
Abstract
In recent years, we have seen antimicrobial resistance rapidly emerge at a global scale and spread from one country to the other faster than previously thought. Superbugs and multidrug-resistant bacteria are endemic in many parts of the world. There is no question that the widespread use, overuse, and misuse of antimicrobials during the last 80 years have been associated with the explosion of antimicrobial resistance. On the other hand, the molecular pathways behind the emergence of antimicrobial resistance in bacteria were present since ancient times. Some of these mechanisms are the ancestors of current resistance determinants. Evidently, there are plenty of putative resistance genes in the environment, however, we cannot yet predict which ones would be able to be expressed as phenotypes in pathogenic bacteria and cause clinical disease. In addition, in the presence of inhibitory and sub-inhibitory concentrations of antibiotics in natural habitats, one could assume that novel resistance mechanisms will arise against antimicrobial compounds. This review presents an overview of antimicrobial resistance mechanisms, and describes how these have evolved and how they continue to emerge. As antimicrobial strategies able to bypass the development of resistance are urgently needed, a better understanding of the critical factors that contribute to the persistence and spread of antimicrobial resistance may yield innovative perspectives on the design of such new therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Antibiotic resistance occurs when bacteria have or develop the ability to circumvent the mechanisms, which drugs use against them. Infections caused by antibiotic-resistant pathogens are usually more difficult to treat, and can relapse and cause significant morbidity and mortality. Epidemiological surveillance networks in Europe and Asia [European Antimicrobial Resistance Surveillance Network -EARS-Net, Central Asia and Eastern European Surveillance of Antimicrobial Resistance-(CAESAR)] have documented that antibiotic-resistant bacteria have become much more prevalent during the last decade (European Centre for Disease Prevention and Control 2018; World Health Organization 2017). Also, according to the Centers for Disease Control and Prevention (CDC), each year in the US, at least 2 million people get infected with antibiotic-resistant bacteria, and at least 23,000 people die as a result of these infections (CDC 2013). It is estimated that globally approximately 700,000 deaths are attributed annually to antimicrobial resistance and this could rise to 10 million deaths per year by 2050 (O’Neill 2014). Additionally, infections due to antimicrobial-resistant bacteria like those caused by multidrug-resistant Acinetobacter baumannii, Klebsiella pneumoniae, and methicillin-resistant Staphylococcus aureus (MRSA) result in longer duration of hospitalization and pose a significant economic burden on national healthcare systems.
Antimicrobial resistance, however, is associated with the widespread use and misuse of antibiotics, in humans, agriculture, animal farming, and industry (Harbarth et al. 2015) and thus needs to be viewed under a One-Health approach, as human health is inextricably linked to the health of animals and the viability of ecosystems. This article aims to highlight the most important mechanisms of antimicrobial resistance as well as describe how these have emerged and evolved and what drives their persistence over time. A better understanding of the evolutionary drivers of antimicrobial resistance may offer novel perspectives on ways to tackle this critical public health threat.
Antimicrobials and Antimicrobial Resistance: A Brief History
Penicillin, discovered by Alexander Fleming in 1928, was the first antibiotic used successfully to control bacterial infections in soldiers during World War II. However, even before the use of penicillin, in 1940, the first penicillin-resistant Staphylococcus strains had already been described. To counteract the first penicillinases, methicillin was introduced in 1959 and one year later, in 1960, a methicillin-resistant Staphylococcus strain was reported (Sengupta et al. 2013). Vancomycin was introduced in 1958 for the treatment of methicillin-resistant staphylococci. A couple of decades later, in 1979, coagulase-negative staphylococci resistant to vancomycin were reported and ten years later resistance in enterococci was described (Courvalin 2006), followed by, in 1997, the report of less-susceptible S. aureus (vancomycin-intermediate S. aureus, VISA) strains in Japan (Levine 2005). Another historical example is tetracycline, which was introduced in 1950 followed by tetracycline-resistant Shigella strains being reported in 1959. Furthermore, levofloxacin was introduced into clinical practice in 1996 and in the same year levofloxacin-resistant pneumococcus was reported (Sengupta et al. 2013).
For two decades, between 1960 and 1980, there was a seemingly adequate production of new antimicrobials by the pharmaceutical industry. However, after the 1980s, the rate of discovery of new antibiotic classes had dramatically decreased, until recently, when a new interest has sparked (Parmar et al. 2018). As a consequence of increasing antimicrobial resistance and a thin new antimicrobial pipeline, bacterial infections due to multidrug-resistant or extensively drug-resistant pathogens have become a major concern in clinical practice worldwide.
Antimicrobial Resistance: Intrinsic, Acquired, and Adaptive
Antibiotic resistance exhibited by bacteria can be intrinsic, acquired, or adaptive (Joon-Hee 2019).
Intrinsic resistance is defined as the resistance exhibited due to the inherent properties of the bacterium. Examples of intrinsic resistance include the glycopeptide resistance exhibited by Gram-negative bacteria due to the impermeability of the outer membrane present in the Gram-negative bacterial cell envelope.
Acquired resistance is defined as the resistance exhibited when a previously sensitive bacterium acquires a resistance mechanism by either a mutation or the acquisition of new genetic material from an exogenous source (horizontal gene transfer). Horizontal gene transfer can occur through three main mechanisms (Holmes et al. 2016; Munita and Arias 2016).
Transformation: This is a form of genetic recombination in which free DNA fragments from a dead bacterium enter a recipient bacterium and are incorporated into its chromosome. Only a few bacteria are naturally transformable.
Transduction: Transduction involves the transfer of genetic material between a donor and a recipient bacterium by a bacteriophage.
Conjugation: This is probably the most important mechanism of horizontal gene transfer. It involves the transfer of genetic material from one bacterial cell to another by direct physical contact between the cells. A sex pillus forms between the two bacterial cells through which a plasmid is transferred from the donor cell to the recipient cell. Multiple resistance genes are often present on a single plasmid enabling the transfer of multidrug resistance in a single conjugation event. The assembly of multiple resistance genes on a single plasmid is mediated by mobile genetic elements (transposons, integrons, and Insertion Sequence Common Region- ISCR-elements).
Adaptive resistance is defined as the resistance to one or more antibiotics induced by a specific environmental signal (e.g., stress, growth state, pH, concentrations of ions, nutrient conditions, sub-inhibitory levels of antibiotics). In contrast to intrinsic and acquired resistance, adaptive resistance is transient. Adaptive resistance, which allows bacteria to respond more rapidly to antibiotic challenge, generally reverts to the original state once the inducing signal is removed (Fernández et al. 2011; Joon-Hee 2019; Motta et al. 2015; Salimiyan Rizi et al. 2018).
Adaptive resistance seems to be the result of modulations in gene expression as a response to environmental changes. Rather than being caused by genetic alterations, which usually produce irreversible phenotypes, adaptive resistance is possibly the result of epigenetic changes. Specifically, it has been suggested that DNA methylation by the DAM methylase could be responsible for the presence of different gene expression profiles in a bacterial population and could provide the heterogeneity and epigenetic heredity of gene expression essential for adaptive resistance to occur (Motta et al. 2015; Salimiyan Rizi et al. 2018). In particular, modulation of the expression of efflux pumps and porins have been implicated in the emergence of adaptive resistance (Motta et al. 2015).
The phenomenon of adaptive resistance may be responsible for the differences observed when comparing the in vitro and in vivo effectiveness of an antibiotic and could be involved in the clinical failure of antibiotic treatments. Moreover, the increase in resistance in response to environmental stimuli may not completely revert upon removal of the stimulus leading to a gradual increase in MIC over time (Fernández et al. 2011). Finally, it has been proposed that the ability of bacterial populations to proliferate in the presence of sub-inhibitory levels of antibiotics through adaptive resistance may eventually allow for more effective and permanent mechanisms of resistance to develop (Fernández et al. 2011; Salimiyan Rizi et al. 2018).
Mechanisms of Antibiotic Resistance
Resistance to antibiotics is typically the result of antibiotic destruction or modification, target alterations (target replacement, target site mutations, target site enzymatic alterations, target site protection, target overproduction or target bypass), and reduced antibiotic accumulation due to either decreased permeability and/or increased efflux. Alternatively, antibiotic resistance can be the result of a global adaptation of the bacterial cell (Table 1). We discuss the main antibiotic resistance mechanisms with their clinically relevant impact.
Antibiotic Destruction
β-Lactamases are the best example of antibiotic resistance mediated by the destruction of the antibiotic molecule. These enzymes destroy the amide bond of the β-lactam ring essentially rendering the antimicrobial ineffective. The first β-lactamases were described in 1940 (Abraham and Chain 1940), 1 year before penicillin was introduced into clinical practice. More than 1000 β-lactamases have been reported up to date produced by diverse bacteria (www.lahey.org/studies) and are considered to be the most common resistance mechanism leading to β-lactam resistance among Gram-negative bacteria. Genes encoding β-lactamases can be found in the chromosome or in mobile genetic elements (MGEs), which has facilitated their dissemination among bacteria. TEM-1, a plasmid-encoded β-lactamase, was described in Gram-negative bacteria in the 1960s. From then on, the introduction of new β-lactams was followed by the identification of new β-lactamases capable of destroying the novel compound. The introduction of third-generation cephalosporins, for example, in the early 1980s was quickly followed by the identification of plasmid-encoded β-lactamases capable of hydrolyzing third-generation cephalosporins (Extended-Spectrum β-Lactamases-ESBLs) in 1983 (Knothe et al. 1983). β-lactamase groups of great clinical importance in Gram-negative bacteria include ESBLs (enzymes conferring resistance to penicillins, first-, second-, third-generation cephalosporins, and aztreonam but not cephamycins or carbapenems and which are inhibited by β-lactamase inhibitors) (Paterson and Bonomo 2005), AmpC enzymes (enzymes conferring resistance to penicillins, first-, second-, third-generation cephalosporins, aztreonam, and cephamycins but not carbapenems and which are not inhibited by β-lactamase inhibitors) (Jacoby 2009) and carbapenemases (a diverse group of enzymes conferring carbapenem resistance with many conferring resistance to almost all hydrolyzable β-lactams) (Queenan and Bush 2007).
Antibiotic Modification
Enzymatic modification of the antibiotic molecule is the commonest mechanism of clinically relevant aminoglycoside resistance. Aminoglycoside modifying enzymes (AMEs) mediate aminoglycoside acetylation, phosphorylation, or adenylation with the resulting modified antibiotic having a decreased avidity for its target. The genes encoding AMEs are usually located in MGEs enabling them to efficiently disseminate among bacteria. As a result, virtually all medically important bacteria can demonstrate aminoglycoside resistance through this mechanism (Ramirez and Tolmasky 2010). Enzymatic acetylation of the antibiotic molecule is the commonest mechanism of chloramphenicol resistance. Many chloramphenicol acetyltransferases (CATs) have been described in a wide range of bacterial species (Schwarz et al. 2004).
Modifications of Antibiotic-Activating Enzymes
Activation of nitrofurantoin by bacterial reductases resulting in the formation of toxic intermediate compounds is required for nitrofurantoin antimicrobial activity. Mutations in the nitroreductase genes nfsA and nfsB comprise the principal mechanism of nitrofurantoin resistance (Osei Sekyere 2018; Whiteway et al. 1998). Mutations in the ribE gene have also been implicated in nitrofurantoin resistance. The ribE gene encodes a lumazine synthase, an enzyme required for the biosynthesis of riboflavin (an important co-factor of nfsA and nfsB) (Osei Sekyere 2018).
Target Replacement or Target Bypass
Replacement of the bacterial Penicillin-Binding Proteins (PBP) is the mechanism underlying β-lactam resistance in Streptococcus pneumoniae and methicillin resistance in Staphylococcus aureus.
β-lactam resistance among Streptococcus pneumoniae is the result of the production of mosaic PBP genes. These genes are produced by the recombination of native DNA and foreign DNA originating from β-lactam-resistant streptococci (transformation). Methicillin resistance in Staphylococcus aureus is the result of the acquisition of the mecA gene and its incorporation into the bacterial chromosomal DNA. The mecA gene is located on a mobile genetic element known as staphylococcal chromosomal cassette mec (SCC mec). The mecA gene encodes Penicillin-Binding Protein 2a (PBP2a), a unique PBP with a very low affinity for all β-lactams (penicillins, cephalosporins except for last-generation compounds and carbapenems). As a result, it allows effective cell wall synthesis to continue even in the presence of β-lactams (Chambers 1999; Hiramatsu et al. 2013; Moellering 2012).
Glycopeptide resistance in enterococci involves the acquisition of a set of genes (van gene clusters) that lead to the replacement of the glycopeptide target (the terminal d-Alanine-d-Alanine moiety of peptidoglycan precursors) reducing the binding affinity of the antibiotic molecule. The change of the terminal d-Alanine-d-Alanine moiety to d-alanine-d-lactate leads to high-level resistance while the change to d-alanine-d-serine leads to low-level resistance (Miller et al. 2014). Development of high-level vancomycin resistance in Staphylococcus aureus (VRSA) due to the acquisition of the vanA gene cluster form vancomycin-resistant enterococci has been reported (Sievert et al. 2008) but fortunately continues to be a rare phenomenon.
Acquisition of dihydrofolate reductase (DHFR) and dihydropteroate synthase (DHPS) genes coding for trimethoprim-resistant DHFR enzymes and sulfonamide-resistant DHPS enzymes, respectively, has been reported to result in transferable resistance to these antimicrobial agents (Eliopoulos and Huovinen 2001). The ability of enterococci to utilize exogenous folinic acid may increase the MIC to trimethoprim-sulfamethoxazole in vivo resulting in therapeutic failure when treating enterococcal infections with trimethoprim-sulfamethoxazole (Zervos and Schaberg 1985).
Target Site Alteration (By Mutation or Enzymatic Alteration)
Mutations in genes encoding the domain V of the 23SrRNA are the commonest mechanism of linezolid resistance. Since bacteria carry multiple copies of the 23SrRNA genes, the number of the mutated alleles correlates with the increase in MIC. Mutations in multiple alleles need to occur in order for clinically relevant resistance to manifest. Mutations in the ribosomal proteins L3 and L4 which border the linezolid binding site have also been associated with linezolid resistance (Miller et al. 2014). 23SrRNA mutations have also been implicated in the development of macrolide, lincosamide, and streptogramin B resistance (Leclercq 2002).
Quinolone resistance is most often the result of chromosomal mutations in the bacterial gyrase and/or topoisomerase IV genes (Aldred et al. 2014), while rifampicin resistance is usually the result of mutations in the RNA polymerase β subunit gene (Goldstein 2014). Mutational or recombinational changes in the dihydrofolate reductase (DHFR) gene or the dihydropteroate synthase (DHPS) gene have been associated with resistance to trimethoprim and sulfonamides, respectively, in a number of clinically important bacteria (Eliopoulos and Huovinen 2001).
Methylation of the 23SrRNA by enzymes, encoded by a variety of erm (erythromycin ribosome methylase) genes, confers cross-resistance to macrolides, lincosamides, and streptogramin B (a phenomenon called the MLSB phenotype) (Leclercq 2002), while methylation of the 23SrRNA by an enzyme encoded by the cfr gene has been identified as a cause of resistance to linezolid, chloramphenicol, and clindamycin (Kehrenberg et al. 2005; Morales et al. 2010; Schwarz et al. 2004).
Target Site Protection
Ribosomal protection proteins (RPPs) are an example of antimicrobial resistance through target site protection and they been described in both Gram-positive and Gram-negative bacteria (Connell et al. 2003; Roberts 2005). Qnr proteins mediate quinolone resistance by acting as a DNA analogue and reducing the interaction of the bacterial gyrase and topoisomerase IV with DNA. In doing so, they reduce the available quinolone binding sites (Aldred et al. 2014). Qnr genes can be chromosomal or plasmid-mediated (Jacoby et al. 2014).
Target Overproduction
Massive overproduction of the antibiotic target can lead to resistance by essentially overwhelming the antibiotic. Overproduction of Dihydrofolate Reductase (DHFR), for example, has been reported as a cause of resistance to trimethoprim in Eschericia Coli (Eliopoulos and Huovinen 2001; Flensburg and Sköld 1987).
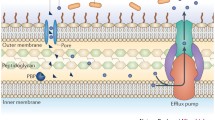
Decreased Permeability of the Bacterial Outer Membrane
Gram-negative bacteria are surrounded by the outer membrane, a permeability barrier for many substances including antibiotics. The low permeability of the bacterial outer membrane to specific antibiotic agents is responsible for the intrinsic resistance of some Gram-negative bacteria to those antibiotics. Moreover, changes in the outer membrane permeability can contribute to the development of acquired resistance (Nikaido 1989).
Porins are the major route of entry of hydrophilic antibiotics (such as β-lactams, fluoroquinolones, tetracyclines, and chloramphenicol) through the bacterial outer membrane. The number and type of porins expressed on the outer membrane will affect the entry of hydrophilic antibiotics and, therefore, the susceptibility of the bacterial cell to them (Fernández and Hancock 2012). Mutations affecting the expression or the function of porins can lead to acquired antibiotic resistance. These mutations can have different effects such as porin loss, the modification of the size or conductance of the porins or the reduced expression of porins. Changes in porin expression generally lead to low-level antibiotic resistance. However, it is common to observe bacterial strains in which the effect of changes in porin expression is enhanced by the co-existence of other resistance mechanisms. In essence, the reduced uptake of the antibiotic due to changes in porin expression enhances the effect of co-existent resistance mechanisms such us efflux pumps or antibiotic degrading enzymes with the result being high-level resistance (Fernández and Hancock 2012).
Efflux Pumps
Efflux pumps are energy-dependent complex bacterial systems present on the cytoplasmic membrane which are capable of pumping toxic molecules out of the cell. The first efflux pump pumping tetracycline out of the bacterial cell was described in Eschericia Coli in 1980 and was plasmid-encoded (Ball et al. 1980; McMurry et al. 2006). Since then numerous examples of efflux systems involved in antibiotic resistance have been identified in both Gram-positive and Gram-negative bacteria. Most efflux systems can transport multiple unrelated substances and can, therefore, result in multidrug resistance (Nikaido and Pagès 2012; Piddock 2006a, b), although there are some examples of drug-specific efflux pumps (Piddock 2006a, b).
Multidrug efflux mechanisms are almost invariably chromosomally encoded and in some cases can explain the intrinsic resistance of bacteria to specific antibiotics. However, despite the fact that multidrug efflux mechanisms are broadly conserved in bacteria, only a few confer clinically relevant antibiotic resistance. Clinical resistance is usually the result of mutational events leading to increased pump expression or improved pump effectiveness (Piddock 2006a, b). Genes encoding substrate-specific efflux pumps, on the other hand, tend to be located on mobile genetic elements (Fernández and Hancock 2012; Keith 2005). Examples of substrate-specific efflux pumps include those specific for tetracyclines, macrolides, and chloramphenicol (Keith 2005).
Global Cell Adaptation (Changes in Cell Regulation)
Resistance to antibiotics can occur due to a global adaptive response in the bacterial cell (changes in cell regulation) as opposed to single changes. The best clinically relevant examples of this type of resistance mechanism are daptomycin resistance in enterococci and Staphylococcus aureus and low-level vancomycin resistance in Staphylococcusaureus.
Daptomycin kills the bacterial cell by altering cell membrane homeostasis. In enterococci, mutations in genes encoding regulatory systems controlling cell envelope homeostasis and genes involved in phospholipid metabolism have been associated with the development of daptomycin resistance (Miller et al. 2014). In Staphylococcus aureus, progressive accumulation of mutations in genes involved in key cell membrane events and modifications of the cell wall have been associated with the development of daptomycin resistance (Bayer et al. 2013).
Intermediate susceptibility of Staphylococcus aureus to vancomycin (MIC 4–8 μg/ml) does not seem to result from the acquisition of a resistance gene (such as the acquisition of the vanA gene cluster seen in VRSA) but from changes that usually involve genes forming part of regulatory systems controlling cell envelope homeostasis. The specific mechanisms involved remain unclear but appear to result in reduced cell wall turnover and autolytic activity and in some cases increased cell wall synthesis, which prevents vancomycin from reaching its target site at the cell wall division septum (Howden et al. 2010).
Epistasis
A single bacterial cell may contain multiple resistance mutations. The resistance phenotype as well as the fitness cost of the resistance mutations are not the expected additive effect of the different mutations but depend on the interaction between the different mutations, a phenomenon known as epistasis (Borrell et al. 2013; Levin-Reisman et al. 2019; Trindade et al. 2009). It has been found that in an antibiotic-free environment, the cost of multiple resistance is smaller than that expected due to positive epistasis between antibiotic resistance mutations (Borrell et al. 2013; Trindade et al. 2009). This, obviously, facilitates the development of multidrug resistance. Moreover, it has been shown that low-level antibiotic exposure can lead to high-level resistance through the accumulation of several small-effect mutations. The resulting resistance level is significantly higher than the expected additive resistance, suggesting strong epistatic interactions between the different resistance mutations (Wistrand-Yuen et al. 2018).
Evolution of Antimicrobial Resistance
The Ancient and Diverse Resistome: A Potential Source of Clinically Relevant Resistance Genes
In recent times, we have seen antimicrobial resistance rapidly emerge at a global scale and spread from one country to the other faster than previously thought. Superbugs and multidrug-resistant bacteria are endemic in many parts of the world. There is no question that the widespread use, overuse, and misuse of antimicrobials during the last 80 years have been associated with the explosion of antimicrobial resistance. On the other hand, the molecular pathways behind the emergence of antimicrobial resistance in bacteria have been present since ancient times (Dcosta et al. 2011). Commensals and soil bacteria have been capable of producing compounds and small molecules with antimicrobial potential throughout evolution. For example, the emergence of the biosynthetic pathway for erythromycin may date as back as 800 million years (Baltz 2006). What is recently better understood is that these inhibitors did not solely evolve to help bacteria survive but also served other functions like cell signaling or contributed in natural degradation processes, quorum sensing, and biofilm formation (Davies et al. 2006; Sengupta et al. 2013).
Moreover, we know that bacterial penicillinases were present before the widespread use of penicillin, while genes encoding β-lactamases have been recovered from very remote environmental habitats (Allen et al. 2009; Bhullar et al. 2012) and from ancient bacteria (Dcosta et al. 2011; Wright 2007). Likewise, it has been found that antibiotic resistance genes exist in the gut of people who live in isolated areas and who have been minimally or never exposed to antimicrobials (Bartoloni et al. 2009; Pallecchi et al. 2007).
Thus, there is an increasing body of evidence showing that resistance genes are naturally occurring and in abundance in microbial populations (Dantas et al. 2008; D’Costa et al. 2006). The antimicrobial resistance genes found in different bacterial species in nature comprise the environmental resistome (Wright 2007). Sequencing and genomic analysis of extensive bacterial libraries (D’Costa 2006; Dantas et al. 2008) has led to the creation of large databases that list thousands of potential resistance genes (Lakin et al. 2017; McArthur et al. 2013).
The resistome consists of known and likely novel genetic resistance determinants, which carry the potential of causing clinically relevant resistance, if successfully transferred and/or expressed in pathogens causing human disease. Species like soil bacteria, which produce antimicrobial products, are obviously intrinsically resistant to those molecules. For example, most antibiotic-producing actinomycetes possess genes encoding resistance to the compounds that they produce (Ogawara et al. 1999). Such resistance mechanisms include genes encoding for β-lactamase production or efflux pumps (Cantón 2009; Forsman et al. 2009; Piddock 2006a, b). Some of these resistance mechanisms have been the ancestors of those found in clinically relevant antibiotic-resistant pathogens, as evidenced by their similar biochemical and genetic profiles (Benveniste and Davies 1973; Cantón 2009).
In addition, old or novel β-lactamases and other resistance determinants can arise as new threats under conditions that will facilitate their transfer to pathogenic bacteria or their enhanced expression. As Kluyvera spp were found to be the source of CTX-M genes, species like Streptomyces known to produce β-lactamases could be the source of clinically important resistance genes (Livermore et al. 2007; Rossolini et al. 2008). Interestingly, Kluyvera spp do not exhibit intrinsic resistance to extended-spectrum β-lactams nor produce antimicrobial compounds (Cantón 2009). It seems that the blaCTX-M genes in Kluyvera spp have originated from a common ancestor, which was incorporated after horizontal transfer into the Kluyvera chromosome, in ancient times (Rossolini et al. 2008). Other examples of such genetic transfers have been documented for genes encoding ribosomal methylases affecting aminoglycosides (armA, rtmB), methyltransferases affecting linezolid (cfr), or plasmid-mediated efflux pumps conferring low-level fluoroquinolone resistance (qepA), all of which were associated with antibiotic-producing bacteria (Cantón 2009). However, we need more phylogenetic studies and metagenomics data in order to define the exact role of the environmental resistome in the emergence of antimicrobial resistance in human and animal pathogens.
Emergence of Antimicrobial Resistance: Random Events, Genetic Exchange, and Selection Pressure
Resistance in bacteria or bacterial communities arises via two major mechanisms, either pick-up/transfer of resistance genes from other bacteria or gene mutation of pre-existing or acquired genes. Strong selection of these genetic events is believed to be enhanced in the presence of antimicrobials (Cantón 2009; Gillings et al. 2017). Under inhibitory and sub-inhibitory concentrations of antimicrobials, bacteria that can survive will be selected (Davies and Davies 2010; Gillings et al. 2017). Nonetheless, some antibiotics can modulate the expression of virulence factors or upregulate genes involved in the SOS response and potentially contribute to increased mutation frequency, via the latter (Sengupta et al. 2013). It has been hypothesized that in the antibiotic era, naturally occurring selection occurred and spread more rapidly compared to the pre-antibiotic era (Cantón 2009; Davies and Davies 2010; Gillings et al. 2017). This can occur either in the environment or within the host.
The emergence of a multitude of antimicrobial-resistant microbes and their now established presence not only in the clinical setting but also in the environment has been facilitated by the widespread use of antibiotics in human health, agriculture, aquaculture, animal farming, veterinary medicine, pest control, and pharmaceutical industry. Nonetheless, it is well documented that increased antibiotic use is correlated with higher resistance rates, as countries with low rates of antimicrobial resistance also report lower rates of antimicrobial consumption (Costelloe et al. 2010).
Pathogens can exchange pathogenic, virulence, and resistance elements. Transmission by conjugation occurs extensively both in nature and within the host, most abundantly in the intestinal tract (Sommer et al. 2010). Horizontal gene transfer is most commonly mediated through plasmids, which have a key role in the spread of resistance between bacterial strains (Davies and Davies 2010; Norman et al. 2009). For example, resistance genes, which encode modified β-lactamases, can be transferred rapidly and efficiently horizontally. One of the original types of β-lactamases known to confer resistance to gram-negative bacteria is TEM. However, nowadays, plenty of plasmid-encoded TEM enzymes, stemming from random mutations (rapid radiation), have occurred and circulate globally (Gniadkowski 2008). Also, extended-spectrum β-lactamases (ESBL) now encoded by more than 100 CTX-M variant genes have shown an enormous ability to transmit and spread all over the world (Rossolini et al. 2008). Interestingly, however, plasmid sequencing of pre-antibiotic era bacterial pathogens has shown that resistance determinants on plasmids were not common (Datta and Hughes 1983).
Resistance acquisition through genetic exchange via plasmids, transposons, and integrons, is favored in dense communities like sewage treatment plants, considered as “hotspots” for bacterial genetic exchange. Plasmids carrying multiple resistant genes have been recovered from samples taken from sewage (Schlüter et al. 2007), where indigenous bacteria and those derived from humans and animals co-exist. For example, Aeromonas cultured from aquaculture environments has been shown to carry multiple resistance genes on plasmids and integrons (Penders and Stobberingh 2008). The human gut is considered another hotspot for such genetic exchanges between pathogens and commensals (Sommer et al. 2010).
In recent years, there has been an increasing interest on the contribution of integrons in bacterial genome evolution, due to their role in promoting genetic diversity in bacteria (Boucher et al. 2007; Mazel 2006). Integrons were first described in 1987 (Stokes and Hall 1989) and are found on the chromosomes of approximately 15% of bacterial species (Boucher et al. 2007; Mazel 2006). However, it was later demonstrated that they were linked to plasmid-mediated resistance encountered in Shigella in the 1950s in Japan (Liebert et al. 1999). The main role of integrons is gene cassette capture followed by recombination into an attachment site (a process catalyzed by integrase) and gene expression via activation of the SOS system. Integrons are a major source of transferable resistance determinants, both in Gram-negative and Gram-positive bacteria (Gillings 2014). Especially class 1 integrons are widely present in pathogens and gut commensals and overall, they have accumulated over 130 gene cassettes conferring resistance to most antibiotic classes (Partridge et al. 2009).
Another important evolutionary mechanism exhibited by bacteria is direct DNA acquisition from the environment, like, for example, in Acinetobacter spp, an organism that has evolved rapidly accumulating, in some cases, more than 20 genomic islands encoding antibiotic resistance determinants and virulence functions (Barbe et al. 2004). In addition, development of resistance may be facilitated in mixed bacterial communities, like polymicrobial infections or biofilms, due to cell–cell fusion, larger populations with lower rates of turnover, and elevated mutation rates (Brockhurst et al. 2019; Driffield et al. 2008). Lastly, physiological tolerance to antibiotics can allow or precede the emergence of resistant phenotypes (Levin-Reisman et al. 2017).
Whole genome sequencing can help tremendously in reconstructing the evolutionary history of resistance in bacterial species. For example, using WGS and phylogenetic analysis of the first MRSA isolates, scientists were able to discover that the early MRSA lineage arose in the mid-1940s (long before the use of methicillin) after the acquisition of an ancestral type I SCCmec element. Therefore, contrary to what was previously thought, the driver for MRSA evolution was not methicillin but the widespread use of first-generation β-lactams. Such data could inform the assessment of evolutionary risk and the design of novel antimicrobial therapies (Brockhurst et al. 2019; Harkins et al. 2017).
Resistance genes can cross species and similar resistance genes have been found in humans and animals. Whole genome sequencing of certain strain lineages like S. aureus CC398 has provided evidence of many livestock-to-human jumps and less frequently, human-to-livestock jumps during its evolutionary history (Ward et al. 2014). In another landmark study and contrary to what was previously believed, in 2015, Liu et al. (Liu et al. 2016) first described transferable colistin resistance in E. coli and K. pneumoniae isolates from animals and humans, mediated by the mcr-1 gene (mobile colistin resistance), located in a plasmid. Since then, more mcr variants have been discovered, in several bacteria in different countries (Dalmolin et al. 2018). These examples highlight the importance of a one-health approach in trying to address the antimicrobial resistance crisis.
Drivers of Antimicrobial Resistance Persistence
Although it is indisputable that the use of antibiotics in clinical and veterinary practices as well as animal husbandry is strongly correlated with the development and spread of antibiotic resistance, it is less evident that reducing the use of antibiotics will result in significant reductions in antibiotic resistance.
There are, of course, several reports of antibiotic resistance decreases as a result of a reduction in antibiotic use such as the decrease in penicillin resistance in Streptococcus pneumoniae in Hungary following a drastic reduction in the prescription of penicillin (Nowak 1994). However, there are also reports of antibiotic resistance persistence despite drastic reductions in antibiotic use. No significant reduction of resistance was observed following drastic reductions in the use of trimethoprim and sulphonamides in Sweden and the United Kingdom, respectively (Andersson et al. 2009; Enne et al. 2001). A possible explanation could be that antibiotic exposure extends beyond therapeutic use in humans, which accounts for less than half of the total antibiotic consumption (Davies and Davies 2010). It does, therefore, seem that antibiotic selection pressure may not be the only driver of resistance.
The acquisition of antibiotic resistance mechanisms is usually associated with a fitness cost for the bacterial cell. Resistance due to chromosomal mutations often involves modification of essential bacterial genes while resistance due to the acquisition of plasmids imposes a burden on the host cell due to the metabolic load of plasmid replication and the expression of plasmid genes, disruptions in the expression of host genes, and other metabolic effects. As a result, antibiotic-resistant bacteria tend to have a reduced growth rate and competitive ability compared to their susceptible counterparts (Carroll and Wong 2018; Vogwill and Maclean 2015).
One would, therefore, expect that the absence of antibiotic selection pressure would lead to a reduction in antibiotic resistance with sensitive bacteria displacing their less fit resistant counterparts. However, antibiotic resistance genes seem to persist for long periods of time in the absence of antibiotic selection pressure. Antibiotic resistance stabilization in bacterial populations and persistence in environments with reduced antibiotic use can occur via the following mechanisms.
Fitness Restoring Compensatory Mutations
Compensatory mutations in other loci, which restore the fitness of antibiotic-resistant bacteria, can allow the maintenance of antibiotic-resistant bacteria even in the absence of antibiotic selection. In vitro and in vivo experiments with antibiotic-resistant bacteria have shown that bacterial evolution in the absence of antibiotics is more likely to result in compensatory mutations improving the fitness of antibiotic-resistant bacteria rather than in reversion to highly fit antibiotic-sensitive bacteria (Levin et al. 2000; Schrag et al. 1997). In the case of conjugative plasmids, these compensatory adaptive mutations have been described to occur on the bacterial chromosome, the plasmid or both (Dahlberg and Chao 2003; Harrison et al. 2016).
The Occurrence of Fitness Cost-Free or Fitness Increasing Resistance Mechanisms
Although resistance mechanisms are usually associated with a fitness cost, this is by no means always the case. There are reports of antibiotic resistance plasmid acquisitions which are either fitness cost-free (Carroll and Wong 2018; Enne et al. 2005) or are associated with an increase in the fitness of the bacterium (Carroll and Wong 2018; Enne et al. 2004). The fitness effect of a plasmid does not seem to be constant but instead is influenced by the host genetic background (Carroll and Wong 2018; Di Luca et al. 2017). Fitness enhancing chromosomally encoded antibiotic resistance mutations has also been described (Luo et al. 2005).
Genetic Linkage and Co-selection with Other Genes
Genetic linkage of multiple antibiotic resistance genes allows for their co-selection. Consequently, reducing the use of one antibiotic may not result in the expected reduction in resistance to this agent if the use of other antibiotics continues to be high (Andersson and Hughes 2011). This is particularly important for plasmid-encoded antibiotic resistance since, as mentioned previously, multiple resistance genes often accumulate on a single plasmid. Alternatively, an antibiotic resistance gene can be co-selected with other genes such those encoding virulence factors (Andersson and Hughes 2011; Salyers and Amábile-Cuevas 1997) or genes encoding resistance to biocides and metals (Pal et al. 2015).
Plasmid Persistence Mechanisms
Apart from the mechanisms mentioned above which can result in the persistence of both chromosomally encoded and plasmid-encoded antibiotic resistance mechanisms, additional mechanisms specifically ensuring plasmid persistence exist. Plasmid stabilization systems include the multimer resolution system, active partitioning, and various types of post-segregational killing systems (which kill individual cells lacking the plasmid) (Andersson and Hughes 2011; Bahl et al. 2009). Infectious growth (meaning that the plasmid transfer rate is sufficiently high to ensure plasmid maintenance during host replication despite fitness cost and segregational loss) may also contribute to plasmid persistence (Andersson and Hughes 2011; Carroll and Wong 2018; Lili et al. 2007). Moreover, Wein et al. have showed that plasmid stability can evolve under non-selective conditions, given that plasmid gene transcription may hinder plasmid replication and inheritance and can be affected by environmental conditions. These findings could offer an explanation, for the ubiquity of plasmids in nature (Wein et al. 2019).
Novel Ways to Tackle Antimicrobial Resistance
Antimicrobial resistance is not only one of the most important global health threats but also one with no easy solution. To date, efforts to combat antimicrobial resistance focus on concerted attempts to improve diagnosis, antibiotic-prescribing practices, and infection prevention strategies. Few new antimicrobial compounds are in the process of clinical development, however, most of them do not represent new antibiotic classes. Moreover, new antimicrobials harbor the risk of a short life due to the microorganisms’ significant capacity for fast adaptation. Therefore, novel treatment strategies are undoubtedly needed in the fight against established and emerging antimicrobial resistance.
Some novel strategies under investigation include either sophisticated forms of antimicrobial delivery, like nanocarriers, which can increase drug bioavailability and decrease treatment duration (Giau et al. 2019), or involve new ways to increase effective antimicrobial concentration within the bacterial cell. One way to accomplish this is via potentiation, which involves manipulating a typically non-essential part of the bacteria (i.e., efflux pump inhibitors) or via active uptake by employing membrane transporters, like iron transporters, to facilitate antibiotic entry into the cell when the former is bound to an iron-binding siderophore mimetic group (i.e., catechol). Recently, a catechol-linked cephalosporin (cefiderocol) has shown to be effective in complicated urinary tract infections (Portsmouth et al. 2018). On the other hand, using fast changes between currently available antibiotics, scientists have managed to reduce selective pressure on bacteria and to limit the emergence of resistance via a mechanism called cellular hysteresis, which entails a persistent change in bacterial cellular physiology induced by one antibiotic, which then sensitizes bacteria to another subsequently administered antibiotic (Roemhild et al. 2018). Though challenging, it would be interesting to see if such an approach could be validated in a clinical setting.
Other non-antibiotic approaches are the development of monoclonal antibodies against bacterial strains and phage therapy. Also, vaccines apart from decreasing antimicrobial use and preventing infections, they can also help limit the antimicrobial resistance potential of bacteria (Rappuoli et al. 2017) with the help of new technologies (Baker et al. 2018). Targeted immunomodulation of host responses against specific pathogens is an area of increasing research interest. Also, attempts to modulate the human microbiome aiming to decolonize the gut of susceptible patients from multidrug-resistant pathogens or to identify a combination of bacterial strains that will prevent colonization and/or infection with antibiotic-resistant pathogens or Clostridium difficile have already shown some promising results (Aroniadis and Brandt 2013; Austin et al. 2014; Biliński et al. 2016; Jouhten et al. 2016).
Conclusion
Antimicrobial resistance emergence may be inevitable in the evolutionary process, whereas the mechanisms that safeguard its persistence, even in the absence of antibiotic selection pressure, are not fully elucidated. We still have a lot to learn regarding the dynamics of antimicrobial-resistant determinants within microbial communities. Together with limiting unnecessary antibiotic use under a “one-health approach,” achieving earlier microbiologic diagnosis and strengthening infection prevention interventions, novel host- or pathogen-targeted therapies are urgently needed in order to combat the multifaceted resistance potential of bacteria.
References
Abraham EP, Chain E (1940) An enzyme from bacteria able to destroy penicillin [1]. Nature. https://doi.org/10.1038/146837a0
Aldred KJ, Kerns RJ, Osheroff N (2014) Mechanism of quinolone action and resistance. Biochemistry. https://doi.org/10.1021/bi5000564
Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J (2009) Functional metagenomics reveals diverse Β-lactamases in a remote Alaskan soil. ISME J. https://doi.org/10.1038/ismej.2008.86
Andersson DI, Hughes D (2011) Peristence of antibiotic resistance in bacterial populations. FEMS Microbiol Rev 35:901–911
Andersson DI, Cars O, Runehagen A, Sjolund-Karlsson M, Cars H, Sundqvist M et al (2009) Little evidence for reversibility of trimethoprim resistance after a drastic reduction in trimethoprim use. J Antimicrob Chemother 65(2):350–360. https://doi.org/10.1093/jac/dkp387
Aroniadis OC, Brandt LJ (2013) Fecal microbiota transplantation: past, present and future. Curr Opin Gastroenterol 29(1):79–84. https://doi.org/10.1097/MOG.0b013e32835a4b3e
Austin M, Mellow M, Tierney WM (2014) Fecal microbiota transplantation in the treatment of clostridium difficile infections. Am J Med. https://doi.org/10.1016/j.amjmed.2014.02.017
Bahl MI, Hansen LH, Sørensen SJ (2009) Persistence mechanisms of conjugative plasmids. Methods Mol Biol 532:73–102. https://doi.org/10.1007/978-1-60327-853-9_5
Baker SJ, Payne DJ, Rappuoli R, De Gregorio E (2018) Technologies to address antimicrobial resistance. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1717160115
Ball PR, Shales SW, Chopra I (1980) Plasmid-mediated tetracycline resistance in escherichia coli involves increased efflux of the antibiotic. Biochem Biophys Res Commun 93(1):74–81. https://doi.org/10.1016/S0006-291X(80)80247-6
Baltz RH (2006) Marcel faber roundtable: is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol. https://doi.org/10.1007/s10295-005-0077-9
Barbe V, Vallenet D, Fonknechten N, Kreimeyer A, Oztas S, Labarre L et al (2004) Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium. Nucleic Acids Res 1:1. https://doi.org/10.1093/nar/gkh910
Bartoloni A, Pallecchi L, Rodríguez H, Fernandez C, Mantella A, Bartalesi F et al (2009) Antibiotic resistance in a very remote Amazonas community. Int J Antimicrob Agents. https://doi.org/10.1016/j.ijantimicag.2008.07.029
Bayer AS, Schneider T, Sahl HG (2013) Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 1277(1):139–158. https://doi.org/10.1111/j.1749-6632.2012.06819.x
Benveniste R, Davies J (1973) Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc Natl Acad Sci USA 70:2276–2280
Bhullar K, Waglechner N, Pawlowski A, Koteva K, Banks ED, Johnston MD et al (2012) Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE. https://doi.org/10.1371/journal.pone.0034953
Biliński J, Grzesiowski P, Muszyński J, Wróblewska M, Mądry K, Robak K et al (2016) Fecal microbiota transplantation inhibits multidrug-resistant gut pathogens: preliminary report performed in an immunocompromised host. Arch Immunol Ther Exp. https://doi.org/10.1007/s00005-016-0387-9
Borrell S, Teo Y, Giardina F, Streicher EM, Klopper M, Feldmann J et al (2013) Epistasis between antibiotic resistance mutations drives the evolution of extensively drug-resistant tuberculosis. Evol Med Public Health 2013(1):65–74. https://doi.org/10.1093/emph/eot003
Boucher Y, Labbate M, Koenig JE, Stokes HW (2007) Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. https://doi.org/10.1016/j.tim.2007.05.004
Brockhurst MA, Harrison F, Veening J-W, Harrison E, Blackwell G, Iqbal Z, Maclean C (2019) Assessing evolutionary risks of resistance for new antimicrobial therapies. Nat Ecol Evol. https://doi.org/10.1038/s41559-019-0854-x
Cantón R (2009) Antibiotic resistance genes from the environment: a perspective through newly identified antibiotic resistance mechanisms in the clinical setting. Clin Microbiol Infect. https://doi.org/10.1111/j.1469-0691.2008.02679.x
Carroll AC, Wong A (2018) Plasmid persistence: costs, benefits and the plasmid paradox. Can J Microbiol 64:293–304. https://doi.org/10.1139/cjm-2017-0609
CDC (2013) Centers for disease control and prevention (CDC). Antibiotic resistance threats in the United States, 2013. https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf. Current. https://doi.org/CS239559-B
Chambers HF (1999) Penicillin-binding protein-mediated resistance in Pneumococci and Staphylococci. J Infect Dis. https://doi.org/10.1086/513854
Connell SR, Tracz DM, Nierhaus KH, Taylor DE (2003) Ribosomal protection proteins and their mechanism of Tetracycline resistance. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.47.12.3675-3681.2003
Costelloe C, Metcalfe C, Lovering A, Mant D, Hay AD (2010) Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: systematic review and meta-analysis. BMJ. https://doi.org/10.1136/bmj.c2096
Courvalin P (2006) Vancomycin resistance in gram-positive cocci. Clin Infect Dis. https://doi.org/10.1086/491711
D’Costa VM, McGrann KM, Hughes DW, Wright GD (2006) Sampling the antibiotic resistome. Science 311:374–377
Dahlberg C, Chao L (2003) Amelioration of the cost of conjugative plasmid carriage in Eschericha coli K12. Genetics 165(4 PG-1641–9):1641–1649
Dalmolin TV, De Lima-Morales D, Barth AL (2018) Plasmid-mediated colistin resistance: what do we know? J Infectiol Mini Rev 1:16–22
Dantas G, Sommer MOA, Oluwasegun RD, Church GM (2008) Bacteria subsisting on antibiotics. Science. https://doi.org/10.1126/science.1155157
Datta N, Hughes VM (1983) Plasmids of the same Inc groups in enterobacteria before and after the medical use of antibiotics. Nature. https://doi.org/10.1038/306616a0
Davies J, Davies D (2010) Origins and evolution of antibiotic resistance O. Microbiol Mol Biol Rev. https://doi.org/10.1128/MMBR.00016-10
Davies J, Spiegelman GB, Yim G (2006) The world of subinhibitory antibiotic concentrations. Curr Opin Microbiol. https://doi.org/10.1016/j.mib.2006.08.006
Dcosta VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C et al (2011) Antibiotic resistance is ancient. Nature. https://doi.org/10.1038/nature10388
Di Luca MC, Sørum V, Starikova I, Kloos J, Hülter N, Naseer U et al (2017) Low biological cost of carbapenemase-encoding plasmids following transfer from Klebsiella pneumoniae to Escherichia coli. J Antimicrob Chemother 72(1):85–89. https://doi.org/10.1093/jac/dkw350
Driffield K, Miller K, Bostock JM, O’neill AJ, Chopra I (2008) Increased mutability of Pseudomonas aeruginosa in biofilms. J Antimicrob Chemother. https://doi.org/10.1093/jac/dkn044
Eliopoulos GM, Huovinen P (2001) Resistance to trimethoprim-sulfamethoxazole. Clin Infect Dis 32(11):1608–1614. https://doi.org/10.1086/320532
Enne VI, Livermore DM, Stephens P, Hall LMC (2001) Persistence of sulphonamide resistance in Escherichia coli in the UK despite national prescribing restriction. Lancet 357(9265):1325–1328. https://doi.org/10.1016/S0140-6736(00)04519-0
Enne VI, Bennett PM, Livermore DM, Hall LMC (2004) Enhancement of host fitness by the sul2-coding plasmid p9123 in the absence of selective pressure. J Antimicrob Chemother 53(6):958–963. https://doi.org/10.1093/jac/dkh217
Enne VI, Delsol AA, Davis GR, Hayward SL, Roe JM, Bennett PM (2005) Assessment of the fitness impacts on Escherichia of acquisition of antibiotic resistance genes encoded by different types of genetic element. J Antimicrob Chemother 56(3):544–551. https://doi.org/10.1093/jac/dki255
European Centre for Disease Prevention and Control (2018) Surveillance of antimicrobial resistance in Europe Annual report of the European Antimicrobial Resistance Surveillance Network (EARS-Net) 2017. ECDC: Surveill Rep. https://doi.org/10.2900/230516
Fernández L, Hancock REW (2012) Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin Microbiol Rev 25(4):661–681. https://doi.org/10.1128/CMR.00043-12
Fernández L, Breidenstein EBM, Hancock REW (2011) Creeping baselines and adaptive resistance to antibiotics. Drug Resist Updates 14(1):1–21. https://doi.org/10.1016/j.drup.2011.01.001
Flensburg J, Sköld O (1987) Massive overproduction of dihydrofolate reductase in bacteria as a response to the use of trimethoprim. Eur J Biochem 162(3):473–476. https://doi.org/10.1111/j.1432-1033.1987.tb10664.x
Forsman M, Haggstrom B, Lindgren L, Jaurin B (2009) Molecular analysis of β-lactamases from four species of streptomyces: comparison of amino acid sequences with those of other β-clactamases. J Gen Microbiol. https://doi.org/10.1099/00221287-136-3-589
Gillings MR (2014) Integrons: past, present, and future. Microbiol Mol Biol Rev. https://doi.org/10.1128/MMBR.00056-13
Gillings MR, Paulsen IT, Tetu SG (2017) Genomics and the evolution of antibiotic resistance. Ann N Y Acad Sci. https://doi.org/10.1111/nyas.13268
Gniadkowski M (2008) Evolution of extended-spectrum β-lactamases by mutation. Clin Microbiol Infect. https://doi.org/10.1111/j.1469-0691.2007.01854.x
Goldstein BP (2014) Resistance to rifampicin: a review. J Antibiot. https://doi.org/10.1038/ja.2014.107
Harbarth S, Balkhy HH, Goossens H, Jarlier V, Kluytmans J, Laxminarayan R et al (2015) Antimicrobial resistance: one world, one fight! Antimicrob Resist Infect Control. https://doi.org/10.1186/s13756-015-0091-2
Harkins CP, Pichon B, Doumith M, Parkhill J, Westh H, Tomasz A et al (2017) Methicillin-resistant Staphylococcus aureus emerged long before the introduction of methicillin into clinical practice. Genome Biol. https://doi.org/10.1186/s13059-017-1252-9
Harrison E, Dytham C, Hall JPJ, Guymer D, Spiers AJ, Paterson S, Brockhurst MA (2016) Rapid compensatory evolution promotes the survival of conjugative plasmids. Mob Genet Elem 6(3):2034–2039
Hiramatsu K, Ito T, Tsubakishita S, Sasaki T, Takeuchi F, Morimoto Y et al (2013) Genomic basis for methicillin resistance in Staphylococcus aureus. Infect Chemother. https://doi.org/10.3947/ic.2013.45.2.117
Holmes AH, Moore LSP, Sundsfjord A, Steinbakk M, Regmi S, Karkey A et al (2016) Understanding the mechanisms and drivers of antimicrobial resistance. The Lancet. https://doi.org/10.1016/S0140-6736(15)00473-0
Howden BP, Davies JK, Johnson PDR, Stinear TP, Grayson ML (2010) Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. https://doi.org/10.1128/CMR.00042-09
Jacoby GA (2009) AmpC β-Lactamases. Clin Microbiol Rev. https://doi.org/10.1128/CMR.00036-08
Jacoby GA, Strahilevitz J, Hooper D (2014) Plasmid-mediated quinolone resistance. NIH Public Access Microbiol Spectr 2(2):1–15
Joon-Hee Lee (2019) Perspectives towards antibiotic resistance: from molecules to population. J Microbiol 57(3):181–184
Jouhten H, Mattila E, Arkkila P, Satokari R (2016) Reduction of antibiotic resistance genes in intestinal microbiota of patients with recurrent clostridium difficile infection after fecal microbiota transplantation. Clin Infect Dis 63(5):710–711
Kehrenberg C, Schwarz S, Jacobsen L, Hansen LH, Vester B (2005) A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Mol Microbiol 57(4):1064–1073. https://doi.org/10.1111/j.1365-2958.2005.04754.x
Keith P (2005) Efflux-mediated antimicrobial resistance. J Antimicrob Chemother 56:20–51
Knothe H, Shah P, Krcmery V, Antal M, Mitsuhashi S (1983) Transferable resistance to cefotaxime, cefoxitin, cefamandole and cefuroxime in clinical isolates of Klebsiella pneumoniae and Serratia marcescens. Infection 11(6):315–317. https://doi.org/10.1007/BF01641355
Lakin SM, Dean C, Noyes NR, Dettenwanger A, Ross AS, Doster E et al (2017) MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw1009
Leclercq R (2002) Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications. Clin Infect Dis 34(4):482–492. https://doi.org/10.1086/324626
Levin BR, Perrot V, Walker N (2000) Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 154(3):985–997. https://doi.org/10.1534/genetics.110.124628
Levine DP (2005) Vancomycin: a history. Clin Infect Dis. https://doi.org/10.1086/491709
Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ (2017) Antibiotic tolerance facilitates the evolution of resistance. Science. https://doi.org/10.1126/science.aaj2191
Levin-Reisman I, Brauner A, Ronin I, Balaban NQ (2019) Epistasis between antibiotic tolerance, persistence, and resistance mutations. Proc Natl Acad Sci USA 116(29):14734–14739. https://doi.org/10.1073/pnas.1906169116
Liebert CA, Hall RM, Summers AO (1999) Transposon Tn21, flagship of the floating genome. Microbiol Mol Biol Rev 63:507–522
Lili LN, Britton NF, Feil EJ (2007) The persistence of parasitic plasmids. Genetics 177(1):399–405. https://doi.org/10.1534/genetics.107.077420
Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J et al (2016) Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. https://doi.org/10.1016/S1473-3099(15)00424-7
Livermore DM, Canton R, Gniadkowski M, Nordmann P, Rossolini GM, Arlet G et al (2007) CTX-M: changing the face of ESBLs in Europe. J Antimicrob Chemother 59:165–174
Luo N, Pereira S, Sahin O, Lin J, Huang S, Michel L, Zhang Q (2005) Enhanced in vivo fitness of fluoroquinolone-resistant Campylobacter jejuni in the absence of antibiotic selection pressure. Proc Natl Acad Sci 102(3):541–546. https://doi.org/10.1073/pnas.0408966102
Mazel D (2006) Integrons: agents of bacterial evolution. Nat Rev Microbiol. https://doi.org/10.1038/nrmicro1462
McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ et al (2013) The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. https://doi.org/10.1128/aac.00419-13
McMurry L, Petrucci RE, Levy SB (2006) Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc Natl Acad Sci 77(7):3974–3977. https://doi.org/10.1073/pnas.77.7.3974
Miller WR, Munita JM, Arias CA (2014) Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther. https://doi.org/10.1586/14787210.2014.956092
Moellering RC (2012) MRSA: the first half century. J Antimicrob Chemother 67(1):4–11. https://doi.org/10.1093/jac/dkr437
Morales G, Picazo JJ, Baos E, Candel FJ, Arribi A, Peláez B et al (2010) Resistance to linezolid is mediated by the cfr gene in the first report of an outbreak of linezolid-resistant Staphylococcus aureus. Clin Infect Dis 50(6):821–825. https://doi.org/10.1086/650574
Motta SS, Cluzel P, Aldana M (2015) Adaptive resistance in bacteria requires epigenetic inheritance, genetic noise, and cost of efflux pumps. PLoS ONE 10(3):1–18. https://doi.org/10.1371/journal.pone.0118464
Munita JM, Arias CA (2016) Mechanisms of antibiotic resistance. HHPS Public Access Microbiol Spectr 4(2):1–37. https://doi.org/10.1128/microbiolspec.VMBF-0016-2015.Mechanisms
Nikaido H (1989) Outer membrane barrier as a mechanism of antimicrobial resistance. Antimicrob Agents Chemother 33(11):1831–1836
Nikaido H, Pagès JM (2012) Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol Rev. https://doi.org/10.1111/j.1574-6976.2011.00290.x
Norman A, Hansen LH, Sørensen SJ (2009) Conjugative plasmids: vessels of the communal gene pool. Philos Trans R Soc B. https://doi.org/10.1098/rstb.2009.0037
Nowak R (1994) Hungary sees an improvement in penicillin resistance. Science 264(5157):364. https://doi.org/10.1126/science.8153619
O’Neill J (2014) Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Rev Antimicrob Resis 20:1–16
Ogawara H, Kawamura N, Kudo T, Suzuki KI, Nakase T (1999) Distribution of β-lactamases in actinomycetes. Antimicrob Agents Chemother 43:3014–3017
Osei Sekyere J (2018) Genomic insights into nitrofurantoin resistance mechanisms and epidemiology in clinical Enterobacteriaceae. Future Sci OA 4(5):293. https://doi.org/10.4155/fsoa-2017-0156
Pal C, Bengtsson-Palme J, Kristiansson E, Larsson DGJ (2015) Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genom 16(1):964. https://doi.org/10.1186/s12864-015-2153-5
Pallecchi L, Lucchetti C, Bartoloni A, Bartalesi F, Mantella A, Gamboa H et al (2007) Population structure and resistance genes in antibiotic-resistant bacteria from a remote community with minimal antibiotic exposure. Antimicrob Agents Chemother 51(4):1179–1184. https://doi.org/10.1128/AAC.01101-06
Parmar A, Lakshminarayanan R, Iyer A, Mayandi V, Leng Goh ET, Lloyd DG et al (2018) Design and syntheses of highly potent teixobactin analogues against Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA), and vancomycin-resistant enterococci (VRE) in vitro and in vivo. J Med Chem. https://doi.org/10.1021/acs.jmedchem.7b01634
Partridge SR, Tsafnat G, Coiera E, Iredell JR (2009) Gene cassettes and cassette arrays in mobile resistance integrons: review article. FEMS Microbiol Rev. https://doi.org/10.1111/j.1574-6976.2009.00175.x
Paterson DL, Bonomo RA (2005) Extended-spectrum β-lactamases: a clinical update. Clin Microbiol Rev. https://doi.org/10.1128/CMR.18.4.657-686.2005
Penders J, Stobberingh EE (2008) Antibiotic resistance of motile aeromonads in indoor catfish and eel farms in the southern part of The Netherlands. Int J Antimicrob Agents. https://doi.org/10.1016/j.ijantimicag.2007.10.002
Piddock LJV (2006a) Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev 19(2):382–402
Piddock LJV (2006b) Multidrug-resistance efflux pumps—not just for resistance. Nat Rev Microbiol. https://doi.org/10.1038/nrmicro1464
Portsmouth S, van Veenhuyzen D, Echols R, Machida M, Ferreira JCA, Ariyasu M et al (2018) Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: a phase 2, randomised, double-blind, non-inferiority trial. Lancet Infect Dis. https://doi.org/10.1016/S1473-3099(18)30554-1
Queenan AM, Bush K (2007) Carbapenemases: the versatile β-lactamases. Clin Microbiol Rev. https://doi.org/10.1128/CMR.00001-07
Ramirez MS, Tolmasky ME (2010) Aminoglycoside modifying enzymes. Drug Resist Updates 13(6):151–171. https://doi.org/10.1016/j.drup.2010.08.003
Rappuoli R, Bloom DE, Black S (2017) Deploy vaccines to fight superbugs. Nature. https://doi.org/10.1038/d41586-017-08323-0
Roberts MC (2005) Update on acquired tetracycline resistance genes. FEMS Microbiol Lett. https://doi.org/10.1016/j.femsle.2005.02.034
Roemhild R, Gokhale CS, Dirksen P, Blake C, Rosenstiel P, Traulsen A et al (2018) Cellular hysteresis as a principle to maximize the efficacy of antibiotic therapy. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1810004115
Rossolini GM, D’Andrea MM, Mugnaioli C (2008) The spread of CTX-M-type extended-spectrum β-lactamases. Clin Microbiol Infect. https://doi.org/10.1111/j.1469-0691.2007.01867.x
Salimiyan Rizi K, Ghazvini K, Noghondar M Kouhi (2018) Adaptive antibiotic resistance: overview and perspectives. J Infect Dis Ther. https://doi.org/10.4172/2332-0877.1000363
Salyers AA, Amábile-Cuevas CF (1997) MINIREVIEW why are antibiotic resistance genes so resistant to elimination? Antimicrob Agents Chemother 41(11):2321–2325
Schlüter A, Szczepanowski R, Kurz N, Schneiker S, Krahn I, Pühler A (2007) Erythromycin resistance-conferring plasmid pRSB105, isolated from a sewage treatment plant, harbors a new macrolide resistance determinant, an integron-containing Tn402-like element, and a large region of unknown function. Appl Environ Microbiol. https://doi.org/10.1128/AEM.02159-06
Schrag SJ, Perrot V, Levin BR (1997) Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc R Soc B 264(1386):1287–1291. https://doi.org/10.1098/rspb.1997.0178
Schwarz S, Kehrenberg C, Doublet B, Cloeckaert A (2004) Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol Rev. https://doi.org/10.1016/j.femsre.2004.04.001
Sengupta S, Chattopadhyay MK, Grossart HP (2013) The multifaceted roles of antibiotics and antibiotic resistance in nature. Front Microbiol. https://doi.org/10.3389/fmicb.2013.00047
Sievert DM, Rudrik JT, Patel JB, Wilkins MJ, McDonald LC, Hageman JC (2008) Vancomycin-resistant Staphylococcus aureus in the United States, 2002-2006. Clin Infect Dis 46(5):668–674. https://doi.org/10.1086/527392
Sommer MOA, Church GM, Dantas G (2010) The human microbiome harbors a diverse reservoir of antibiotic resistance genes. Virulence 1(4):299–303. https://doi.org/10.4161/viru.1.4.12010
Stokes HW, Hall RM (1989) A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol Microbiol. https://doi.org/10.1111/j.1365-2958.1989.tb00153.x
Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I (2009) Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. https://doi.org/10.1371/journal.pgen.1000578
Van Giau V, An SSA, Hulme J (2019) Recent advances in the treatment of pathogenic infections using antibiotics and nano-drug delivery vehicles. Drug Des Dev Ther. https://doi.org/10.2147/DDDT.S190577
Vogwill T, Maclean RC (2015) The genetic basis of the fitness costs of antimicrobial resistance: a meta-analysis approach. Evol Appl 8(3):284–295. https://doi.org/10.1111/eva.12202
Ward MJ, Gibbons CL, McAdam PR, van Bunnik BAD, Girvan EK, Edwards GF et al (2014) Time-scaled evolutionary analysis of the transmission and antibiotic resistance dynamics of Staphylococcus aureus clonal complex 398. Appl Environ Microbiol. https://doi.org/10.1128/AEM.01777-14
Wein T, Hülter NF, Mizrahi I, Dagan T (2019) Emergence of plasmid stability under non-selective conditions maintains antibiotic resistance. Nature Commun. https://doi.org/10.1038/s41467-019-10600-7
Whiteway J, Koziarz P, Veall J, Sandhu N, Kumar P, Hoecher B, Lambert IB (1998) Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli. J Bacteriol 180(21):5529–5539
Wistrand-Yuen E, Knopp M, Hjort K, Koskiniemi S, Berg OG, Andersson DI (2018) Evolution of high-level resistance during low-level antibiotic exposure. Nat Commun 9(1):1–12. https://doi.org/10.1038/s41467-018-04059-1
World Health Organization (2017) Central Asian and Eastern European Surveillance of Antimicrobial Resistance (CAESAR). http://www.euro.who.int/en/health-topics/disease-prevention/antimicrobial-resistance/about-amr/central-asian-and-eastern-european-surveillance-of-antimicrobial-resistance-caesar
Wright GD (2007) The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. https://doi.org/10.1038/nrmicro1614
Zervos MJ, Schaberg DR (1985) Reversal of the in vitro susceptibility of enterococci to trimethoprim-sulfamethoxazole by folinic acid. Antimicrob Agents Chemother 28(3):446–448. https://doi.org/10.1128/AAC.28.3.446
Funding
None to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Konstantinos Voskarides.
Rights and permissions
About this article

Cite this article
Christaki, E., Marcou, M. & Tofarides, A. Antimicrobial Resistance in Bacteria: Mechanisms, Evolution, and Persistence. J Mol Evol 88, 26–40 (2020). https://doi.org/10.1007/s00239-019-09914-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-019-09914-3