNF-α) and the Th17 cytokines interleukin 17 and 22 (IL-17, IL-22) have been identified in MS lesions (Maddur et al. 2012; Miossec 2009) and are thought to be involved in the disease progression. Opposing the pro-inflammatory actions of Th1 and Th17 lymphocytes, regulatory T cells (Treg) play an inhibitory role in MS especially in the later phases of the disease.
Reactive oxygen and nitrogen species including superoxide anion, hydrogen peroxide, nitric oxide, and peroxynitrite, are produced in response to inflammatory stimuli and have a critical role in tissue damage in MS and experimental autoimmune encephalomyelitis (EAE). Studies from our group and others have previously demonstrated the production and accumulation of high levels of nitric oxide, superoxide and peroxynitrite in spinal cord white matter of EAE mice (Li et al. 2011b; Vana et al. 2011) and in cerebral spinal fluid of MS patients (Calabrese et al. 2002). The increased induction of inducible nitric oxide synthase (iNOS) and activation of NADPH oxidases are thought to be the major sources of nitric oxide and superoxide, which contribute to loss of oligodendrocytes and demyelination (Lin et al. 1993). Melatonin has been shown to reduce oxidative stress, for example it was reported that melatonin stimulates antioxidant enzymes thus acting as an indirect antioxidant (Bonnefont-Rousselot and Collin 2010).
Metabolites in the melatonin and kynurenine pathways of tryptophan catabolism have potent effects on immune system activity and inflammation. In the melatonin pathway, N-acetylserotonin (NAS) and melatonin possess anti-inflammatory and antioxidant actions. Tryptophan derivatives in the kynurenine pathway can have excitotoxic (e.g., quinolinic acid) or anti-inflammatory (e.g., hydroxyanthranilate) actions in the nervous system (Darlington et al. 2010; Stone et al. 2012). The regulation of tryptophan metabolism via melatonin and kynurenine pathways can have significant effects on the progression and remission of inflammatory CNS disorders including MS. Conversion of tryptophan to kynurenine and other downstream metabolites via the rate limiting enzyme indoleamine 2,3-dioxygenase (IDO) is an important regulatory pathway in inflammatory disorders including MS (Kwidzinski et al. 2005; Mancuso et al. 2015). IDO is strongly upregulated by IFN-γ resulting in the production of both immunosuppressive (Sakurai et al. 2002) and neurotoxic (Sundaram et al. 2014) metabolites in the kynurenine pathway of tryptophan catabolism.
Tryptophan enters the melatonin pathway when the enzyme tryptophan hydroxylase converts this essential amino acid to 5-hydroxytryptophan, which can then be sequentially converted to serotonin, NAS and melatonin. In addition to their antioxidant and anti-inflammatory properties, melatonin and NAS are known to act as immune signaling agents that play important roles in MS (Anderson and Rodriguez 2015). In view of the safety and effectiveness via multiple biochemical mechanisms, melatonin has attracted widespread attention in a variety of disease conditions involving inflammation and oxidative conditions, including MS (Chen et al. 2016). As a potent anti-inflammatory and antioxidant agent NAS was found to be effective in experimental models of ischemic injury (Zhou et al. 2014). However, the effectiveness of NAS in the animal model of MS has not been investigated. Here we show that NAS and melatonin have therapeutic potential in EAE with NAS being as effective as melatonin for preventative treatment under our experimental conditions.
Methods
Materials
Fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Invitrogen (Carlsbad, CA). 2′,7′-dichlorofluorescein diacetate (DCF) was purchased from Molecular Probes, Inc. (Eugene, OR). NAS, melatonin and other chemicals and reagents were purchased from Sigma (St. Louis, MO), unless stated otherwise.
Animals
Female 7-week-old C57BL/6 J mice were from Jackson Laboratory (Bar Harbor, ME). Animal care and experimental procedures were carried out in accordance with NIH guidelines and approved by the Uniformed Services University Animal Care and Use Committee. Animals were housed in an environmentally controlled room (20–23 °C, ~44 % humidity, 12 h light/dark cycle, 350–400 lx, lights on at 6:00 am), with food and water available continuously. Animal handling was minimized to reduce animal stress.
Induction of EAE and Clinical Score
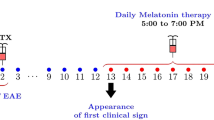
EAE was induced in C57BL/6 mice as we previously reported (Vana et al. 2011; Li et al. 2011b). In brief, following a sterile preparation of the skin, female 8-week-old C57BL/6 J mice were injected subcutaneously with 200 μg MOG 35–55 peptide emulsified in complete Freund’s adjuvant containing 500 μg Mycobacterium tuberculosis (DIFCO). Immediately and 24 h after immunization, mice were administered 200 ng pertussis toxin intraperitoneally (List Biological Labs). At 1 week after induction, EAE mice received a booster of 200 μg of MOG in incomplete Freund’s adjuvant. Animals were monitored blindly and daily by two independent observers and neurological signs were assessed as follows: 0, normal; 1, piloerection, tail weakness; 2, tail paralysis; 3, tail paralysis plus hind limb weakness/paralysis; 4, tail, hind limb and fore limb paralysis; 5, moribund/death, using increment of 0.5 points for intermediate clinical findings. The onset of EAE was defined as the first day when an animal showed a clinical score ≥ 0.5.
Drug Treatment
NAS or melatonin (20 mg/kg i.p.) were administered to one of two groups of mice starting at day 3 after MOG injection (pre-treatment) or at the disease onset at around day 10 after EAE induction (post-treatment) and then once a day until day 28. All drugs were dissolved in 1 % DMSO, which was used as a vehicle control.
Immunostaining
Animals were euthanized using a combination of ketamine and xylazine solution (90 mg ketamine/10 mg xylazine per ml, i.p.), then transcardially perfused with ice cold PBS followed by 4 % paraformaldehyde in the same buffer. Spinal cords were dissected and kept in 4 % paraformaldehyde at 4 °C overnight. Tissues were then transferred into PBS containing 30 % sucrose at 4 °C overnight and stored at −80 °C after being embedded in OCT (Tissue Tek). Transverse sections of lumbar spinal cord were cut at 14 μm using a cryostat (Leica model CM1900) and mounted onto Superfrost Plus slides for immunohistochemistry. Primary antibodies including anti-mouse F4/80 (1:300; eBioscience; Cat# E04273–1635), anti-rabbit iNOS (1:50; Abcam; Cat# ab15323), anti-mouse CD4 (1:100; BD Pharmingen, San Jose, CA; Cat# E04273–1635), anti-mouse APC monoclonal (CC1, 1:100; Calbiochem; Cat# OP80), anti-fluoromyelin (1:1000; Thermo Fisher; Cat# F34651), anti-rabbit NF-200 (1:250; Calbiochem; Cat# AB1989) and anti-p67phox (1:100; BD Pharmingen; Cat# 610,913) were used for immunofluorescence studies. Briefly, the slides were washed twice with PBS and then incubated in PBS containing 1 % donkey serum and 0.3 % Triton X-100 for 30 min at room temperature, followed by respective primary antibody incubation in the same buffer overnight. The slides were then washed with PBS containing 0.2 % Triton X-100 and incubated with Alexa fluor 488 or Alexa fluor 594 conjugated donkey anti-rabbit, mouse or rat secondary antibodies (1:750; Invitrogen, Grand Island, NY) for 1 h. The slides were again washed with PBS twice and covered with Fluoroshield mounting medium including DAPI for nuclear staining. Immunofluorescence images shown in Figs. 2-4 were obtained with a fluorescence microscope (Nikon Eclipse TE-2000 U).
The cells with both DAPI and primary antibody fluorescence were defined as positively stained cells. All immunofluorescence data were obtained in a minimum of 5–7 serial sections from the lumbar spinal cord of the same animal. Negative controls were performed in which the primary antibodies were omitted. For cell counting, the image of whole spinal cord cross-section was assembled from separate images taken under 20× objective and the positively stained cells in spinal cord white matter were counted in each serial section. The data shown in the figures represents the average of the positively stained cells over the entire white matter area as expressed as cell number/mm2.
Cell Culture
Forebrains of postnatal day 2 rats were dissected, dissociated and plated in 75 cm2 flasks containing DMEM plus 20 % FBS and 1 % penicillin/streptomycin. After 10 days, the mixed cultures containing microglia, astrocytes and oligodendrocytes were shaken on an orbital shaker for 1 h at 200 rpm and 37 °C. The loosely attached microglial cells were detached into the medium while astrocytes and oligodendrocytes remain attached to the flask. Microglia in the medium were collected, centrifuged and then plated into 24-well plates at the density of 2 x 105 cells/well and 96-well plates at a density of 5 x 104 cells/well.
Western Blot Analysis
Microglial cultures grown in 96 well plates were pretreated with 0.3 and 3 mM NAS or melatonin for 15 min before the addition of LPS (0.1 μg/ml), and were incubated for an additional 3 h. Cells were washed, and the whole cell lysates from the cultured primary microglial cells were obtained by using ice cold protein lysis buffer (containing 1× Tris buffered saline (TBS), 1 % Nonidet P-40, 0.5 % sodium deoxycholate, 0.1 % SDS, 0.004 % sodium azide) with freshly added protease inhibitor cocktail. The lysates were subject to centrifugation at 10,000 g for 10 min at 4 °C. Five μg of the whole cell lysates were mixed with one-third volume of 4× electrophoresis sample buffer and boiled for 5 min. The samples were separated on Novex 4–12 % Bis-tris gel and protein was transferred to PVDF membrane using a Bio-Rad mini-trans-blot cell. Transferred blots were then blocked by incubating the membrane with 5 % BSA for 1 h at room temperature to reduce non-specific binding. The blocked membrane was incubated with a rabbit anti-iNOS antibody overnight. After washing with TBS containing 1 % Tween 20, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody and washed three times in the same buffer. Finally, the membrane was incubated using a chemiluminescence western blot detection kit from Pierce (Rockford, IL) for 1 min and protein was visualized using Image Reader LAS-3000 software.
Measurement of Reactive Oxygen Species (ROS)
Microglia were grown in 96-well plates and were pre-treated with NAS or melatonin at 0.5 and 2.5 mM for 15 min and then lipopolysaccharide (LPS, 100 ng/ml) was added. Six hours later, wells were washed with Earle’s balanced salt solution (EBSS) three times and incubated with 20 μM DCF in the incubator at 37 °C for 30 min. The generation of ROS was quantified using the Spectra MAX Gemini XS microplate reader with excitation wavelength at 485 nm and emission wavelength at 530 nm at room temperature.
Statistical Analysis
Statistical analyses were performed using GraphPad Instat 3 software (GraphPad Software). Analysis of variance (ANOVA) together with Bonferroni’s multiple comparison post-test was used to compare the differences among the multiple groups. Results were quantified and expressed as means ± standard error of mean (SEM). Statistical difference was defined as p ≤ 0.05.
Results
Tryptophan Metabolites NAS and Melatonin Reduce Clinical Severity of EAE
Mice were observed daily for 28 days after induction of EAE. We observed significant attenuation of the clinical signs in the experimental groups given melatonin or NAS starting on day 3, prior to disease onset (Fig. 1a). Mice treated with NAS showed a significant delay in the appearance of the symptoms compared to the untreated EAE group (p < 0.01). The clinical symptoms in EAE vehicle group mice reached peak on day 17 with the average highest score of 2.7, which remained at the same level until the end of the experiment on day 28. The NAS treated group reached peak clinical scores on day 20 with the average score of 1.4. The highest score was only maintained for three days and then was slightly reduced. From days 12 to 28, the clinical scores in the NAS treated group were significantly lower compared to those in the EAE vehicle group. Consistently, EAE mice treated with melatonin displayed dampened clinical symptoms. From days 15 to 18 and days 22 to 28, treated mice had significantly reduced clinical scores with the highest average score of 2. In the pre-treatment group NAS was somewhat more effective at delaying and reducing clinical signs as compared with melatonin. The therapeutic effect of NAS and melatonin was also observed in mice treated after the onset of clinical signs (Fig. 1b). Treatment with NAS and melatonin after symptom onset (day 10 after induction) improved motor function, but the effect was not as great as seen with the groups pre-treated with the two compounds. The clinical scores were significantly reduced from days 22 to 28 after EAE induction in groups treated with both melatonin and NAS with an average score difference between vehicle and drug treated groups of 1 (p < 0.05).

Both NAS and melatonin ameliorated clinical severity of EAE. Melatonin or NAS were administrated on day 3 after EAE induction and the clinical scores were recorded based on the reported criteria. Data for clinical score are means ± SEM for 12 mice per group. The score in the pre-treatment melatonin group (blue line) was significantly lower than the untreated (vehicle) EAE group (red line, *p < 0.05). The clinical score in the pre-NAS treated group (black line) was also significantly lower than that in the EAE vehicle group (*p < 0.05, **p < 0.01, ***p < 0.001) (a). After the onset of clinical signs on day 10, melatonin or NAS were administrated. The clinical scores in the post-treatment of melatonin (blue line) and NAS (black line) were significantly lower than that in the EAE vehicle group, (*p < 0.05 NAS treated group from day 23 to 28; melatonin treated group from day 22 to 28) (b)
Inflammatory Infiltrates within Spinal Cord during EAE Are Diminished in the NAS and Melatonin Treated Mice
The progression of MS, as well as that of EAE, is mediated by the infiltration of T- and B-lymphocytes and the activation of microglia and macrophages. The peripherally expanded cells infiltrate into CNS and release inflammatory cytokines, ROS and nitric oxide that contribute to demyelination and axonal degeneration. We investigated the effectiveness of post-treatment with NAS or melatonin (beginning after symptom onset) in EAE mice on reducing lymphocyte infiltration and microglial activation in spinal cord. We examined lumbar spinal cord sections from 28 day control mice and vehicle and drug treated EAE mice with immunofluorescence histochemistry using an anti-F4/80 antibody that recognizes reactive microglia and an anti-CD4 antibody that labels CD4+ T lymphocytes. We found increased activated microglia and CD4+ T-cells in the untreated EAE spinal cord white matter in comparison to the control mice (Fig. 2a). The cell counts for F4/80 positive cells in the lumber spinal cord white matter of EAE vehicle group were 359 ± 43 cells/mm2 (Fig. 2b) and the number of CD4+ T cells was 477 ± 80 cells/mm2 (Fig. 2a and b). These values were significantly greater than those in the melatonin and NAS treated groups. The number of F4/80 positive microglia in the melatonin and NAS treated EAE groups was 83 ± 28 cells/mm2, 102 ± 26 cells/mm2, respectively. The positive stained CD4+ T cell in melatonin and NAS treated group was also significantly decreased, with 33 ± 17 cells/mm2 and 23 ± 14 cells/mm2 respectively (Fig. 2a and c). Overall, post-treatment with NAS or melatonin significantly reduced microglial activation and lymphocyte infiltration in EAE mouse spinal cord.

Melatonin or NAS post-treatment reduced microglial activation and lymphocyte infiltrates in the spinal cord white matter of EAE mice. Immunofluorescence staining was done on transverse sections from mouse lumbar spinal cord 28 days after EAE induction (a). The micron bar =100 μm. The numbers of F4/80 and CD4+ positive cells in the vehicle-treated EAE mice were greater than that in melatonin and NAS treatment groups. The F4/80 and CD4+ positive cell counts are given in (***p < 0.001, n = 12) (b, c)
NAS and Melatonin Are Effective at Reducing Oxidative and Nitrative Stress in EAE
We next examined whether NAS and melatonin can regulate expression of enzymes involved in generating reactive oxygen and nitrogen species. NADPH oxidases generate superoxide anion and are composed of several subunits, including p67phox which acts as the activator subunit (Pick 2014). Staining for p67phox was very low in the spinal cord of control mice. In spinal cord tissues of the experimental EAE mice there was increased immunofluorescence staining with anti-p67phox antibody (Fig. 3a). In the spinal cord white matter of the EAE vehicle group p67phox positively stained cells numbered 183 ± 24 cells/mm2, whereas in the melatonin and NAS treated EAE groups significantly lower numbers of p67phox positive cells were seen, with cell counts of 32 ± 8 cells/mm2 and 43 ± 11 cells/mm2 respectively (Figs. 3a, b). Similarly, the increased expression of iNOS in the spinal cord white matter of EAE mice was also greatly reduced by NAS and melatonin treatments. In the EAE-vehicle group the positively stained iNOS cells with the average number of 90 ± 13 cells/mm2 were much greater than those in the melatonin and NAS treated EAE groups. The numbers of positive iNOS cells in the treated group were 12 ± 5 cells/mm2 in melatonin treated group and 9 ± 4 cells/mm2 in the NAS treated group (Figs. 3a, c). These results show that NAS and melatonin exert inhibitory actions on the expression and activation NADPH oxidase and iNOS in EAE mice.

Melatonin and NAS down-regulated the expression of NADPH oxidase component p67phox and inducible nitric oxide synthase (iNOS) in the spinal cord white matter of EAE mice. Immunohistochemistry was performed with mouse lumbar spinal cord 28 days after EAE induction with antibodies against p67phox and iNOS (a). The micron bar =100 μm. The expression of p67phox and iNOS were both significantly decreased in the melatonin and NAS treated groups, (***, p < 0.001, n = 12) (b, c)
NAS and Melatonin Protect EAE Mice from Neurodegeneration
Having shown that NAS and melatonin exhibit beneficial anti-oxidative and anti-inflammatory effects in the EAE model, we then investigated the neuroprotective effects of NAS and melatonin on axonal damage and inflammatory demyelination. We compared the neurofilament 200 (NF-200) positive profile and the extent of myelination in the spinal cords of treated and untreated EAE mice. We employed anti-NF200 antibody and FluoroMyelin™ Green Fluorescent myelin stain for this purpose. In the EAE-vehicle group, there was approximately 3.5 % NF-200 positive stain area in spinal cord as compared to 10 % NF-200 positive stain area in the control animals. In the melatonin and NAS treated EAE animals increased NF-200 positive staining was observed. With the treatment, the NF-200 positive stain area increased to 8.3 % (melatonin) and 9.6 % (NAS), indicating less axonal injury (Figs. 4a, b). Compared to the 5.1 % of the myelinated spinal cord areas in the EAE vehicle group, melatonin and NAS treated EAE groups presented 11.2 % and 12.8 % myelinated areas, which were significantly greater than the myelinated spinal cord areas in the EAE vehicle group and were closer to the 14.6 % myelinated area observed in the control group (Fig. 4a, c). These results demonstrate the neuroprotective effects of melatonin and NAS on EAE mice.

Administration of melatonin and NAS protected neurons from damage and demyelination in the spinal cord white matter of EAE mice. Immunofluorescence staining was performed with transverse sections of lumbar spinal cord tissue from the mice 28 days after EAE induction. Sections were stained with anti-neurofilament 200 and Myelin-Fluoro (a). The micron bar =100 μm. Axon loss and demyelination were observed in the untreated EAE mice. Greatly reduced axon loss and less demyelination were seen in the EAE mice with melatonin and NAS treatment. Neurofilament 200-positive area and myelinated area were measured and showed in comparison to the whole spinal cord area (**p < 0.01; ***p < 0.001, n = 12) (b, c). The marker of mature oligodendrocyte CC1 was used to evaluate the number of mature oligodendrocytes in the EAE and melatonin and NAS treated groups (A). The positive stained cells in the treated group were significantly more numerous than that in the EAE vehicle group. The number of positive stained cells in the different treated groups is shown, (*p < 0.01, ***p < 0.001, n = 12) (d)
The loss of oligodendrocytes in MS patients leads to demyelination and motor and cognitive deficits. CC1 is a marker to detect mature, myelin producing oligodendrocytes in the CNS. We further investigated the degree of oligodendrocyte loss in the spinal cord white matter with anti-CC1 antibody in the spinal cord tissue from EAE mice. The results showed that compared to the control mice there was remarkably reduced CC1 positive staining in the spinal cord white matter of the vehicle-treated EAE mice (Fig. 4a). Administration of melatonin and NAS attenuated the loss of oligodendrocytes with spinal cord sections from the treated mice showing similar CC1 positive staining to that in the control mice (Fig. 4d). Overall, our immunofluorescence findings indicate that NAS and melatonin have significant anti-inflammatory, anti-oxidant and neuroprotective effects in the EAE mouse model.
NAS and Melatonin Attenuate Oxidative Stress in Primary Microglial Culture
To further explore the mechanism underlying NAS and melatonin anti-oxidative function, primary microglia were activated by LPS in the presence of NAS or melatonin to evaluate their roles on the production of reactive oxidative spices (ROS) and iNOS. We pre-treated primary microglia cells with NAS or melatonin at concentrations of 0.3 and 3 mM starting at 30 min before and then 6 h after the start of LPS exposure. The iNOS production was determined by Western Blot. No iNOS expression was observed in microglial cells that were not treated with LPS (Fig. 5a). Treatment of LPS-stimulated microglia with NAS or melatonin at a concentration of 0.3 mM did not significantly affect iNOS expression. However, treatment with NAS or melatonin at 3 mM significantly inhibited the production of iNOS, and melatonin was more effective at this concentration than NAS. Relative band intensity analysis in LPS-treated microglia showed a 4-fold reduction in iNOS expression with 3 mM melatonin and a 2-fold reduction with 3 mM NAS (Fig. 5b). These observations demonstrate that tryptophan metabolites can modulate microglial iNOS induction.

NAS and melatonin reduced iNOS and ROS expression in primary microglial culture. Primary microglial cells were treated with LPS (0.1 μg/ml) for 6 h in the presence of different concentrations of melatonin or NAS. Western blots showed that melatonin at 3 mM almost completely blocked iNOS induction and 3 mM NAS halved iNOS expression compared to the untreated LPS-activated microglia (a). The ratio of the relative band intensity of iNOS to β-actin from 4 different experiments is shown. Significant reductions in iNOS induction were obtained when the NAS and melatonin treated groups were compared to the LPS group (*p < 0.05, ***p < 0.001). (b). Primary microglial cells cultured in 96-well plates were treated with LPS (0.1 μg/ml) for 3 h in the presence of melatonin or NAS as indicated. The fluorescence from each well was obtained using the Spectra MAX Gemini XS microplate reader with excitation wavelength at 485 nm and emission wavelength at 530 nm. Melatonin and NAS dose dependently blocked ROS generation in reactive microglia. Significant reductions were obtained when the LPS alone treated group were compared to the drug treated groups (*p < 0.05, **p < 0.01, ***p < 0.001). (c). A representative experiment of four that were performed is shown
To determine if ROS generation was also affected by the tryptophan metabolites we used the dichlorofluorescein assay in primary microglial cultures. Microglia were cultured in 96-well plates and treated with different doses of NAS or melatonin for 30 min, followed by 6 h LPS stimulation. Figure 5c shows that with LPS stimulation, primary microglia produced almost three times more ROS than untreated cells. Both NAS and melatonin treatment attenuated ROS generation in a dose dependent manner. At a concentration of 0.5 mM NAS and melatonin decreased ROS generation by ~15 % relative to untreated LPS-stimulated microglia. At a concentration of 2.5 mM NAS and melatonin reduced ROS generation in LPS-activated primary microglia cells by approximately 40 %. These findings indicate that NAS and melatonin reduce both iNOS expression and ROS generation in LPS-activated microglial cultures.
Discussion
MS is the most common inflammatory demyelinating disease of the central nervous system. Increasing evidence has demonstrated that inflammation and ROS play significant roles in the pathogenesis of MS and the murine model, EAE. Thus, treatment with anti-inflammatory and antioxidant biologics should reduce tissue damage and improve neurological outcomes. NAS, the immediate biochemical precursor of melatonin, has previously been shown to have oncostatic, (Blask and Hill 1986; Sanchez-Barcelo et al. 2012) immune modulatory (Guerrero and Reiter 2002; Carrillo-Vico et al. 2005) and anti-inflammatory effects (Cuzzocrea and Reiter 2002). NAS also has neuroprotective properties through the activation the tropomyosin receptor kinase B (TrkB) receptor, whereas melatonin does not activate this pathway (Jang et al. 2010). Melatonin has both immune enhancing and inhibiting effects depending on the state of immune activation and the dose applied (reviewed in Carrillo-Vico et al. 2013). Melatonin has been identified as a powerful direct free radical scavenger (Reiter et al. 2007) and indirect antioxidant (Reiter et al. 2003; Reiter et al. 2004). Melatonin is also effective in reducing infiltration of Th1/Th17 lymphocytes in EAE and increasing Treg frequency and improving clinical scores (Alvarez-Sanchez et al. 2015; Chen et al. 2016), but the efficacy of NAS in the treatment of MS has not been investigated. In order to examine the therapeutic effects of these tryptophan metabolites, we treated MOG peptide-induced EAE mice with NAS or melatonin at a dose of 20 mg/kg and monitored clinical progression and pathological outcomes. This dose was intermediate between the doses of melatonin used in two previous studies. Kashani et al. found that 5 mg/kg melatonin was effective in reducing demyelination in the cuprizone model of MS (Kashani et al. 2014), whereas Chen and colleagues found that melatonin administered at 200 mg/kg was effective at suppressing the expression of pro-inflammatory cytokines such as INF- and IL-17 in the MOG peptide model of MS (Chen et al. 2016). These doses far exceed the physiological concentrations of melatonin recorded from serum in control and MS patients (Farhadi et al. 2014) but considering the well-established safety profile of melatonin, higher doses may be required to elicit the full range of antioxidant and anti-inflammatory actions.
In the present studies we found significant and persistent improvement with melatonin or NAS treatment in EAE clinical scores based on motor function (Fig. 1). Overall, pre-treatment with melatonin or NAS, prior to the onset of symptoms, provided better protection from motor dysfunction. Significantly reduced iNOS and p67phox immunoreactivity was observed in spinal cord white matter in the melatonin and NAS treated as compared to vehicle treated EAE mice (Fig. 3) suggesting an attenuated inflammatory response and reduced reactive nitrogen species formation. Both tryptophan metabolites also significantly increased NF-200, myelin and CC1 staining relative to vehicle treated EAE mice indicating reduced oligodendrocyte and axonal loss (Fig. 4). Melatonin and NAS also significantly reduced staining for nitrotyrosine, a marker of peroxynitrite formation in the EAE spinal cord white matter (data not shown). Consistently we found that both NAS and melatonin treatment attenuated the expression of iNOS and the generation of ROS in microglial cell cultures after LPS treatment. These results show that these two tryptophan metabolites in the serotonin pathway can have therapeutic effects in EAE via anti-oxidative and immunomodulatory mechanisms.
Previously it has been hypothesized that NAS may have utility in the treatment of MS (Anderson and Rodriguez 2015) and our report confirms its effectiveness in an MS model. The NAS pre-treated group had a delayed onset of motor dysfunction and displayed no hind-limb paresis (see Fig. 1). Both melatonin and NAS treated groups had persistently lower clinical scores compared to the untreated EAE group whether administered before or after symptom onset. The effectiveness of melatonin in the EAE model of MS has been reported in the two previous studies noted above (Chen et al. 2016; Kashani et al. 2014). In the more recent study, both severity of the disease and incidence were reduced without altering the onset of the disease. Also, the maximum clinical sign and cumulative scores were significantly lower in the melatonin treated group (Chen et al. 2016). Another study demonstrated that melatonin levels are negatively correlated with MS relapses in humans and the therapeutic effects afforded by melatonin may be due to its suppression of Th17 cell differentiation and the enhanced production of IL-10 from regulatory T cells (Farez et al. 2015). Our observation that NAS showed somewhat greater effectiveness in reducing clinical scores than melatonin when given prior to the disease onset suggests that NAS may have better inhibitory effects on Th17 differentiation. Melatonin given at doses up to 300 mg per day was found to improve primary progressive MS in a 28 year old patient who failed to respond to the standard treatment (Lopez-Gonzalez et al. 2015).
We used CD4 and F4/80 as markers for inflammatory effects and found that both melatonin and NAS suppressed the inflammatory responses (Fig. 2). Melatonin has been found to have anti-inflammatory effects in a variety of both central and peripheral disease conditions involving immune activation and our results are consistent with those observations (Alvarez-Sanchez et al. 2015; Esposito and Cuzzocrea 2010; Carrillo-Vico et al. 2005). An earlier study in the rat model of MS showed that the protective effects of melatonin are associated with reduced inflammatory infiltration due to decreased vascular ICAM-1 expression in the spinal cord (Kang et al. 2001). Melatonin and NAS have also been shown to inhibit leukotriene B4-induced leukocyte endothelial adhesion (Lotufo et al. 2001). A recent study examined the immune-suppressive effects of melatonin in the EAE model and showed that melatonin has multiple effects (Alvarez-Sanchez et al. 2015). For example, melatonin was found to down-regulate the Th1/Th17 effector immune responses by decreasing both the number of these T cell populations as well as the cytokines synthesized by them. Also, melatonin was found to enhance the number of Treg cells in the CNS which have an immune suppressive phenotype via the membrane receptor CD44. While not much is known about the immune suppressive action of NAS, it is reasonable to think that NAS also acts through similar mechanisms.
We employed iNOS as a marker for oxidative stress effects in microglial cultures and found that both melatonin and NAS reduced iNOS expression and ROS generation effectively (Fig. 5). A recent study using in vitro and in vivo models of ischemic injury has shown that NAS provides neuroprotection by inhibiting mitochondrial death pathways and autophagic activation (Ding et al. 2015). In this study NAS did not appear to act via any known receptor, including the TrkB receptor, in the in vivo models of ischemic injury. Recent observations that NAS can protect against oxidative stress injury caused by hydrogen peroxide in a hepatic cell line and acute hepatic ischemia-reperfusion injury in mice are consistent with the receptor independent action of NAS (Yu et al. 2013). A recent theoretical study has compared the effects of NAS, hydroxymelatonin and melatonin against oxidative stress and concluded that NAS and hydroxymelatonin are better peroxyl radical scavengers than melatonin (Alvarez-Diduk et al. 2015). These authors propose that NAS and hydroxymelatonin derived from melatonin via metabolism during melatonin treatment could contribute to the protective effects of melatonin administration. They also stress that the phenolic moiety of NAS and hydroxymelatonin might play a major role in the anti-oxidative stress effects.
As much as 10–15 % of the administered melatonin could be demethylated to NAS and it is therefore likely that part of the protective effects of melatonin administration could be mediated by NAS. It is possible that some NAS and melatonin synthesis occurs throughout the brain, not necessarily only in the pineal, and that the progression of MS could be influenced by their local concentrations. Induction of the kynurenine pathway of tryptophan metabolism in MS can have immunosuppressive, oxidative or excitotoxic actions depending on which metabolites are generated (Stone et al. 2013; Schwarcz 2016; Stone et al. 2012). In fact, IFN-γ strongly induces indoleamine dioxygenase in immune cells and drives tryptophan metabolism via the kynurenine pathway and away from the serotonin pathway, potentially reducing production of serotonin, NAS and melatonin (Fig. 6). Some of the clinical symptoms of MS such as depression are thought to be caused by this shifting of tryptophan metabolism away from serotonin, NAS and melatonin. Melatonin also inhibits demyelination and increases remyelination and therefore its local regulation by serotonin availability in white matter glia could play a role in the etiology, course and treatment of MS (Anderson and Rodriguez 2015).

Pathways of tryptophan catabolism in the CNS. The tryptophan catabolizing enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan hydroxylase-2 (TPH2) are expressed in the CNS, and they can compete for available tryptophan during an inflammatory immune response such as that in MS. IDO converts tryptophan (Trp) to N-formal kynurenine (f-Kyn), which is the first metabolite in the kynurenine pathway. Tryptophan hydroxylase-2 (TPH-2) converts tryptophan to 5-hydroxytryptophan (5-HTP) which is the first metabolite in the melatonin pathway. Both pathways produce anti-inflammatory metabolites. However, the kynurenine pathway also generates quinolinate, which has excitotoxic actions in the CNS
In conclusion, we found that NAS is as effective as melatonin in reducing clinical signs, free radical generation and iNOS expression as well as reducing oligodendrocyte, axonal and myelin loss in the EAE model of MS. It is of important to note that both NAS and melatonin belong to a library of 1040 FDA approved compounds selected by NINDS/NIH for further studies. Based on the lack of toxicity, easy access to the CNS across the blood brain barrier and the demonstrated effects in the EAE model by us and previous investigators and the recent case report of improvement in a primary progressive MS in a 28 year old patient (Lopez-Gonzalez et al. 2015), clinical trials with proper placebo controls are warranted for both NAS and melatonin.
References
Alvarez-Diduk R, Galano A, Tan DX, Reiter RJ (2015) N-acetylserotonin and 6-hydroxymelatonin against oxidative stress: implications for the overall protection exerted by melatonin. J Phys Chem B 119:8535–8543
Alvarez-Sanchez N, Cruz-Chamorro I, Lopez-Gonzalez A, Utrilla JC, Fernandez-Santos JM, Martinez-Lopez A, Lardone PJ, Guerrero JM, Carrillo-Vico A (2015) Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain Behav Immun 50:101–114
Anderson G, Rodriguez M (2015) Multiple sclerosis: the role of melatonin and N-acetylserotonin. Mult Scler Relat Disord 4:112–123
Blask DE, Hill SM (1986) Effects of melatonin on cancer: studies on MCF-7 human breast cancer cells in culture. J Neural Transm Suppl 21:433–449
Bonnefont-Rousselot D, Collin F (2010) Melatonin: action as antioxidant and potential applications in human disease and aging. Toxicology 278:55–67
Calabrese V, Scapagnini G, Ravagna A, Bella R, Foresti R, Bates TE, Giuffrida Stella AM, Pennisi G (2002) Nitric oxide synthase is present in the cerebrospinal fluid of patients with active multiple sclerosis and is associated with increases in cerebrospinal fluid protein nitrotyrosine and S-nitrosothiols and with changes in glutathione levels. J Neurosci Res 70:580–587
Carrillo-Vico A, Guerrero JM, Lardone PJ, Reiter RJ (2005) A review of the multiple actions of melatonin on the immune system. Endocrine 27:189–200
Carrillo-Vico A, Lardone PJ, Alvarez-Sanchez N, Rodriguez-Rodriguez A, Guerrero JM (2013) Melatonin: buffering the immune system. Int J Mol Sci 14:8638–8683
Chen SJ, Huang SH, Chen JW, Wang KC, Yang YR, Liu PF, Lin GJ, Sytwu HK (2016) Melatonin enhances interleukin-10 expression and suppresses chemotaxis to inhibit inflammation in situ and reduce the severity of experimental autoimmune encephalomyelitis. Int Immunopharmacol 31:169–177
Cuzzocrea S, Reiter RJ (2002) Pharmacological actions of melatonin in acute and chronic inflammation. Curr Top Med Chem 2:153–165
Darlington LG, Forrest CM, Mackay GM, Smith RA, Smith AJ, Stoy N, Stone TW (2010) On the biological importance of the 3-hydroxyanthranilic acid: anthranilic acid ratio. Int J Tryptophan Res 3:51–59
Ding K, Xu J, Wang H, Zhang L, Wu Y, Li T (2015) Melatonin protects the brain from apoptosis by enhancement of autophagy after traumatic brain injury in mice. Neurochem Int 91:46–54
Esposito E, Cuzzocrea S (2010) Antiinflammatory activity of melatonin in central nervous system. Curr Neuropharmacol 8:228–242
Farez MF, Mascanfroni ID, Mendez-Huergo SP, Yeste A, Murugaiyan G, Garo LP, Balbuena, Aguirre ME, Patel B, Ysrraelit MC, Zhu C, Kuchroo VK, Rabinovich GA, Quintana FJ, Correale J (2015) Melatonin Contributes to the Seasonality of Multiple Sclerosis Relapses. Cell 162:1338–1352
Farhadi N, Oryan S, Nabiuni M (2014) Serum levels of melatonin and cytokines in multiple sclerosis. Biomed J 37:90–92
Guerrero JM, Reiter RJ (2002) Melatonin-immune system relationships. Curr Top Med Chem 2:167–179
Jang SW, Liu X, Pradoldej S, Tosini G, Chang Q, Iuvone PM, Ye K (2010) N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc Natl Acad Sci U S A 107:3876–3881
Kang JC, Ahn M, Kim YS, Moon C, Lee Y, Wie MB, Lee YJ, Shin T (2001) Melatonin ameliorates autoimmune encephalomyelitis through suppression of intercellular adhesion molecule-1. J Vet Sci 2:85–89
Kashani IR, Rajabi Z, Akbari M, Hassanzadeh G, Mohseni A, Eramsadati MK, Rafiee K, Beyer C, Kipp M, Zendedel A (2014) Protective effects of melatonin against mitochondrial injury in a mouse model of multiple sclerosis. Exp Brain Res 232:2835–2846
Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, Nitsch R, Bechmann I (2005) Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J 19:1347–1349
Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA (2005) Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A 102:9936–9941
Li S, Lin W, Tchantchou F, Lai R, Wen J, Zhang Y (2011a) Protein kinase C mediates peroxynitrite toxicity to oligodendrocytes. Mol Cell Neurosci 48:62–71
Li S, Vana AC, Ribeiro R, Zhang Y (2011b) Distinct role of nitric oxide and peroxynitrite in mediating oligodendrocyte toxicity in culture and in experimental autoimmune encephalomyelitis. Neuroscience 184:107–119
Lin RF, Lin TS, Tilton RG, Cross AH (1993) Nitric oxide localized to spinal cords of mice with experimental allergic encephalomyelitis: an electron paramagnetic resonance study. J Exp Med 178:643–648
Lopez-Gonzalez A, Alvarez-Sanchez N, Lardone PJ, Cruz-Chamorro I, Martinez-Lopez A, Guerrero JM, Reiter RJ, Carrillo-Vico A (2015) Melatonin treatment improves primary progressive multiple sclerosis: a case report. J Pineal Res 58:173–177
Lotufo CM, Lopes C, Dubocovich ML, Farsky SH, Markus RP (2001) Melatonin and N-acetylserotonin inhibit leukocyte rolling and adhesion to rat microcirculation. Eur J Pharmacol 430:351–357
Maddur MS, Miossec P, Kaveri SV, Bayry J (2012) Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol 181:8–18
Mancuso R, Hernis A, Agostini S, Rovaris M, Caputo D, Fuchs D, Clerici M (2015) Indoleamine 2,3 dioxygenase (IDO) expression and activity in relapsing-remitting multiple sclerosis. PLoS One 10:e0130715
Milo R, Kahana E (2010) Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev 9:A387–A394
Miossec P (2009) IL-17 and Th17 cells in human inflammatory diseases. Microbes Infect 11:625–630
Pick E (2014) Role of the rho GTPase Rac in the activation of the phagocyte NADPH oxidase: outsourcing a key task. Small GTPases 5:e27952
Reiter RJ, Tan DX, Mayo JC, Sainz RM, Leon J, Czarnocki Z (2003) Melatonin as an antioxidant: biochemical mechanisms and pathophysiological implications in humans. Acta Biochim Pol 50:1129–1146
Reiter RJ, Tan DX, Gitto E, Sainz RM, Mayo JC, Leon J, Manchester LC, Vijayalaxmi, Kilic E, Kilic U (2004) Pharmacological utility of melatonin in reducing oxidative cellular and molecular damage. Pol J Pharmacol 56:159–170
Reiter RJ, Tan DX, Terron MP, Flores LJ, Czarnocki Z (2007) Melatonin and its metabolites: new findings regarding their production and their radical scavenging actions. Acta Biochim Pol 54:1–9
Sakurai K, Zou JP, Tschetter JR, Ward JM, Shearer GM (2002) Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J Neuroimmunol 129:186–196
Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C, Reiter RJ (2012) Melatonin uses in oncology: breast cancer prevention and reduction of the side effects of chemotherapy and radiation. Expert Opin Investig Drugs 21:819–831
Schwarcz R (2016) Kynurenines and glutamate: multiple links and therapeutic implications. Adv Pharmacol 76:13–37
Stone TW, Forrest CM, Darlington LG (2012) Kynurenine pathway inhibition as a therapeutic strategy for neuroprotection. FEBS J 279:1386–1397
Stone TW, Stoy N, Darlington LG (2013) An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci 34:136–143
Sundaram G, Brew BJ, Jones SP, Adams S, Lim CK, Guillemin GJ (2014) Quinolinic acid toxicity on oligodendroglial cells: relevance for multiple sclerosis and therapeutic strategies. J Neuroinflammation 11:204
Vana AC, Li S, Ribeiro R, Tchantchou F, Zhang Y (2011) Arachidonyl trifluoromethyl ketone ameliorates experimental autoimmune encephalomyelitis via blocking peroxynitrite formation in mouse spinal cord white matter. Exp Neurol 231:45–55
Yu S, Zheng J, Jiang Z, Shi C, Li J, Du X, Wang H, Jiang J, Wang X (2013) Protective effect of N-acetylserotonin against acute hepatic ischemia-reperfusion injury in mice. Int J Mol Sci 14:17680–17693
Zhou H, Wang J, Jiang J, Stavrovskaya IG, Li M, Li W, Wu Q, Zhang X, Luo C, Zhou S, Sirianni AC, Sarkar S, Kristal BS, Friedlander RM, Wang X (2014) N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. J Neurosci 34:2967–2978
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
Grant Support
this work was supported by grants from the National Multiple Sclerosis Society (RG3741 to YZ), the Defense Medical Research and Development Program (0130–15–0003-00002 to YZ), the Uniformed Services University (R0701Z to YZ) and the NINDS at the NIH (NS084206 to AN).
Additional information
Jie Wen, Prasanth S. Ariyannur, Rachel Ribeiro are Equal Contribution
Rights and permissions
About this article

Cite this article
Wen, J., Ariyannur, P.S., Ribeiro, R. et al. Efficacy of N-Acetylserotonin and Melatonin in the EAE Model of Multiple Sclerosis. J Neuroimmune Pharmacol 11, 763–773 (2016). https://doi.org/10.1007/s11481-016-9702-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-016-9702-9