
Abstract
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder with a complex physiopathology whose initiators are poorly defined. Accumulating clinical and experimental evidence suggests a causal role of lifetime stress in AD. This chapter summarizes current knowledge about how chronic stress and its accompanying high levels of glucocorticoid (GC) secretion, trigger the two main pathomechanisms of AD: (i) misprocessing of amyloid precursor protein (APP) and the generation of amyloid beta (Aβ) and (ii) Tau hyperphosphorylation and aggregation. Given that depression is a well-known stress-related illness, and the evidence that depression may precede AD, this chapter also explores neurobiological mechanisms that may be common to depressive and AD pathologies. This review also discusses emerging insights into the role of Tau and its malfunction in disrupting neuronal cascades and neuroplasticity and, thus triggering brain pathology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Introduction
The brain is the most adaptive of all organs. It has a remarkable capacity to respond to a variety of internal and environmental stimuli and to mount pro-survival behavioral responses by orchestrating multiple molecular and biochemical cascades. The latter changes are embraced by the term neural plasticity, the cornerstone of learning and memory [1]. Impairments in neuroplastic mechanisms are commonly found during aging, the primary risk factor for Alzheimer’s disease (AD), a disorder characterized by memory deficits. Over their lifetime, individuals experience both good and adverse (stressful) events and notably, stressful events appear to accelerate brain aging [2]. Accumulating clinical and experimental evidence suggests a causal role of lifetime stress in AD. This chapter summarizes current knowledge about how chronic stress and its accompanying high levels of glucocorticoid (GC) secretion, trigger the two main pathomechanisms of AD: (i) misprocessing of amyloid precursor protein (APP) and the generation of amyloid beta (Aβ) and (ii) Tau hyperphosphorylation and aggregation. Given that depression is a well-known stress-related illness, and the evidence that depression may precede AD, this chapter also explores neurobiological mechanisms that may be common to depressive and AD pathologies. This review also discusses emerging insights into the role of Tau and its malfunction in disrupting neuronal cascades and neuroplasticity and, thus triggering brain pathology.
Stress: A Physiological Tug-of-War – From Adaptive to Maladaptive Responses
Stress is defined as a challenge to homeostasis (physiological and behavioral equilibrium) by physical or psychological events [3]. When challenged by endogenous or exogenous aversive or threatening stimuli (stressors), a series of defensive systems and processes become activated; these include the release of monoamines and GC that initially promote a return to the homeostatic state [4, 5]. The “stress response” normally terminates once homeostasis has been restored, but when the organism is faced with an insurmountable stress (high intensity, contextually inappropriate and/or chronic), it may take inappropriate – maladaptive – actions that result in chronically elevated GC secretion. Besides interfering with normal structural and plastic arrangements within the brain, such inadequate responses can have negative consequences for the immune and visceral systems that may ultimately lead to multiple disorders, including neuropsychiatric and neurological diseases [6,7,8,9].
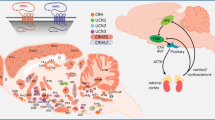
The endocrine response to stress is orchestrated within the so-called hypothalamo–pituitary–adrenal (HPA) axis (Fig. 20.1). Stress, perceived by cortical areas of the brain, triggers the release of corticotropin-releasing hormone (CRH) from the hypothalamic paraventricular nucleus (PVN) which, in turn, induces the secretion of adrenocorticotropic hormone (ACTH) release from the pituitary and GC (cortisol in humans and corticosterone in rodents) from the adrenal glands. This sequence of events is normally curtailed by negative feedback of GC at central sites; however, the nature of the stressor and/or impairments in negative feedback mechanisms (e.g. during aging) may block this crucial feedback loop, resulting in supraphysiological exposure to GC. A key area among the brain regions involved in the regulation of the HPA axis is the hippocampus; this area, which also plays a pivotal role in learning and memory, sends inhibitory projections to the PVN (and other hypothalamic nuclei). Similarly, the frontal cortex mediates GC negative feedback effects on the HPA axis, whereas the GC activation of the amygdala results in a positive drive on this axis (see Fig. 20.1).

The hypothalamo-pituitary-adrenal (HPA) axis and the response to stress in the healthy state. Stressors perceived in higher brain centers trigger the release of corticotropin-releasing hormone (CRH) from neurons in the paraventricular nucleus (PVN). Carried via a portal vein system, CRH reaches the anterior pituitary where it stimulates the secretion of adrenocorticotropin hormone (ACTH) which, in turn, stimulates the production and release of glucocorticoids (GC) [cortisol in humans and corticosterone in rodents] from the adrenal cortex. The secreted GC access peripheral and central tissues via the general circulation where they serve to mount adaptive responses to the initiating stimulus (stress) after binding to glucocorticoid receptors (GR). Eventually, GC secretion and action is restrained by inhibitory feedback of GC on central (chiefly, the frontal cortex, hippocampus, hypothalamus and pituitary) components of the HPA axis
Mechanisms and Consequences of GC Action in the Brain
Corticosteroid actions in the brain are mediated by glucocorticoid (GR) and mineralocorticoid (MR) receptors. The previously-referred feedback effects of GC during stressful experiences primarily depend on activation of GR, expressed throughout the brain, but at highest density in the hippocampus [10]. Indeed, almost all we know about GC actions in the brain are GR-mediated. Less is known about the role of MR, although, they have a ten-fold higher affinity for GC compared to GR and are believed to play an important role in GC feedback under physiological (non-stressful) conditions [11]. In addition, MR have been implicated in “protecting” the brain against GR-mediated cytotoxicity [12, 13] and behavioral maladaptation [14]. Interestingly, the central expression of GR and MR is subject to regulation by stress and age [15, 16].
While GC have been shown to modulate synaptic activity through non-genomic mechanisms [17,18,19], GR and MR are better known as potent ligand-dependent transcriptional regulators, i.e. control gene expression/repression [20,21,22,23]. The unliganded receptors are located in the cytoplasm in association with chaperone proteins (e.g. the heat shock proteins Hsp90 and 70 and the immunophilins FKBP51 and 52) [24]. Ligand binding results in conformational change of the GR-chaperone complex and subsequently, receptor translocation to the nucleus and binding to specific regions of DNA containing glucocorticoid response elements (GRE) within the promoters of target genes [25]. Gene transcription or repression is then determined by the recruitment of co-activators and repressors [26] as well as by post-translational modifications of the receptor [27,28,29].
In the brain, MR and GR differentially regulate the expression of genes, in a site-specific manner; these include genes responsible for the regulation of the HPA axis (CRH and CRH receptors and pro-opiomelanocortin [POMC], from whose gene product ACTH is cleaved) as well as pro- and anti-apoptotic genes [13] and, importantly, genes with roles in neural energy metabolism, structure and synaptic transmission, the synthesis of rate-limiting neurotransmitter enzymes and receptors as well of various neuropeptide, growth factors and cell adhesion molecule [30,31,32,33,34]. While all of these GC-initiated transcriptional events contribute to neural plasticity, stress and GC result in the manifestation of visualizable effects, namely, alterations in neuronal morphology. As reviewed by Lucassen [35], stress and GC lead to changes in the rate of neurogenesis, cell death, and neuronal connectivity as well as astroglia-neuronal interactions. In particular, numerous studies have highlighted how stress, acting through GC, impacts on dendritic arborization and synaptic number; this aspect of GC actions is considered in the following section. First, in the context of AD as a disease that develops as age progresses, it is important to briefly mention the growing view that stress and GC leave long-lasting “memories” of past experiences via epigenetic mechanisms; these are thought to contribute importantly to the organism’s physical and mental health trajectory [36,37,38,39]. Notably, epigenetic mechanisms have recently been implicated in the lasting effects of lifetime adversity in humans [40, 41].
Stress, Glucocorticoids and Neural Plasticity
Functional plasticity in the brain is generally preceded by structural plasticity, typically, dendritic and synaptic remodeling. Basal levels of GC are crucial for maintaining synaptic plasticity in the hippocampus in the form of long-term potentiation (LTP) [42], a well-documented mechanism involved in memory formation [43]. On the other hand, high levels of GC such as those experienced during stress impair LTP induction and facilitate long-term depression (LTD) [44]. An important role for N-methyl-d-aspartate (NMDA) receptors and shifts in calcium flux has been suggested in both, LTP and LTD modulation by stress/GC [45,46,47,48].
One of the best-described forms of stress-evoked structural plasticity is dendritic retraction, with a pioneering study revealing that chronic stress interrupts connectivity between hippocampal CA1 neurons and neurons in the medial prefrontal cortex (PFC) [49, 50]. The latter work followed previous demonstrations that chronic stress causes atrophy of apical (but not basal) dendritic complexity in CA3 pyramidal neurons [51]. Meanwhile, other studies have reported that stress can also increase dendritic length in certain brain regions such as the orbitofrontal cortex, amygdala and bed nucleus of the stria terminalis (BNST, also known as the “extended amygdala”) [52, 53]. Interestingly, chronic stress has also been associated with a loss of mossy fiber synapses, increased surface area of the post-synaptic density, and rearrangements of synaptic mitochondria and vesicles at the presynaptic terminals [54]. Further, dendritic spines, which have an important role in information storage, are severely reduced by stress [55] but can mostly be rapidly reversed after a recovery period or subsequent training ([56, 57]; but see [58] for exceptions).
New work from our labs indicates that Tau, a key factor in AD pathology, is essential for chronic stress to disrupt neuroplasticity. Briefly, we showed that mice in which Tau has been deleted are spared from the deleterious behavioral (e.g. deficits in learning and memory, depressive-like behavior and anxiety) and neurostructural (namely, dendritic atrophy disconnection of the hippocampal-prefrontocortical pathway) of chronic stress and GC [59, 60].
As already alluded to, stress and GC also influence neuroplasticity by modulating the production of new neurons in adult brain [61]. Several studies indicate that stress/GC related effects on neurogenesis have the potential to affect mental health, including susceptibility to depression [62, 63] and AD [64, 65].
Chronic Stress: Etiopathogenic Role and Mechanisms in AD
A sizeable literature suggests that elevated GC and chronic stress – a state that an increasing proportion of the population finds itself in today – may increase the risk for developing AD pathology and related dementias [66, 67] and may even advance the age of onset of the familial form of AD [68,69,70]. Indirect support for the link between high GC exposure and AD includes reports that AD patients produce and secrete higher-than-normal levels of cortisol [67, 71,72,73,74]. Interestingly, transgenic mouse models of AD also display high levels of GC [75, 76]. Nevertheless, while the direction of the cause-effect relationship between AD-like pathology and hypercorticalism remains unclear, it is worth recalling (see previous section) that the hippocampus is responsible for mediating the negative feedback effects of GC on the HPA axis; thus, any damage to this brain region is likely to uncouple this control mechanism and unleash unrestrained GC secretion.
To put these findings into context, it is worth noting that there is evidence that GC levels correlate with the rate of cognitive decline [35, 77] and the extent of neuronal remodeling in AD subjects [78]. Such remodeling is especially marked in the hippocampus, the area in which most studies on stress/GC effects on neuroplasticity have been conducted in rodents, and the brain area which clearly displays the first signs of AD neuropathology – deposits of amyloid β (Aβ) and accumulation and aggregation of hyperphosphorylated Tau [79,80,81]. The hippocampal lesions induced by these deposits correlate with the extent of deficits in declarative, spatial and contextual memory [82].
Consideration Regarding How Chronic Stress and High GC Levels May Contribute to AD Pathology
In this section, we will review some of the evidence for a link between GC/stress and AD and consider some of the possible underlying mechanisms. After briefly considering stress/GC effects on amyloidogenesis, our attention will focus on how chronic exposure to stress or high levels of GC influence Tau biology, culminating in its malfunction and dendro-synaptic toxicity.
As noted earlier, AD neuropathology is characterized by overproduction of Aβ that forms deposits into senile (amyloid) plaques, and by accumulation of hyperphosphorylated forms of Tau protein that becomes insoluble, aggregates and forms neurofibrillary tangles (NFT) [79,80,81]. Aβ is the proteolytic product of amyloid precursor protein (APP), a large transmembrane protein called, that is sequentially cleaved by β-secretase (BACE-1) and γ-secretase (a complex of enzymes, including presenilin) to yield Aβ; this post-translational pathway is often called APP misprocessing. Studies have shown that extended exposure to immobilization stress increases the load of extracellular Aβ deposits and exacerbates memory deficits in mice expressing an aggressive (human) mutant form of APP V717ICT-100 [76, 83]. Similar observations were made when young 3xTg-AD mice (expressing APP Swedish, P301L-Tau, and PSEN1 M146V mutations) were treated with the synthetic GC, dexamethasone [76]. That the effects of stress are most likely transduced by GC was demonstrated by experiments on dexamethasone-treated neural cell lines (N2A [76] and differentiated PC12 cells [84]). Consistent with these reports, our own studies in wildtype rats demonstrated that chronic stress and/or treated with GC increases APP misprocessing along the amyloidogenic pathway by upregulating BACE-1 and Nicastrin (a component of the γ-secretase complex) to produce neurotoxic and cognition-impairing effects [85]; in this regard, it is worth noting that high exogenous levels of GC upregulate the transcription of APP and βACE-1, the promoters of which contain a glucocorticoid response element (GRE) [76]. Lastly, experiments that attempted to mimic intermittent stressful events (the effects of which may be cumulative over the lifetime) showed that GC potentiate the APP misprocessing pathway [85].
In recent years, an increasing amount of attention has turned to Tau pathology, especially its hyperphosphorylated forms, in a range of neurodegenerative diseases. Among the first reports to indicate a relationship between stress/GC and Tau was a study by Stein-Behrens et al. [86] who found that GC exacerbate kainic acid-induced hippocampal neuronal loss with a contemporaneous increase in Tau immunoreactivity. A later study showed that chronic treatment of 3xTg AD mice with dexamethasone leads to the somatodendritic accumulation of Tau in the hippocampus, amygdala and cortex [76]. Our own in vivo studies demonstrated that chronic stress or GC increase the levels of aberrantly hyperphosphorylated Tau in the rat hippocampus and PFC, both in the presence and absence of exogenous Aβ [87]. Importantly, the hyperphosphorylation occurred at certain Tau epitopes that are strongly implicated in cytoskeletal dysfunction and synaptic loss (e.g., pSer262) [88, 89] and hippocampal atrophy (e.g., pThr231) [90] in AD patients. Here, it is pertinent to note that the extent of phosphorylation at Thr231- and Ser262-Tau correlates strongly with severity of memory impairment, speed of mental processing, and executive functioning in AD patients [91,92,93]. Although chronic stress and GC treatment exert similar, but not identical, effects on individual Tau phosphoepitopes in vivo and in vitro [84], the overall evidence points to GC as the key mediator of the AD-like pathology induced by stress. On the other hand, some studies have suggested a role for at least one other stress-related molecule, namely, corticotrophin-releasing hormone (CRH) as deletion of the CRH receptor 1 gene in mice prevents the detrimental effects of stress on Tau phosphorylation [94, 95]. Supporting these links, are the results from in vitro experiments which indicate that the GC effects on Tau involve activation of glycogen synthase kinase 3 (GSK3) and cyclin-dependent kinase 5 (CDK5), two principal Tau kinases [84].
Transgenic mice expressing human P301L-Tau (the most common Tau mutation), also helped strengthen the evidence that chronic stress can exacerbate Tau pathology. Briefly, we found that stress stimulates the aberrant hyperphosphorylation and aggregation of insoluble Tau [96]. Further, we demonstrated in the latter work that chronic stress enhances caspase 3-mediated truncation of Tau at its C-terminal in the hippocampus, with the protein misfolding and adopting a conformation [96] that facilitates its nucleation and recruitment of other Tau molecules into neurotoxic, pre-tangle aggregates of Tau (see [97,98,99,100]. Importantly, experiments also showed that GC contribute to AD pathology by reducing the degradation of Tau, thereby increasing its accumulation [84]. The latter is likely to result from dysregulation of molecular chaperones (e.g. Hsp90 and Hsp70) that are responsible for Tau proteostasis [96]. As noted previously, these same heat shock proteins serve to maintain GR in a high affinity state; thus, they may represent a point at which GC signaling intersects with the cellular machinery that regulates Tau degradation.
While Kobayashi et al. [101] showed Tau may be synthesized de novo in the somato-dendritic compartment, earlier work by Ittner and colleagues [102] demonstrated that hyperphosphorylated Tau is missorted to synapses which subsequently become dysfunctional. The missorting of Tau to synapses is now acknowledged as an early event in AD, preceding the manifestation of detectable neurodegenerative processes [102,103,104]. It is important to note that this series of events depend on Tau hyperphosphorylation [103, 105, 106] and results in the targeting of Fyn (a member of the Src kinase family) to postsynaptic sites [102] where it selectively modulates the function of GluN2B-containing NMDAR (GluN2BR), by phosphorylation of the GluN2B at the Y1472 epitope [102, 107]. The latter stabilizes GluN2B at postsynaptic sites, thus increasing the risk for excitotoxicity [102, 107].
Since NMDAR are known to mediate stress- and GC-driven neurotoxicity [108] and neuronal remodeling [109], we were prompted to examine whether the mechanistic scenario just described also applies to the actions of stress and GC. Indeed, we found that chronic stress and GC also trigger Tau accumulation at synapses with subsequent increases of Fyn at postsynaptic sites [59, 110] (see also Fig. 20.2).

Multiple mechanisms contribute to the induction of Tau pathology and AD by chronic stress. The scheme summarizes the potential mechanisms through which chronic stress and GC activate processes that result Tau accumulation, aggregation and neurotoxicity. Stress leads to increased activation of glucocorticoid receptors (GR) by GC; GR transcriptional activity depends on an interplay of a variety of molecular chaperones (e.g. Hsp90, Hsp70, FKBP51) and HDAC6, a protein that may lead to cytoskeletal instability by reducing the acetylation of tubulin and cortactin. As described in the text, GC induce hyperphosphorylation of Tau and its consequent detachment from microtubules, leading to microtubule destabilization and cytoskeletal disturbances that, together with HDAC6, may contribute to: (i) the formation of stress granules (SG) that promote Tau aggregation and (ii) the inhibition of autophagic process that also contribute to Tau accumulation and aggregation. Interestingly, stress/GC inhibit mTOR, a crucial signaling molecule in the initial phases of autophagy
Other mechanisms that may underlie the ability of stress/GC to contribute to AD pathology have been coming to light in the last few years. One of these is autophagy. As the guardian of cellular homeostasis, autophagy is now seen to play a pivotal role in the pathology of a number of neurodegenerative disorders [111, 112]. Briefly, autophagic mechanisms are responsible for the degradation of misfolded proteins and aggregates such as Tau aggregates; interruption of autophagy leads to the accumulation of protein aggregates, a pathological features shared by a range of neurodegenerative disorders [113, 114]. Our investigations demonstrated (summarized in Fig. 20.3), for the first time, that both, chronic stress and high GC levels inhibit autophagic process, thus explaining how these conditions contribute to the accumulation and aggregation of Tau [115]. In fact, defective autophagy is suggested to be major player in AD pathology [116,117,118]; although Tau itself is a proteosomal substrate [119, 120], it is thought that Tau inclusions and aggregates may be inaccessible to the ubiquitin-proteasome system [121, 122]. Our results showing that chronic stress and GC increase mTOR signaling and reduces the ratio of the autophagic markers LC3II:LC3I and accumulation of p62 [115], indicate that chronic stress inhibits the autophagic process by activating the mTOR pathway; these findings are in line with previous reports that chronic stress stimulates mTOR activity in the hippocampus [123], an event associated with increased total Tau levels in the brains of AD subjects [124, 125]. In addition, support for our interpretation comes from the finding that inhibition of mTOR signaling ameliorates Tau pathology [126, 127] while we demonstrated that inhibition of mTOR blocked the GC-triggered Tau accumulation and aggregation [115].

Cumulative effects of stress and glucocorticoids on normal and pathological aging. In this hypothetical representation of brain aging, cognitive and mood status may decline over time. Chronic exposure to stressful conditions, associated with higher exposure to GC, lead to cumulative effects that accelerate brain aging by imposing an increasing allostatic load on brain function by causing neuronal atrophy and synaptic loss, modified by other factors such as genetics and sex – the latter also influence the magnitude of the stress load by modulating the activity of the hypothalamo-pituitary-adrenal (HPA) axis. The model assumes that elements of the HPA axis serve as part of a threshold-regulator mechanism (represented by thick line). Note that brain areas important for regulation of HPA axis (e.g. hippocampus, prefrontal cortex) also appear to be subject to impairments triggered by presymptomatic AD pathology (e.g. mild cognitive impairment, depression), thus feeding into a vicious cycle that further drives GC secretion and neuronal damage. The shaded grey area represents the threshold-transition area where a subject may progress from depression (with or without MCI symptoms) to Alzheimer’s disease (AD)
New research has implicated the endolysosomal pathway in neurodegenerative diseases such as AD and Parkinson’s disease in which Tau accumulation is a pathological feature [128,129,130]. As shown in Fig. 20.2, Tau has been identified as a substrate of the endolysosomal degradation pathway [131]. We demonstrated that in vitro or in vivo exposure to elevated GC levels block this pathway, accompanied by increases in the build-up of Tau, including that of specific phospho-Tau species. Further, we showed that the involvement of the small GTPase Rab35 and the endosomal sorting complexes required for transport (ESCRT) machinery that delivers Tau to lysosomes via early endosomes and multivesicular bodies (MVBs). The ESCRT system mediates the degradation of membrane-associated proteins such as epidermal growth factor receptor [132], but is also implicated in the degradation of cytosolic proteins GAPDH and aldolase [133]; these findings are of particular relevance for Tau, which has both cytosolic and membrane-associated pools [134, 135], and has been shown to localize to different neuronal sub-compartments, depending on its phosphorylation state [103, 110]. Interestingly, not all phosphorylated Tau species are equally susceptible to degradation in the Rab35/ESCRT pathway. In particular, we found that pSer396/404 and pSer262, but not pSer202, phospho-Tau species undergo Rab35-mediated degradation, indicative of preferential sorting of specific phospho-Tau proteins into the Rab35/ESCRT pathway [131]. Importantly, we demonstrated that high GC levels suppress Rab35 transcription, and thus, result in an accumulation of Tau due impaired degradation of the protein (Fig. 20.2). Further, overexpression of Rab35 reverses GC-induced Tau accumulation and rescues hippocampal neurons from the dystrophic actions of chronic stress [131].
RNA-Binding Proteins and Stress Granules Facilitate Stress-Induced Tau Pathology
Stress granules (SG) have been recently implicated in the Tau pathology that accompanies AD and fronto-temporal dementia with parkinsonism-17 (FTDP-17) in humans as well as in various transgenic mouse models of Tau-related disorders [136]. The eukaryotic stress response involves translational suppression of non-housekeeping proteins and the sequestration of unnecessary mRNA transcripts by RNA-binding proteins (RBP) into SG. These macromolecular complexes constitute a protective mechanism against cellular stress (e.g. oxidative stress) that help protect mRNA species and enable the fast production of cytoprotective proteins [136,137,138]. However, prolonged SG induction can become pathological and neurotoxic; in neurodegenerative diseases such as AD, SG promote the accumulation of Tau aggregates [139,140,141,142]. In fact, SG are suggested to accelerate Tau aggregation in a vicious cycle wherein Tau stimulates SG formation, with the RNA binding protein TIA1 playing a lead role in Tau misfolding and aggregation [143]. Notably, while hyperphosphorylation and aggregation-prone mutations of Tau can enhance SG formation, they are not essential for this event [143].
We recently showed that chronic stress and high GC upregulate various RBP and SG markers in soluble and insoluble fractions in the hippocampus of P301L-Tau Tg animals and primary neuron cultures. Specifically, tissues from animals exposed to chronic stress displayed increased cytoplasmic (soluble and insoluble) levels of several RBPs and SG-associated markers (e.g. TIA-1, PABP, G3BP, FUS, DDX5) that contributed to the formation of insoluble Tau inclusions and Tau accumulation. As noted above, TIA-1 plays a prominent role in Tau aggregation: under stressful conditions, TIA-1 is trafficked the nucleus to the cytospasm where it interacts directly with Tau (and other RBP such as PABP and EWSR1) to stimulate its aggregation and accumulation [143,144,145].
In other recent work, we showed that Tau missorting and accumulation in the dendritic compartment, such as is found in AD pathology [102], is also triggered by chronic stress/GC exposure [59, 110]. This is interesting because Tau missorting is hypothesized to facilitate formation of SG as part of the translational stress response [143]. While the temporal profile and precise mechanisms underlying stress/GC-evoked dysregulation of RBPs and the associated SG cascade remain to be elucidated, Fig. 20.2 illustrates our current working model, designed to explore more about the biology of RNA-protein interactions in stress-related pathologies.
Tau and Its Malfunction in Stress-Related Brain Pathology: Beyond Alzheimer’s Disease
Stress pervades all our lives and most of us will respond to daily life stressors in an adaptive manner. However, as noted by Selye as early as 1936, mounting a transient and adaptive response may not be possible in all circumstances and the stressful experience may become chronic and maladaptive. The negative impact of chronic stress (and the associated rise in circulating GC levels) on brain structure and function (e.g. cognition, mood, emotion) is now well recognized. In addition to the role of chronic stress/GC in the development of AD pathology, chronic stress is causally related to major depression which, as in AD, may reflect defects in neuroplastic mechanisms [1, 5, 146]. As briefly mentioned above, major depressive disorder appears to predispose to AD [147]. The body of evidence supporting the latter clinical observation includes findings of potentially common neurobiological mechanisms in the two disorders [84, 85, 148, 149]. Given this, it is interesting that epidemiological studies implicate depression as a risk factor for the development of AD [5], with support for this coming from the observation that previously depressed subjects have increased amyloid plaque and neurofibrillary tangles (NFT) loads [150]. Indeed, since clinicians are sometimes faced with the challenge of distinguishing between patients suffering from depression and AD, several authors have attempted to develop assays based on the detection of various APP cleavage products that might help such distinction [151,152,153,154], albeit with little success.
We previously referred to neurogenesis as a phenomenon that contributes neuroplasticity, with impaired neurogenesis being implicated in the pathogenesis of depression [155] as well as AD [156]. Neurogenesis declines with age (also in humans [156]) and is disrupted by stress and high GC levels [62]. In light of the previously-referred interactions between stress/GC and Tau, it is therefore interesting that our recent research suggests that Tau plays an essential role in stress-driven suppression of birth of neurons (but not astrocytes) in the adult dentate gyrus (DG, a hippocampal subfield) [62]; specifically, chronic stress is unable to impair the propliferation of neuroblasts and newborn neurons in the DG of mice in which the tau gene is deleted. Interestingly, tau ablation does not interfere with stress-induced suppression of astrocyte proliferation. This finding is likely related to the differential expression of Tau in neuronal vs. astrocytic precursor cells – Tau is expressed in neurons at much higher levels than in astrocytes [157]. These observations suggest a novel mechanism through which stress can remodel the adult brain. Interestingly, our investigations also showed that chronic stress increases the 4R-Tau:3R-Tau isoform ratio in the DG. Given that 3R-Tau has a lower affinity for MT than 4R-tau, neuroblasts may be endowed with greater cytoskeletal plasticity than mature neurons since 3R-Tau is more abundant in the former. Here, it is also relevant to note that it was recently shown that higher levels of 4R-Tau are associated with increased Tau phosphorylation and brain pathology [158]. Moreover, an increased 4R/3R-Tau ratio is associated with cytoskeletal disturbances and tauopathies such as AD [64].
Summary/Conclusions
The evidence reviewed in this chapter suggest that deficits in Tau function and Tau proteostasis may be critical and cooperative mechanisms through which stress (whose actions are executed by GC) remodels neural circuits (cell birth and death, dendritic and synaptic atrophy/connectivity), thus inducing impairments mood and cognition. Importantly, we suggest that Tau lies at the core of a set of common neurobiological mechanisms that link stress with AD and other brain pathologies such as depression.
References
Sousa N, Almeida OFX. Disconnection and reconnection: the morphological basis of (mal)adaptation to stress. Trends Neurosci. 2012;35:742–51. https://doi.org/10.1016/j.tins.2012.08.006.
Carroll BJ. Ageing, stress and the brain. Novartis Found Symp. 2002;242:26–36.. discussion 36–45
McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54:200–7.
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. https://doi.org/10.1152/physrev.00041.2006.
de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–75. https://doi.org/10.1038/nrn1683.
Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry. 2000;57:925–35.
Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21:259–72. https://doi.org/10.1016/j.bbi.2006.11.006.
Sotiropoulos I, Cerqueira JJ, Catania C, Takashima A, Sousa N, Almeida OFX. Stress and glucocorticoid footprints in the brain—the path from depression to Alzheimer’s disease. Neurosci Biobehav Rev. 2008;32:1161–73. https://doi.org/10.1016/j.neubiorev.2008.05.007.
Vyas S, Rodrigues AJ, Silva JM, Tronche F, Almeida OFX, Sousa N, Sotiropoulos I. Chronic stress and glucocorticoids: from neuronal plasticity to neurodegeneration. Neural Plast. 2016;2016:1–15. https://doi.org/10.1155/2016/6391686.
Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–11. https://doi.org/10.1210/endo-117-6-2505.
de Kloet ER, Reul JM, Sutanto W. Corticosteroids and the brain. J Steroid Biochem Mol Biol. 1990;37:387–94.
Hassan AH, von Rosenstiel P, Patchev VK, Holsboer F, Almeida OF. Exacerbation of apoptosis in the dentate gyrus of the aged rat by dexamethasone and the protective role of corticosterone. Exp Neurol. 1996;140:43–52. https://doi.org/10.1006/exnr.1996.0113.
Almeida OF, Condé GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH, Meyer M, Holsboer F, Michaelidis TM. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. FASEB J. 2000;14(5):779–90.
ter Horst JP, van der Mark MH, Arp M, Berger S, de Kloet ER, Oitzl MS. Stress or no stress: mineralocorticoid receptors in the forebrain regulate behavioral adaptation. Neurobiol Learn Mem. 2012;98(1):33–40. https://doi.org/10.1016/j.nlm.2012.04.006.
van Eekelen JA, Rots NY, Sutanto W, de Kloet ER. The effect of aging on stress responsiveness and central corticosteroid receptors in the brown Norway rat. Neurobiol Aging. 1992;13:159–70.
Hassan AH, Patchev VK, von Rosenstiel P, Holsboer F, Almeida OF. Plasticity of hippocampal corticosteroid receptors during aging in the rat. FASEB J. 1999;13:115–22.
Riedemann T, Patchev AV, Cho K, Almeida OFX. Corticosteroids: way upstream. Mol Brain. 2010;3:2. https://doi.org/10.1186/1756-6606-3-2.
Groeneweg FL, Karst H, de Kloet ER, Joëls M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol. 2012;350:299–309. https://doi.org/10.1016/j.mce.2011.06.020.
Yang S, Roselli F, Patchev AV, Yu S, Almeida OFX. Non-receptor-tyrosine kinases integrate fast glucocorticoid signaling in hippocampal neurons. J Biol Chem. 2013;288:23725–39. https://doi.org/10.1074/jbc.M113.470146.
Trapp T, Rupprecht R, Castrén M, Reul JMHM, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptor: a new principle of glucocorticoid action in the CNS. Neuron. 1994;13:1457–62. https://doi.org/10.1016/0896-6273(94)90431-6.
Nishi M, Ogawa H, Ito T, Matsuda K-I, Kawata M. Dynamic changes in subcellular localization of mineralocorticoid receptor in living cells: in comparison with glucocorticoid receptor using dual-color labeling with green fluorescent protein spectral variants. Mol Endocrinol. 2001;15:1077–92. https://doi.org/10.1210/mend.15.7.0659.
Gesing A, Bilang-Bleuel A, Droste SK, Linthorst AC, Holsboer F, Reul JM. Psychological stress increases hippocampal mineralocorticoid receptor levels: involvement of corticotropin-releasing hormone. J Neurosci. 2001;21:4822–9.
Chen Y, Rex CS, Rice CJ, Dube CM, Gall CM, Lynch G, Baram TZ. Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc Natl Acad Sci. 2010;107:13123–8. https://doi.org/10.1073/pnas.1003825107.
Rein T. FK506 binding protein 51 integrates pathways of adaptation: FKBP51 shapes the reactivity to environmental change. Bioessays. 2016;38:894–902. https://doi.org/10.1002/bies.201600050.
Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275:2–12. https://doi.org/10.1016/j.mce.2007.05.018.
Obradović D, Tirard M, Némethy Z, Hirsch O, Gronemeyer H, Almeida OFX. DAXX, FLASH, and FAF-1 modulate mineralocorticoid and glucocorticoid receptor-mediated transcription in hippocampal cells--toward a basis for the opposite actions elicited by two nuclear receptors? Mol Pharmacol. 2004;65:761–9. https://doi.org/10.1124/mol.65.3.761.
Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin L-Y, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–45. https://doi.org/10.1128/MCB.19.6.4535.
Tirard M, Jasbinsek J, Almeida OFX, Michaelidis TM. The manifold actions of the protein inhibitor of activated STAT proteins on the transcriptional activity of mineralocorticoid and glucocorticoid receptors in neural cells. J Mol Endocrinol. 2004;32:825–41.
Tirard M, Almeida OFX, Hutzler P, Melchior F, Michaelidis TM. Sumoylation and proteasomal activity determine the transactivation properties of the mineralocorticoid receptor. Mol Cell Endocrinol. 2007;268:20–9. https://doi.org/10.1016/j.mce.2007.01.010.
Datson NA, Van Der Perk J, De Kloet ER, Vreugdenhil E. Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression: corticosteroid-responsive genes in hippocampus. Eur J Neurosci. 2001;14:675–89. https://doi.org/10.1046/j.0953-816x.2001.01685.x.
Schaaf MJM, de Jong J, de Kloet ER, Vreugdenhil E. Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813:112–20. https://doi.org/10.1016/S0006-8993(98)01010-5.
Hansson AC, Cintra A, Belluardo N, Sommer W, Bhatnagar M, Bader M, Ganten D, Fuxe K. Gluco- and mineralocorticoid receptor-mediated regulation of neurotrophic factor gene expression in the dorsal hippocampus and the neocortex of the rat: GR- and MR-mediated gene expression. Eur J Neurosci. 2000;12:2918–34. https://doi.org/10.1046/j.1460-9568.2000.00185.x.
Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5:917–30. https://doi.org/10.1038/nrn1555.
Sabban EL, Liu X, Serova L, Gueorguiev V, Kvetnansky R. Stress triggered changes in gene expression in adrenal medulla: transcriptional responses to acute and chronic stress. Cell Mol Neurobiol. 2006;26:843–54. https://doi.org/10.1007/s10571-006-9069-1.
Lucassen PJ, Pruessner J, Sousa N, Almeida OFX, Van Dam AM, Rajkowska G, Swaab DF, Czéh B. Neuropathology of stress. Acta Neuropathol. 2014;127:109–35. https://doi.org/10.1007/s00401-013-1223-5.
Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav. 2011;59:279–89. https://doi.org/10.1016/j.yhbeh.2010.06.007.
Seckl JR. Prenatal glucocorticoids and long-term programming. Eur J Endocrinol. 2004;151(Suppl 3):U49–62.
Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: mechanisms. Nat Rev Endocrinol. 2014;10:403–11. https://doi.org/10.1038/nrendo.2014.74.
Patchev AV, Rodrigues AJ, Sousa N, Spengler D, Almeida OFX. The future is now: early life events preset adult behaviour. Acta Physiol (Oxf). 2014;210:46–57. https://doi.org/10.1111/apha.12140.
McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–8. https://doi.org/10.1038/nn.2270.
Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106.
Diamond DM, Bennett MC, Fleshner M, Rose GM. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus. 1992;2:421–30. https://doi.org/10.1002/hipo.450020409.
Martin SJ, Grimwood PD, Morris RGM. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. https://doi.org/10.1146/annurev.neuro.23.1.649.
Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–62. https://doi.org/10.1038/nrn849.
Kim JJ, Foy MR, Thompson RF. Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc Natl Acad Sci U S A. 1996;93:4750–3.
Oitzl MS, de Kloet ER. Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci. 1992;106:62–71.
Sandi C, Woodson JC, Haynes VF, Park CR, Touyarot K, Lopez-Fernandez MA, Venero C, Diamond DM. Acute stress-induced impairment of spatial memory is associated with decreased expression of neural cell adhesion molecule in the hippocampus and prefrontal cortex. Biol Psychiatry. 2005;57:856–64. https://doi.org/10.1016/j.biopsych.2004.12.034.
Cacucci F, Wills TJ, Lever C, Giese KP, O’Keefe J. Experience-dependent increase in CA1 place cell spatial information, but not spatial reproducibility, is dependent on the autophosphorylation of the alpha-isoform of the calcium/calmodulin-dependent protein kinase II. J Neurosci. 2007;27(29):7854–9. https://doi.org/10.1523/JNEUROSCI.1704-07.2007.
Cerqueira JJ, Taipa R, Uylings HBM, Almeida OFX, Sousa N. Specific configuration of dendritic degeneration in pyramidal neurons of the medial prefrontal cortex induced by differing corticosteroid regimens. Cereb Cortex. 2007;17(9):1998–2006. https://doi.org/10.1093/cercor/bhl108.
Sousa N, Lukoyanov NV, Madeira MD, Almeida OFX, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97:253–66. https://doi.org/10.1016/S0306-4522(00)00050-6.
Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531:225–31.
Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, Morrison JH, McEwen BS. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26(30):7870–4. https://doi.org/10.1523/JNEUROSCI.1184-06.2006.
Schwabe L, Wolf OT. Stress prompts habit behavior in humans. J Neurosci. 2009;29:7191–8. https://doi.org/10.1523/JNEUROSCI.0979-09.2009.
Sandi C, Davies HA, Cordero MI, Rodriguez JJ, Popov VI, Stewart MG. Rapid reversal of stress induced loss of synapses in CA3 of rat hippocampus following water maze training. Eur J Neurosci. 2003;17:2447–56.
Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic–pituitary–adrenal axis. Stress. 2011;14:398–406. https://doi.org/10.3109/10253890.2011.586446.
Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, Morrison JH. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164:798–808. https://doi.org/10.1016/j.neuroscience.2009.08.053.
Radley JJ, Morrison JH. Repeated stress and structural plasticity in the brain. Ageing Res Rev. 2005;4:271–87. https://doi.org/10.1016/j.arr.2005.03.004.
Shansky RM, Morrison JH. Stress-induced dendritic remodeling in the medial prefrontal cortex: effects of circuit, hormones and rest. Brain Res. 2009;1293:108–13. https://doi.org/10.1016/j.brainres.2009.03.062.
Lopes S, Vaz-Silva J, Pinto V, Dalla C, Kokras N, Bedenk B, Mack N, Czisch M, Almeida OFX, Sousa N, Sotiropoulos I. Tau protein is essential for stress-induced brain pathology. Proc Natl Acad Sci. 2016;113:E3755–63. https://doi.org/10.1073/pnas.1600953113.
Lopes S, Teplytska L, Vaz-Silva J, Dioli C, Trindade R, Morais M, Webhofer C, Maccarrone G, Almeida OFX, Turck CW, Sousa N, Sotiropoulos I, Filiou MD. Tau deletion prevents stress-induced dendritic atrophy in prefrontal cortex: role of synaptic mitochondria. Cereb Cortex. 2016;27(4):2580–91. https://doi.org/10.1093/cercor/bhw057.
Heine VM, Maslam S, Zareno J, Joëls M, Lucassen PJ. Suppressed proliferation and apoptotic changes in the rat dentate gyrus after acute and chronic stress are reversible. Eur J Neurosci. 2004;19:131–44.
Dioli C, Patrício P, Trindade R, Pinto LG, Silva JM, Morais M, Ferreiro E, Borges S, Mateus-Pinheiro A, Rodrigues AJ, Sousa N, Bessa JM, Pinto L, Sotiropoulos I. Tau-dependent suppression of adult neurogenesis in the stressed hippocampus. Mol Psychiatry. 2017;22:1110–8. https://doi.org/10.1038/mp.2017.103.
Kempermann G, Krebs J, Fabel K. The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr Opin Psychiatry. 2008;21:290–5. https://doi.org/10.1097/YCO.0b013e3282fad375.
Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6:85. https://doi.org/10.1186/1750-1326-6-85.
Demars M, Hu Y-S, Gadadhar A, Lazarov O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J Neurosci Res. 2010;88:2103–17. https://doi.org/10.1002/jnr.22387.
O’Brien JT, Ames D, Schweitzer I, Mastwyk M, Colman P. Enhanced adrenal sensitivity to adrenocorticotrophic hormone (ACTH) is evidence of HPA axis hyperactivity in Alzheimer’s disease. Psychol Med. 1996;26:7–14.
Rasmuson S, Andrew R, Näsman B, Seckl JR, Walker BR, Olsson T. Increased glucocorticoid production and altered cortisol metabolism in women with mild to moderate Alzheimer’s disease. Biol Psychiatry. 2001;49:547–52. https://doi.org/10.1016/S0006-3223(00)01015-5.
Simard M, Hudon C, van Reekum R. Psychological distress and risk for dementia. Curr Psychiatry Rep. 2009;11:41–7.
Mejía S, Giraldo M, Pineda D, Ardila A, Lopera F. Nongenetic factors as modifiers of the age of onset of familial Alzheimer’s disease. Int Psychogeriatr. 2003;15:337–49.
Rothman SM, Mattson MP. Adverse stress, hippocampal networks, and Alzheimer’s disease. Neuromolecular Med. 2010;12:56–70. https://doi.org/10.1007/s12017-009-8107-9.
Hatzinger M, Z’Brun A, Hemmeter U, Seifritz E, Baumann F, Holsboer-Trachsler E, Heuser IJ. Hypothalamic-pituitary-adrenal system function in patients with Alzheimer’s disease. Neurobiol Aging. 1995;16:205–9.
Peskind ER, Wilkinson CW, Petrie EC, Schellenberg GD, Raskind MA. Increased CSF cortisol in AD is a function of APOE genotype. Neurology. 2001;56:1094–8.
Hoogendijk WJG, Meynen G, Endert E, Hofman MA, Swaab DF. Increased cerebrospinal fluid cortisol level in Alzheimer’s disease is not related to depression. Neurobiol Aging. 2006;27:780.e1–2. https://doi.org/10.1016/j.neurobiolaging.2005.07.017.
Hartmann A, Veldhuis JD, Deuschle M, Standhardt H, Heuser I. Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol Aging. 1997;18:285–9.
Touma C, Ambrée O, Görtz N, Keyvani K, Lewejohann L, Palme R, Paulus W, Schwarze-Eicker K, Sachser N. Age- and sex-dependent development of adrenocortical hyperactivity in a transgenic mouse model of Alzheimer’s disease. Neurobiol Aging. 2004;25:893–904. https://doi.org/10.1016/j.neurobiolaging.2003.09.004.
Green KN. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–56. https://doi.org/10.1523/JNEUROSCI.2797-06.2006.
Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NPV, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73. https://doi.org/10.1038/271.
Huang C-W, Lui C-C, Chang W-N, Lu C-H, Wang Y-L, Chang C-C. Elevated basal cortisol level predicts lower hippocampal volume and cognitive decline in Alzheimer’s disease. J Clin Neurosci. 2009;16:1283–6. https://doi.org/10.1016/j.jocn.2008.12.026.
Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. https://doi.org/10.1038/nrd3505.
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019–31. https://doi.org/10.1016/S0140-6736(10)61349-9.
Duyckaerts C, Delatour B, Potier M-C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol (Berl). 2009;118:5–36. https://doi.org/10.1007/s00401-009-0532-1.
Dede AJO, Wixted JT, Hopkins RO, Squire LR. Hippocampal damage impairs recognition memory broadly, affecting both parameters in two prominent models of memory. Proc Natl Acad Sci. 2013;110:6577–82. https://doi.org/10.1073/pnas.1304739110.
Jeong YH, Park CH, Yoo J, Shin KY, Ahn S-M, Kim H-S, Lee SH, Emson PC, Suh Y-H. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APP V717I-CT100 transgenic mice, an Alzheimer’s disease model. FASEB J. 2006;20:729–31. https://doi.org/10.1096/fj.05-4265fje.
Sotiropoulos I, Catania C, Riedemann T, Fry JP, Breen KC, Michaelidis TM, Almeida OFX. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau: glucocorticoids, human tau and Alzheimer’s disease. J Neurochem. 2008;107:385–97. https://doi.org/10.1111/j.1471-4159.2008.05613.x.
Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OFX. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry. 2009;14:95–105. https://doi.org/10.1038/sj.mp.4002101.
Stein-Behrens BA, Elliott EM, Miller CA, Schilling JW, Newcombe R, Sapolsky RM. Glucocorticoids exacerbate kainic acid-induced extracellular accumulation of excitatory amino acids in the rat hippocampus. J Neurochem. 1992;58:1730–5.
Sotiropoulos I, Catania C, Pinto LG, Silva R, Pollerberg GE, Takashima A, Sousa N, Almeida OFX. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J Neurosci. 2011;31:7840–7. https://doi.org/10.1523/JNEUROSCI.0730-11.2011.
Callahan LM, Vaules WA, Coleman PD. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:384–95.
Lauckner J, Frey P, Geula C. Comparative distribution of tau phosphorylated at Ser262 in pre-tangles and tangles. Neurobiol Aging. 2003;24:767–76.
Hampel H, Bürger K, Pruessner JC, Zinkowski R, DeBernardis J, Kerkman D, Leinsinger G, Evans AC, Davies P, Möller H-J, Teipel SJ. Correlation of cerebrospinal fluid levels of tau protein phosphorylated at threonine 231 with rates of hippocampal atrophy in Alzheimer disease. Arch Neurol. 2005;62(5):770–3. https://doi.org/10.1001/archneur.62.5.770.
Augustinack JC, Schneider A, Mandelkow E-M, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol (Berl). 2002;103:26–35.
Ewers M, Buerger K, Teipel SJ, Scheltens P, Schroder J, Zinkowski RP, Bouwman FH, Schonknecht P, Schoonenboom NSM, Andreasen N, Wallin A, DeBernardis JF, Kerkman DJ, Heindl B, Blennow K, Hampel H. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurology. 2007;69:2205–12. https://doi.org/10.1212/01.wnl.0000286944.22262.ff.
van der Vlies AE, Verwey NA, Bouwman FH, Blankenstein MA, Klein M, Scheltens P, van der Flier WM. CSF biomarkers in relationship to cognitive profiles in Alzheimer disease. Neurology. 2009;72:1056–61. https://doi.org/10.1212/01.wnl.0000345014.48839.71.
Carroll JC, Iba M, Bangasser DA, Valentino RJ, James MJ, Brunden KR, Lee VM-Y, Trojanowski JQ. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J Neurosci. 2011;31(40):14436–49. https://doi.org/10.1523/JNEUROSCI.3836-11.2011.
Rissman RA, Lee K-F, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27:6552–62. https://doi.org/10.1523/JNEUROSCI.5173-06.2007.
Sotiropoulos I, Silva J, Kimura T, Rodrigues AJ, Costa P, Almeida OFX, Sousa N, Takashima A. Female hippocampus vulnerability to environmental stress, a precipitating factor in Tau aggregation pathology. J Alzheimers Dis. 2015;43:763–74. https://doi.org/10.3233/JAD-140693.
de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73:685–97. https://doi.org/10.1016/j.neuron.2011.11.033.
Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow E-M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci. 2007;104:10252–7. https://doi.org/10.1073/pnas.0703676104.
Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–30. https://doi.org/10.1172/JCI20640.
Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging. 2000;21:719–27.
Kobayashi S, Tanaka T, Soeda Y, Almeida OFX, Takashima A. Local somatodendritic translation and hyperphosphorylation of tau protein triggered by AMPA and NMDA receptor stimulation. EBioMedicine. 2017;20:120–6. https://doi.org/10.1016/j.ebiom.2017.05.012.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–97. https://doi.org/10.1016/j.cell.2010.06.036.
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan L-L, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–81. https://doi.org/10.1016/j.neuron.2010.11.030.
McKinney RA. Excitatory amino acid involvement in dendritic spine formation, maintenance and remodelling: glutamate and dendritic spines. J Physiol. 2010;588:107–16. https://doi.org/10.1113/jphysiol.2009.178905.
Mondragon-Rodriguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012;287:32040–53. https://doi.org/10.1074/jbc.M112.401240.
Miller EC, Teravskis PJ, Dummer BW, Zhao X, Huganir RL, Liao D. Tau phosphorylation and tau mislocalization mediate soluble Aβ oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci. 2014;39:1214–24. https://doi.org/10.1111/ejn.12507.
Nong Y, Huang Y-Q, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–7. https://doi.org/10.1038/nature01497.
Yang C-H. Behavioral stress enhances hippocampal CA1 long-term depression through the blockade of the glutamate uptake. J Neurosci. 2005;25:4288–93. https://doi.org/10.1523/JNEUROSCI.0406-05.2005.
Magariños AM, McEwen BS, Flügge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci. 1996;16:3534–40.
Pinheiro S, Silva J, Mota C, Vaz-Silva J, Veloso A, Pinto V, Sousa N, Cerqueira J, Sotiropoulos I. Tau mislocation in glucocorticoid-triggered hippocampal pathology. Mol Neurobiol. 2016;53:4745–53. https://doi.org/10.1007/s12035-015-9356-2.
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91. https://doi.org/10.1242/jcs.019265.
Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–9. https://doi.org/10.1016/j.tins.2010.09.001.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. https://doi.org/10.1083/jcb.200412022.
Frake RA, Ricketts T, Menzies FM, Rubinsztein DC. Autophagy and neurodegeneration. J Clin Invest. 2015;125:65–74. https://doi.org/10.1172/JCI73944.
Silva JM, Rodrigues S, Sampaio-Marques B, Gomes P, Neves-Carvalho A, Dioli C, Soares-Cunha C, Mazuik BF, Takashima A, Ludovico P, Wolozin B, Sousa N, Sotiropoulos I. Dysregulation of autophagy and stress granule-related proteins in stress-driven Tau pathology. Cell Death Differ. 2018;26(8):1411–27. https://doi.org/10.1038/s41418-018-0217-1.
Hamano T, Gendron TF, Causevic E, Yen S-H, Lin W-L, Isidoro C, DeTure M, Ko L. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–30. https://doi.org/10.1111/j.1460-9568.2008.06084.x.
Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow E-M, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–70. https://doi.org/10.1093/hmg/ddp367.
Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, Komatsu M, Abeliovich A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener. 2012;7:48. https://doi.org/10.1186/1750-1326-7-48.
Brown MR, Bondada V, Keller JN, Thorpe J, Geddes JW. Proteasome or calpain inhibition does not alter cellular tau levels in neuroblastoma cells or primary neurons. J Alzheimers Dis. 2005;7:15–24.
Feuillette S, Blard O, Lecourtois M, Frébourg T, Campion D, Dumanchin C. Tau is not normally degraded by the proteasome: tau not normally degraded by the proteasome. J Neurosci Res. 2005;80:400–5. https://doi.org/10.1002/jnr.20414.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. https://doi.org/10.1038/nature04724.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926–37. https://doi.org/10.1523/JNEUROSCI.0800-08.2008.
Polman JAE, Hunter RG, Speksnijder N, van den Oever JME, Korobko OB, McEwen BS, de Kloet ER, Datson NA. Glucocorticoids modulate the mTOR pathway in the hippocampus: differential effects depending on stress history. Endocrinology. 2012;153:4317–27. https://doi.org/10.1210/en.2012-1255.
An W-L, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal I-G, Winblad B, Pei J-J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol. 2003;163:591–607. https://doi.org/10.1016/S0002-9440(10)63687-5.
Pei J-J, Hugon J. mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med. 2008;12:2525–32. https://doi.org/10.1111/j.1582-4934.2008.00509.x.
Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010;133:93–104. https://doi.org/10.1093/brain/awp292.
Jiang T, Yu J-T, Zhu X-C, Zhang Q-Q, Cao L, Wang H-F, Tan M-S, Gao Q, Qin H, Zhang Y-D, Tan L. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology. 2014;85:121–30. https://doi.org/10.1016/j.neuropharm.2014.05.032.
Rivero-Ríos P, Gómez-Suaga P, Fernández B, Madero-Pérez J, Schwab AJ, Ebert AD, Hilfiker S. Alterations in late endocytic trafficking related to the pathobiology of LRRK2-linked Parkinson’s disease. Biochem Soc Trans. 2015;43:390–5. https://doi.org/10.1042/BST20140301.
Kett LR, Dauer WT. Endolysosomal dysfunction in Parkinson’s disease: recent developments and future challenges: Endolysosomal dysfunction in PD. Mov Disord. 2016;31:1433–43. https://doi.org/10.1002/mds.26797.
Small SA, Simoes-Spassov S, Mayeux R, Petsko GA. Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer’s disease. Trends Neurosci. 2017;40:592–602. https://doi.org/10.1016/j.tins.2017.08.003.
Vaz-Silva J, Gomes P, Jin Q, Zhu M, Zhuravleva V, Quintremil S, Meira T, Silva J, Dioli C, Soares-Cunha C, Daskalakis NP, Sousa N, Sotiropoulos I, Waites CL. Endolysosomal degradation of Tau and its role in glucocorticoid-driven hippocampal malfunction. EMBO J. 2018;37:e99084. https://doi.org/10.15252/embj.201899084.
Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–52. https://doi.org/10.1038/nature07961.
Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–9. https://doi.org/10.1016/j.devcel.2010.12.003.
Pooler AM, Hanger DP. Functional implications of the association of tau with the plasma membrane. Biochem Soc Trans. 2010;38:1012–5. https://doi.org/10.1042/BST0381012.
Georgieva ER, Xiao S, Borbat PP, Freed JH, Eliezer D. Tau binds to lipid membrane surfaces via short amphipathic helices located in its microtubule-binding repeats. Biophys J. 2014;107:1441–52. https://doi.org/10.1016/j.bpj.2014.07.046.
Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 2012;7:56. https://doi.org/10.1186/1750-1326-7-56.
Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10:1324–32. https://doi.org/10.1038/ncb1791.
Arimoto-Matsuzaki K, Saito H, Takekawa M. TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat Commun. 2016;7:10252. https://doi.org/10.1038/ncomms10252.
Kampers T, Friedhoff P, Biernat J, Mandelkow E-M, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–9. https://doi.org/10.1016/S0014-5793(96)01386-5.
Liu-Yesucevitz L, Bilgutay A, Zhang Y-J, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5(10):e13250. https://doi.org/10.1371/journal.pone.0013250.
Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu Y-F, Kobrin K, Murphy GJ, Petrucelli L, Wolozin B. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci. 2014;34:4167–74. https://doi.org/10.1523/JNEUROSCI.2350-13.2014.
Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012;32:8270–83. https://doi.org/10.1523/JNEUROSCI.1592-12.2012.
Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, Ivanov P, Abisambra JF, Steffen M, Li H, Petrucelli L, Wolozin B. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 2016;15:1455–66. https://doi.org/10.1016/j.celrep.2016.04.045.
Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A, Ferragud A, Botelho E, Ballance HI, Dhawan U, Boudeau S, Cruz AL, Kashy D, Wong A, Goldberg LR, Yazdani N, Zhang C, Ung CY, Tripodis Y, Kanaan NM, Ikezu T, Cottone P, Leszyk J, Li H, Luebke J, Bryant CD, Wolozin B. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci. 2018;21:72–80. https://doi.org/10.1038/s41593-017-0022-z.
Maziuk B, Ballance HI, Wolozin B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Front Mol Neurosci. 2017;10:89. https://doi.org/10.3389/fnmol.2017.00089.
McEwen BS, Gray JD, Nasca C. 60 years of neuroendocrinology: redefining neuroendocrinology: stress, sex and cognitive and emotional regulation. J Endocrinol. 2015;226:T67–83. https://doi.org/10.1530/JOE-15-0121.
Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63:530. https://doi.org/10.1001/archpsyc.63.5.530.
Heun R, Kockler M, Ptok U. Depression in Alzheimer’s disease: is there a temporal relationship between the onset of depression and the onset of dementia? Eur Psychiatry. 2002;17:254–8.
Kim HK, Nunes PV, Oliveira KC, Young LT, Lafer B. Neuropathological relationship between major depression and dementia: a hypothetical model and review. Prog Neuropsychopharmacol Biol Psychiatry. 2016;67:51–7. https://doi.org/10.1016/j.pnpbp.2016.01.008.
Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, Purohit DP, Gorman JM, Haroutunian V. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry. 2006;63:161–7. https://doi.org/10.1001/archpsyc.63.2.161.
Mayeux R, Honig LS, Tang M-X, Manly J, Stern Y, Schupf N, Mehta PD. Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–90.
Andreasen N, Blennow K. CSF biomarkers for mild cognitive impairment and early Alzheimer’s disease. Clin Neurol Neurosurg. 2005;107:165–73. https://doi.org/10.1016/j.clineuro.2004.10.011.
Evered L, Scott DA, Silbert B, Maruff P. Postoperative cognitive dysfunction is independent of type of surgery and anesthetic. Anesth Analg. 2011;112:1179–85. https://doi.org/10.1213/ANE.0b013e318215217e.
Sun X, Steffens DC, Au R, Folstein M, Summergrad P, Yee J, Rosenberg I, Mwamburi DM, Qiu WQ. Amyloid-associated depression: a prodromal depression of Alzheimer disease? Arch Gen Psychiatry. 2008;65:542. https://doi.org/10.1001/archpsyc.65.5.542.
Mateus-Pinheiro A, Pinto L, Sousa N. Epigenetic (de)regulation of adult hippocampal neurogenesis: implications for depression. Clin Epigenetics. 2011;3:5. https://doi.org/10.1186/1868-7083-3-5.
Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, Ávila J, Llorens-Martín M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25:554–60. https://doi.org/10.1038/s41591-019-0375-9.
Gu Y, Oyama F, Ihara Y. Tau is widely expressed in rat tissues. J Neurochem. 1996;67:1235–44.
Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF, Dawson HN, Bennett CF, Rigo F, Miller TM. Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron. 2016;90:941–7. https://doi.org/10.1016/j.neuron.2016.04.042.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sotiropoulos, I., Silva, J.M., Gomes, P., Sousa, N., Almeida, O.F.X. (2019). Stress and the Etiopathogenesis of Alzheimer’s Disease and Depression. In: Takashima, A., Wolozin, B., Buee, L. (eds) Tau Biology. Advances in Experimental Medicine and Biology, vol 1184. Springer, Singapore. https://doi.org/10.1007/978-981-32-9358-8_20
Download citation
DOI: https://doi.org/10.1007/978-981-32-9358-8_20
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9357-1
Online ISBN: 978-981-32-9358-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)