
Abstract
Antiphospholipid syndrome (APS) is an autoimmune thrombophilia propelled by circulating antiphospholipid antibodies that herald vascular thrombosis and obstetrical complications. Antiphospholipid antibodies recognize phospholipids and phospholipid-binding proteins and are not only markers of disease but also key drivers of APS pathophysiology. Thrombotic events in APS can be attributed to various conspirators including activated endothelial cells, platelets, and myeloid-lineage cells, as well as derangements in coagulation and fibrinolytic systems. Furthermore, recent work has especially highlighted the role of neutrophil extracellular traps (NETs) and the complement system in APS thrombosis. Beyond acute thrombosis, patients with APS can also develop an occlusive vasculopathy, a long-term consequence of APS characterized by cell proliferation and infiltration that progressively expands the intima and leads to organ damage. This review will highlight known pathogenic factors in APS and will also briefly discuss similarities between APS and the thrombophilic coagulopathy of COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antiphospholipid syndrome (APS) is a thrombo-inflammatory disease that complicates up to one-third of cases of systemic lupus erythematosus (referred to as “lupus” going forward) where it portends the acquisition of more organ damage over time [1,2,3,4,5,6]. Meanwhile, the standalone form of APS (primary APS) is even more common, affecting at least 1 in 2000 Americans [7]. APS is propelled by circulating antiphospholipid antibodies (aPL) that cause vascular thrombosis and obstetrical complications [8]. Thrombosis in APS may affect vascular beds of all sizes including arterial, venous, and microvascular circuits. Lower extremity deep veins and cerebral arteries are the most frequent sites of venous and arterial thrombosis, respectively [9]. Thrombi may also form in sites uncommonly seen in the general population including arteries that supply the viscera and venous sinuses surrounding the brain. Patients with APS are additionally at risk for microvascular thrombosis in the skin, eyes, heart, lungs, kidneys, and other organs. A minority of patients develop catastrophic APS (CAPS), characterized by microvascular thrombosis in at least three organs, typically all emerging within 1 week [10, 11]. Beyond thrombosis and pregnancy loss, APS is also associated with a variety of extra-criteria manifestations, including livedo reticularis and racemosa, neurologic pathology (cognitive dysfunction, choreiform movements, seizures), valvular heart disease, occlusive vasculopathy, pulmonary hypertension, nephropathy, and thrombocytopenia, among others [12, 13].
In addition to a history of at least one morbid thrombotic or obstetric event, APS classification criteria (Table 1) seek the stable presence of anticardiolipin or anti-beta-2 glycoprotein I (β2GPI) antibodies [8]. Furthermore, the “lupus anticoagulant” test—a functional assay screening for aPL based on prolongation of clotting times—is part of the classification criteria where it detects a variety of species of aPL including anti-phosphatidylserine/prothrombin antibodies [14]. Modern anticardiolipin assays are designed to recognize anti-β2GPI antibodies, as β2GPI protein present in the sample diluent provides a bridge between antibody and cardiolipin [15,16,17]. Furthermore, some anti-β2GPI antibodies clearly have lupus anticoagulant activity [18,19,20]. It should therefore be recognized that a single antibody can potentially turn all three criteria lab tests positive, and the information provided by these different assays is therefore not as granular as one may initially assume.
APS is present if at least one of the clinical criteria and one of the laboratory criteria are met.
It is now recognized that the term “antiphospholipid” is something of a misnomer since the best characterized aPL do not recognize isolated anionic phospholipids such as cardiolipin and phosphatidylserine as originally surmised, but rather specific phospholipid-binding proteins, with aPL targeting the abundant plasma protein β2GPI particularly pathogenic [21,22,23]; it is also conceivable that these antibodies detect heterotypic complexes of phospholipids and phospholipid-binding proteins. Intriguingly, the isotype of aPL immunoglobulin abnormality may vary by patient subset. For example, an early study observed that IgA was the most prevalent isotype among Black patients with SLE [24], although the potential pathogenic role of IgA and implications for APS remain to be firmly established.
Life-long anticoagulation is so far the only treatment that has been proven to reduce the vascular complications of APS. However, while anticoagulation regimens are relatively effective in restraining large-vessel events such as deep vein thrombosis and thromboembolic stroke, they do not combat many extra-criteria manifestations such as livedoid vasculopathy, seizures, cognitive decline, alveolar hemorrhage, and thrombocytopenia. Furthermore, anticoagulants do not mitigate the chronic occlusive vasculopathy and progressive organ deterioration that afflict many patients over time. This unmet need is emphasized by an international cohort of more than 800 aPL-positive patients in which 56% of patients had at least one non-thrombotic/non-obstetric manifestation of APS [25]. Notably, more than 25% of these patients were identified as having either white matter brain lesions or premature cognitive dysfunction, and 20% were found to have microvascular disease involving either the kidney or skin. Strategies to combat the long-term, anticoagulant-resistant manifestations of APS are unknown and will likely require new immunomodulatory approaches.
The development of a consensus, unified explanation of APS pathophysiology has unfortunately been hindered by the heterogeneity of aPL profiles (only a fraction of which is likely revealed by standard clinical laboratory testing) and diversity of potential aPL effector functions. As will be discussed below, numerous and wide-ranging “bad actors” have been implicated to date, including blood and immune cells, complement proteins, and coagulation/fibrinolytic systems.
Thrombosis
Notably, aPL are not only markers of disease but also key drivers of APS pathogenesis. Indeed, many manifestations of APS can be reproduced experimentally via transfer of patient serum or immunoglobulins into animals [26,27,28]. Although the infusion of pathogenic aPL does not cause spontaneous thrombosis in animals, the introduction of some type of disruption to the vasculature (such as injury to the vessel wall; alteration of blood flow; or infusion of lipopolysaccharide, histones, or other immune stimulants) unmasks the exaggerated thromboinflammatory state. The “two-hit” concept of APS (in animal models as well as in patients) posits that aPL provide the first hit, creating a generalized procoagulant state. Subsequently, a second hit (sometimes cryptic) such as a vascular injury or inflammatory stimulus then tips circulating blood toward coagulation. Although this triggering stimulus is not obvious in many cases of thrombotic APS, a precipitating factor such as surgery, infection, pregnancy, or anticoagulation withdrawal has been identified in 50 to 80% of CAPS episodes [29].
Attention should also be paid to additional risk factors that further increase thrombotic risk in aPL-positive patients [30, 31]. Some potential factors include a concomitant diagnosis of lupus, pregnancy, receipt of estrogen-containing contraceptives, immobilization after surgery, active cancer, heritable thrombophilias, and traditional cardiovascular risk factors such as smoking, hypertension, hypercholesterolemia, and obesity. As an example, one large population-based case–control study found that the odds ratio of ischemic stroke in lupus anticoagulant-positive females was 43.1 (95% confidence interval 12.2 to 152.0), further increasing to 87 (95% confidence interval 14.5 to 523.0) in individuals who smoked and to a remarkable 201 (95% confidence interval 1.9 to 242) in individuals using estrogen-containing oral contraceptives [32].
The earliest identified prothrombotic effects of aPL were via interference with natural anticoagulant systems regulating coagulation and fibrinolysis. However, subsequent studies eventually revealed that a key role of aPL (arguably the key role) is to induce activation of various blood and immune cells, as well as the complement system, with procoagulant and proinflammatory consequences. Major pathogenic mechanisms are summarized in Table 2. The relative importance of these factors to a particular thrombotic event is likely dependent on the vascular bed being considered, a concept that will benefit from further mechanistic research.
Endothelial cells. Given its constant confrontation with whole blood, the endothelium necessarily has properties that counter thrombosis and inflammation [62]. For example, heparanoid proteoglycans, prostacyclins, ectonucleotidases such as CD39 and CD73, protein C receptor, and tissue factor pathway inhibitor all help promote an antithrombotic surface [63]. The endothelium is also a barrier that selectively permits molecular and cellular transit from the blood compartment into tissue. When activated, the normally quiescent endothelium sheds its antithrombotic profile and acquires a phenotype that promotes an inflammatory response. Leukocyte-endothelial interactions and extravasation are orchestrated by selectins and cell adhesion molecules that facilitate rolling at the endothelial surface, followed by stronger integrin-mediated interactions that promote adhesion and eventual exodus of leukocytes from vessels [64]. Translational studies have detected endothelium-derived microparticles in the circulation of APS patients as a surrogate for endothelial activation, suggesting the vessel wall may be primed for leukocyte interactions [35, 36].
In vitro, aPL activate healthy cultured endothelial cells to express adhesion molecules and tissue factor [33, 34]. Mechanistically, aPL co-opt pathways normally associated with non-autoimmune inflammatory stimuli. aPL engage apolipoprotein E receptor 2 and possibly other surface receptors on endothelial cells [65,66,67,68,69,70] with subsequent activation of NF-κB and p38 MAPK, and suppression of vasculo-protective Krüppel-like factors [71,72,73]. Modulation of these pathways leads to suppression of anti-inflammatory transcription factors, reduction in nitric oxide synthesis, and increased tissue factor synthesis [33, 34, 69, 70, 72,73,74,75,76]. In a mouse model, aPL increased tissue factor activity in carotid homogenates [77]. Meanwhile, aPL administration to mice also results in increased leukocyte-endothelium interactions [78, 79]. Concordantly, mice can be protected from aPL-mediated thrombosis by disrupting the function of E-selectin and P-selectin (the key selectins expressed on endothelium), P-selectin glycoprotein ligand-1 (PSGL-1, a key selectin ligand), or endothelial integrin ligands VCAM-1 and ICAM-1 [76, 79, 80].
Platelets. Platelets are being increasingly recognized for their roles that extend beyond hemostasis and thrombosis. Platelet-leukocyte interactions result in bidirectional immune crosstalk and transactivation, with downstream effects on vascular inflammation. Circulating platelet-leukocyte aggregates are detected at increased levels in patients with APS, consistent with persistent, low-grade platelet activation [38]. The unstimulated platelet surface resists binding by β2GPI protein and anti-β2GPI antibodies; however, under shear stress, β2GPI engages surface ApoER2 and GPIb, creating a platform by which anti-β2GPI antibodies can then trigger platelet activation [37]. Meanwhile, aPL also activate platelets primed by low levels of thrombin in a mitogen-activated protein kinase (MAP kinase)-dependent fashion [81]. In a mouse model of APS, aPL-activated platelets are preferentially recruited to injured endothelium where they are required for fibrin generation in the expanding thrombus [82]. As discussed above, thrombocytopenia commonly complicates the course of APS. The extent to which this thrombocytopenia of APS is attributable to low-grade platelet activation and subsequent clearance, or to autoimmune-mediated removal via anti-platelet glycoprotein antibodies likely varies from patient to patient [83,84,85,86].
Monocytes. The relative ease of monocyte isolation from peripheral blood has led to deeper characterization of monocytes than with endothelial cells or platelets. For example, it was demonstrated 20 years ago that in patients with lupus, the presence of aPL is associated with enhanced monocyte tissue factor production [87]. Similar findings have been appreciated in patients with primary APS [46,47,48]. Beyond tissue factor, APS monocytes also express high levels of VEGF and its receptor Flt-1 [88]. Unbiased transcriptomic profiling has demonstrated upregulation of proinflammatory genes including TLR8, CD14, and genes associated with oxidative stress [89, 90]. APS monocytes have also been shown to upregulate certain protease-activated receptors [91], best known for their response to activated coagulation factors such as thrombin but also now appreciated for their immune signaling functions. Monocyte-derived microparticles are found at increased levels in APS circulation [92, 93], where they are potentially an important source of tissue factor [36].
Experimentally, aPL trigger monocytes to express tissue factor [39,40,41,42] and pro-inflammatory cytokines including TNF-α and IL-1β in vitro [43,44,45]. Concordantly, neutralizing tissue factor in mice with a blocking antibody protects against aPL-mediated venous thrombosis [94]. Although circulating monocytes have not, for the most part, been specifically characterized in animal studies, one interesting report demonstrated that the introduction of a Nox2 (NADPH oxidase) mutation into bone marrow-derived cells (e.g., myeloid cells but not endothelial cells) protects against venous thrombosis [94].
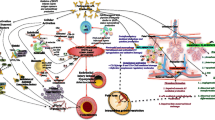
Neutrophils. Neutrophils are the most abundant leukocytes in circulation where they patrol the bloodstream waiting to be recruited to sites of inflammation. Until recently, phagocytosis was thought to be the dominant mechanism by which neutrophils neutralized invading pathogens [95, 96]. In 2004, Brinkmann and colleagues described a process whereby neutrophils eject webs of chromatin into the extracellular space [97, 98]. These neutrophil extracellular traps (NETs) are tangles of decondensed extracellular DNA and histones decorated with microbicidal proteins derived from neutrophil granules and cytoplasm. NETs are released in response to both infectious and sterile stimuli including bacteria, fungi, protozoa, and viruses, as well as activated platelets and endothelial cells, complement proteins, cytokines, autoantibodies, and immune complexes [99]. While NETs likely evolved to trap pathogens, they are also now well recognized to be prothrombotic (Fig. 1) [100]. NETs activate platelets and clotting factors and can be found in both deep vein thrombi [101,102,103,104] and arterial clots [105,106,107]. Indeed, studies by various groups have shown that disrupting neutrophil-endothelium interactions, preventing NET formation, and dissolving NETs are all strategies that can mitigate thrombosis in animal models [104, 108,109,110,111,112,113,114,115].

Neutrophil extracellular promote thrombosis. Activated neutrophils release decondensed chromatin decorated with nuclear (histones), granule (proteases that degrade antithrombotic molecules such as TFPI and antithrombin), and cytoplasmic proteins that promote inflammation and coagulation (tissue factor, factor XI and XII). Together, NETs form a scaffold for cell aggregation and thrombus formation. TFPI = tissue factor pathway inhibitor. Illustration credit: Ethan Tyler (NIH)
In the 1990s, prior to the first descriptions of NETs, it was found that mouse monoclonal antibodies against human β2GPI activated neutrophils, stimulating degranulation and hydrogen peroxide production [116]. In the early 2000s, an important series of experiments characterized pregnancy models of APS and found that neutrophils and complement were important mediators of fetal injury [117,118,119]. In vitro experiments have demonstrated that various human monoclonal aPL induce neutrophil activation as measured by oxidative burst, phagocytosis, and shedding of L-selectin [120]; these phenotypes were potentiated by lipopolysaccharide and Pam3Cys-Ser-(Lys)4, demonstrating the potential for synergy with Toll-like receptor signaling [120]. Another study found increased tissue factor expression by control neutrophils cultured with APS serum; in this system, complement activation, and specifically the C5a receptor, was required for maximum tissue factor expression [121].
In one of the first studies to evaluate a potential role for NETs in APS, pre-formed NETs were exposed to APS patient serum [122]. As compared with healthy serum, the authors found that approximately 13% of APS serum samples (both primary and secondary) were defective in NET degradation [122]. In the same study, “anti-NET antibodies” were detected by adding APS serum to preformed NETs and then visualizing IgG deposition [122]. The concept of anti-NET antibodies has been more comprehensively examined in a recent study [50]. In a cohort of 76 patients with primary APS, the authors found IgG and IgM anti-NET antibodies to be markedly elevated as compared with healthy controls. Anti-NET antibodies did not correlate with anti-β2GPI antibodies but did associate with impaired NET degradation by patient serum as well as a clinical history of recurrent venous thrombosis [50]. The extent to which anti-NET antibodies recognize similar antigens as the anti-chromatin antibodies previously described in primary APS is an intriguing question worthy of further study [123].
A study in 2015 was the first to show an association between aPL and NET release [49], a finding that has since been replicated by independent groups [124, 125]. The authors of the original study found high levels of NETs in circulation even in the absence of active thrombosis and that freshly isolated neutrophils from patients with APS released more NETs than neutrophils from healthy patients [49]. Moreover, human monoclonal anti-β2GPI antibodies promoted NET release, while patients with triple-positive APS (presence of anticardiolipin, anti-β2GPI, and lupus anticoagulant) tended to have the highest levels of circulating NET remnants [49]. Mechanistically, aPL-stimulated NET release depended on ROS generation by the NADPH oxidase and TLR4 signaling [49], with a role for Mac-1-mediated adhesion [126]. In vivo experiments used a flow restriction model of venous thrombosis to characterize aPL-mediated thrombosis in mice [127]. Mice administered APS IgG formed large thrombi that were enriched for NETs [127]. Meanwhile, both neutrophil depletion and deoxyribonuclease administration reduced thrombosis in APS mice to levels observed in control mice [127]. Other NET-disrupting strategies that mitigate aPL-mediated thrombosis in mouse models include PSGL-1 deficiency or inhibition [79], activation of cell surface adenosine receptors by drugs such as dipyridamole and defibrotide [128, 129], and even administration of ginger-derived phenolic substances, which function as phosphodiesterase inhibitors [130]. In patients, administration of the antioxidant coenzyme Q10 has been suggested as a complementary strategy for inhibiting NETs [131, 132].
Low-density granulocytes (LDGs), a subset of neutrophils best characterized in lupus, are proinflammatory and have a low threshold for releasing NETs [133,134,135,136,137]. van den Hoogen and colleagues recently found higher frequency of LDGs in APS patients whether or not the patients had coexisting lupus [138]. Notably, they also observed that anti-β2GPI-positivity was predictive of APS patients who would have more LDGs in circulation.
A few studies have examined gene expression in APS neutrophils. In one, transcriptomic analysis of APS neutrophils by RNA sequencing revealed increase expression of pro-inflammatory genes, particularly with regard to type I interferon signaling, Toll-like receptor signaling, and metabolic reprogramming [79]. IFIT1, a type I interferon responsive gene, was most significantly upregulated (8.5-fold) in APS neutrophils [79]. In another study, genome-wide DNA methylation analysis of APS neutrophils did not find notable demethylation of interferon genes as has been previously reported for lupus neutrophils, suggesting divergent epigenomic signatures [139]. Gene ontology analysis of hypomethylated genes in APS neutrophils demonstrated an enrichment of ETS1, EMP2, OXT, and DPPA3, all genes associated with mammalian pregnancy [139]. The physiologic consequences of epigenetic changes in APS neutrophils, including their potential relevance to APS-associated pregnancy morbidity, remain to be elucidated.
Taken together, these studies suggest exaggerated NET formation and impaired NET degradation in APS, both mechanisms that could amplify the impact of NETs on thrombosis. However, numerous questions remain, including the extent to which NETs might contribute to obstetric and extra-criteria manifestations of APS. Given that β2GPI is a recognized DNA-binding protein, future studies may also ask whether anti-β2GPI antibodies provide the possibility for epitope expansion to traditional lupus autoantigens such as double-stranded DNA and chromatin in some patients [140].
Complement. Complement is a system of over 50 proteins of the innate immune system that interact via protease activity to promote inflammatory cell recruitment, opsonization and clearance of pathogens, and sometimes cell death. Complement also links inflammatory responses to coagulation pathways [141]. The system can be activated by different stimuli with eventual convergence at the level of C5a generation (a chemotactic and pro-inflammatory protein) and assembly of the so-called membrane-attack complex (inclusive of C5b, C6, C7, C8, and C9) [142].
There is evidence of smoldering complement activation in APS [52,53,54], via both the alternative pathway [143,144,145] and the classical pathway [146,147,148]. Mechanistically, an important recent study used sera and purified anti-β2GPI antibodies to demonstrate C5b-9 deposition and complement-mediated cell death via what the authors described as a “modified Ham test” [51]; importantly, complement activation as measured by this novel test correlated clinically with both triple-positive status and recurrent thrombosis.
Animal models provide strong evidence linking the complement system to APS. After early work demonstrated that antagonizing complement could protect against pregnancy loss [149], attention turned to its potential role in aPL-accelerated thrombosis. In a femoral vein injury model of thrombosis, disrupting complement C3, C5, and C6 were all individually protective against thrombosis [150,151,152,153]. Similarly, antagonizing either C5 or C6 was protective in a mesenteric thrombosis model triggered by lipopolysaccharide [154]. A deeper understanding of the complement pathway in APS is now needed, including mechanisms by which it integrates inflammation and coagulation.
Clinical reports of the complement inhibitor eculizumab effectively treating thrombosis in APS and CAPS [155, 156] are intriguing. Although there is a paucity of therapeutics available to mitigate the high mortality associated with CAPS, well-designed, randomized clinical trials are needed to develop a stronger evidence basis for complement inhibition and appropriate patient selection in APS.
Coagulation. The complex of β2GPI and anti-β2GPI disrupts the annexin A5 “anticoagulant shield” whereby annexin A5 normally binds to and neutralizes procoagulant phospholipids such as phosphatidylserine on cell surfaces [157,158,159]. In addition, anti-β2GPI antibodies have been reported to impair the natural ability of β2GPI to blunt von Willebrand factor-dependent platelet aggregation [160].
As discussed above, aPL represent a broader repertoire of antigenic targets than β2GPI and cardiolipin, and the effects of aPL on specific components of the coagulation system remain an area of investigation. For example, aPL contribute to so-called activated protein C resistance which occurs when activated protein C is unable to inactivate coagulation factors V and VIII [55, 161]. Some aPL have been found to antagonize antithrombin activity by inhibiting the heparin binding that is required for full activation of antithrombin [56]; meanwhile, aPL with activity against thrombin may further protect thrombin from inactivation by antithrombin [162]. Similarly, aPL targeting factors IX [163] and X [164] appear to prevent their negative regulation by antithrombin. Elevated levels of factor XI are a known risk factor for thrombosis in the general population [165], and as compared with age- and sex-matched controls, APS patients carry higher-than-expected circulating levels of the active free thiol form of factor XI [57]. The activity of tissue factor may also be potentiated in APS via aPL-mediated inhibition of tissue factor pathway inhibitor (TFPI), or disassembly of a normally inhibited TF complex at the cell surface [166,167,168].
Fibrinolysis. Impaired fibrinolysis has been found in APS patients with thrombotic as well as obstetric manifestations [169]. Some aPL may inhibit fibrinolysis by neutralizing the ability of β2GPI to stimulate tissue plasminogen activator (tPA)-mediated plasminogen activation and fibrinolysis [58]. Furthermore, there are reports of APS-associated autoantibodies that directly antagonize various pro-fibrinolytic factors (e.g., anti-annexin-A2, anti-tissue-type plasminogen activator/tPA, anti-plasmin) [169,170,171]. Small studies have also demonstrated upregulation of natural anti-fibrinolytic proteins [59,60,61], most notably plasminogen activator inhibitor-1 (PAI-1, the physiologic inhibitor of both tPA, and urokinase plasminogen activator). Mechanistically, PAI-1 is upregulated in human umbilical vein endothelial cells upon exposure to anti-β2GPI antibodies from APS patients [172]. Interestingly, PAI-1 appears to have diverse functions beyond its role in restraining fibrinolysis, as elevated PAI-1 levels have regularly been associated with chronic disease states including fibrosis (of lung, liver, and kidney) and atherosclerosis [173,174,175,176] raising the possibility that PAI-1 might also play a role in the chronic occlusive APS vasculopathy that will be discussed below.
Catastrophic APS
CAPS is characterized by rapidly developing and widespread microvascular thrombosis causing ischemic injury (Table 3) [10].
Current standard of care treatment for CAPS includes anticoagulation (typically with unfractionated heparin), immunosuppression (with high-dose corticosteroids), and plasmapheresis to emergently reduce the circulating aPL burden [11]. Given the relative rarity of CAPS, few studies have had the opportunity to pursue deep, mechanistic studies. There is some suggestion of endothelial and/or platelet activation based on high levels of von Willebrand factor and P-selectin in circulation [177]. Complement activation has also been indirectly implicated in the pathogenesis of CAPS as complement regulatory gene variants have been found in 60% of patients, perhaps contributing to uncontrolled complement activation [51]. Most patients with CAPS have an identifiable precipitating event such as surgery, infection, or pregnancy, which may serve as a complement-stimulating “second hit” in the setting of germline variants that may have reduced capacity to restrain complement amplification.
The potential role of complement in CAPS is further supported by reports of successful use (as mentioned above) of eculizumab in patients refractory to standard therapies. One series reported that 5 of 11 patients with CAPS responded to treatment with eculizumab [156]; individuals who had a response were more likely to have microangiopathic hemolytic anemia and thrombocytopenia, while those who already had dialysis-dependent kidney failure were less likely to respond. In another report, eculizumab allowed successful kidney transplantation in two patients with CAPS [178]. Based on these and other reports, the enthusiasm in the clinic for complement-inhibiting approaches to CAPS remains high, and additional evidence for this is eagerly anticipated.
APS vasculopathy
Distinct from APS-associated thrombotic events that acutely close vessels, the chronic occlusive vasculopathy of APS is characterized by cell proliferation and infiltration that progressively expands the intima [179,180,181]. These lesions are reminiscent of those seen following vascular interventions such as angioplasty and stent deployment in which the intima becomes thickened due to proliferation of vascular smooth muscle cells and production of proteoglycan-rich extracellular matrix between the endothelium and the internal elastic lamina [182]. Although this pathology was initially reported—and is still best defined—in the small vessels of APS kidneys [183,184,185,186], occlusive APS vasculopathy has also been observed in small- and medium-sized vessels of the brain, heart, and mesentery [179,180,181].
The molecular pathways that license these lesions are for the most part unknown, although one yet-to-be-reproduced report posited that the mTOR/Akt pathway is an important mediator of APS nephropathy and therefore a potential pharmacologic target via clinically available agents such as sirolimus [187]. mTOR is a kinase that integrates a variety of signaling pathways to regulate cellular growth, proliferation, and survival. In individuals with aPL-associated nephropathy, the vascular endothelium of intrarenal vessels was found to display molecular markers consistent with activation of mTOR and downstream signaling [187]. Furthermore, patients with aPL-associated nephropathy who required transplantation and were receiving sirolimus had minimal recurrence of vascular lesions, which contrasted with matched patients with aPL who were not receiving sirolimus [187].
The signaling pathways by which aPL trigger neointimal hyperplasia and occlusive vasculopathy may occur through direct or indirect interactions with the endothelium and smooth muscle of the vessel wall. Endothelial activation by aPL may create a dysfunctional, nitric oxide-depleted state in addition to facilitating leukocyte and platelet adhesion. Proliferation of endothelial and smooth muscle cells may be supported by mTOR signaling, complement activation, macrophage foam cell formation, canonical cell adhesion molecules, and repression of pro-resolving factors, among other possible mechanisms. As mechanisms and new treatment approaches are investigated for the acute and catastrophic complications of APS, it will be important to keep in view the chronic sequelae of multi-system vasculopathy that also leads to progressive organ dysfunction in patients with APS.
COVID-19
Like APS, coronavirus disease 2019 (COVID-19) is associated with a high incidence of thrombosis in arterial, venous, and microcirculatory vascular beds [188, 189]. Notably, studies of COVID-19 patient samples demonstrate some similarities with APS, including evidence for aberrant activation of neutrophils [190, 191], endothelial cells [192], platelets [193], and complement [194]. A report from early in the pandemic detected aPL in three patients with COVID-19 who experienced cerebrovascular accidents [195]. This was soon followed by a study of 56 hospitalized in whom lupus anticoagulant was detected in 25; five of the patients also had either anticardiolipin or anti-β2GPI antibodies [196]. Studies in COVID-19 patients have detected both traditional aPL and various “non-criteria” aPL (anti-phosphatidylserine/prothrombin IgG and IgM as well as anticardiolipin and anti-β2GPI IgA). Studies have demonstrated significant heterogeneity in terms of prevalence of aPL (some as high as 50%) and which aPL species are most detected [197,198,199]. At the present time, it is mostly unknown whether these are transient aPL, as have been reported in other viral infections [200], or persistent aPL that herald long-term thrombotic risk.
Most studies have not found a clear association of aPL with macrovascular thrombotic events in COVID-19. Furthermore, functional assays such as lupus anticoagulant should be interpreted with caution in severely ill patients due to potential confounding by high levels of C-reactive protein and administration of anticoagulation. Despite these caveats, the relationship between aPL and COVID-19 is an emerging area deserving of further research. There is some evidence that IgG fractions isolated from the serum of patients with COVID-19 with high titers of aPL have prothrombotic properties in vitro and in mice [199, 201]. Future studies are required to determine persistence of these antibodies and identify mechanistic connections that can further clarify the extent to which aPL-like antibodies in patients with COVID-19 mimic those seen in patients with traditional APS.
Summary
Like other systemic autoimmune diseases such as lupus, systemic sclerosis, and autoimmune vasculitis, there is significant person-to-person heterogeneity in individuals presenting with APS. One individual may present with heart valve lesions and thrombocytopenia, another with recurrent venous thrombosis, and another with livedo racemosa and white matter hyperintensities. The potential mechanisms covered above are myriad (Fig. 2) and the extent to which each mechanism manifests in a particular individual may help explain disease heterogeneity. One potential model is that aPL profiles are relatively consistent, while heterogeneity is best explained by comorbid genetic and acquired risk factors. Alternatively, aPL profiles may vary more than we realize as only a handful of types of aPL can be routinely tested for clinically. In that scenario, we will not be able to fully explain APS pathophysiology until the full autoantigenome of a particular individual has been defined; this is an important area for future research.

Potential mechanisms contributing to thrombotic APS. A Endothelial cells increase expression of tissue factor (TF) and adhesion molecules. Complement damages the endothelium via the membrane attack complex (MAC) and acts as a chemoattractant via C5a. Monocytes express TF and cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and type I interferons (IFNs), and release microparticles. Neutrophils produce reactive oxygen species and release neutrophil extracellular traps (NETs). B NETs form an intravascular scaffold that promotes thrombus accretion. C Chronic activation of the endothelium by aPL can result in progressively occlusive vasculopathy. aPL = antiphospholipid antibodies; ApoER2 = apolipoprotein E receptor 2; β2GPI = beta-2 glycoprotein I; NF-κB = nuclear factor kappa B; KLFs = Kruppel-like factors. Illustration credit: Ethan Tyler (NIH)
While the thrombophilia of COVID-19 does not appear to be explained by the best characterized aPL such as anti-β2GPI antibodies, it does seem possible that less refined aPL-like antibodies do contribute to the COVID thrombotic burden, especially in the microvasculature. The ongoing global pandemic emphasizes the importance of more deeply defining understudied disease states such as APS. Meanwhile, the hope is that the relative spotlight APS has received during the pandemic will build momentum for significant discovery over the next decade in pursuit of the personalized proactive approaches to diagnosis and treatment that our patients deserve.
References
Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C (2004) High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 164(1):77–82
Ruiz-Irastorza G, Egurbide MV, Martinez-Berriotxoa A, Ugalde J, Aguirre C (2004) Antiphospholipid antibodies predict early damage in patients with systemic lupus erythematosus. Lupus 13(12):900–905
Unlu O, Zuily S, Erkan D (2016) The clinical significance of antiphospholipid antibodies in systemic lupus erythematosus. Eur J Rheumatol 3(2):75–84
L. Riancho-Zarrabeitia, V. Martinez-Taboada, I. Rua-Figueroa, F. Alonso, M. Galindo-Izquierdo, J. Ovalles, A. Olive-Marques, A. Fernandez-Nebro, J. Calvo-Alen, R. Menor-Almagro, E. Tomero-Muriel, E. Uriarte-Isacelaya, A. Botenau, M. Andres, M. Freire-Gonzalez, G. Santos Soler, E. Ruiz-Lucea, M. Ibanez-Barcelo, I. Castellvi, C. Galisteo, V. Quevedo Vila, E. Raya, J. Narvaez-Garcia, L. Exposito, J.A. Hernandez-Beriain, L. Horcada, E. Aurrecoechea, J.M. Pego-Reigosa, Antiphospholipid syndrome (APS) in patients with systemic lupus erythematosus (SLE) implies a more severe disease with more damage accrual and higher mortality, Lupus 29(12) (2020) 1556–1565.
Levine JS, Branch DW, Rauch J (2002) The antiphospholipid syndrome. N Engl J Med 346(10):752–763
Pons-Estel GJ, Andreoli L, Scanzi F, Cervera R, Tincani A (2017) The antiphospholipid syndrome in patients with systemic lupus erythematosus. J Autoimmun 76:10–20
Duarte-Garcia A, Pham MM, Crowson CS, Amin S, Moder KG, Pruthi RK, Warrington KJ, Matteson EL (2019) The Epidemiology of antiphospholipid syndrome: a population-based study. Arthritis Rheumatol 71(9):1545–1552
S. Miyakis, M.D. Lockshin, T. Atsumi, D.W. Branch, R.L. Brey, R. Cervera, R.H. Derksen, D.E.G. PG, T. Koike, P.L. Meroni, G. Reber, Y. Shoenfeld, A. Tincani, P.G. Vlachoyiannopoulos, S.A. Krilis, International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS), J Thromb Haemost 4(2) (2006) 295–306.
R. Cervera, R. Serrano, G.J. Pons-Estel, L. Ceberio-Hualde, Y. Shoenfeld, E. de Ramon, V. Buonaiuto, S. Jacobsen, M.M. Zeher, T. Tarr, A. Tincani, M. Taglietti, G. Theodossiades, E. Nomikou, M. Galeazzi, F. Bellisai, P.L. Meroni, R.H. Derksen, P.G. de Groot, M. Baleva, M. Mosca, S. Bombardieri, F. Houssiau, J.C. Gris, I. Quere, E. Hachulla, C. Vasconcelos, A. Fernandez-Nebro, M. Haro, Z. Amoura, M. Miyara, M. Tektonidou, G. Espinosa, M.L. Bertolaccini, M.A. Khamashta, G. Euro-phospholipid project, morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients, Ann Rheum Dis 74(6) (2015) 1011–8.
R.A. Asherson, R. Cervera, P.G. de Groot, D. Erkan, M.C. Boffa, J.C. Piette, M.A. Khamashta, Y. Shoenfeld, G. Catastrophic Antiphospholipid Syndrome Registry Project, Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines, Lupus 12(7) (2003) 530–4.
Kazzaz NM, McCune WJ, Knight JS (2016) Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 28(3):218–227
Linnemann B (2018) Antiphospholipid syndrome - an update. Vasa 47(6):451–464
Bertolaccini ML, Amengual O, Andreoli L, Atsumi T, Chighizola CB, Forastiero R, de Groot P, Lakos G, Lambert M, Meroni P, Ortel TL, Petri M, Rahman A, Roubey R, Sciascia S, Snyder M, Tebo AE, Tincani A, Willis R (2014) 14th International Congress on Antiphospholipid Antibodies Task Force. Report on antiphospholipid syndrome laboratory diagnostics and trends, Autoimmun Rev 13(9):917–930
Shi H, Zheng H, Yin YF, Hu QY, Teng JL, Sun Y, Liu HL, Cheng XB, Ye JN, Su YT, Wu XY, Zhou JF, Norman GL, Gong HY, Shi XM, Peng YB, Wang XF, Yang CD (2018) Antiphosphatidylserine/prothrombin antibodies (aPS/PT) as potential diagnostic markers and risk predictors of venous thrombosis and obstetric complications in antiphospholipid syndrome. Clin Chem Lab Med 56(4):614–624
Levy RA, de Meis E, Pierangeli S (2004) An adapted ELISA method for differentiating pathogenic from nonpathogenic aPL by a beta 2 glycoprotein I dependency anticardiolipin assay. Thromb Res 114(5–6):573–577
T. McDonnell, C. Wincup, I. Buchholz, C. Pericleous, I. Giles, V. Ripoll, H. Cohen, M. Delcea, A. Rahman, The role of beta-2-glycoprotein I in health and disease associating structure with function: More than just APS, Blood Rev 39 (2020) 100610.
Jones JV, James H, Tan MH, Mansour M (1992) Antiphospholipid antibodies require beta 2-glycoprotein I (apolipoprotein H) as cofactor. J Rheumatol 19(9):1397–1402
Arnout J (2000) The role of beta 2-glycoprotein I-dependent lupus anticoagulants in the pathogenesis of the antiphospholipid syndrome. Verh K Acad Geneeskd Belg 62(5):353–372
de Laat HB, Derksen RH, Urbanus RT, Roest M, de Groot PG (2004) beta2-glycoprotein I-dependent lupus anticoagulant highly correlates with thrombosis in the antiphospholipid syndrome. Blood 104(12):3598–3602
Galli M, Finazzi G, Bevers EM, Barbui T (1995) Kaolin clotting time and dilute Russell’s viper venom time distinguish between prothrombin-dependent and beta 2-glycoprotein I-dependent antiphospholipid antibodies. Blood 86(2):617–623
V. Pengo, A. Ruffatti, M. Tonello, S. Cuffaro, A. Banzato, E. Bison, G. Denas, S. Padayattil Jose, Antiphospholipid syndrome: antibodies to Domain 1 of beta2-glycoprotein 1 correctly classify patients at risk, J Thromb Haemost 13(5) (2015) 782–7.
Andreoli L, Chighizola CB, Nalli C, Gerosa M, Borghi MO, Pregnolato F, Grossi C, Zanola A, Allegri F, Norman GL, Mahler M, Meroni PL, Tincani A (2015) Clinical characterization of antiphospholipid syndrome by detection of IgG antibodies against beta2 -glycoprotein i domain 1 and domain 4/5: ratio of anti-domain 1 to anti-domain 4/5 as a useful new biomarker for antiphospholipid syndrome. Arthritis Rheumatol 67(8):2196–2204
McDonnell TCR, Willis R, Pericleous C, Ripoll VM, Giles IP, Isenberg DA, Brasier AR, Gonzalez EB, Papalardo E, Romay-Penabad Z, Jamaluddin M, Ioannou Y, Rahman A (2018) PEGylated domain I of beta-2-glycoprotein I inhibits the binding, coagulopathic, and thrombogenic properties of IgG from patients with the antiphospholipid syndrome. Front Immunol 9:2413
Cucurull E, Gharavi AE, Diri E, Mendez E, Kapoor D, Espinoza LR (1999) IgA anticardiolipin and anti-beta2-glycoprotein I are the most prevalent isotypes in African American patients with systemic lupus erythematosus. Am J Med Sci 318(1):55–60
E. Sevim, D. Zisa, D. Andrade, S. Sciascia, V. Pengo, M.G. Tektonidou, A. Ugarte, M. Gerosa, H.M. Belmont, M.A. Aguirre Zamorano, P.R. Fortin, L. Ji, M. Efthymiou, H. Cohen, D.W. Branch, G.R. de Jesus, L. Andreoli, M. Petri, E. Rodriguez, R. Cervera, J.S. Knight, T. Atsumi, R. Willis, R. Roubey, M.L. Bertolaccini, D. Erkan, M. Barbhaiya, A.A. Investigators, Characteristics of antiphospholipid antibody positive patients in antiphospholipid syndrome alliance for clinical trials and international networking, Arthritis Care Res (Hoboken) (2020).
Branch DW, Dudley DJ, Mitchell MD, Creighton KA, Abbott TM, Hammond EH, Daynes RA (1990) Immunoglobulin G fractions from patients with antiphospholipid antibodies cause fetal death in BALB/c mice: a model for autoimmune fetal loss. Am J Obstet Gynecol 163(1 Pt 1):210–216
Blank M, Cohen J, Toder V, Shoenfeld Y (1991) Induction of anti-phospholipid syndrome in naive mice with mouse lupus monoclonal and human polyclonal anti-cardiolipin antibodies. Proc Natl Acad Sci U S A 88(8):3069–3073
Pierangeli SS, Barker JH, Stikovac D, Ackerman D, Anderson G, Barquinero J, Acland R, Harris EN (1994) Effect of human IgG antiphospholipid antibodies on an in vivo thrombosis model in mice. Thromb Haemost 71(5):670–674
R. Cervera, S. Bucciarelli, M.A. Plasin, J.A. Gomez-Puerta, J. Plaza, G. Pons-Estel, Y. Shoenfeld, M. Ingelmo, G. Espinos, G. Catastrophic antiphospholipid syndrome registry project, catastrophic antiphospholipid syndrome (CAPS): descriptive analysis of a series of 280 patients from the "CAPS Registry", J Autoimmun 32(3–4) (2009) 240–5.
A. Danowski, M.N. de Azevedo, J.A. de Souza Papi, M. Petri, Determinants of risk for venous and arterial thrombosis in primary antiphospholipid syndrome and in antiphospholipid syndrome with systemic lupus erythematosus, J Rheumatol 36(6) (2009) 1195–9.
Matyja-Bednarczyk A, Swadzba J, Iwaniec T, Sanak M, Dziedzina S, Cmiel A, Musial J (2014) Risk factors for arterial thrombosis in antiphospholipid syndrome. Thromb Res 133(2):173–176
Urbanus RT, Siegerink B, Roest M, Rosendaal FR, de Groot PG, Algra A (2009) Antiphospholipid antibodies and risk of myocardial infarction and ischaemic stroke in young women in the RATIO study: a case-control study. Lancet Neurol 8(11):998–1005
Simantov R, LaSala JM, Lo SK, Gharavi AE, Sammaritano LR, Salmon JE, Silverstein RL (1995) Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest 96(5):2211–2219
N. Del Papa, L. Guidali, A. Sala, C. Buccellati, M.A. Khamashta, K. Ichikawa, T. Koike, G. Balestrieri, A. Tincani, G.R. Hughes, P.L. Meroni, Endothelial cells as target for antiphospholipid antibodies. Human polyclonal and monoclonal anti-beta 2-glycoprotein I antibodies react in vitro with endothelial cells through adherent beta 2-glycoprotein I and induce endothelial activation, Arthritis Rheum 40(3) (1997) 551–61.
Dignat-George F, Camoin-Jau L, Sabatier F, Arnoux D, Anfosso F, Bardin N, Veit V, Combes V, Gentile S, Moal V, Sanmarco M, Sampol J (2004) Endothelial microparticles: a potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb Haemost 91(4):667–673
Chaturvedi S, Cockrell E, Espinola R, Hsi L, Fulton S, Khan M, Li L, Fonseca F, Kundu S, McCrae KR (2015) Circulating microparticles in patients with antiphospholipid antibodies: characterization and associations. Thromb Res 135(1):102–108
Pennings MT, Derksen RH, van Lummel M, Adelmeijer J, VanHoorelbeke K, Urbanus RT, Lisman T, de Groot PG (2007) Platelet adhesion to dimeric beta-glycoprotein I under conditions of flow is mediated by at least two receptors: glycoprotein Ibalpha and apolipoprotein E receptor 2’. J Thromb Haemost 5(2):369–377
Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ (2001) Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol 115(2):451–459
Kornberg A, Blank M, Kaufman S, Shoenfeld Y (1994) Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J Immunol 153(3):1328–1332
Amengual O, Atsumi T, Khamashta MA, Hughes GR (1998) The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb Haemost 79(2):276–281
Reverter JC, Tassies D, Font J, Khamashta MA, Ichikawa K, Cervera R, Escolar G, Hughes GR, Ingelmo M, Ordinas A (1998) Effects of human monoclonal anticardiolipin antibodies on platelet function and on tissue factor expression on monocytes. Arthritis Rheum 41(8):1420–1427
Zhou H, Wolberg AS, Roubey RA (2004) Characterization of monocyte tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood 104(8):2353–2358
Sorice M, Longo A, Capozzi A, Garofalo T, Misasi R, Alessandri C, Conti F, Buttari B, Rigano R, Ortona E, Valesini G (2007) Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum 56(8):2687–2697
Xie H, Zhou H, Wang H, Chen D, Xia L, Wang T, Yan J (2013) Anti-beta(2)GPI/beta(2)GPI induced TF and TNF-alpha expression in monocytes involving both TLR4/MyD88 and TLR4/TRIF signaling pathways. Mol Immunol 53(3):246–254
Muller-Calleja N, Kohler A, Siebald B, Canisius A, Orning C, Radsak M, Stein P, Monnikes R, Lackner KJ (2015) Cofactor-independent antiphospholipid antibodies activate the NLRP3-inflammasome via endosomal NADPH-oxidase: implications for the antiphospholipid syndrome. Thromb Haemost 113(5):1071–1083
Cuadrado MJ, Lopez-Pedrera C, Khamashta MA, Camps MT, Tinahones F, Torres A, Hughes GR, Velasco F (1997) Thrombosis in primary antiphospholipid syndrome: a pivotal role for monocyte tissue factor expression. Arthritis Rheum 40(5):834–841
Dobado-Berrios PM, Lopez-Pedrera C, Velasco F, Aguirre MA, Torres A, Cuadrado MJ (1999) Increased levels of tissue factor mRNA in mononuclear blood cells of patients with primary antiphospholipid syndrome. Thromb Haemost 82(6):1578–1582
Lopez-Pedrera C, Buendia P, Cuadrado MJ, Siendones E, Aguirre MA, Barbarroja N, Montiel-Duarte C, Torres A, Khamashta M, Velasco F (2006) Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappaB/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis Rheum 54(1):301–311
Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez C, Hernandez-Ramirez D, Bockenstedt PL, Liaw PC, Cabral AR, Knight JS (2015) Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol 67(11):2990–3003
Y. Zuo, S. Yalavarthi, K. Gockman, J.A. Madison, J.E. Gudjonsson, J.M. Kahlenberg, W. Joseph McCune, P.L. Bockenstedt, D.R. Karp, J.S. Knight, Anti-neutrophil extracellular trap antibodies and impaired neutrophil extracellular trap degradation in antiphospholipid syndrome, Arthritis Rheumatol 72(12) (2020) 2130–2135.
Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, Gavriilaki E, Alluri R, Streiff MB, Petri M, Crowther MA, McCrae KR, Brodsky RA (2020) Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 135(4):239–251
Devreese KM, Hoylaerts MF (2010) Is there an association between complement activation and antiphospholipid antibody-related thrombosis? Thromb Haemost 104(6):1279–1281
E. Sarmiento, J. Dale, M. Arraya, A. Gallego, N. Lanio, J. Navarro, J. Carbone, CD8+DR+ T-cells and C3 complement serum concentration as potential biomarkers in thrombotic antiphospholipid syndrome, Autoimmune diseases 2014 (2014) 868652.
Davis WD, Brey RL (1992) Antiphospholipid antibodies and complement activation in patients with cerebral ischemia. Clin Exp Rheumatol 10(5):455–460
Wahl D, Membre A, Perret-Guillaume C, Regnault V, Lecompte T (2009) Mechanisms of antiphospholipid-induced thrombosis: effects on the protein C system. Curr Rheumatol Rep 11(1):77–81
Shibata S, Harpel PC, Gharavi A, Rand J, Fillit H (1994) Autoantibodies to heparin from patients with antiphospholipid antibody syndrome inhibit formation of antithrombin III-thrombin complexes. Blood 83(9):2532–2540
Giannakopoulos B, Gao L, Qi M, Wong JW, Yu DM, Vlachoyiannopoulos PG, Moutsopoulos HM, Atsumi T, Koike T, Hogg P, Qi JC, Krilis SA (2012) Factor XI is a substrate for oxidoreductases: enhanced activation of reduced FXI and its role in antiphospholipid syndrome thrombosis. J Autoimmun 39(3):121–129
Bu C, Gao L, Xie W, Zhang J, He Y, Cai G, McCrae KR (2009) beta2-glycoprotein i is a cofactor for tissue plasminogen activator-mediated plasminogen activation. Arthritis Rheum 60(2):559–568
Singh NK, Gupta A, Behera DR, Dash D (2013) Elevated plasminogen activator inhibitor type-1 (PAI-1) as contributing factor in pathogenesis of hypercoagulable state in antiphospholipid syndrome. Rheumatol Int 33(9):2331–2336
Jurado M, Paramo JA, Gutierrez-Pimentel M, Rocha E (1992) Fibrinolytic potential and antiphospholipid antibodies in systemic lupus erythematosus and other connective tissue disorders. Thromb Haemost 68(5):516–520
Ames PR, Tommasino C, Iannaccone L, Brillante M, Cimino R, Brancaccio V (1996) Coagulation activation and fibrinolytic imbalance in subjects with idiopathic antiphospholipid antibodies–a crucial role for acquired free protein S deficiency. Thromb Haemost 76(2):190–194
P.G. de Groot, R.T. Urbanus, Antiphospholipid syndrome--not a noninflammatory disease, seminars in thrombosis and hemostasis 41(6) (2015) 607–14.
Colling ME, Tourdot BE, Kanthi Y (2021) Inflammation, Infection and Venous Thromboembolism. Circ Res 128(12):2017–2036
McEver RP (2015) Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res 107(3):331–339
Sacharidou A, Chambliss KL, Ulrich V, Salmon JE, Shen YM, Herz J, Hui DY, Terada LS, Shaul PW, Mineo C (2018) Antiphospholipid antibodies induce thrombosis by PP2A activation via apoER2-Dab2-SHC1 complex formation in endothelium. Blood 131(19):2097–2110
C. Mineo, L. Lanier, E. Jung, S. Sengupta, V. Ulrich, A. Sacharidou, C. Tarango, O. Osunbunmi, Y.M. Shen, J.E. Salmon, R.A. Brekken, X. Huang, P.E. Thorpe, P.W. Shaul, Identification of a monoclonal antibody that attenuates antiphospholipid syndrome-related pregnancy complications and thrombosis, PLoS One 11(7) (2016) e0158757.
Ulrich V, Gelber SE, Vukelic M, Sacharidou A, Herz J, Urbanus RT, de Groot PG, Natale DR, Harihara A, Redecha P, Abrahams VM, Shaul PW, Salmon JE, Mineo C (2016) ApoE receptor 2 mediation of trophoblast dysfunction and pregnancy complications induced by antiphospholipid antibodies in mice. Arthritis Rheumatol 68(3):730–739
Urbanus RT, Pennings MT, Derksen RH, de Groot PG (2008) Platelet activation by dimeric beta2-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2’. J Thromb Haemost 6(8):1405–1412
Ramesh S, Morrell CN, Tarango C, Thomas GD, Yuhanna IS, Girardi G, Herz J, Urbanus RT, de Groot PG, Thorpe PE, Salmon JE, Shaul PW, Mineo C (2011) Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J Clin Invest 121(1):120–131
Romay-Penabad Z, Aguilar-Valenzuela R, Urbanus RT, Derksen RH, Pennings MT, Papalardo E, Shilagard T, Vargas G, Hwang Y, de Groot PG, Pierangeli SS (2011) Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood 117(4):1408–1414
S. Dunoyer-Geindre, P. de Moerloose, B. Galve-de Rochemonteix, G. Reber, E.K.O. Kruithof, NF kappa B is an essential intermediate in the activation of endothelial cells by anti-beta(2)-glycoprotein 1 antibodies, thrombosis and haemostasis 88(5) (2002) 851–857.
Vega-Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS (2005) Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum 52(5):1545–1554
Allen KL, Hamik A, Jain MK, McCrae KR (2011) Endothelial cell activation by antiphospholipid antibodies is modulated by Kruppel-like transcription factors. Blood 117(23):6383–6391
Ramesh S, Morrell CN, Tarango C, Thomas GD, Yuhanna IS, Girardi G, Herz J, Urbanus RT, de Groot PG, Thorpe PE, Salmon JE, Shaul PW, Mineo C (2011) Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via beta2GPI and apoER2. J Clin Invest 121(1):120–131
S. Dunoyer-Geindre, P. de Moerloose, B. Galve-de Rochemonteix, G. Reber, E.K. Kruithof, NFkappaB is an essential intermediate in the activation of endothelial cells by anti-beta(2)-glycoprotein 1 antibodies, Thromb Haemost 88(5) (2002) 851–7.
Pierangeli SS, Espinola RG, Liu X, Harris EN (2001) Thrombogenic effects of antiphospholipid antibodies are mediated by intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1, and P-selectin. Circ Res 88(2):245–250
Pierangeli SS, Vega-Ostertag ME, Raschi E, Liu X, Romay-Penabad Z, De Micheli V, Galli M, Moia M, Tincani A, Borghi MO, Nguyen-Oghalai T, Meroni PL (2007) Toll-like receptor and antiphospholipid mediated thrombosis: in vivo studies. Ann Rheum Dis 66(10):1327–1333
Pierangeli SS, Colden-Stanfield M, Liu X, Barker JH, Anderson GL, Harris EN (1999) Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation 99(15):1997–2002
J.S. Knight, H. Meng, P. Coit, S. Yalavarthi, G. Sule, A.A. Gandhi, R.C. Grenn, L.F. Mazza, R.A. Ali, P. Renauer, J.D. Wren, P.L. Bockenstedt, H. Wang, D.T. Eitzman, A.H. Sawalha, Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target, JCI Insight 2(18) (2017).
Espinola RG, Liu X, Colden-Stanfield M, Hall J, Harris EN, Pierangeli SS (2003) E-Selectin mediates pathogenic effects of antiphospholipid antibodies. Journal of thrombosis and haemostasis : JTH 1(4):843–848
Vega-Ostertag M, Harris EN, Pierangeli SS (2004) Intracellular events in platelet activation induced by antiphospholipid antibodies in the presence of low doses of thrombin. Arthritis Rheum 50(9):2911–2919
Proulle V, Furie RA, Merrill-Skoloff G, Furie BC, Furie B (2014) Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood 124(4):611–622
Cuadrado MJ, Mujic F, Munoz E, Khamashta MA, Hughes GR (1997) Thrombocytopenia in the antiphospholipid syndrome. Ann Rheum Dis 56(3):194–196
Diz-Kucukkaya R, Hacihanefioglu A, Yenerel M, Turgut M, Keskin H, Nalcaci M, Inanc M (2001) Antiphospholipid antibodies and antiphospholipid syndrome in patients presenting with immune thrombocytopenic purpura: a prospective cohort study. Blood 98(6):1760–1764
Fanelli A, Bergamini C, Rapi S, Caldini A, Spinelli A, Buggiani A, Emmi L (1997) Flow cytometric detection of circulating activated platelets in primary antiphospholipid syndrome. Correlation with thrombocytopenia and anticardiolipin antibodies, Lupus 6(3):261–267
Vreede AP, Bockenstedt PL, McCune WJ, Knight JS (2019) Cryptic conspirators: a conversation about thrombocytopenia and antiphospholipid syndrome. Curr Opin Rheumatol 31(3):231–240
Martini F, Farsi A, Gori AM, Boddi M, Fedi S, Domeneghetti MP, Passaleva A, Prisco D, Abbate R (1996) Antiphospholipid antibodies (aPL) increase the potential monocyte procoagulant activity in patients with systemic lupus erythematosus. Lupus 5(3):206–211
Cuadrado MJ, Buendia P, Velasco F, Aguirre MA, Barbarroja N, Torres LA, Khamashta M, Lopez-Pedrera C (2006) Vascular endothelial growth factor expression in monocytes from patients with primary antiphospholipid syndrome. Journal of thrombosis and haemostasis : JTH 4(11):2461–2469
Bernales I, Fullaondo A, Marin-Vidalled MJ, Ucar E, Martinez-Taboada V, Lopez-Hoyos M, Zubiaga AM (2008) Innate immune response gene expression profiles characterize primary antiphospholipid syndrome. Genes Immun 9(1):38–46
Perez-Sanchez C, Barbarroja N, Messineo S, Ruiz-Limon P, Rodriguez-Ariza A, Jimenez-Gomez Y, Khamashta MA, Collantes-Estevez E, Cuadrado MJ, Aguirre MA, Lopez-Pedrera C (2015) Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann Rheum Dis 74(7):1441–1449
Lopez-Pedrera C, Aguirre MA, Buendia P, Barbarroja N, Ruiz-Limon P, Collantes-Estevez E, Velasco F, Khamashta M, Cuadrado MJ (2010) Differential expression of protease-activated receptors in monocytes from patients with primary antiphospholipid syndrome. Arthritis Rheum 62(3):869–877
Nagahama M, Nomura S, Kanazawa S, Ozaki Y, Kagawa H, Fukuhara S (2003) Significance of anti-oxidized LDL antibody and monocyte-derived microparticles in anti-phospholipid antibody syndrome. Autoimmunity 36(3):125–131
Vikerfors A, Mobarrez F, Bremme K, Holmstrom M, Agren A, Eelde A, Bruzelius M, Antovic A, Wallen H, Svenungsson E (2012) Studies of microparticles in patients with the antiphospholipid syndrome (APS). Lupus 21(7):802–805
D. Manukyan, N. Muller-Calleja, S. Jackel, K. Luchmann, R. Monnikes, K. Kiouptsi, C. Reinhardt, K. Jurk, U. Walter, K.J. Lackner, Cofactor independent human antiphospholipid antibodies induce venous thrombosis in mice, Journal of thrombosis and haemostasis : JTH (2016).
Mayadas TN, Cullere X, Lowell CA (2014) The multifaceted functions of neutrophils. Annu Rev Pathol 9:181–218
Grayson PC, Schauer C, Herrmann M, Kaplan MJ (2016) Review: neutrophils as invigorated targets in rheumatic diseases. Arthritis Rheumatol 68(9):2071–2082
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535
B.E. Steinberg, S. Grinstein, Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death, Sci STKE 2007(379) (2007) pe11.
Brinkmann V (2018) Neutrophil extracellular traps in the second decade. J Innate Immun 10(5–6):414–421
Engelmann B, Massberg S (2013) Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 13(1):34–45
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107(36):15880–15885
Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 10(1):136–144
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16(8):887–896
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S (2012) Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209(4):819–835
Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Ten Cate H, Hofstra L, Crijns HJ, Wagner DD, Kietselaer BL (2013) Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol 33(8):2032–2040
Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT, Kaplan MJ (2014) Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 114(6):947–956
Farkas AZ, Farkas VJ, Gubucz I, Szabo L, Balint K, Tenekedjiev K, Nagy AI, Sotonyi P, Hidi L, Nagy Z, Szikora I, Merkely B, Kolev K (2019) Neutrophil extracellular traps in thrombi retrieved during interventional treatment of ischemic arterial diseases. Thromb Res 175:46–52
McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, Jenne CN (2017) Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 129(10):1357–1367
Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, Tsung A (2015) Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 62(2):600–614
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD (2012) Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A 109(32):13076–13081
Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS (2014) Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 123(24):3818–3827
Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR (2012) Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 122(7):2661–2671
Yago T, Liu Z, Ahamed J, McEver RP (2018) Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood 132(13):1426–1437
Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD (2015) P-selectin promotes neutrophil extracellular trap formation in mice. Blood 126(2):242–246
Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD (2013) Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A 110(21):8674–8679
Arvieux J, Jacob MC, Roussel B, Bensa JC, Colomb MG (1995) Neutrophil activation by anti-beta 2 glycoprotein I monoclonal antibodies via Fc gamma receptor II. J Leukoc Biol 57(3):387–394
Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, Hollmann TJ, Casali P, Caroll MC, Wetsel RA, Lambris JD, Holers VM, Salmon JE (2003) Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest 112(11):1644–1654
Redecha P, Tilley R, Tencati M, Salmon JE, Kirchhofer D, Mackman N, Girardi G (2007) Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood 110(7):2423–2431
Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G (2008) Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest 118(10):3453–3461
G. Gladigau, P. Haselmayer, I. Scharrer, M. Munder, N. Prinz, K. Lackner, H. Schild, P. Stein, M.P. Radsak, A role for Toll-like receptor mediated signals in neutrophils in the pathogenesis of the anti-phospholipid syndrome, PLoS One 7(7) (2012) e42176.
Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, Rafail S, Kartalis G, Sideras P, Lambris JD (2006) A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol 177(7):4794–4802
Leffler J, Stojanovich L, Shoenfeld Y, Bogdanovic G, Hesselstrand R, Blom AM (2014) Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol 32(1):66–70
Andreoli L, Pregnolato F, Burlingame RW, Allegri F, Rizzini S, Fanelli V, Radice A, Corace C, Sinico RA, Meroni PL, Tincani A (2008) Antinucleosome antibodies in primary antiphospholipid syndrome: a hint at systemic autoimmunity? J Autoimmun 30(1–2):51–57
Zha C, Zhang W, Gao F, Xu J, Jia R, Cai J, Liu Y (2018) Anti-β2GPI/β2GPI induces neutrophil extracellular traps formation to promote thrombogenesis via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology 138:140–150
T. Foret, V. Dufrost, L. Salomon du Mont, P. Costa, C. Lakomy, J. Lagrange, P. Lacolley, V. Regnault, S. Zuily, D. Wahl, A new pro-thrombotic mechanism of neutrophil extracellular traps in antiphospholipid syndrome: impact on activated protein C resistance, Rheumatology (Oxford) (2021).
Sule G, Kelley WJ, Gockman K, Yalavarthi S, Vreede AP, Banka AL, Bockenstedt PL, Eniola-Adefeso O, Knight JS (2020) increased adhesive potential of antiphospholipid syndrome neutrophils mediated by β2 integrin mac-1. Arthritis Rheumatol 72(1):114–124
Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, Pinsky DJ, Henke PK, Knight JS (2017) In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol 69(3):655–667
Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, Palmer OR, Bockenstedt PL, Pinsky DJ, Greve JM, Diaz JA, Kanthi Y, Knight JS (2019) Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun 10(1):1916
R.A. Ali, S.K. Estes, A.A. Gandhi, S. Yalavarthi, C.K. Hoy, H. Shi, Y. Zuo, D. Erkan, J.S. Knight, Defibrotide inhibits antiphospholipid antibody-mediated NET formation and venous thrombosis, Arthritis Rheumatol (2021).
R.A. Ali, A.A. Gandhi, L. Dai, J. Weiner, S.K. Estes, S. Yalavarthi, K. Gockman, D. Sun, J.S. Knight, Antineutrophil properties of natural gingerols in models of lupus, JCI Insight 6(3) (2021).
Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, Bertolaccini ML, Khamashta MA, Rodriguez-Ariza A, Segui P, Collantes-Estevez E, Barbarroja N, Khraiwesh H, Gonzalez-Reyes JA, Villalba JM, Velasco F, Cuadrado MJ, Lopez-Pedrera C (2012) Mitochondrial dysfunction in antiphospholipid syndrome: implications in the pathogenesis of the disease and effects of coenzyme Q(10) treatment. Blood 119(24):5859–5870
C. Pérez-Sánchez, M. Aguirre, P. Ruiz-Limón, M.C. Ábalos-Aguilera, Y. Jiménez-Gómez, I. Arias-de la Rosa, A. Rodriguez-Ariza, L. Fernández-Del Río, J.A. González-Reyes, P. Segui, E. Collantes-Estévez, N. Barbarroja, F. Velasco, S. Sciascia, I. Cecchi, M.J. Cuadrado, J.M. Villalba, C. López-Pedrera, Ubiquinol effects on antiphospholipid syndrome prothrombotic profile: a randomized, placebo-controlled trial, Arterioscler Thromb Vasc Biol 37(10) (2017) 1923–1932.
Hacbarth E, Kajdacsy-Balla A (1986) Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum 29(11):1334–1342
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ (2010) A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol 184(6):3284–3297
Carmona-Rivera C, Kaplan MJ (2013) Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol 35(4):455–463
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ (2011) Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187(1):538–552
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22(2):146–153
van den Hoogen LL, Fritsch-Stork RD, van Roon JA, Radstake TR (2016) Low-Density Granulocytes are increased in antiphospholipid syndrome and are associated With Anti-β2 -glycoprotein I antibodies: comment on the article by Yalavarthi et al. Arthritis Rheumatol 68(5):1320–1321
Weeding E, Coit P, Yalavarthi S, Kaplan MJ, Knight JS, Sawalha AH (2018) Genome-wide DNA methylation analysis in primary antiphospholipid syndrome neutrophils. Clin Immunol 196:110–116
Brehm SP, Hoch SO, Hoch JA (1975) DNA-binding proteins in human serum. Biochem Biophys Res Commun 63(1):24–31
Foley JH, Conway EM (2016) Cross talk pathways between coagulation and inflammation. Circ Res 118(9):1392–1408
Weitz IC (2014) Complement the hemostatic system: an intimate relationship. Thromb Res 133(Suppl 2):S117–S121
Breen KA, Seed P, Parmar K, Moore GW, Stuart-Smith SE, Hunt BJ (2012) Complement activation in patients with isolated antiphospholipid antibodies or primary antiphospholipid syndrome. Thromb Haemost 107(3):423–429
K.A. Breen, D.C. Kilpatrick, A.S. Swierzko, M. Cedzynski, B.J. Hunt, Lack of association of serum mannose/mannan binding lectin or ficolins with complement activation in patients with antiphospholipid antibodies, blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis 25(6) (2014) 644–5.
Watanabe H, Sugimoto M, Asano T, Sato S, Suzuki E, Takahashi A, Katakura K, Kobayashi H, Ohira H (2015) Relationship of complement activation route with clinical manifestations in Japanese patients with systemic lupus erythematosus: a retrospective observational study. Modern rheumatology / the Japan Rheumatism Association 25(2):205–209
Munakata Y, Saito T, Matsuda K, Seino J, Shibata S, Sasaki T (2000) Detection of complement-fixing antiphospholipid antibodies in association with thrombosis. Thromb Haemost 83(5):728–731
Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, Yasuda S, Koike T (2009) Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis 68(6):1030–1035
Peerschke EI, Yin W, Alpert DR, Roubey RA, Salmon JE, Ghebrehiwet B (2009) Serum complement activation on heterologous platelets is associated with arterial thrombosis in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus 18(6):530–538
Salmon JE, Girardi G (2008) Antiphospholipid antibodies and pregnancy loss: a disorder of inflammation. J Reprod Immunol 77(1):51–56
Z. Romay-Penabad, A.L. Carrera Marin, R. Willis, W. Weston-Davies, S. Machin, H. Cohen, A. Brasier, E.B. Gonzalez, Complement C5-inhibitor rEV576 (coversin) ameliorates in-vivo effects of antiphospholipid antibodies, Lupus 23(12) (2014) 1324–6.
Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J (2005) Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum 52(7):2120–2124
Romay-Penabad Z, Liu XX, Montiel-Manzano G, Papalardo De Martinez E, Pierangeli SS (2007) C5a receptor-deficient mice are protected from thrombophilia and endothelial cell activation induced by some antiphospholipid antibodies. Ann N Y Acad Sci 1108:554–566
Carrera-Marin A, Romay-Penabad Z, Papalardo E, Reyes-Maldonado E, Garcia-Latorre E, Vargas G, Shilagard T, Pierangeli S (2012) C6 knock-out mice are protected from thrombophilia mediated by antiphospholipid antibodies. Lupus 21(14):1497–1505
Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, Bossi F, Ziller F, Sblattero D, Meroni P, Tedesco F (2005) Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood 106(7):2340–2346
Meroni PL, Macor P, Durigutto P, De Maso L, Gerosa M, Ferraresso M, Borghi MO, Mollnes TE, Tedesco F (2016) Complement activation in antiphospholipid syndrome and its inhibition to prevent rethrombosis after arterial surgery. Blood 127(3):365–367
Yelnik CM, Miranda S, Mekinian A, Lazaro E, Quemeneur T, Provot F, Frimat M, Morell-Dubois S, Le Guern V, Hachulla E, Costedoat-Chalumeau N, Lambert M (2020) Patients with refractory catastrophic antiphospholipid syndrome respond inconsistently to eculizumab. Blood 136(21):2473–2477
Irman S, Miha S, Igor M, Rozman B, Bozic B (2009) In vitro model of annexin A5 crystallization on natural phospholipid bilayers observed by atomic force microscopy. Autoimmunity 42(5):414–423
Rand JH, Wu XX, Quinn AS, Chen PP, McCrae KR, Bovill EG, Taatjes DJ (2003) Human monoclonal antiphospholipid antibodies disrupt the annexin A5 anticoagulant crystal shield on phospholipid bilayers: evidence from atomic force microscopy and functional assay. Am J Pathol 163(3):1193–1200
Rand JH, Wu XX, Quinn AS, Chen PP, Hathcock JJ, Taatjes DJ (2008) Hydroxychloroquine directly reduces the binding of antiphospholipid antibody-beta2-glycoprotein I complexes to phospholipid bilayers. Blood 112(5):1687–1695
Hulstein JJ, Lenting PJ, de Laat B, Derksen RH, Fijnheer R, de Groot PG (2007) beta2-Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood 110(5):1483–1491
Marciniak E, Romond EH (1989) Impaired catalytic function of activated protein C: a new in vitro manifestation of lupus anticoagulant. Blood 74(7):2426–2432
Hwang KK, Grossman JM, Visvanathan S, Chukwuocha RU, Woods VL Jr, Le DT, Hahn BH, Chen PP (2001) Identification of anti-thrombin antibodies in the antiphospholipid syndrome that interfere with the inactivation of thrombin by antithrombin. J Immunol 167(12):7192–7198
Yang YH, Chien D, Wu M, FitzGerald J, Grossman JM, Hahn BH, Hwang KK, Chen PP (2009) Novel autoantibodies against the activated coagulation factor IX (FIXa) in the antiphospholipid syndrome that interpose the FIXa regulation by antithrombin. J Immunol 182(3):1674–1680
Yang YH, Hwang KK, FitzGerald J, Grossman JM, Taylor M, Hahn BH, Chen PP (2006) Antibodies against the activated coagulation factor X (FXa) in the antiphospholipid syndrome that interfere with the FXa inactivation by antithrombin. J Immunol 177(11):8219–8225
Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR (2000) High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med 342(10):696–701
Muller-Calleja N, Hollerbach A, Ritter S, Pedrosa DG, Strand D, Graf C, Reinhardt C, Strand S, Poncelet P, Griffin JH, Lackner KJ, Ruf W (2019) Tissue factor pathway inhibitor primes monocytes for antiphospholipid antibody-induced thrombosis. Blood 134(14):1119–1131
Liestol S, Sandset PM, Jacobsen EM, Mowinckel MC, Wisloff F (2007) Decreased anticoagulant response to tissue factor pathway inhibitor type 1 in plasmas from patients with lupus anticoagulants. Br J Haematol 136(1):131–137
Adams M, Breckler L, Stevens P, Thom J, Baker R, Oostryck R (2004) Anti-tissue factor pathway inhibitor activity in subjects with antiphospholipid syndrome is associated with increased thrombin generation. Haematologica 89(8):985–990
Antovic A, Bruzelius M (2021) Impaired fibrinolysis in the antiphospholipid syndrome. Semin Thromb Hemost 47(5):506–511
Cugno M, Cabibbe M, Galli M, Meroni PL, Caccia S, Russo R, Bottasso B, Mannucci PM (2004) Antibodies to tissue-type plasminogen activator (tPA) in patients with antiphospholipid syndrome: evidence of interaction between the antibodies and the catalytic domain of tPA in 2 patients. Blood 103(6):2121–2126
Yang CD, Hwang KK, Yan W, Gallagher K, FitzGerald J, Grossman JM, Hahn BH, Chen PP (2004) Identification of anti-plasmin antibodies in the antiphospholipid syndrome that inhibit degradation of fibrin. J Immunol 172(9):5765–5773
Rikarni R, Dharma KL, Tambunan H, Isbagyo BE, Dewi N, Acang R, Setiabudy AK (2015) Aman, Prothrombotic effect of anti-beta-2 glycoprotein-1 antibodies on the expression of tissue factor, thrombomodulin, and plasminogen activator inhibitor-1 in endothelial cells. Acta Med Indones 47(1):31–37
Ghosh AK, Vaughan DE (2012) PAI-1 in tissue fibrosis. J Cell Physiol 227(2):493–507
Sobel BE, Taatjes DJ, Schneider DJ (2003) Intramural plasminogen activator inhibitor type-1 and coronary atherosclerosis. Arterioscler Thromb Vasc Biol 23(11):1979–1989
Flevaris P, Vaughan D (2017) The role of plasminogen activator inhibitor type-1 in fibrosis. Semin Thromb Hemost 43(2):169–177
Diaz JA, Ballard-Lipka NE, Farris DM, Hawley AE, Wrobleski SK, Myers DD, Henke PK, Lawrence DA, Wakefield TW (2012) Impaired fibrinolytic system in ApoE gene-deleted mice with hyperlipidemia augments deep vein thrombosis. J Vasc Surg 55(3):815–822
Bontadi A, Ruffatti A, Falcinelli E, Giannini S, Marturano A, Tonello M, Hoxha A, Pengo V, Punzi L, Momi S, Gresele P (2013) Platelet and endothelial activation in catastrophic and quiescent antiphospholipid syndrome. Thromb Haemost 109(5):901–908
Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, Singer AL, Carter-Monroe N, Nazarian SM, Segev DL, Streiff MB, Montgomery RA (2014) Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant 14(2):459–465
B.R. Long, F. Leya, The role of antiphospholipid syndrome in cardiovascular disease, Hematol Oncol Clin North Am 22(1) (2008) 79–94, vi-vii.
Alarcon-Segovia D, Cardiel MH, Reyes E (1989) Antiphospholipid arterial vasculopathy. J Rheumatol 16(6):762–767
Hughson MD, McCarty GA, Brumback RA (1995) Spectrum of vascular pathology affecting patients with the antiphospholipid syndrome. Hum Pathol 26(7):716–724
Davies MG, Hagen PO (1994) Pathobiology of intimal hyperplasia. Br J Surg 81(9):1254–1269
Nochy D, Daugas E, Droz D, Beaufils H, Grunfeld JP, Piette JC, Bariety J, Hill G (1999) The intrarenal vascular lesions associated with primary antiphospholipid syndrome. J Am Soc Nephrol 10(3):507–518
Daugas E, Nochy D, Huong DLT, Duhaut P, Beaufils H, Caudwell V, Bariety J, Piette JC, Hill G (2002) Antiphospholipid syndrome nephropathy in systemic lupus erythematosus. J Am Soc Nephrol 13(1):42–52
Tektonidou MG, Sotsiou F, Nakopoulou L, Vlachoyiannopoulos PG, Moutsopoulos HM (2004) Antiphospholipid syndrome nephropathy in patients with systemic lupus erythematosus and antiphospholipid antibodies: prevalence, clinical associations, and long-term outcome. Arthritis Rheum 50(8):2569–2579
Tektonidou MG (2014) Identification and treatment of APS renal involvement. Lupus 23(12):1276–1278
Canaud G, Bienaime F, Tabarin F, Bataillon G, Seilhean D, Noel LH, Dragon-Durey MA, Snanoudj R, Friedlander G, Halbwachs-Mecarelli L, Legendre C, Terzi F (2014) Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med 371(4):303–312
Connors JM, Levy JH (2020) COVID-19 and its implications for thrombosis and anticoagulation. Blood 135(23):2033–2040
Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, Kucher N, Studt JD, Sacco C, Bertuzzi A, Sandri MT, Barco S, Humanitas C-TF (2020) Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res 191:9–14
Y. Zuo, S. Yalavarthi, H. Shi, K. Gockman, M. Zuo, J.A. Madison, C. Blair, A. Weber, B.J. Barnes, M. Egeblad, R.J. Woods, Y. Kanthi, J.S. Knight, Neutrophil extracellular traps in COVID-19, JCI Insight 5(11) (2020).
Shi H, Zuo Y, Yalavarthi S, Gockman K, Zuo M, Madison JA, Blair C, Woodward W, Lezak SP, Lugogo NL, Woods RJ, Lood C, Knight JS, Kanthi Y (2021) Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J Leukoc Biol 109(1):67–72
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418
Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, Weyrich AS, Yost CC, Rondina MT, Campbell RA (2020) Platelet gene expression and function in patients with COVID-19. Blood 136(11):1317–1329
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus A, Laurence J (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res 220:1–13
Y. Zhang, M. Xiao, S. Zhang, P. Xia, W. Cao, W. Jiang, H. Chen, X. Ding, H. Zhao, H. Zhang, C. Wang, J. Zhao, X. Sun, R. Tian, W. Wu, D. Wu, J. Ma, Y. Chen, D. Zhang, J. Xie, X. Yan, X. Zhou, Z. Liu, J. Wang, B. Du, Y. Qin, P. Gao, X. Qin, Y. Xu, W. Zhang, T. Li, F. Zhang, Y. Zhao, Y. Li, S. Zhang, Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19, N Engl J Med 382(17) (2020) e38.
Harzallah I, Debliquis A, Drenou B (2020) Lupus anticoagulant is frequent in patients with Covid-19. J Thromb Haemost 18(8):2064–2065
Xiao M, Zhang Y, Zhang S, Qin X, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H, Zhang H, Wang C, Zhao J, Sun X, Tian R, Wu W, Wu D, Ma J, Chen Y, Zhang D, Xie J, Yan X, Zhou X, Liu Z, Wang J, Du B, Qin Y, Gao P, Lu M, Hou X, Wu X, Zhu H, Xu Y, Zhang W, Li T, Zhang F, Zhao Y, Li Y, Zhang S (2020) Antiphospholipid antibodies in critically ill patients with COVID-19. Arthritis Rheumatol 72(12):1998–2004
Devreese KMJ, Linskens EA, Benoit D, Peperstraete H (2020) antiphospholipid antibodies in patients with COVID-19: a relevant observation? J Thromb Haemost 18(9):2191–2201
Y. Zuo, S.K. Estes, R.A. Ali, A.A. Gandhi, S. Yalavarthi, H. Shi, G. Sule, K. Gockman, J.A. Madison, M. Zuo, V. Yadav, J. Wang, W. Woodard, S.P. Lezak, N.L. Lugogo, S.A. Smith, J.H. Morrissey, Y. Kanthi, J.S. Knight, Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19, Sci Transl Med 12(570) (2020).
Abdel-Wahab N, Talathi S, Lopez-Olivo MA, Suarez-Almazor ME (2018) Risk of developing antiphospholipid antibodies following viral infection: a systematic review and meta-analysis. Lupus 27(4):572–583
Hollerbach A, Muller-Calleja N, Pedrosa D, Canisius A, Sprinzl MF, Falter T, Rossmann H, Bodenstein M, Werner C, Sagoschen I, Munzel T, Schreiner O, Sivanathan V, Reuter M, Niermann J, Galle PR, Teyton L, Ruf W, Lackner KJ (2021) Pathogenic lipid-binding antiphospholipid antibodies are associated with severity of COVID-19. J Thromb Haemost 19(9):2335–2347
Funding
JSK was supported by grants from the NIH (R01HL115138), Burroughs Wellcome Fund, Lupus Research Alliance, and Rheumatology Research Foundation. YK was supported by the Intramural Research Program of the NHLBI and NIAID, and the Lasker Foundation. YK & JSK have received funding support from the A. Alfred Taubman Medical Research Institute. Illustrations credited to Ethan Tyler (NIH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
JSK received support from Jazz Pharmaceuticals for preclinical research. YK is an inventor on a pending patent (US20180369278A1) filed by the University of Michigan on the use of biogases in vascular disease.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the special issue on:Inflammation in vascular diseases - Guest Editors: Mariana Kaplan & Peter Grayson
Rights and permissions
About this article

Cite this article
Knight, J.S., Kanthi, Y. Mechanisms of immunothrombosis and vasculopathy in antiphospholipid syndrome. Semin Immunopathol 44, 347–362 (2022). https://doi.org/10.1007/s00281-022-00916-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-022-00916-w