
Abstract
Background
It has been proposed that the prion, the infectious agent of transmissible spongiform encephalopathies, is PrPSc, a post-translationally modified form of the normal host protein PrPC. We showed previously that mice devoid of PrPC (Prn-p0/0) are completely resistant to scrapie. We now report on the unexpected response of heterozygous (Prn-p0/+) mice to scrapie infection.
Materials and Methods
Prn-p0/+, Prn-p0/0 and Prn-p+/+ mice were obtained from crosses of Prn-p0/+ mice. Mice were inoculated intracerebrally with mouse-adapted scrapie agent and the clinical progression of the disease recorded. Mice were sacrificed at intervals, PrPSc was determined as protease-resistant PrP and the prion titer by the incubation time assay.
Results
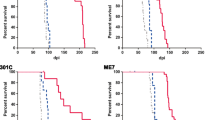
Prn-p0/+ mice, which have about half the normal level of PrPC in their brains, show enhanced resistance to scrapie, as manifested by a significant delay in onset and progression of clinical disease. However, while in wild type animals an increase in prion titer and PrPSc levels is followed within weeks by scrapie symptoms and death, heterozygous Prn-p0/+ mice remain free of symptoms for many months despite similar levels of scrapie infectivity and PrPSc.
Conclusions
Our findings extend previous reports showing an inverse relationship between PrP expression level and incubation time for scrapie. However, contrary to expectation, overall accumulation of PrPSc and prions to a high level do not necessarily lead to clinical disease. These findings raise the question whether high titers of prion infectivity could also persist for long periods under natural circumstances in the absence of clinical symptoms.





Similar content being viewed by others

References
Prusiner SB. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136–144.
Prusiner SB. (1989) Scrapie prions. Annu. Rev. Microbiol. 43: 345–374.
Prusiner SB, Scott M, Foster D, et al. (1990) Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63: 673–686.
Prusiner SB. (1993) Transgenetic investigations of prion diseases of humans and animals. Philos. Trans. R. Soc. Lond. Biol. 339: 239–254.
Weissmann C, Büeler H, Fischer M, Aguet M. (1993) Role of the PrP gene in transmissible spongiform encephalopathies. In: Zinkernagel RM, Stauffacher W (eds). Viruses and Virus-like Agents in Disease. Karger, Basel, pp. 164–175.
Oesch B, Westaway D, Walchli M, et al. (1985) A cellular gene encodes scrapie PrP 27–30 protein. Cell 40: 735–746.
Chesebro B, Race R, Wehrly K, et al. (1985) Identification of scrapie prion protein-specific messenger RNA in scrapie-infected and uninfected brain. Nature 315: 331–333.
Basler K, Oesch B, Scott M, et al. (1986) Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46: 417–428.
Stahl N, Borchelt DR, Hsiao K, Prusiner SB. (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51: 229–240.
Stahl N, Baldwin MA, Hecker R, Pan KM, Burlingame AL, Prusiner SB. (1993) Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 31: 5043–5053.
Manson J, West JD, Thomson V, Mcbride P, Kaufman MH, Hope J. (1992) The prion protein gene: A role in mouse embryogenesis? Dev. Camb. 115: 117–122.
Bendheim PE, Brown HR, Rudelli RD, et al. (1992) Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 42: 149–156.
Scott M, Foster D, Mirenda C, et al. (1989) Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59: 847–857.
McKinley MP, Taraboulos A, Kenaga L, et al. (1991) Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Invest. 65: 622–630.
Taraboulos A, Rogers M, Borchelt DR, et al. (1990) Acquisition of protease resistance by prion proteins in scrapie-infected cells does not require asparagine-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 87: 8262–8266.
Taraboulos A, Serban D, Prusiner SB. (1990) Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J. Cell Biol. 110: 2117–2132.
Turk E, Teplow DB, Hood LE, Prusiner SB. (1988) Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176: 21–30.
Stahl N, Baldwin MA, Teplow DB, et al. (1993) Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 32: 1991–2002.
Bolton DC, Bendheim PE. (1988) A modified host protein model of scrapie. In: Bock G, Marsh J (eds). Novel Infectious Agents and the Central Nervous System. John Wiley & Sons, Chichester, U.K., pp. 164–177.
Prusiner SB. (1991) Molecular biology of prion diseases. Science 252: 1515–1522.
Oesch B, Groth DF, Prusiner SB, Weissmann C. (1988) Search for a scrapie-specific nucleic acid: A progress report. In: Bock G, Marsh J (eds). Novel Infectious Agents and the Central Nervous System. John Wiley & Sons, Chichester, U.K. pp. 209–223.
Pan KM, Baldwin M, Nguyen J, et al. (1993) Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U.S.A. 90: 10962–10966.
Dickinson AG, Outram GW. (1988) Genetic aspects of unconventional virus infections: The basis of the virino hypothesis. Ciba Found. Symp. 135: 63–83.
Bruce ME, Dickinson AG. (1987) Biological evidence that scrapie agent has an independent genome. J. Gen. Virol 68: 79–89.
Weissmann C, Büeler H, Sailer A, Fischer M, Aguet M, Aguzzi A. (1993) Role of PrP in prion diseases. Brit. Med. Bull. 49: 995–1011.
Prusiner SB. (1992) Molecular biology and genetics of neurodegenerative diseases caused by prions. Adv. Virus Res. 41: 241–280.
Sailer A, Büeler H, Fischer M, Aguzzi A, Weissmann C. (1994) No propagation of prions in mice devoid of PrP. Cell 77: 967–968.
Büeler H, Aguzzi A, Sailer A, et al. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73: 1339–1347.
Prusiner SB, Groth D, Serban A, et al. (1993) Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. U.S.A. 90: 10608–10612.
Carlson GA, Ebeling C, Yang S-L, et al. (1994) Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 91: 5690–5694.
Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. (1982) Measurement of the scrapie agent using an incubation time interval assay. Ann. Neurol. 11: 353–358.
Büeler H, Fischer M, Lang Y, et al. (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356: 577–582.
Collinge J, Whittington MA, Sidle KCL, et al. (1994) Prion protein is necessary for normal synaptic function. Nature 370: 295–297.
Brown P, Kaur P, Sulima MP, Goldfarb LG, Gibbs CJ, Gajdusek D. (1993) Real and imagined clinicopathological limits of “prion dementia.” Lancet 341: 127–129.
Chandler RL. (1961) Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet 1: 1378–1379.
Brown P, Wolff A, Gajdusek DC. (1990) A simple and effective method for inactivating virus infectivity in formalin-fixed tissue samples from patients with Creutzfeldt-Jakob disease. Neurology 40: 887–890.
Lämmli UK. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685.
Serban D, Taraboulos A, DeArmond SJ, Prusiner SB. (1990) Rapid detection of Creutzfeldt-Jakob disease and scrapie prion proteins. Neurology 40: 110–117.
Acknowledgments
We thank S. Prusiner for the R073 antiserum and for the tg(SHaPrP)81 mice and P. Autenried for providing animal facilities. This work was supported by grants from the Kanton of Zürich, the Schweizerische Nationalfonds, and NIH NS22786.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Büeler, H., Raeber, A., Sailer, A. et al. High Prion and PrPSc Levels but Delayed Onset of Disease in Scrapie-Inoculated Mice Heterozygous for a Disrupted PrP Gene. Mol Med 1, 19–30 (1994). https://doi.org/10.1007/BF03403528
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF03403528