
Abstract
Multiple sclerosis (MS) is a devastating chronic autoimmune demyelinating disease of the central nervous system (CNS), thought to affect more than 2.5 million people worldwide. Regulation of the sleep-wake cycle might influence disease activity and the frequency of relapses in patients. As melatonin (or sleep hormone) involves the regulation of circadian rhythms, much attention has been paid to the management of MS symptoms with melatonin. This review describes the pharmacological mechanisms underlying the neuroprotective effects of melatonin and recent clinical evidence from MS patients. Apparent risks and benefits of melatonin therapies are also discussed. Various in vivo and clinical data presented in this up-to-date review suggest that melatonin may possibly possess a protective role against the behavioral deficits and neuropathological characteristics of MS. Multiple mechanisms of the neuroprotective effects of melatonin such as mitochondrial protection and antioxidant, anti-inflammatory, and anti-apoptotic properties, as well as its anti-demyelinating function are also discussed. A large body of evidence shows that melatonin potently regulates the immune system, demyelination, free radical generation, and inflammatory responses in neural tissue, which are mediated by multiple signal transduction cascades. In the present article, we focus on different pathways that are targeted by melatonin to prevent the development and progression of MS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Multiple sclerosis (MS) is a disabling neuroinflammatory disease affecting the central nervous system (CNS), particularly in young adults. |
Melatonin is the main secretory production of the pineal gland, which activates the intracellular pathways and changes several gene expressions; it has shown noticeable effects on reducing development of MS. |
Alterations of melatonin in cellular pathways lead to the suppression of oxidative damage, inflammatory responses, apoptosis, and demyelination; melatonin is also able to pass through the blood–brain barrier, suggesting a potential role for melatonin as a safe agent for protecting neurons and their supportive cells against MS. |
1 Introduction
Multiple sclerosis (MS) is a chronic neuroinflammatory demyelinating disease of the human central nervous system (CNS) that is characterized by episodic reversible symptomatic attacks of demyelinating neurological deficits, causing disruption in neuronal signaling, axonal function, and physical disability in young to middle-aged individuals, particularly women [1, 2]. MS commonly shows seasonality in the incidence of flare-ups—its symptoms are more likely to occur in spring/summer than in fall/winter, and also are more frequent in northern than in southern climates. MS is supposed to be an immune-mediated disorder, yet the antigen specificity of the immune response has not been identified [3]. The precise etiology of MS is unknown; however, epidemiological data confirmed that both environmental and genetic factors are major contributors [3]. A series of studies demonstrated that the formation of lesions in the CNS, inflammation, and the destruction of myelin sheaths are the main hallmarks of MS. The pathophysiology of MS can be divided into two subsets: the inflammatory processes and the degenerative processes that affect both white and gray matter [4]. In brief, MS lesions are formed by immune-mediated dysregulation of the blood–brain barrier, facilitating the transmission of activated inflammatory cells into the brain and spinal cord in which focal lymphocytic infiltration allows inflammation, demyelination, gliosis, and neuronal signaling disruption, and eventually causes neuronal and axonal degeneration within the CNS [5,6,7]. The pineal gland is a neuroendocrine transducer that receives photoperiodic information from the retina and circadian superchiasmatic nucleus oscillator, enabling mammals to respond to the annual changes in photoperiods by the activation/inactivation of the reproductive axis [8,9,10,11,12,13,14]. Melatonin as the most important secretory product of the pineal gland has radical scavenging and antioxidant properties; it is well tolerated and safe, thus it has been proposed as a neuroprotective agent against brain ischemia [15]. Examination of possible beneficial effects of melatonin, or melatonin analogs, in treating or preventing MS flare-ups opens a new chapter in melatonin neuropharmacology, in which it could be prescribed either in lower doses given as a “replacement therapy” or in larger doses as a medicine. Although melatonin is available on the market, it has noteworthy drawbacks, including short-term feelings of depression, unwanted drowsiness, and stomach cramps. Melatonin stimulates the activities of superoxide dismutase (SOD) and glutathione peroxidase (GPx), but inhibits the pro-oxidant enzyme nitric oxide synthase (NOS), thus it could be a very powerful antioxidant molecule. The production of melatonin decreases with age [16], and maintaining high levels of melatonin could slow age- and cancer-related changes [17, 18].
2 Methodology of the Literature Search
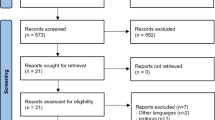
Databases including the Cochrane library, PubMed, and Scopus were searched with the keywords ‘Multiple sclerosis’, ‘MS’, and ‘Neurodegenerative disease’ in the title/abstract and ‘Melatonin’ in the whole text in order to find relevant citations. Data were collected from the year 1964 until 2018 (December), and only English-language articles were included. A total number of 4638 records were collected, and 25 relevant studies assessing the efficacy of melatonin in clinical trials in patients with MS and animal models of MS were used in the main structure of this review (Fig. 1).

PRISMA diagram. Of 4102 articles evaluated by group consensus, 4077 articles were excluded because they did not meet the inclusion criteria
3 Melatonin Biosynthesis and Bioavailability
In mammalians, melatonin is produced in the pineal gland in the dark phase of the light/dark cycle and it is promptly distributed to the tissues via the blood circulation. It acts as a circadian signal used by the superchiasmatic nucleus clock to send the circadian message to the melatonin target structures (containing melatonin receptors). Melatonin also has a chronobiotic effect, in that it directly influences the superchiasmatic nucleus that contains melatonin-R [19] to affect the circadian clock [20]. Melatonin is also synthesized peripherally in the retina, Harderian gland, and the gut [21,22,23,24,25,26,27,28,29,30,31]. It was shown that pinealectomy reduces the plasma concentration of melatonin in structures containing melatonin-R [32, 33], indicating that melatonin plays an auto/paracrine role. Melatonin can change the release of dopamine, 5-HT, noradrenalin, and acetylcholine, and thus bias the postsynaptic response [34,35,36,37].
Melatonin is able to enhance the vasoconstriction of the rat caudal artery [37], and it may modulate the function of the gamma-aminobutyric acid receptor type A (GABA-A) [38] through the activation of several receptor subtypes. Interaction of exogenous melatonin in humans with the GABAergic system [39] provoked sedative, analgesic, anticonvulsive, and anxiolytic effects. High doses of melatonin administered in vivo generally led to stimulation of the immune system by increasing T-cell activity and lymphocytes, producing several humoral responses, and inhibiting the age-related thymus involution. Administration of melatonin in an in vitro model also enhanced the T-helper and natural killer (NK) cell activities, production of interleukin 2 (IL-2) and interferon gamma (IFNγ), and expression of IL-1 mRNA in human monocytes. Melatonin has a revitalizing effect on transplanted pineal functionality in old mice [40], thus functioning as an antiaging/antioxidant bioactive substance, probably linked to lipophilic melatonin diffusion into the cell (cytosol and nucleuses) [41], and exerts a protective radical scavenger effect on cellular macromolecules [17, 18].
Micromolar doses of melatonin are able to deactivate the free-radical molecules and the hydroxyl anion radical, producing a cyclic 3-hydroxy-melatonin-derivative, which is eliminated by the kidneys. The anti-carcinogenic effect of melatonin has also been found in in vivo and in vitro models, in particular with estrogen-responsive breast cancers [42,43,44,45,46,47]. In vitro, 1 nM to 0.1 μM of melatonin was found to produce a 40–60% reduction in human breast tumor cells, due to the association with oxidative phosphorylation, which leads to cell alteration and finally to autophagic response. Melatonin can stimulate the transcriptional activity of estrogen receptors in the signal transduction pathway, and thus may be described as a potent coadjutant in the treatment of tumors, at least at micromolar doses. In fact, it helps to protect the cells from carcinogenic compounds by acting together with retinoic acid to strongly reduce breast cancer development [48, 49]. Melatonin may also be useful for the synchronization of important biological functions and can prevent circadian clock alterations [50]. One of the most important metabolic pathways of the amino acid tryptophan are the kynurenine and serotine pathways, which produce some of the kynurenine metabolites, e.g., kynurenic acid, 3-OH-kynurenine, quinolinic acid, NAD, and some other neuroactive compounds like serotonin (5-HT) and melatonin. These two metabolic pathways are known to be involved in the control of certain immune and nervous diseases [51].
However, very little is known about the links between these two tryptophan metabolisms. Melatonin is synthesized from tryptophan, and is then hydroxylated and decarboxylated, which leads to the production of 5-HT. Serotonin is subsequently acetylated on the amine group and methylated on the hydroxyl group in position 5. In addition, other metabolites involved in the tryptophan catabolism could have different biological effects such as on the immune system and as inflammation modulators. N-Acetyl serotonin and melatonin also possess anti-inflammatory and antioxidant activities, quinolinic acid has excitotoxic activity, while hydroxyanthranilate exhibits anti-inflammatory activity, and these effects may be exerted directly on the CNS [52, 53].
The enzyme indole-amine-2,3-dioxygenase (IDO) has an important regulatory function, modulating the tryptophan conversion to kynurenine and other metabolites. This enzyme is involved in several inflammatory diseases including MS [54, 55]. Indeed, IFN-γ is able to up-regulate IDO, producing more immunosuppressive [56] and neurotoxic [57] by-products in tryptophan catabolism, following the kynurenine pathway.
Tryptophan is metabolized by the melatonin pathway only when the enzyme tryptophan hydroxylase produces 5-hydroxytryptophan, which, as mentioned above, will be converted to 5-HT, N-acetyl serotonin, and melatonin. Furthermore, melatonin and N-acetyl serotonin may also function as immune-signaling messengers, thus playing a crucial role in MS [58]. N-Acetyl serotonin has been shown to be effective in experimental models of ischemic injury, and also has potent anti-inflammatory and antioxidant effects [59]; however, these positive properties have not yet been investigated in MS.
In this pathway, the limiting step has been shown to be the acetylation of 5-HT by the enzyme aryl alkyl amine N-acetyl transaminase (AANAT) in the pineal gland and in the mucosal enterochromaffin cells, where AANAT is expressed [60]. Moreover, melatonin is largely present in the gut, where it can be produced by the O-methylation of N-acetyl-5-HT. The enzyme AANAT has been recently cloned from humans, but ovine AANAT is more frequently used by researchers, in a truncated version that has better solubility [61, 62].
Brain melatonin production is thus strongly controlled by the AANAT enzyme through its phosphorylation by protein kinase-A, and other kinases such as Rho kinase and the checkpoint protein 1 (CHK1) [63], as well as by ethero-dimerization between the phosphorylated AANAT and the chaperone-like 14.3.3 protein [64]. This complex provides a protective barrier against proteasome-mediated destruction. AANAT catabolism takes place mainly in the pineal gland; on the other hand, retinal AANAT is protected against catabolism during the day time [65]. Melatonin catabolism is less understood than its metabolism, but it is known that a portion of melatonin is eliminated unmodified; IDO is responsible for the remaining catabolism [66], being a ubiquitous enzyme. It has been reported that IDO is not able to work on melatonin or N-acetyl-tryptophan [67]; indeed, IDO catalyzes the conversion of tryptophan to kynurenine and a peroxidase-like reaction, which is independent of the presence of hydrogen peroxide [67].
H2O2 is a co-substrate of many heme-containing enzymes, but several reports [68, 69] show that myeloperoxidase (MPO) could also be responsible for the oxidative catabolism of melatonin due to its affinity to IDO for melatonin in a micromolar range. Melatonin is mainly cleaved by a pathway mediated by MPO, but under tryptophan breakdown, melatonin is not attached to IDO, but becomes a substrate for MPO [70].
Oxidative catabolism forms N1-acetyl-N2-formyl-5-methoxykynurenine (AFMK), a kynurenine derivative, which is then spontaneously or through kynurenine formamidase action transformed to N1-acetyl-5-methoxy-kynurenine (AMK). These substances may be associated with some in vivo functions of melatonin. The existence of two pathways has been validated in rat adrenal pheochromocytoma (PC12) cells [71]. AANAT expression is up-regulated in stable IDO1 knockdown cells and its over-expression is inhibited by IDO1 expression. Correspondingly, the expressions of IDO1 and AANAT are dose- and time-dependent, once melatonin is administered, demonstrating that melatonin enhances the kynurenine pathway, but inhibits the serotonin pathway. Phosphorylation of Janus kinase 2 (JAK2) and activation of transcription 1 (STAT1) are also improved by melatonin in a time-dependent manner; this process may be reversed by the inhibitor AG490. Melatonin promotes the nuclear translocation of STAT1 and nuclear factor kappa light-chain enhancer of activated B cells (NF-kB), both involved in IDO1 up-regulation [71, 72].
4 Pharmacokinetics and Oral Bioavailability of Melatonin
The pharmacokinetics of melatonin have been investigated in humans following intravenous [73] and oral [74] administration in rats, dogs, and monkeys. The apparent half-life of melatonin was 34.2 min after intravenous administration of 3 mg/kg. The doses have been normalized to oral bioavailability, where following a 10 mg/kg oral dose it was over 100% in monkeys; in vitro permeability studies on CaCo-2 cells revealed that melatonin is adequately absorbed in humans; on the contrary intrinsic clearance in humans is lower than that of rats, as shown in studies performed in rat and human livers [75]. Fourtillan and collaborators described the first cross-over study on melatonin bioavailability in males and females, through the quantification of exogenous and endogenous melatonin. After intravenous administration, melatonin reaches a plasma level < 165 pg/ml in males and 200 pg/ml in females; however, after 1 h amounts dropped to < 70 pg/ml, indicating that supra-physiological levels may be present after the consumption of melatonin supplements. Melatonin is transformed to 6-hydroxy-melatonin in the liver and eliminated in the urine as sulphatoxy-melatonin [76]. Following oral administration, the maximum plasma concentration (Cmax) was threefold higher for females than for males, but no gender difference was detected for total body clearance. Moderate tissue distribution is positively correlated with the volume of distribution at steady state (Vd,ss) and the elimination half-life (t½) is estimated to be 40 min for each administration.
Melatonin can exert sleep-inducing effects with no relevant collateral effects, similar to common hypnoinduction, but after oral administration, the bioavailability reaches below 20%, mainly due to first-pass metabolism in the liver; however, transdermal delivery systems have lag time and depot-effects limiting their usefulness [76].
Recently Flo et al. described the first pharmacokinetic study of melatonin administered transdermally, in which pharmacokinetic parameters were shown to be affected by the time of administration during the day in rats [77]. This circadian variation could also be expected in humans since melatonin levels interfere with glucose metabolism [78, 79]. The low solubility of melatonin in water also represents a problem for buccal delivery, thus, a nasal formulation has been proposed; however, this delivery system is ineffective for clinical use in light of serious local irritation and pain [79]. In 2004 Mao et al. developed melatonin starch microspheres for intranasal administration. It has been shown that > 80% of them are present in the nose tissues 2 h after administration, compared to 30% obtained with a solution. Absorption was fast, with a time taken to reach the maximum concentration (Tmax) of 7.8 min and bioavailability calculated to be near 84% [80]. Critically, patients lost circadian rhythms and had low endogenous melatonin; exogenous melatonin produces supra-physiological ephemeral concentrations, resulting in an inconsistent therapeutic effect on sleep. Bellapart et al. studied the oral administration of exogenous melatonin with a 3-mg dose, followed by a 0.5-mg dose after 1 h, and reported that this protocol produced sustained concentrations of serum melatonin over 12 h overnight in ill patients [81].
5 Melatonin and Neuroprotection
Neurons are very sensitive to injuries, with subsequent cell death, probably due to their high-energy demand and chemical composition. Neuronal death or damage has profound effects on a person’s behavior and physiological functions, affecting normal living. A number of acute events, such as hypoxia, stroke, mechanical trauma, prolonged hypoglycemia, neurotoxins, some types of viruses, and radiation are able to produce serious damage to the brain. Some mechanisms are also involved in neurodegenerative disorders, which are typically chronic and progressive disorders, characterized by selective and symmetric neuronal death, in different areas such as the motor, sensory, or cognitive systems. The causes of neurodegenerative diseases are still largely unknown. Glutamate cytotoxicity, injuries evoked by the action of free radicals, and dysfunction of the mitochondria are processes that have been recognized to be among the most common physiopathological mechanisms behind neuronal loss. Melatonin has been proposed to be a scavenger for oxygen radicals and a lipid antioxidant. It also exerts strong anti-excitatory and sedative effects at higher doses. Several reactive species, e.g. hydroxyl radical, carbonate radical (CO3·–), and peroxyl radical (ROO·, ONOO–), as well as damaged lipid, protein, and DNA, can induce neuronal death. The superoxide anion (O2·–) is formed by mitochondrial electron leakage, whereas nitric oxide (·NO) is recognized to be a neurotransmitter. These substances are very labile and several enzymes are able to scavenge them, preventing cell injury. Physiologic defense against radicals and oxidants includes the enzyme family of SOD, able to transform O2·– to H2O2 and O2. The cycle is completed by the enzymes GPx and catalase (CAT), which can metabolize H2O2. Pathological conditions in which there is an unfavorable balance between free radicals and elimination procedures generate so-called “oxidative stress”, which enhances oxidative damage to the biomolecules. The CNS is highly sensitive to oxidative stress due to its elevated oxygen utilization, which, per se, may increase the generation of free reactive oxygen species (ROS). The presence of specific enzymes like SOD, GPx, glutathione reductase (GR), and CAT, together with substances like glutathione (GSH), are lower in the CNS than in other parts of the body. In addition, a higher amount of iron and ascorbate was detected in the specific part of the CNS that may furnish the pro-oxidative state and could generate free radicals. The study of neurodegenerative diseases is a major topic of research. It has been well accepted that the consumption of a large amount of fresh fruit and vegetables may help to prevent the occurrence of neurodegenerative diseases. Adequate consumption of vitamins C and E has also been assumed to be important in preventing age-associated neurodegenerative diseases. In this regard, melatonin may play a significant role. As this product is synthesized in the pineal gland, its production gradually reduces with the age, and this may be related to the insurgence of the neurodegenerative diseases with aging, which is probably related to the increase of oxidative stress. Furthermore, melatonin deficiency has been related to neuronal degeneration in various experimental models of neurodegenerative diseases, and melatonin intake has a positive effect in preventing and maintaining neuronal degeneration. Pyramidal neurons of the hippocampus are vulnerable to ischemia, which evokes a series of physiologic events, including the formation of oxygen free radicals (ROS). Pharmacokinetic and structural properties of melatonin, for example amphiphilicity, allow its presence and protective action in the brain [82]. Continuous intravenous administration of melatonin for 6 h prevented the damage of these neurons in CA1-4 areas of the hippocampus after 15 min of acute cerebral ischemia. This neuroprotective effect can be explained by the increase of GPx and mitochondrial respiratory complex I and IV activities [18]. In therapy, the melatonin hormone could be directly applied into the ischemic tissue or melatonin-secreting cells could be implanted in stroke-induced areas [83]. The administration of 5 mg/kg melatonin intraperitoneally (i.p.) in rat models of stroke, where their pineal glands were removed, decreased the white and gray matter in ischemic areas and reduced inflammatory responses, but also caused cerebral edema [84]. Several in vivo and in vitro studies reported that melatonin may also protect the glial cells, as they secrete trophic factors such as glial cell-line-derived neurotrophic factor (GDNF) with neuroprotective effects [85]. Rats exposed to chronic immobilization stress (CIS) for 6 weeks showed increases in the adrenal size and the level of corticosterone; in addition the presence of an anxiety-like behavior was observed. Furthermore, the levels of serotonin, noradrenaline, and oxytocin were decreased in the frontal cortex [86]. Chronic administration of melatonin mitigated these biochemical and histological alterations, partly mediated by its effect to produce the release of oxytocin and monoamines, and down-regulated the expression of CgA [86]. Melatonin is able to suppress NOS, causing the down-regulation of NO formation against Ca2+-dependent excitotoxicity and peroxynitrite-dependent radicals. This aspect makes melatonin a good candidate for therapy in the treatment of amyotrophic lateral sclerosis (ASL) [87]. In a pilot program in Gottingen, three ASL patients entered into a study to investigate the possible adverse effects of chronically administered melatonin at a high dose. This study demonstrated that melatonin was largely tolerated during the time of the study, without the insurgence of the adverse effects in subjects with an onset of ASL from 2 to 4 years [87]. In another study that included children with muscular dystrophy, a dose of 70 mg/day of melatonin decreased the cytokines and lipid peroxidase [88]. Melatonin is reported to reduce the formation of amyloid beta (Aβ) (the main neurotoxin involved in Alzheimer disease, AD), aggregation, and neurotoxicity in patients treated for AD [89]. Melatonin also inhibited the secretion of amyloid precursor protein (APP) in diverse cell lines. Melatonin blocks the formation of β-sheets and amyloid fibrils, interacting with Aβ proteins [90]. It is worth noting that the protective activity of melatonin could also be mediated through the GABAergic system [91]. In addition, it has been demonstrated that melatonin is able to prevent the neurotoxicity produced by the over-activation of the kainite glutamate receptors, supporting the theory that melatonin can prevent cell death due to glutamate toxicity [92,93,94]. As mentioned, MLD may reduce the damage of the hippocampal region CA1 after brain ischemia [95, 96]. Indeed, in rats deficient in MLF, following a stroke or excitotoxic seizure, brain injuries are more marked than in normal rats [94], suggesting that the lack of melatonin results in more widespread damage of neuronal cells. This effect also enhanced peptide clearance, through proteolytic degradation produced by the insulin-degrading enzyme (IDE). There was an attenuation in tau hyper-phosphorylation, once neuroblastoma cells treated with melatonin were exposed to N2a and SH-SYS5Y; this was mediated by affecting protein kinases and antagonized the oxidative stress [97]. Melatonin attenuates the production of pro-inflammatory cytokines and NO in rat brain [98] and inhibits the NF-κB binding to DNA [99], indicating a neuroprotective effect of melatonin on the cholinergic neurons. In fact, melatonin can prevent the inhibition of the choline transport and choline acetyl transferase (ChAT) induced by peroxynitrite in the synaptic vesicles of hippocampus in AD patients [100]. The reduction of melatonin in the cerebrospinal fluid and plasma could also be a marker for early detection of AD [92]. In AD, melatonin improved sleep quality and reduced progression of cognitive decay in several open-label study case reports [101]. Considering its binding properties and potency, melatonin could be used in combination with other treatments currently available [102], which could be beneficial for subjects with AD; however, melatonin receptor agonists showed limited benefit in laboratory animal models of AD [103]. Several studies showed that patients with Parkinson disease (PD) have modifications in melatonin production and in the expression of melatonin receptors MT1 and MT2 in the SNC [104]. Exogenous administration of melatonin induced neuroprotective effects in animal models of PD induced by different toxins, due to its antioxidant activity promoting reduced levels of free radicals [105]. Melatonin was also able to prevent hydroxyl radical production by DA auto-oxidation in vitro. Free radical damage is an essential aspect of neonatal hemorrhagic brain injury, leading to cerebral dysfunction. In neonatal mice, melatonin attenuates the development of white matter cysts, and following intrauterine asphyxia melatonin administration to pre- and near-term fetal sheep reduced oxidative stress and cell death. Melatonin ameliorated the cognitive and sensory-motor dysfunction in juvenile rats. Furthermore, administration of melatonin to a pregnant female in an animal model reduced fetal brain oxidative stress, and restored myelination of the nerve, white matter, and motor functions [106].
Patients with mild cognitive impairment treated with melatonin exhibited significant improvement following the application of neuropsychological challenges consisting of Mattis’ test and the Digit-symbol test [107] since in neurodegenerative processes, the disorganization of circadian rhythm is responsible for sun-downing and behavioral problems. Hence, positive results derived from experimental studies make pharmacological doses of melatonin highly desirable for patients with mild cognitive impairment [108].
6 Multiple Sclerosis (MS): Pathophysiology and Current Treatments
MS is a chronic neuroinflammatory demyelinating disease of the human CNS [1, 2]. Depending on the disease courses, MS patients may experience relapsing/remitting MS (RRMS), secondary progressive MS (SPMS), or primary progressive MS (PPMS). Upon relapsing, exacerbations of neurologic dysfunction decline, and, thereafter, intermittent clinical remission takes place. In the next stage, SPMS occurs in a deteriorating manner, with or without acute exacerbations during the progressive course, and the disability is seriously progressed [109]; notably, neurodegenerative processes were found to be the prime underlying mechanism. PPMS is another pathologic form of MS, with less inflammatory signs, where the neurologic disability occurs from the early onset of the disease without regular or occasional relapses. Pathophysiologically, PPMs is not different from relapsing forms of MS that have progressed to SPMS [109,110,111].
MS usually starts with inflammatory demyelination triggered by an inflammatory attack on the myelin components and destruction of the myelin sheath [4] and with deterioration of oligodendrocyte cells, which are attributed with the maintenance or repair of the myelin sheath and neuronal signaling transmission [111]. It has long been believed that MS has its roots in inflammation and T cells (also called autoreactive lymphocytes) activated outside the CNS. These are known to play key roles in such events by disrupting the blood–brain barrier, activating macrophages, and attacking the myelin [111]. T cells are reactivated by local antigen-presenting cells by releasing the relevant antigens to CD4+ T-helper cells in the periphery and by producing autoreactive pro-inflammatory T helpers (Th 1 and Th 17). Within the CNS, target antigens are recognized, T cells are reactivated, and macrophages become functional. Together, these immune cells induce the CNS cascade of inflammatory events via pro-inflammatory cytokines (e.g., IL-12, IL-23, IFNγ, tumor-necrosis factor-α (TNF-α)) and other destructive stressors (e.g., proteases, free radicals, antibodies), which later stimulate additional inflammatory cells, along with microglial and astrocyte cells. Accordingly, the myelin-directed inflammatory responses result in demyelination, axonal loss, and irreversible neurological deficits [3, 112, 113]. Collectively, inflammation strictly interplays MS by contributing to a set of different pathways. For instance, it has been found that the intestinal microbiome, part of the brain-gut axis, is also involved in the pathophysiology of MS mainly through CNS demyelination, where the gastrointestinal barrier is broken down (intestinal barrier dysfunction) and the content of the intestinal microbiome reaches the CNS microglia through the blood circulatory pathway and modifies its function [114]. Cerebral cortical pathology (e.g., cortical inflammation, demyelination, neurodegeneration, and atrophy) was also shown to be correlated with disease progression and disability [115].
Mechanistic experiments and clinical evidence suggest that MS disease-modifying therapies should aim at manipulating the inflammatory state, and promoting nervous system regeneration and neuroprotection [116, 117]. At least ten disease-modifying therapies (DMTs) have obtained approval for use in suppressing RRMS, including first-generation injectable drugs (glatiramer acetate and interferon β), oral drugs (e.g., fingolimod, teriflunomide, dimethyl fumarate), and second-generation injectable humanized monoclonals (e.g., natalizumab, alemtuzumab) [117]. Current predictive biomarkers and pharmacogenomics in conjunction with remarkable advances in imaging techniques are rapidly moving in the direction of individual therapeutic strategies within the context of sequential monotherapy, escalation therapy (immunomodulatory and immunosuppressive drugs), induction and maintenance therapies, and combination strategies (for a comprehensive review, see [117]). Studies targeting the symptomatic features of MS, remyelination and repair, and oligodendrocyte precursor cell inhibitors would be of great interest.
Indeed, complimentary medicine appears to be helpful. Clinical documentations have suggested that several medicinal plant species (e.g., Cannabis sativa, Boswellia papyrifera, Ginkgo biloba, Camellia sinensis, Panax ginseng) could also help to manage the main complications of MS, including spasticity, fatigue, scotoma, incontinence, tremor, memory performance, and functional ability, in a way that is more tolerated with less adverse effects [118]. More recently, dietary patterns were also demonstrated to be potentially important in MS treatment, although their definitive impacts require further discussion [119].
7 Melatonin and MS: Neuropharmacological Mechanisms and Clinical Evidence
7.1 Animal Studies
MS exclusively affects human life; non-human primates are resistant to MS [120]. Due to the difficulty of accessing the CNS of MS patients, a number of animal models have been established for translational research in order to understand the pathophysiological details of MS and for development of treatments. Due to the high similarity between the clinical and histopathological features of experimental autoimmune encephalomyelitis (EAE) and human MS, EAE is the most commonly used model for human inflammatory demyelinating disease, particularly MS. In EAE, the interaction between some immune-neuropathological mechanisms leads to a condition that mimics the key features of MS pathogenesis, such as inflammation, demyelination, axonal loss, etc. [121]. Another known model is the cuprizone toxic demyelination model. In this model, young animals are fed with cuprizone for specific oligodendrocyte death induction and subsequent reversible demyelination, making this excellent for studying the factors that can prevent demyelination and stimulate remyelination [122]. Here, we present the reliable animal studies that evaluated the effects of melatonin on MS. Figure 2 depicts the most relevant neuropharmacological signaling cascades involved in the protective effect of melatonin against the development of MS.

Neuropharmacological targets involved in preventing and therapeutic effects of melatonin against multiple sclerosis (MS) development. Mfn mitofusin, NF nuclear factor, ICAM intracellular adhesion molecule, IFN interferon, Treg T regulatory, Th T helper, IL Interleukin, Nrf2 nuclear factor E2-related factor, ARE antioxidant response elements, HO-1 heme oxygenase-1, GSH glutathione, ROS reactive oxygen species, CAT catalase, SOD superoxide dismutase, NADPH nicotinamide adenine dinucleotide phosphate, NQO nicotinamide quinone oxidoreductase, MOG myelin oligodendrocyte glycoprotein
7.1.1 Behavioral Effects of Melatonin
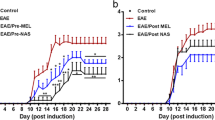
It was shown that 5 weeks of administration of cuprizone significantly declined the distance moved and velocity of mice in an open field test. Intraperitoneal administration of melatonin in doses of 50 and 100 mg/kg during the last week of cuprizone treatment restored their ability, and increased the velocity and distance moved dose dependently, while melatonin alone did not show any changes in a healthy control group [123]. The enhancement of nociception latency in cuprizone-treated animals after melatonin administration was noted, suggesting the anti-nociceptive effect of melatonin [123]. In an EAE-induced model, mice treated with N-acetyl serotonin and melatonin prior to or after the onset of symptoms showed that they were both able to decrease the symptom severity and delay symptom onset. Although N-acetyl serotonin was more effective than melatonin, pre-treatment of N-acetyl serotonin and melatonin showed more prominent effects than when given post administration [124]. Such effects were also confirmed in a study where melatonin was administered from day 0 after EAE induction, which was representative of the prophylactic/therapeutic effect of melatonin in mice. Melatonin decreased the severity of clinical symptoms without changing its onset time [125]. Generally, paralysis is considered another clinical manifestation of EAE induction in rats. In Kang’s study, although both melatonin-treated and vehicle-treated groups showed paralysis to some extent, the duration and severity of the disease were significantly decreased in the melatonin group [126]. The maximum clinical scores were lower, the latent period was postponed, and the advanced stage was shorter in melatonin-treated mice compared with the untreated group [127].
7.1.2 Anti-oxidative Effects of Melatonin
It is well established that inflammation-induced oxidative stress can contribute to the pathogenesis of MS and its animal model, EAE [128]. Activated macrophages and astrocytes release large amounts of ROS or reactive nitrogen species (RNS) [129]. The main cellular components like lipids, proteins, and nucleic acids (e.g., RNA, DNA) are damaged with ROS, leading to cell death and tissue damage by necrosis or apoptosis [128]. Stress response against ROS-mediated toxicity is mediated by the activation of genes responsible for anti-oxidative action [129]. Normally, the nuclear factor-E2-related factor (Nrf2) links to Keap1 in the cytoplasm, but under stress conditions, Nrf2 is evaded and translocated to the nucleus and binds to the antioxidant response elements (ARE), inducing the transcription of ROS-protective genes such as SOD, CAT, heme oxygenase-1 (HO-1), nicotinamide adenine dinucleotide phosphate (NADPH), and quinone oxidoreductase 1 (NQO1) [130].
In an EAE animal model, melatonin treatment resulted in higher levels of antioxidant agents including SOD and CAT, and decreased the thiobarbituric acid reactive substances (TBARS) and ROS concentrations versus untreated ones. Overall, Nrf2/ARE pathway up-regulation following melatonin administration could contribute to the HO-1 and NQO1 expressions in the spinal cord of mice and anti-oxidative action [127]. Such results were also observed in another study when melatonin was administered alone or in combination with baclofen in EAE-model mice; in both groups, the levels of the antioxidant enzymes like SOD and CAT increased and malondialdehyde (MDA; the lipid peroxidation marker) reduced in comparison with that of the control group; however, the combined group (melatonin + baclofen) exhibited stronger antioxidant activity [131]. In Kashani’s study, in order to assess the free radical-mediated activity of melatonin following cuprizone-induced demyelination, estimations of MDA and GSH tissue levels were carried out in the corpus callosum of mice on the cuprizone diet. Melatonin antagonized the effect of cuprizone and increased the GSH level significantly, and decreased the MDA level [132]. In EAE mice, pre- and post-treatment of melatonin and N-acetyl serotonin reduced the number of p67phox, NADPH oxidases activator subunit, and the iNOS positive cells in the spinal cord of treated mice versus untreated mice [124]. Furthermore, free radicals can activate NF-κB, leading to the up-regulation of a set of genes that play crucial roles in the pathogenesis of MS, such as TNF-α, intracellular adhesion molecule 1 (ICAM-1) and vascular-cell adhesion molecule 1 (VCAM-1) [128]. The administration of melatonin in cuprizone-treated mice enhanced NF-κB and reduced the HO-1 level dose dependently [123].
7.1.3 Effects of Melatonin on Mitochondrial Function
The diverse functions of the mitochondria make it one of the crucial components of the body, and any mitochondrial abnormality such as mitochondrial enzyme function failure, gene expression defect, or an imbalance of the mitochondrial fusion and fission can contribute to the development and progression of MS lesions [133]. The mitochondrial density, length, and number within the cross sections of axons in the corpus callosum of the cuprizone-induced mice was reduced after melatonin administration, while there was no detectable change in their size before and after melatonin treatment. Melatonin significantly increased the levels of all four mitochondrial subunits, which were down-regulated after cuprizone induction. In addition, the expression of mitofusin mfn1, an important mediator of mitochondrial fusion, increased following melatonin administration [132].
7.1.4 Anti-inflammatory Effects of Melatonin
The fundamental units of the nervous system, the neurons, are the target of inflammatory processes in MS [134]. The progression of MS, as well as that of EAE, is mediated by the infiltration of T- and B-lymphocytes, in addition to the activation of microglia and macrophages [124]. N-Acetyl serotonin or melatonin administration (following the onset of symptoms) significantly reduced the number of activated microglial and infiltrated lymphocytes in the EAE mouse spinal cord [124]. Another immune response that was affected by melatonin was reported in the study by Alvarez, where melatonin decreased the peripheral and central Th1/Th17 ratio and increased the regulatory responses such as IL10 synthesis and T regulatory (Treg) frequency. On the other hand, there was a reduction of the T-effector memory population and CD44 expression. CD44 is widely used as a marker of antigen-experienced T cells (Ag-Tcells), the principal ligand of which is hyaluronan (HA). CD44 is involved in the recruitment of T cells to sites of inflammation; adhesion of T cells to the endothelium; in Th cells survival, polarization, and differentiation; as well as in Treg cells’ maintenance and suppression. The surface expression of CD44 is elevated in circulating T cells of MS patients during the relapse phase. It has been demonstrated that melatonin down-regulates the expression of CD44 in T-effector cells, and up-regulates the CD44 level in Treg cells [125]. IFN-γ secretion produced by Th-1 cells leads to the destruction of myelin and oligodendrocytes through activating phagocytic macrophages and inhibiting developmental myelination or remyelination of the demyelinated lesions in EAE [135].
In an attempt to find the mechanism underlying the reduction of the clinical severity of the EAE model following the administration of melatonin, mononuclear cells were driven from the spleens and CNS of mice treated with either melatonin or solvent. Flow cytometry analysis of the lymphocyte subsets showed a significant reduction in the Th17 population of the CD4 T-cell subset of the CNS in the melatonin-treated group, mainly in correlation with the IL-17 reduction in the CNS. IL-10 increased in the splenic Treg cells of the melatonin-treated mice, while the percentages of splenic Tregs were not different in the solvent- versus the melatonin-treated group. Following melatonin treatment, the expressions of IFN-γ, IL-17, IL-6, and CCL20 were also suppressed [136]. Melatonin or N-acetyl serotonin treatment increased the number of activated microglia and CD4+ T-cells in the white matter of EAE mice reduced near to normal levels [124]. Subsequent to oral administration of melatonin in EAE-induced Lewis rats, histological analysis of the spinal cord showed less inflammatory cell infiltration versus the vehicle group. In addition, immunohistochemical detection showed a significant enhancement (about twofold) of the proportion of NK+ cells/total inflammatory cells in melatonin-treated animals’ versus the vehicle-treated animals. Adhesion molecule intensity was also compared in the two groups; the ICAM-1 immunoreactivity was reduced in the melatonin group but the LFA-1 immunoreactivity was the same in the two groups [126]. Melatonin treatment could reduce the numbers of inflammatory cells (mainly single nucleus) infiltrated in the spinal cord of EAE mice [127]. In contrast to the previous studies, in a study conducted on young Lewis EAE model rats, the administration of melatonin enhanced the ratio of IFN-γ/IL-4 (as an indicator of the ratio of Th-1/Th-2), although the increase was observed in both pro- and anti-inflammatory cytokines; however, the increase in IFN-γ was more prominent than in IL-4 [137].
7.1.5 Anti-apoptotic Effects of Melatonin
Oligodendrocytes, known as myelinating cells of the CNS, have been shown to play a key role in the pathophysiology of MS [138]. In the cuprizone-induced model of demyelination, the inhibition of copper-dependent mitochondrial enzymes caused oligodendrocyte degeneration. Evaluation of apoptotic markers in cuprizone-treated mice after melatonin administration showed that in cuprizone-treated mice, melatonin treatment reduced the mean amount of apoptotic cells by reducing the caspase-3 and Bax (pro-apoptotic protein), as well as by increasing the Bcl-2 (anti-apoptotic protein) level and therefore reducing the Bax/Bcl-2 ratio [123] in a dose-dependent manner.
7.1.6 Anti-demyelinating Effects of Melatonin
Demyelination has a pivotal role in the pathogenesis of MS. In a study by Kashani, in the corpus callosum sections of the cuprizone-induced mice, following melatonin treatment, the myelin loss and the number of oligodendrocytes were nearly restored to normal, according to both staining parameters luxol fast blue (LFB) and proteolipid protein (PLP) [132]. In contrast, administration of melatonin did not show any initiation of myelin production in a cuprizone model of demyelination [123]. Myelin oligodendrocyte glycoprotein (MOG), located at the outer surface of myelin sheaths, and the oligodendrocyte membranes are considered to have great importance in myelination. Due to the late postnatal developmental expression, MOG is known as a marker for oligodendrocyte maturation [139]. Melatonin treatment with or without baclofen, a muscle relaxant, indicated an elevation in the expression of MOG, compared to untreated samples in EAE-induced mice [131]. A significant reduction in the loss of mature oligodendrocytes, demyelination, and axonal injury were observed in the spinal cord of EAE mice after melatonin and N-acetyl serotonin administration [124].
7.1.7 Effects of Melatonin on Bone Health
Although physical inactivity is likely the most important contributing factor to low bone mineral density (BMD) in MS, the higher chance of developing osteoporosis in MS patients than in the other non-inflammatory disabling diseases, besides the presence of similar inflammatory cytokines in both osteoporosis and MS, suggests a stronger correlation between these diseases and their pathogenesis. Additional factors that may play role in the incidence of osteoporosis in MS include a low level of vitamin D, and the use of some medications (e.g., glucocorticoids and anticonvulsants) [140]. Procalcitonin, a precursor of calcitonin involved in calcium homeostasis, also has been suggested to be a marker of systemic inflammation and autoimmune diseases. Melatonin treatment in EAE mice showed enhanced serum levels of 25-hydroxyvitamin D and osteocalcin a marker of bone matrix synthesis. In addition, melatonin reduced the procalcitonin level [141]. Table 1 presents a summary of melatonin intervention in animal models of MS.
7.2 Clinical Studies
7.2.1 Effects of Melatonin on MS Symptoms
In a case-control study, the association between the polymorphisms in the tryptophan hydroxylase2 (TPH2) and melatonin receptor 1B (MTNR1B) gene with PPMS was investigated. The disability and dysregulation of the melatonin pathway were assumed to be the main cause of the disease progression [142]. In another study, the clinical correlation between serum melatonin and procalcitonin in MS patients was investigated. Forty-six volunteers including 23 MS patients (19 women and four men), and 23 healthy subjects (19 women and four men) were participated. A significant increase (p < 0.05) in the serum level of procalcitonin was observed in MS patients compared with the healthy participants. The melatonin serum level decreased significantly (p < 0.05) in MS patients, in comparison with the healthy controls [141].
The effect of IFN-β-1b therapy on melatonin secretion, fatigue, and sleep features was investigated by Melamud et al. [143]. In this study, 25 volunteers including 12 healthy subjects and 13 female RRMS patients participated. Patients were evaluated prior (6 weeks) and after treatment with IFN-β-1b for 4 months. Fatigue was calculated based on the Fatigue Impact Scale (FIS); the day/night levels of 6-sulphatoxy-melatonin (6-SMT) were also assessed. Sleep efficiency was significantly decreased in the MS group (p < 0.05), and the urine melatonin metabolite level was decreased in IFN-β-1b-pretreated patients (p < 0.001). In addition, after 4 months of IFN-1β therapy, total fatigue significantly decreased (p = 0.068) compared with controls. Moreover, the urine melatonin metabolite level in the pretreated patient group reduced significantly [143].
Adamczyk-sowa et al. [144] determined the effect of melatonin supplementation on chronic fatigue syndrome in MS patients. 102 MS patients participated in the treatment, which involved taking oral melatonin (5 mg per day) over 3 months. This research showed that the lipid hydroxyperoxide (LHP) concentrations were significantly higher in all the studied MS groups (p = 0.000003). However, a significant decrease was seen in the plasma LHP concentration in MS patients, while in the RRMS-relapse group the plasma level of homocysteine was significantly higher compared with the RRMS-pretreated group (p = 0.02). No significant difference was reported in the plasma homocysteine concentration before and after melatonin therapy. The fatigue score was significantly lower in the RRMS pretreated group. The degree of fatigue on the MFIS score decreased significantly in the RRMS pretreated group [144].
A relationship between the tolerability and the efficacy of supplemental therapy with melatonin, motor cognitive and neuroimaging in MS patients was investigated in RRMS patients. Twenty-three RRMS patients received melatonin (3 mg/day) and INF-1β once weekly for 12 months. No significant effect on the extended disability status scale (EDSS), MS functional composite (MSFC), and Beck Depression Inventory-second edition 17 (BDI-II) was reported for the treatment with melatonin on the clinical and functional debility and development of brain lesions [145]. In a case-control study of an Iranian MS population, Gholipour et al. showed the urinary level of melatonin decreased in MS patients compared with the control group [146].
7.2.2 Anti-oxidative Effects of Melatonin
Miller et al. [147] studied the correlation between melatonin and the decrease of oxidative stress in the erythrocytes of MS patients. SPMS patients (n = 16) including 11 females and five males were exposed to melatonin (10 mg daily/30 days) and 13 subjects were included as controls. Melatonin caused a significant increase in SOD and GPx, and decreased the MDA level in erythrocytes of SPMS patients [147]. In another experiment, the correlation between melatonin supplementation and oxidative stress parameters, especially oxidative protein modifications of the blood serum, for example the levels of N′-formyl kynurenine, kynurenine, dityrosine, carbonyl groups, advanced glycation products (AGEs), advanced oxidation protein products (AOPP), and MDA, was investigated. Thirty-six patients with RRMS received 250 μg IFN-β-1b subcutaneously (SC) every day, and 25 RRMS patients received IFN-β-1b plus 5 mg melatonin supplementation orally over a period of 3 months. The total levels of oxidative proteins were elevated in the blood serum in the control group. The N′-formyl kynurenine (p < 0.001), kynurenine (p < 0.01), AGEs, and carbonyl (p < 0.05) levels were decreased only in the group treated with IFN-β plus melatonin, whereas the dityrosine (p < 0.001) and AOPP levels were reduced in both groups of patients treated with IFN-β and IFN-β-1b plus melatonin [148].
In another study, the total oxidant status, total anti-oxidant capacity, and their influence on the sleep quality and depression level of MS were evaluated. In this study, melatonin (5 mg) was administrated daily over 3 months to volunteers including 102 MS patients (69 women, 33 men) and 20 healthy controls (13 women and seven men). The serum level of total oxidant decreased significantly in all MS patients (p < 0.05). The total anti-oxidant capacity level was significantly lower only in the mitoxantrone-treated group and increased after melatonin supplementation (p < 0.05).
It was shown that melatonin can act as an antioxidant and can recover reduced sleep quality in MS patients [147]. Following melatonin therapy, INF-β and glatiramer acetate were reduced in the treated group, and an increase in SOD activity was observed (p < 0.05) [149]. In another study, the correlation between anti-oxidative melatonin therapy and ceruloplasmin concentration, as an immune-modifying agent, was assessed in MS patients. In this study, 117 volunteers including 102 MS patients and 15 healthy controls participated. MS patients were supplemented with melatonin for 3 months. Melatonin caused a decline in ceruloplasmin level (p < 0.05), making ceruloplasmin a valuable serum marker for the chronic demyelinating process that occurs in oxidative stress mechanisms as well as a neurodegenerative marker, but not a marker of acute-phase MS [150].
In a study conducted in 14 healthy subjects and 12 female patients with RRMS treated with melatonin, melatonin therapy significantly increased the sirtuin1 (SIRT1) (p < 0.001), manganese-dependent-SOD (MNSOD) (p = 0.003) and CAT (p < 0.001) mRNA levels of the peripheral blood mononuclear cells (PBMCs) in RRMS patients. In MS patients, SIRT1 activity was not correlated with CAT and MNSOD activities before melatonin treatment, whereas after melatonin therapy, a significant correlation was detected between SIRT1 and CAT activities in PBMCs patients (p = 0.039). The mRNA expression of CAT in untreated PBMCs in RRMS patients was significantly high (p = 0.01) [151]. In a case-control study the correlation between melatonin and the pathogenesis of MS was investigated. In this process, 35 patients with MS and 35 healthy subjects were evaluated. There was no difference between the melatonin level in patients with dissimilar clinical features such as disease duration, first attack, and treatment type, and no correlation was observed between the melatonin level and sleep quality. Melatonin can have a broad range of actions in neurogenesis immunomodulation, improvement of the immune defense, removal of free radicals, prevalence of lipid metabolism, and neuroprotection [152].
Mrowicka et al. [153] investigated the influence of melatonin on the cellular Cu Zn-SOD in red blood cells of MS patients. The experiment included 33 subjects who were immobilized and 17 subjects with normal physical activity. Melatonin was given at a dose of 5 mg daily, 1 h before sleep. A slight increase was reported in Cu Zn-SOD activity (+ 3.1%) 10 days after alloplasty and melatonin administration, whereas a considerable rise in the enzyme activity (+ 23.3%) was noted 30 days after restoration and melatonin supplementation in the treatment group. The average Cu Zn-SOD activity was lower in the controls. These results indicated that short- and long-lasting hypokinesis leads to an increase in ROS generation, which is shown by an increase in Cu Zn-SOD activity. The results of the study on SOD activity demonstrated that the oral administration of melatonin for a period of 30 days has a more satisfactory impact on anti-oxidative courses than 10-day’s melatonin consumption [153].
7.2.3 Anti-inflammatory Effects of Melatonin
Farez et al. [154, 155] showed that melatonin levels are modulated by seasonal variations overnight. The study was conducted on 139 RRMS patients. Melatonin caused a significant increase of expression of the repressor transcription factor Nfil3, blocked the differentiation of the pathogenic Th17 cells, increased the generation of protective Tr1 cells through Erk1/2, and enhanced the trans-activating of the IL-10 promoter by ROR-α. A higher serum melatonin level was associated with the lower expression of IL17. A positive relationship was found between higher IL10 expression in peripheral CD4+T cells and melatonin in serum (p = 0.003). Melatonin modified various human Th17 and Tr1cells in vitro, and endogenous melatonin levels are related to the expression levels of IL17 and IL10 in peripheral CD4+T cells in RRMS patients [154, 155].
8 Conclusion
Oxidative stress and inflammation are important factors underlying the process of demyelination. Many studies have confirmed that melatonin has significant protective effects against redox signaling in both MS animal models and clinical studies. Redox signaling was potentially regulated by melatonin through reduction of ROS, increasing the GSH and NADPH content, as well as activating the antioxidant enzymes (e.g., HO-1, SOD, CAT, and NQO1) mediated by Nrf2/ARE pathway. It has been revealed that melatonin has a highly anti-inflammatory effect in both animal models (six studies) and MS patients (two studies). Basically, melatonin protects neurons by reducing the neuroinflammatory mediators (e.g., IFN-γ, IL-17, IL-4, IL-6, and CCL20), endothelial activation of ICAM-1, microgliosis and induction of IL-10, as well as by increasing the population of Treg cells instead of Th1 cells. According to the results of five studies, it could be concluded that melatonin possibly decreases the severity of clinical symptoms and behavioral deficits in animal models of MS. Of six clinical studies that evaluated the effects of melatonin on MS symptoms, a number of studies only measured the level of melatonin in patients and some others used melatonin as supplementary therapy; thus, the differences in the results make it difficult to form conclusions. Although there are no reports of risks regarding melatonin supplementation from human studies, it seems that adherence to risk-assessment plans outlined by standard protocols is necessary. Since melatonin is usually used as a supplement and its properties are shared by the effects of routine therapies, stringent follow-up of patients, besides the utilization of proper control individuals, is recommended. Several factors including bone health (one study), apoptosis (one study), mitochondrial function (one study), and anti-demyelinating effects (two studies) were evaluated in animal models, and the clinical monitoring of MS patients are also suggested. Melatonin supplementation may modulate the apoptosis pathway by decreasing the expression of pro-apoptotic genes (Bax and caspase3) and increasing the level of anti-apoptotic gene Bcl-2 as well as decreasing mitochondrial noxious enzymes. Another signaling pathway involved in neuronal function, which was found to be modulated by melatonin, is mature oligodendrocyte degeneration and myelin loss. Notably, melatonin promotes the oligodendrogenesis and repopulation of mature oligodendrocytes in vivo, which has substantial implications for remyelination in MS.
The conclusions of this article are derived from a comprehensive review of the literature and indicate that there is an urgent need for detailed screening of molecular mechanisms that mediate the neuroprotective effects of melatonin in both animal and human subjects. Though it seems that melatonin alone cannot resolve the therapeutic challenges of MS, it possibly may reveal new therapeutic interventions or new functions. Further evaluation of cellular and molecular mechanisms with an emphasis on the effects of melatonin may lead to the development of new management schemes for MS.
References
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545.
Ransohoff RM, Hafler DA, Lucchinetti CF. Multiple sclerosis—a quiet revolution. Nat Rev Neurol. 2015;11(3):134.
Kamm CP, Uitdehaag BM, Polman CH. Multiple sclerosis: current knowledge and future outlook. Eur Neurol. 2014;72(3–4):132–41.
Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55(4):458–68.
Rahmanzadeh R, Moghadasi AN, Navardi S, Minagar A, Sahraian MA. Multiple sclerosis: clinical features, pathophysiology, diagnosis, and management. In: Minagar A, editor. Neuroinflammation. New York: Elsevier; 2018. p. 1–20.
Ortiz GG, Pacheco-Moisés FP, Macías-Islas MÁ, Flores-Alvarado LJ, Mireles-Ramírez MA, González-Renovato ED, et al. Role of the blood–brain barrier in multiple sclerosis. Arch Med Res. 2014;45(8):687–97.
Kamphuis WW, Derada Troletti C, Reijerkerk A, Romero IA, de Vries HE. The blood–brain barrier in multiple sclerosis: microRNAs as key regulators. CNS Neurol Disord Drug Targets (Curr Drug Targets CNS Neurol Disord). 2015;14(2):157–67.
Hoffmann K. Photoperiod, pineal, melatonin and reproduction in hamsters. Prog Brain Res. 1979;52:397–415.
Reiter RJ. The pineal and its hormones in the control of reproduction in mammals. Endocr Rev. 1980;1(2):109–31.
Goldman BD, Darrow JM. The pineal gland and mammalian photoperiodism. Neuroendocrinology. 1983;37(5):386–96.
Bittman EL, Karsch FJ. Nightly duration of pineal melatonin secretion determines the reproductive response to inhibitory day length in the ewe. Biol Reprod. 1984;30(3):585–93.
Tamarkin L, Baird CJ, Almeida O. Melatonin: a coordinating signal for mammalian reproduction? Science. 1985;227(4688):714–20.
Pévet P. The role of the pineal gland in the photoperiodic control of reproduction in different hamster species. Reprod Nutr Dev. 1988;28(2B):443–58.
Goldman BD. Mammalian photoperiodic system: formal properties and neuroendocrine mechanisms of photoperiodic time measurement. J Biol Rhythms. 2001;16(4):283–301.
Letechipı́a-Vallejo G, González-Burgos I, Cervantes M. Neuroprotective effect of melatonin on brain damage induced by acute global cerebral ischemia in cats. Arch Med Res. 2001;32(3):186–92.
Kennaway DJ, Lushington K, Dawson D, Lack L, van den Heuvel C, Rogers N. Urinary 6-sulfatoxymelatonin excretion and aging: new results and a critical review of the literature. J Pineal Res. 1999;27(4):210–20.
Reiter RJ. The pineal gland and melatonin in relation to aging: a summary of the theories and of the data. Exp Gerontol. 1995;30(3–4):199–212.
Reiter RJ, Tan D-X, Osuna C, Gitto E. Actions of melatonin in the reduction of oxidative stress. J Biomed Sci. 2000;7(6):444–58.
Vaněček J, Pavlík A, Illnerová H. Hypothalamic melatonin receptor sites revealed by autoradiography. Brain Res. 1987;435(1–2):359–62.
Pevet P, Bothorel B, Slotten H, Saboureau M. The chronobiotic properties of melatonin. Cell Tissue Res. 2002;309(1):183–91.
Quay W. Circadian and estrous rhythms in pineal melatonin and 5-hydroxy indole-3-acetic acid. Proc Soc Exp Biol Med. 1964;115(3):710–3.
Cardinali DP, Larin F, Wurtman RJ. Control of the rat pineal gland by light spectra. Proc Natl Acad Sci. 1972;69(8):2003–5.
Brammer GL, Yumiler A, Wetterberg L. N-Acetyltransferase activity of the rat Harderian gland. Biochim Biophys Acta. 1978;526(1):93–9.
Vivien-Roels B, Pevet P, Dubois M, Arendt J, Brown G. Immunohistochemical evidence for the presence of melatonin in the pineal gland, the retina and the Harderian gland. Cell Tissue Res. 1981;217(1):105–15.
Roseboom P, Coon S, Baler R, McCune S, Weller J, Klein D. Melatonin synthesis: analysis of the more than 150-fold nocturnal increase in serotonin N-acetyltransferase messenger ribonucleic acid in the rat pineal gland. Endocrinology. 1996;137(7):3033–44.
Ribelayga C, Gauer F, Pévet P, Simonneaux V. Distribution of hydroxyindole-O-methyltransferase mRNA in the rat brain: an in situ hybridisation study. Cell Tissue Res. 1998;291(3):415–21.
Djeridane Y, Vivien-Roels B, Simonneaux V, Miguez JM, Pévet P. Evidence for melatonin synthesis in rodent Harderian gland: a dynamic in vitro study. J Pineal Res. 1998;25(1):54–64.
Djeridane Y, Pitrosky B, Vivien-Roels B, Simonneaux V, Kirsch R, Pévet P. Long-term daily melatonin infusion induces a large increase in N-acetyltransferase activity, hydroxyindole-O-methyltransferase activity, and melatonin content in the Harderian gland and eye of pinealectomized male Siberian hamsters (Phodopus sungorus). J Pineal Res. 2000;29(2):65–73.
Quay W, Ma Y. Demonstration of gastrointestinal hydroxyindole-O-methyltransferase. IRCS Med Sci. 1976;4:563.
Gauer F, Craft CM. Circadian regulation of hydroxyindole-O-methyltransferase mRNA levels in rat pineal and retina. Brain Res. 1996;737(1–2):99–109.
Pévet P, Balemans M, Legerstee W, Vivien-Roels B. Circadian rhythmicity of the activity of hydroxyindole-O-methyl transferase (HIOMT) in the formation of melatonin and 5-methoxytryptophol in the pineal, retina, and harderian gland of the golden hamster. J Neural Transm. 1980;49(4):229–45.
Dubocovich ML, Takahashi JS. Use of 2-[125I] iodomelatonin to characterize melatonin binding sites in chicken retina. Proc Natl Acad Sci. 1987;84(11):3916–20.
Lopez-Gonzalez M, Calvo J, Rubio A, Goberna R, Guerrero J. Characterization of melatonin binding sites in the Harderian gland and median eminence of the rat. Life Sci. 1991;48(12):1165–71.
Cardinali D, Nagle C, Freire F, Rosner J. Effects of melatonin on neurotransmitter uptake and release by synaptosome-rich homogenates of the rat hypothalamus. Neuroendocrinology. 1975;18(1):72–85.
Carneiro RC, Toffoleto O, Cipolla-Neto J, Marcus RP. Modulation of sympathetic neurotransmission by melatonin. Eur J Pharmacol. 1994;257(1–2):73–7.
Markus RP, Zago WM, Carneiro R. Melatonin modulation of presynaptic nicotinic acetylcholine receptors in the rat vas deferens. J Pharmacol Exp Ther. 1996;279(1):18–22.
Bucher B, Gauer F, Pévet P, Masson-Pévet M. Vasoconstrictor effects of various melatonin analogs on the rat tail artery in the presence of phenylephrine. J Cardiovasc Pharmacol. 1999;33(2):316–22.
Wan Q, Man H-Y, Liu F, Braunton J, Niznik HB, Pang SF, et al. Differential modulation of GABA A receptor function by Mel 1a and Mel 1b receptors. Nat Neurosci. 1999;2(5):401.
Golombek DA, Pévet P, Cardinali DP. Melatonin effects on behavior: possible mediation by the central GABAergic system. Neurosci Biobehav Rev. 1996;20(3):403–12.
Pierpaoli W, Regelson W. Pineal control of aging: effect of melatonin and pineal grafting on aging mice. Proc Natl Acad Sci. 1994;91(2):787–91.
Menendez-Pelaez A, Reiter RJ. Distribution of melatonin in mammalian tissues: the relative importance of nuclear versus cytosolic localization. J Pineal Res. 1993;15(2):59–69.
Tamarkin L, Cohen M, Roselle D, Reichert C, Lippman M, Chabner B. Melatonin inhibition and pinealectomy enhancement of 7,12-dimethylbenz(a) anthracene-induced mammary tumors in the rat. Cancer Res. 1981;41(11 Pt 1):4432–6.
Blask D, Hill S. Effects of melatonin on cancer: studies on MCF-7 human breast cancer cells in culture. J Neural Transm Suppl. 1986;21:433–49.
Hill SM, Blask DE. Effects of the pineal hormone melatonin on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res. 1988;48(21):6121–6.
Teplitzky S, Kiefer T, Cheng Q, Dwivedi P, Moroz K, Myers L, et al. Chemoprevention of NMU-induced rat mammary carcinoma with the combination of melatonin and 9-cis-retinoic acid. Cancer Lett. 2001;168(2):155–63.
Kiefer T, Ram P, Yuan L, Hill S. Melatonin inhibits estrogen receptor transactivation and cAMP levels in breast cancer cells. Breast Cancer Res Treat. 2002;71(1):37–45.
Scott AE, Cosma GN, Frank AA, Wells RL, Gardner HS Jr. Disruption of mitochondrial respiration by melatonin in MCF-7 cells. Toxicol Appl Pharmacol. 2001;171(3):149–56.
Pozo D, Reiter RJ, Calvo JR, Guerrero JM. Physiological concentrations of melatonin inhibit nitric oxide synthase in rat cerebellum. Life Sci. 1994;55(24):PL455–60.
Benot S, Gobema R, Reiter RJ, Garcia-Mauriño S, Osuna C, Guerrero JM. Physiological levels of melatonin contribute to the antioxidant capacity of human serum. J Pineal Res. 1999;27(1):59–64.
Armstrong S, Redman J. Melatonin: a chronobiotic with anti-aging properties? Med Hypotheses. 1991;34(4):300–9.
Reiter R, Tan D, Mayo J, Sainz R, Leon J, Bandyopadhyay D. Neurally-mediated and neurally-independent beneficial. J Physiol Pharmacol. 2003;54(4):113–25.
Darlington LG, Forrest CM, Mackay GM, Smith RA, Smith AJ, Stoy N, et al. On the biological importance of the 3-hydroxyanthranilic acid: anthranilic acid ratio. Int J Tryptophan Res. 2010;3:51–9.
Stone TW, Forrest CM, Stoy N, Darlington LG. Involvement of kynurenines in Huntington’s disease and stroke-induced brain damage. Neural Transm. 2012;119(2):261–74.
Kwidzinski E, Bunse J Jr, Aktas O, Richter D, Mutlu L, Zipp F, et al. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005;19(10):1347–9.
Mancuso R, Hernis A, Agostini S, Rovaris M, Caputo D, Fuchs D, et al. Indoleamine 2,3 dioxygenase (IDO) expression and activity in relapsing-remitting multiple sclerosis. PLoS ONE. 2015;10(6):e0130715.
Sakurai K, Zou J-P, Tschetter JR, Ward JM, Shearer GM. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;129(1–2):186–96.
Sundaram G, Brew BJ, Jones SP, Adams S, Lim CK, Guillemin GJ. Quinolinic acid toxicity on oligodendroglial cells: relevance for multiple sclerosis and therapeutic strategies. J Neuroinflamm. 2014;11(1):204.
Anderson G, Rodriguez M. Multiple sclerosis, seizures, and antiepileptics: role of IL-18, IDO, and melatonin. Eur J Neurol. 2011;18(5):680–5.
Zhou H, Wang J, Jiang J, Stavrovskaya IG, Li M, Li W, et al. N-Acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. J Neurosci. 2014;34(8):2967–78.
Klein DC, Coon SL, Roseboom PH, Weller J, Bernard M, Gastel JA, et al. The melatonin rhythm-generating enzyme: molecular regulation of serotonin N-acetyltransferase in the pineal gland. Recent Prog Hormone Res. 1997;52:307–58.
Khalil EM, De Angelis J, Ishii M, Cole PA. Mechanism-based inhibition of the melatonin rhythm enzyme: pharmacologic exploitation of active site functional plasticity. Proc Natl Acad Sci. 1999;96(22):12418–23.
Boutin JA, Audinot V, Ferry G, Delagrange P. Molecular tools to study melatonin pathways and actions. Trends Pharmacol Sci. 2005;26(8):412–9.
Ferry G, Mozo J, Ubeaud C, Berger S, Bertrand M, Try A, et al. Characterization and regulation of a CHO cell line stably expressing human serotonin N-acetyltransferase (EC 2.3. 1.87). Cell Mol Life Sci CMLS. 2002;59(8):1395–405.
Ganguly S, Coon SL, Klein DC. Control of melatonin synthesis in the mammalian pineal gland: the critical role of serotonin acetylation. Cell Tissue Res. 2002;309(1):127–37.
Garbarino-Pico E, Carpentieri AR, Contin MA, Sarmiento MIK, Brocco M, Panzetta P, et al. Retinal ganglion cells are autonomous circadian oscillators synthesizing N-acetylserotonin during the day. J Biol Chem. 2004;279(49):51172–81.
Hirata F, Hayaishi O, Tokuyama T, Senoh S. In vitro and in vivo formation of two new metabolites of melatonin. J Biol Chem. 1974;249(4):1311–3.
Takikawa O, Yoshida R, Hayaishi O. Monooxygenase activities of dioxygenases. Benzphetamine demethylation and aniline hydroxylation reactions catalyzed by indoleamine 2,3-dioxygenase. J Biol Chem. 1983;258(11):6808–15.
Allegra M, Furtmüller PG, Regelsberger G, Turco-Liveri ML, Tesoriere L, Perretti M, et al. Mechanism of reaction of melatonin with human myeloperoxidase. Biochem Biophys Res Commun. 2001;282(2):380–6.
de Oliveira Silva S, Ximenes VF, Catalani LH, Campa A. Myeloperoxidase-catalyzed oxidation of melatonin by activated neutrophils. Biochem Biophys Res Commun. 2000;279(2):657–62.
Ferry G, Ubeaud C, Lambert P-H, Bertin S, Francis C, Chomarat P, et al. Molecular evidence that melatonin is enzymatically oxidized in a different manner than tryptophan: investigations with both indoleamine 2,3-dioxygenase and myeloperoxidase. Biochem J. 2005;388(1):205–15.
Li Y, Hu N, Yang D, Oxenkrug G, Yang Q. Regulating the balance between the kynurenine and serotonin pathways of tryptophan metabolism. FEBS J. 2017;284(6):948–66.
Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S, et al. JAK 2/STAT 3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res. 2013;55(3):275–86.
Iguchi H, Kato K-I, Ibayashi H. Age-dependent reduction in serum melatonin concentrations in healthy human subjects. J Clin Endocrinol Metab. 1982;55(1):27–9.
Vakkuri O, Leppäluoto J, Kauppila A. Oral administration and distribution of melatonin in human serum, saliva and urine. Life Sci. 1985;37(5):489–95.
Yeleswaram K, McLaughlin LG, Knipe JO, Schabdach D. Pharmacokinetics and oral bioavailability of exogenous melatonin in preclinical animal models and clinical implications. J Pineal Res. 1997;22(1):45–51.
Fourtillan J, Brisson A, Gobin P, Ingrand I, Decourt JP, Girault J. Bioavailability of melatonin in humans after day-time administration of D7 melatonin. Biopharm Drug Dispos. 2000;21(1):15–22.
Flo A, Cambras T, Díez-Noguera A, Calpena A. Melatonin pharmacokinetics after transdermal administration changes according to the time of the day. Eur J Pharm Sci. 2017;96:164–70.
Morgan L, Arendt J, Owens D, Folkard S, Hampton S, Deacon S, et al. Effects of the endogenous clock and sleep time on melatonin, insulin, glucose and lipid metabolism. J Endocrinol. 1998;157(3):443–51.
Sharma S, Singh H, Ahmad N, Mishra P, Tiwari A. The role of melatonin in diabetes: therapeutic implications. Arch Endocr Metab. 2015;59(5):391–9.
Mao S, Chen J, Wei Z, Liu H, Bi D. Intranasal administration of melatonin starch microspheres. Int J Pharm. 2004;272(1–2):37–43.
Bellapart J, Roberts JA, Appadurai V, Wallis SC, Nuñez-Nuñez M, Boots RJ. Pharmacokinetics of a novel dosing regimen of oral melatonin in critically ill patients. Clin Chem Lab Med (CCLM). 2016;54(3):467–72.
Reiter RJ, Guerrero JM, Escames G, Pappolla MA, Acuña‐Castroviejo D. Prophylactic actions of melatonin in oxidative neurotoxicity. Ann N Y Acad Sci. 1997;825(1):70–8.
Leker R, Teichner A, Lavie G, Shohami E, Lamensdorf I, Ovadia H. The nitroxide antioxidant tempol is cerebroprotective against focal cerebral ischemia in spontaneously hypertensive rats. Exp Neurol. 2002;176(2):355–63.
Watson N, Diamandis T, Gonzales-Portillo C, Reyes S, Borlongan CV. Melatonin as an antioxidant for stroke neuroprotection. Cell Transplant. 2016;25(5):883–91.
Lee MY, Kuan YH, Chen HY, Chen TY, Chen ST, Huang CC, et al. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J Pineal Res. 2007;42(3):297–309.
Gomaa AM, Galal HM, Abou-Elgait AT. Neuroprotective effects of melatonin administration against chronic immobilization stress in rats. Int J Physiol Pathophysiol Pharmacol. 2017;9(2):16.
Jacob S, Poeggeler B, Weishaupt JH, Sirén AL, Hardeland R, Bähr M, et al. Melatonin as a candidate compound for neuroprotection in amyotrophic lateral sclerosis (ALS): high tolerability of daily oral melatonin administration in ALS patients. J Pineal Res. 2002;33(3):186–7.
Chahbouni M, Escames G, Venegas C, Sevilla B, García JA, López LC, et al. Melatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from Duchenne muscular dystrophy. J Pineal Res. 2010;48(3):282–9.
Cardinali DP, Vigo DE, Olivar N, Vidal MF, Brusco LI. Melatonin therapy in patients with Alzheimer’s disease. Antioxidants. 2014;3(2):245–77.
Pappolla M, Bozner P, Soto C, Shao H, Robakis NK, Zagorski M, et al. Inhibition of Alzheimer β-fibrillogenesis by melatonin. J Biol Chem. 1998;273(13):7185–8.
Lima ACP, Louzada PR, De Mello FG, Ferreira ST. Neuroprotection against Aβ and glutamate toxicity by melatonin: Are GABA receptors involved? Neurotox Res. 2003;5(5):323–7.
Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res. 2003;35(2):125–30.
Giusti P, Lipartiti M, Franceschini D, Schiavo N, Floreani M, Manev H. Neuroprotection by melatonin from kainate-induced excitotoxicity in rats. FASEB J. 1996;10(8):891–6.
Manev H, Uz T, Kharlamov A, Joo J. Increased brain damage after stroke or excitotoxic seizures in melatonin-deficient rats. FASEB J. 1996;10(13):1546–51.
Cho S, Joh TH, Baik HH, Dibinis C, Volpe BT. Melatonin administration protects CA1 hippocampal neurons after transient forebrain ischemia in rats. Brain Res. 1997;755(2):335–8.
Kilic E, Öuzdemir YG, Bolay H, Keleştimur H, Dalkara T. Pinealectomy aggravates and melatonin administration attenuates brain damage in focal ischemia. J Cereb Blood Flow Metab. 1999;19(5):511–6.
Deng Y-Q, Xu G-G, Duan P, Zhang Q, Wang J-Z. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol Sin. 2005;26(5):519.
Lau WW, Ng JK, Lee MM, Chan AS, Wong YH. Interleukin-6 autocrine signaling mediates melatonin MT1/2 receptor-induced STAT3 Tyr705 phosphorylation. J Pineal Res. 2012;52(4):477–89.
Chuang JI, Mohan N, Meltz ML, Reiter RJ. Effect, of melatonin, on NF-κb dna-binding activity in the rat spleen. Cell Biol Int. 1996;20(10):687–92.
Feng Z, Chang Y, Cheng Y, Zhang BL, Qu ZW, Qin C, et al. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer’s disease. J Pineal Res. 2004;37(2):129–36.
Cardinali DP, Furio AM, Brusco LI. Clinical aspects of melatonin intervention in Alzheimer’s disease progression. Curr Neuropharmacol. 2010;8(3):218–27.
Rajaratnam SM, Polymeropoulos MH, Fisher DM, Roth T, Scott C, Birznieks G, et al. Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials. Lancet. 2009;373(9662):482–91.
McKenna JT, Christie MA, Jeffrey BA, McCoy JG, Lee E, Connolly NP, et al. Chronic ramelteon treatment in a mouse model of Alzheimer’s disease. Arch Ital Biol. 2012;150(1):5.
Mack JM, Schamne MG, Sampaio TB, Pértile RAN, Fernandes PACM, Markus RP, et al. Melatoninergic system in Parkinson’s disease: from neuroprotection to the management of motor and nonmotor symptoms. Oxid Med Cell Longev. 2016;2016:3472032.
Singhal NK, Srivastava G, Patel DK, Jain SK, Singh MP. Melatonin or silymarin reduces maneb- and paraquat-induced Parkinson’s disease phenotype in the mouse. J Pineal Res. 2011;50(2):97–109.
Wilkinson D, Shepherd E, Wallace EM. Melatonin for women in pregnancy for neuroprotection of the fetus. Cochrane Libr. 2016;3:CD010527.
Cardinali DP, Vigo DE, Olivar N, Vidal MF, Furio AM, Brusco LI. Therapeutic application of melatonin in mild cognitive impairment. Am J Neurodegener Dis. 2012;1(3):280.
Pandi-Perumal SR, BaHammam AS, Brown GM, Spence DW, Bharti VK, Kaur C, et al. Melatonin antioxidative defense: therapeutical implications for aging and neurodegenerative processes. Neurotox Res. 2013;23(3):267–300.
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014. https://doi.org/10.1212/wnl.0000000000000560.
Graetz C, Groppa S, Zipp F, Siller N. Preservation of neuronal function as measured by clinical and MRI endpoints in relapsing-remitting multiple sclerosis: how effective are current treatment strategies? Expert Rev Neurother. 2018;18(3):203–19.
Compston A, Coles A. Multiple sclerosis. Lancet (London, England). 2008;372(9648):1502–17 (Epub 2008/10/31).
Tullman MJ. Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am J Manag Care. 2013;19(2 Suppl):S15–20.
Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17(2):210–8.
Camara-Lemarroy CR, Metz L, Meddings JB, Sharkey KA, Wee Yong V. The intestinal barrier in multiple sclerosis: implications for pathophysiology and therapeutics. Brain. 2018;141(7):1900–16.
Bannerman P. Cortical involvement in multiple sclerosis. In: Cechetto D, Weishaupt N, editors. The cerebral cortex in neurodegenerative and neuropsychiatric disorders: New York: Elsevier; 2017. pp. 243–73.
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183–93.
Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014;89(2):225–40.
Farzaei MH, Shahpiri Z, Bahramsoltani R, Najafi F, Rahimi R. Efficacy and tolerability of phytomedicines in multiple sclerosis patients: a review. CNS Drugs. 2017;31(10):867–89.
Sand IK. The role of diet in multiple sclerosis: mechanistic connections and current evidence. Curr Nutr Rep. 2018;7(3):150–60.
Hart BA. Why does multiple sclerosis only affect human primates? Multip Scler. (Houndmills, Basingstoke, England). 2016;22(4):559–63 (Epub 2015/11/06).
Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079–106 (Epub 2011/03/05).
Torkildsen O, Brunborg LA, Myhr KM, Bo L. The cuprizone model for demyelination. Acta Neurol Scand Suppl. 2008;188:72–6 (Epub 2008/06/18).
Vakilzadeh G, Khodagholi F, Ghadiri T, Ghaemi A, Noorbakhsh F, Sharifzadeh M, et al. The effect of melatonin on behavioral, molecular, and histopathological changes in cuprizone model of demyelination. Mol Neurobiol. 2016;53(7):4675–84 (Epub 2015/08/28).
Wen J, Ariyannur PS, Ribeiro R, Tanaka M, Moffett JR, Kirmani BF, et al. Efficacy of N-acetylserotonin and melatonin in the EAE model of multiple sclerosis. J Neuroimmune Pharmacol. 2016;11(4):763–73.
Alvarez-Sanchez N, Cruz-Chamorro I, Lopez-Gonzalez A, Utrilla JC, Fernandez-Santos JM, Martinez-Lopez A, et al. Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain Behav Immun. 2015;50:101–14 (Epub 2015/07/03).
Kang JC, Ahn M, Kim YS, Moon C, Lee Y, Wie MB, et al. Melatonin ameliorates autoimmune encephalomyelitis through suppression of intercellular adhesion molecule-1. J Vet Sci. 2001;2(2):85–9 (Epub 2003/11/14).
Long T, Yang Y, Peng L, Li Z. Neuroprotective effects of melatonin on experimental allergic encephalomyelitis mice via anti-oxidative stress activity. J Mol Neurosci MN. 2018;64(2):233–41 (Epub 2018/02/17).
Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251(3):261–8 (Epub 2004/03/12).
van Horssen J, Schreibelt G, Bo L, Montagne L, Drukarch B, van Muiswinkel FL, et al. NAD(P)H:quinone oxidoreductase 1 expression in multiple sclerosis lesions. Free Radic Biol Med. 2006;41(2):311–7 (Epub 2006/07/04).
Arnold P, Mojumder D, Detoledo J, Lucius R, Wilms H. Pathophysiological processes in multiple sclerosis: focus on nuclear factor erythroid-2-related factor 2 and emerging pathways. Clin Pharmacol Adv Appl. 2014;6:35–42 (Epub 2014/03/05).
Ghareghani M, Zibara K, Sadeghi H, Farhadi N. Spasticity treatment ameliorates the efficacy of melatonin therapy in experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis. Cell Mol Neurobiol. 2018;38(5):1145–51 (Epub 2018/03/03).
Kashani IR, Rajabi Z, Akbari M, Hassanzadeh G, Mohseni A, Eramsadati MK, et al. Protective effects of melatonin against mitochondrial injury in a mouse model of multiple sclerosis. Exp Brain Res. 2014;232(9):2835–46 (Epub 2014/05/07).
Campbell GR, Mahad DJ. Mitochondria as crucial players in demyelinated axons: lessons from neuropathology and experimental demyelination. Autoimmune Dis. 2011;2011:262847.
Campbell G, Mahad DJ. Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis. FEBS Lett. 2018;592(7):1113–21 (Epub 2018/02/18).
Lin W, Lin Y. IFN-γ inhibits central nervous system myelination through both STAT1-dependent and STAT1-independent pathways. J Neurosci Res. 2010;88(12):2569–77.
Chen SJ, Huang SH, Chen JW, Wang KC, Yang YR, Liu PF, et al. Melatonin enhances interleukin-10 expression and suppresses chemotaxis to inhibit inflammation in situ and reduce the severity of experimental autoimmune encephalomyelitis. Int Immunopharmacol. 2016;31:169–77 (Epub 2016/01/07).
Ghareghani M, Dokoohaki S, Ghanbari A, Farhadi N, Zibara K, Khodadoust S, et al. Melatonin exacerbates acute experimental autoimmune encephalomyelitis by enhancing the serum levels of lactate: a potential biomarker of multiple sclerosis progression. Clin Exp Pharmacol Physiol. 2017;44(1):52–61 (Epub 2016/10/04).
Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):37–53.
Peschl P, Bradl M, Hoftberger R, Berger T, Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529 (Epub 2017/05/24).
Gupta S, Ahsan I, Mahfooz N, Abdelhamid N, Ramanathan M, Weinstock-Guttman B. Osteoporosis and multiple sclerosis: risk factors, pathophysiology, and therapeutic interventions. CNS Drugs. 2014;28(8):731–42 (Epub 2014/05/30).
Ghareghani M, Scavo L, Arnoult D, Zibara K, Farhadi N. Melatonin therapy reduces the risk of osteoporosis and normalizes bone formation in multiple sclerosis. Fundam Clin Pharmacol. 2018;32(2):181–7.
Natarajan R, Einarsdottir E, Riutta A, Hagman S, Raunio M, Mononen N, et al. Melatonin pathway genes are associated with progressive subtypes and disability status in multiple sclerosis among Finnish patients. J Neuroimmunol. 2012;250(1–2):106–10.
Melamud L, Golan D, Luboshitzky R, Lavi I, Miller A. Melatonin dysregulation, sleep disturbances and fatigue in multiple sclerosis. J Neurol Sci. 2012;314(1–2):37–40.
Adamczyk-Sowa M, Sowa P, Adamczyk J, Niedziela N, Misiolek H, Owczarek M, et al. Effect of melatonin supplementation on plasma lipid hydroperoxides, homocysteine concentration and chronic fatigue syndrome in multiple sclerosis patients treated with interferons-beta and mitoxantrone. J Physiol Pharmacol. 2016;67:235–42.
Roostaei T, Sahraian MA, Hajeaghaee S, Gholipour T, Togha M, Siroos B, et al. Impact of melatonin on motor, cognitive and neuroimaging indices in patients with multiple sclerosis. Iran J Allergy Asthma Immunol. 2015;14(6):589–95.
Gholipour T, Ghazizadeh T, Babapour S, Mansouri B, Ghafarpour M, Siroos B, et al. Decreased urinary level of melatonin as a marker of disease severity in patients with multiple sclerosis. Iran J Allergy Asthma Immunol. 2015;14(1):91–7.
Miller E, Walczak A, Majsterek I, Kedziora J. Melatonin reduces oxidative stress in the erythrocytes of multiple sclerosis patients with secondary progressive clinical course. J Neuroimmunol. 2013;257(1–2):97–101 (Epub 2013/03/23).
Adamczyk-Sowa M, Galiniak S, Żyracka E, Grzesik M, Naparło K, Sowa P, et al. Oxidative modification of blood serum proteins in multiple sclerosis after interferon beta and melatonin treatment. Oxid Med Cell Longev. 2017;2017:7905148.
Adamczyk-Sowa M, Pierzchala K, Sowa P, Polaniak R, Kukla M, Hartel M. Influence of melatonin supplementation on serum antioxidative properties and impact of the quality of life in multiple sclerosis patients. J Physiol Pharmacol. 2014;65(4):543–50.
Adamczyk-Sowa M, Sowa P, Mucha S, Zostawa J, Mazur B, Owczarek M, et al. Changes in serum ceruloplasmin levels based on immunomodulatory treatments and melatonin supplementation in multiple sclerosis patients. Med Sci Monit Int Med J Exp Clin Res. 2016;22:2484.
Emamgholipour S, Hossein-nezhad A, Sahraian MA, Askarisadr F, Ansari M. Evidence for possible role of melatonin in reducing oxidative stress in multiple sclerosis through its effect on SIRT1 and antioxidant enzymes. Life Sci. 2016;145:34–41.
Ghorbani A, Salari M, Shaygannejad V, Norouzi R. The role of melatonin in the pathogenesis of multiple sclerosis: a case-control study. Int J Prev Med. 2013;4(Suppl 2):S180.
Mrowicka M, Garncarek P, Miller E, Kedziora J, Smigielski J, Malinowska K, et al. Effect of melatonin on activity of superoxide dismutase (CuZn-SOD) in erythrocytes of patients during short- and long-term hypokinesis. Wiadomosci Lekarskie (Warsaw, Poland: 1960). 2010;63(1):3–9.
Farez MF, Mascanfroni ID, Méndez-Huergo SP, Yeste A, Murugaiyan G, Garo LP, et al. Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell. 2015;162(6):1338–52.
Alvarez-Sanchez N, Cruz-Chamorro I, Diaz-Sanchez M, Sarmiento-Soto H, Medrano-Campillo P, Martinez-Lopez A, et al. Melatonin reduces inflammatory response in peripheral T helper lymphocytes from relapsing-remitting multiple sclerosis patients. J Pineal Res. 2017;63(4):e12442 (Epub 2017/08/10).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No source funding was used to prepare this review.
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Yeganeh Salehpour, M., Mollica, A., Momtaz, S. et al. Melatonin and Multiple Sclerosis: From Plausible Neuropharmacological Mechanisms of Action to Experimental and Clinical Evidence. Clin Drug Investig 39, 607–624 (2019). https://doi.org/10.1007/s40261-019-00793-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-019-00793-6